

Abstract
Objectives
To determine the proportion of patients with tumor response, the proportion who survived progression-free for at least six months (PFS ≥ 6 months) and the frequency and severity of toxicities of patients with recurrent squamous cell carcinoma of the uterine cervix treated with erlotinib.
Methods
This was a multicenter, open-label single arm trial evaluating the toxicity and efficacy of oral erlotinib at an initial dose of 150 mg daily until progressive disease or adverse effects prohibited further therapy.
Results
Twenty-eight patients with squamous cell carcinoma were enrolled onto this trial. Twenty-five patients were evaluable. There were no objective responses with four (16%) achieving stable disease; only one patient had a PFS ≥ 6 months (4%). The one-sided 90% confidence interval (CI) for response was 0.0%–8.8%. The two-sided 90% CI for the proportion of patients surviving progression-free for at least 6 months is 0.2%–17.6%. Erlotinib was well tolerated with the most common drug-related adverse events being gastrointestinal toxicities, fatigue and rash.
Conclusion
Erlotinib is inactive as monotherapy in patients with recurrent squamous cell carcinoma of the uterine cervix.
Keywords: cervical cancer, EGFR, erlotinib
Introduction
Cervical cancer remains a major health issue for women worldwide. It ranks second only to breast cancer in incidence and cancer related mortality. In parts of the developing world, it is the major cause of death in women of reproductive age(1). In the United States, there were approximately 11,000 new cases of cervical cancer with 3700 deaths estimated for 2007(2). The vast majority of cases are of squamous cell histology. Cisplatin is the single most active agent with a response rate reported between 13–19% in recent phase III trials. In combination with other agents such as paclitaxel or topotecan, the response rate increases to 36% and 27%, respectively(3,4). The responses tend to be partial in nature and of short duration. Therefore, the development of new anti-neoplastic compounds that could be combined with cisplatin with or without radiation is vital to improve primary therapy for advanced disease.
Erlotinib is an oral selective inhibitor of the epidermal growth factor receptor (EGFR) tyrosine kinase. Its administration reduced the level of EGFR autophosphorylation in mice bearing human tumor xenografts by greater than 70% over 24 hours after a single dose(5). EGFR is a transmembrane glycoprotein that promotes cell growth in a variety of normal and transformed tissues when activated. Binding of ligands such as transforming growth factor-alpha (TGFα) or EGF to EGFR activates a number of signal transducing pathways, such as MAP-MEK-ERK and PI3K-AKT, which are important for cell growth and survival, respectively(6,7). EGFR is known to be overexpressed in both normal squamous epithelium and squamous cell cancers, and it plays a key role in HPV-16 mediated transformation of normal keratinocytes. EGFR is expressed in greater than 75% of squamous cell cancers of the cervix although the impact of EGFR overexpression on the prognosis of patients with squamous cell carcinomas of the cervix is controversial(8–11). Inhibition of EGFR by either monoclonal antibodies or tyrosine kinase inhibitors (TKI) leads to growth arrest of squamous cell cancers(12,13). Based on randomized phase III trials, erlotinib is now indicated for use in patients with non-small cell lung cancer as second or third line treatment and in patients with pancreatic cancer as the front-line therapy in combination with gemcitabine(14,15). Based on these data, erlotinib was selected by the Gynecologic Oncology Group (GOG) for evaluation in patients with recurrent squamous cell cancer of the cervix.
Materials and methods
Patients were required to have recurrent or persistent squamous cell carcinoma of the cervix that was histologically confirmed by central review performed by the GOG Pathology Committee. EGFR expression was not a eligibility criterion nor determined since approximately 75% of squamous cell carcinomas of the cervix express EGFR(16). Written informed consent was obtained from all patients according to all institutional, state and federal regulations. Prior to study entry, patients must have received at least one, but not more than two, prior systemic cytotoxic chemotherapeutic regimens for the management of advanced, metastatic or recurrent squamous cell cervical cancer and were required to have measurable disease as defined by Response Evaluation Criteria in Solid Tumors (RECIST)(17). (Patients must not have received any prior EGFR inhibitor). Tumors within a previously irradiated field were designated as non target lesions, unless clear progression was documented or a biopsy was obtained to confirm disease persistence at least 90 days following the completion of radiation therapy. Chemotherapy administered as a radiosensitizer in conjunction with primary radiation was not considered as a systemic chemotherapy regimen. Patients who had only one prior cytotoxic chemotherapy regimen were required to have a GOG performance status of 0 to 2; those patients who had two prior regimens were required to have a GOG performance status of 0 or 1. Patients were required to have adequate bone marrow (absolute neutrophil count ≥ 1500/μl, platelet count ≥ 100,000/μl), renal (serum creatinine ≤ 1.5 × the upper limit of normal), hepatic function (total bilirubin ≤ the upper limit of normal and both transaminases and alkaline phosphatase ≤ 2.5 × the upper limit of normal) and neurologic function (sensory and motor neuropathy < Common Toxicity Criteria (CTC)-grade 1). Toxicities were assessed at each office visit every 21 days.
This trial was a multicenter, open-label, single arm study evaluating the efficacy and toxicity of oral erlotinib administered at a dose of 150 mg daily until progressive disease or toxicity prohibited further administration. A cycle was defined as 28 days. Febrile neutropenia, grade 4 neutropenia lasting ≥ 7 days or grade 4 thrombocytopenia required the drug be held until patients recovered from the infectious episode and the respective blood counts recovered to CTC grade ≤ 1. Prophylactic use of granulocyte-colony stimulating factor (G-CSF) and/or thrombopoietic agents were not permitted since erlotinib was administered daily. Chemo-protective agents such as amifostine also were prohibited. Patients were removed from the study if their blood counts had not recovered by two weeks off treatment.
Grade 1 or 2 non-hematologic toxicity required no dose adjustments except for grade 2 keratitis for which one dose level reduction was permitted down to 100 mg daily for the first reduction and 50 mg daily for the second reduction. Patients requiring further dose reductions stopped study therapy. Grade 2 skin rash was managed with minocycline, topical agents, or a short course of steroids but no dose reduction unless perceived as intolerable by the patient. Grade 3 or 4 non-hematologic toxicity including skin rash, required a dose reduction by one level once toxicity resolved to grade ≤ 1. Two dose reductions were allowed; a third dose reduction (25 mg/day) could occur after discussion with the Cancer Therapy Evaluation Program (CTEP) monitor if the patient was still benefiting from therapy. Toxicity was reported according to CTC v. 2 (http://ctep.cancer.gov/reporting/ctc.html).
Statistics
Erlotinib was not expected to act through cytotoxic mechanisms which ordinarily result in cell death. Instead, it was anticipated that this agent would inhibit cell growth, division, or metastasis. Therefore, tumor response, which requires an actual reduction of tumor burden, was not considered an appropriate outcome for identifying drug activity in this class of agents. Therefore, the primary endpoint selected was progression-free survival (PFS). In particular, this study estimated the distribution of the failure times, which was used to estimate the proportion of patients who survived progression-free for at least 6 months (PFS at 6 months). Time to progression or death (PFS endpoint) was assessed from the date of enrollment to the date of first clinical progression or death, whichever occurred first. If neither event was observed, then the patient’s event status was considered censored, and the time to progression was measured from the date of enrollment to the date of last contact. Patients with times to progression or death of at least 6 months were considered to be surviving progression-free for at least 6 months. Patients with censored PFS times of less than 6 months would have been deemed treatment failures.
Under the hypothesis that Erlotinib is effective, a greater proportion of patients would be expected to survive progression-free in a fixed period of time, regardless of whether it acted in a cytotoxic or cytostatic mechanism. An analysis of historical phase II trials (18–23) indicates that agents with a proportion of patients surviving progression-free at six months (PFS >6 months) of 10% or less was uninteresting. Increasing this proportion to 25% or more was considered clinically significant. [See Monk et al. for further discussion (24)]. The sample size for the current trial was targeted to detect this effect with 90% power with the probability of a type I error equal to 10%. A flexible and nearly optimal two-stage design was utilized with a method provided by Chen and Ng(25). Specifically, the targeted size for the first stage of accrual was 22 eligible and evaluable patients, but in practice the sample size was permitted to range from 19 to 26 patients for administrative reasons. If there were more than two (i.e., >2) out of 19–25, or more than three (i.e., >3) out of 26 patients alive and progression-free for at least six months and medical judgment indicated, accrual to a second stage of the trial was to be initiated. Otherwise, the accrual would be stopped, and the treatment regimen would be considered clinically uninteresting. If the study advanced to the second stage, then an overall study accrual of 47 eligible and evaluable patients was targeted but permitted to range from 44 to 51. If there were no more than six (i.e., ≤6) out of 44–45 or seven (i.e., ≤7) out of 46–51 patients that were alive and progression-free after six months, the regimen would have been considered clinically uninteresting.
The frequency and severity of adverse events were tabulated according to the descriptions provided by NCI’s Common Toxicity Criteria version 2.
Secondary analyses included a description of overall survival (OS) and PFS with quartile estimates of the distribution for the time to event (progression/death) along with Kaplan-Meier plots. The number of patients with tumor responses was provided along with confidence intervals using exact methods assuming a binomial distribution. Specifically, for a two-sided (1−α) confidence interval, the bounds of the interval, [πL, πU] were found by solving the set of polynomial equations:
and
where the value, , is the usual binomial coefficient, n is the sample size, and r is the number of responses. Data were analyzed in SAS version 9.1.
The period of active accrual was 41.2 months. Patients were followed for median of 13 months after study closure.
Results
Twenty-eight patients were entered into the trial from 16 centers between April 2002 and October 2005. One patient had an ineligible primary (lung) cancer, one patient was never treated, and one patient had inadequate data collected leaving 25 evaluable patients. Patient characteristics are listed in Table 1. Eighteen patients had one prior chemotherapy regimen and 24 had prior radiotherapy.
Table 1.
Patient Characteristics and Response
| Age (median, years) | 47 |
| (range) | (27–83) |
| Performance Status | |
| 0 | 9 |
| 1 | 12 |
| 2 | 4 |
| Prior Chemotherapy | |
| 1 regimen | 18 |
| 2 regimens | 7 |
| Prior Radiation | 24 |
| Prior Surgery | 19 |
| Grade | |
| 1 | 0 |
| 2 | 13 |
| 3 | 12 |
| Courses | |
| Median | 2 |
| (range) | (1–5) |
| Response | |
| CR/PRa (%) | 0 (0) |
| Stable and PFSa ≥6 months | 1 (4) |
| Stable and PFS < 6 months | 3 (12) |
| Progression | 17(68) |
| Indeterminate | 4 (16) |
| Progression-Free Survival (months) | |
| Median | 1.87 |
| 1st quartile | 1.48 |
| 3rd quartile | 2.33 |
| Range | 0.79–19.06 |
| Overall Survival (months) | |
| Median | 4.96 |
| 1st quartile | 3.65 |
| 3rd quartile | 8.12 |
| Range | 0.79–19.06 |
–CR: complete response; PR- partial response; PFS- progression-free survival
There were no objective responses. Assuming a binomial distribution, the exact one-sided 90% confidence interval for the probability of a tumor response is 0.0%–8.8%. Sixteen percent of patients had stable disease, but only one patient survived progression-free for more than six months (4%). Because the total number of patients with PFS > 6 months was less than three, the trial did not open to a second stage. The exact two-sided 90% confidence interval assuming a binomial distribution for the proportion of patients surviving progression-free for at least six months is 0.2%–17.6%. This confidence interval excludes the minimally significant proportion of 25%. The median PFS was 1.87 months (first quartile 1.48; third quartile 2.33) with a median OS of 4.96 months (first quartile 3.65, third quartile 8.12) (Table 2). Figure 1 provides a plot of the Kaplan-Meier estimates of OS and PFS.
Table 2.
Grade 3 or 4 Toxicities
| Toxicity | Grade (n=25) | |
|---|---|---|
| 3 | 4 | |
| Fatigue | 2 | - |
| Nausea | 2 | - |
| Emesis | 2 | - |
| Diarrhea | 3 | - |
| Dehydration | 1 | - |
| Anorexia | 1 | - |
| Anemia | 3 | 1 |
| Rash | 2 | - |
| Renal* | - | 1 |
| Infection without neutropenia | 2 | - |
Reversible
Figure 1.
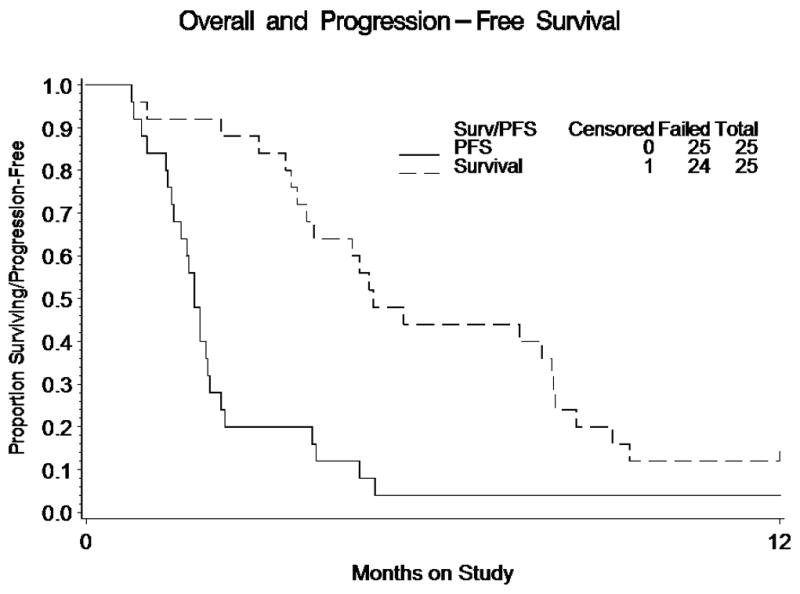
Plot of the Kaplan-Meier Estimates of Overall Survival and Progression-Free Survival. Note that 22, 10, 5, and 3 patients were at risk for progression or death at 1, 2, 3 and 4 months respectively. One patient remained progression-free at 12 months. Also note that 24, 23, 21, 16, 12, 11, 11, 9, 5, and 3 patients were at risk of death at 1,2,3,4, 5, 6, 7, 8, 9, and 10 months respectively. One patient was censored near 12 months, and 2 patients survived for more than 12 months.
The median number of cycles administered was two (range, 1–5). Grade 3 or 4 toxicities are summarized in Table 3. Grade 3 diarrhea occurred in only three patients. Other grade 3 gastrointestinal related toxicities included nausea (2), emesis (2), dehydration (1) and anorexia (1). Anemia was the only grade 3 or 4 hematologic toxicity observed. Only two patients had grade 3 rash, although 15 patients had milder forms. One patient had grade 4 renal toxicity that reversed with bilateral percutaneous nephrostomy tube placements, suggesting that the ureteral obstruction was due to progressive disease rather than secondary to drug effect. There were 9 patients that required dose modifications. Four patients were delayed for at least seven days on at least on occasion.
Table 3.
Grade 3 or 4 Toxicities
| Toxicity | Grade (n=25) | |
|---|---|---|
| 3 | 4 | |
| Fatigue | 2 | - |
| Nausea | 2 | - |
| Emesis | 2 | - |
| Diarrhea | 3 | - |
| Dehydration | 1 | - |
| Anorexia | 1 | - |
| Anemia | 3 | 1 |
| Rash | 2 | - |
| Renal* | - | 1 |
| Infection without neutropenia | 2 | - |
Reversible
Discussion
The EGFR is known to be highly expressed on the surface of cervical carcinoma cells. Many of the early studies that elucidated the biology of the EGFR employed the human cervical squamous carcinoma cell line, A431(26,27). Thus, cervical cancer was a logical disease in which to evaluate an EGFR inhibitor. However, we observed no objective responses nor were there many patients who sustained a prolonged period of stable disease. Our data using erlotinib were similar to the data obtained by Goncalves and colleagues using gefitinib in recurrent or metastatic cervical carcinoma (28). They also did not observe any objective responses. They reported a stable disease rate of 20% with a median duration of 111.5 days (range, 77–188 days). The median PFS and OS were 37 days and 107 days, respectively, which are similar to our own observations of 1.87 and 4.96 months, respectively. In their study, disease control did not correlate with EGFR expression. Arias-Pulido and co-investigators analyzed 89 patient samples for EGFR mutations in exons 19–21(29). In addition, nine cervical cancer cell lines were similarly evaluated for mutations in exons 18–21 similar to what had been previously done in lung cancer specimens. No mutations were detected in any sample in either group. In a separate study, no amplification of the EGFR gene was detected(30). These findings may explain the lack of activity of EGFR TKI despite the high level of expression of EGFR. Similar results have been seen in EGFR TK1 monotherapy trials in breast, head and neck, renal cell, prostate, ovarian and endometrial cancers(31).
A recent study evaluating the mechanism of acquired resistance to erlotinib in A431 cells demonstrated that the extent of gene amplification or mutation was not altered as erlotinib resistance was induced in these cells(32). Erlotinib similarly reduced the levels of phosphorylated EGFR in both sensitive and resistant cell lines. However, mutated in multiple advanced cancers 1/phosphatase and tensin homologue (MMAC1/PTEN) was induced and phosphorylated Akt was suppressed cells in sensitive cells but not resistant cells. Overexpression of MMAC/PTEN by transfection with Ad.MMAC1/PTEN or by pharmacological suppression of Akt activity restored erlotinib sensitivity in resistant A431 cells. Conversely, transfection of parentalA431 cells with constitutively active Akt was sufficient to induce resistance to erlotinib. Thus, erlotinib resistance is, at least in part, mediated by MMAC1/PTEN down regulation and/or increased Akt activation and may be reversed in the presence of inhibitors of signaling through the phosphatidlylinositol-3-kinase pathway.
Monoclonal antibodies against EGFR have a different spectrum of clinical activity. They are active in chemotherapy refractory colorectal cancer and also may be more active than EGFR TKIs in head and neck cancer(33–38). These antibodies, including cetuximab and panitumumab, have a different mechanism of action compared with the TKIs and also combine well with chemotherapy or radiation.
In in vitro systems, cervical cancer cell lines were demonstrated to be highly sensitive to cetuximab mediated antibody dependent cell mediated cytotoxicity. These reactions were further augmented by the presence of complement(9). The combination of EGFR monoclonal antibody and cisplatin inhibits tumor growth unresponsive to either agent as monotherapy in mice bearing human squamous cell xenografts and the antitumor activity appears to be independent of the level of EGFR expression(39).
The negative results of TKI therapy targeting EGFR in the treatment of metastatic cervical cancer do not necessarily carry over to the monoclonal antibodies. There are ongoing trials within the GOG evaluating cetuximab in combination with cisplatin as front-line treatment for patients with metastatic squamous cell carcinoma of the cervix in a phase II trial and in a phase I trial of cetuximab in combination with radiation and weekly cisplatin for front-line treatment of locally advanced disease. The results of these trials will assist in determining how to further develop EGFR inhibitors in the treatment of cervical cancer.
There have been concerns raised about the potential for bias introduced by dichotomizing a time-to-event endpoint. More specifically, whether the statistic is an unbiased estimate of the parameter it is proposing to estimate. In particular, whether the proportion of patients deemed progression-free at six months according to protocol procedures and methods using a Kaplan-Meier (KM) estimate is an unbiased estimate of the probability of truly being progression-free at six months. The strict answer to this question is obvious. The statistic is not an unbiased estimate of this probability. One source of bias can result from informative censoring. For example, if censored observations are associated with debilitated patients not being able to obtain CT scans to document disease progression, then the KM estimate is positively biased for the probability of being progression-free at 6 months. This problem is countered in two ways. First, the maturity of the data in the historical controls is quite advanced, so there are relatively few cases that are censored. Second, when evaluating a new agent against the historical record, cases that are censored before 6 months are considered treatment failures, yielding a conservative (perhaps negatively biased) estimate in this regard.
Another potential source of bias stems from the timing of the radiological/imaging exams (scans). The date of disease progression is often reported as the date confirmed by these scans. However, the date of actual progression occurred some time before the scan, so the data are technically interval censored. Treatment delays caused by drug toxicities can cause delays in the scanning for disease progression. Therefore, more toxic drugs could appear to be more active simply by causing greater delays in the timing of the scans. This problem could be corrected to some extent if all surviving patients were required to be scanned at exactly 6 months. Then a greater proportion would be determined to have progressed at that time, providing a more accurate estimate of the parameter of interest. However, the magnitude of bias is expected to be fairly small with inactive agents. Additionally, the historical controls were not assessed in such a manner. The best information available to us is a Kaplan-Meier estimate at 6 months, which was used to determine the baseline level of activity to assess regimens in future studies that were assumed to be conducted in a similar fashion. Requiring a scan at exactly 6 months (or nearly so) may provide a reduced biased estimate for the parameter specified but could adversely disrupt the operating characteristics of design by yielding one with lower than expected type I error and statistical power. Furthermore, a 6 month scan cannot correct systematic bias caused by patient treatment patterns. For example, more toxic drugs can cause patients to withdraw from the study for toxicity. These patients can subsequently move onto a more active regimen thereby giving an inflated estimate of being progression-free at 6 months. Only a large, randomized phase III study would be able to assure of a fair comparison with a high level of confidence. Like most phase II studies, this one has inferential limitations. Therefore, it is important to keep in mind that the primary objective of the study is to screen agents, with as few patients as possible, for sufficient activity to deem them worthy of further study in a fully powered phase III study. Implicit in that objective is the tolerance of some acceptable sacrifice to scientific validity of the study such as possible bias in the estimate of the primary endpoint.
Acknowledgments
This study was supported by National Cancer Institute grants to the Gynecologic Oncology Group (GOG) Administrative Office (CA 27469) and the GOG Statistical Office (CA 37517). The following member institutions participated in this study: Roswell Park Cancer Institute, Abington Memorial Hospital, University of Pennsylvania Cancer Center, Milton S. Hershey Medical Center, University of Texas Southwestern Medical Center at Dallas, Wake Forest University School of Medicine, SUNY Downstate Medical Center, The Cleveland Clinic Foundation, Fox Chase Cancer Center, University of Oklahoma, Tacoma General Hospital, Case Western Reserve University, and Community Clinical Oncology Program.
References
- 1.Janicek MF, Averette HE. Cervical cancer: prevention, diagnosis and therapeutics. CA Cancer J Clin. 2001;51:92–114. doi: 10.3322/canjclin.51.2.92. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, et al. Cancer Statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 3.Moore DH, Blessing JA, McQuellon RP, et al. Phase III study of cisplatin with or without paclitaxel in stage IVB, recurrent or persistent squamous cell carcinoma of the cervix: a Gynecologic Oncology Group study. J Clin Oncol. 2004;15:3113–9. doi: 10.1200/JCO.2004.04.170. [DOI] [PubMed] [Google Scholar]
- 4.Long HJ, 3rd, Bundy BN, Grendys EC, Jr, et al. Randomized phase III trial of cisplatin with or without topotecan in carcinoma of the uterine cervix: a Gynecologic Oncology Group Study. J Clin Oncol. 2005;23:4626–33. doi: 10.1200/JCO.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 5.Pollack VA, Savage DM, Baker DA, et al. Inhibition of epidermal growth factor receptor-associated tyrosine phosphorylation in human carcinomas with CP-358, 774: dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther. 1999;291:739–48. [PubMed] [Google Scholar]
- 6.Carpenter G, Cohen S. Epidermal growth factor. J Biol Chem. 1990;265:7709–12. [PubMed] [Google Scholar]
- 7.Okano J, Gaslightwala I, Birnbaum MJ, et al. Akt/protein kinase B isoforms are differentially regulated by epidermal growth factor stimulation. J Biol Chem. 2000;275:30934–42. doi: 10.1074/jbc.M004112200. [DOI] [PubMed] [Google Scholar]
- 8.Kim JW, Kim YT, Kim DK, et al. Expression of epidermal growth factor receptor in carcinoma of the cervix. Gynecol Oncol. 1996;60:283–7. doi: 10.1006/gyno.1996.0039. [DOI] [PubMed] [Google Scholar]
- 9.Bellone S, Frera G, Landolfi G, et al. Overexpression of epidermal growth factor type-1 receptor (EGF-R1) in cervical cancer: implications for Cetuximab-mediated therapy in recurrent/metastatic disease. Gynecol Oncol. 2007;106:513–20. doi: 10.1016/j.ygyno.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 10.Scambia G, Ferrandina G, Distefano M, et al. Epidermal growth factor receptor (EGFR) is not related to the prognosis of cervical cancer. Cancer Lett. 1998;123:135–9. doi: 10.1016/s0304-3835(97)00421-7. [DOI] [PubMed] [Google Scholar]
- 11.Kersemaekers AM, Fleuren GJ, Kenter GG, et al. Oncogene alterations in carcinomas of the uterine cervix: overexpression of the epidermal growth factor receptor is associated with poor prognosis. Clin Cancer Res. 1999;5:577–86. [PubMed] [Google Scholar]
- 12.Fan Z, Shang BY, Lu Y, et al. Reciprocal changes in p27(Kipl) and p21 (Cipl) in growth inhibition mediated by blockage or overstimulation of epidermal growth factor receptors. Clin Cancer Res. 1997;3:1943–8. [PubMed] [Google Scholar]
- 13.Ciardiello F, Caputo R, Bianco R, et al. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. 2000;6:2053–63. [PubMed] [Google Scholar]
- 14.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 15.Normanno N, De Luca A. Erlotinib in pancreatic cancer: are tumor cells the (only) target? J Clin Oncol. 2007;25:5836–7. doi: 10.1200/JCO.2007.14.6258. [DOI] [PubMed] [Google Scholar]
- 16.Kim GE, Kim YB, Cho NH, et al. Synchronous coexpression of epidermal growth factor receptor and cyclooxygenase-2 in carcinoms of the uterine cervix: a potential predictor of poor survival. Clin Cancer Res. 2004;10:1366–74. doi: 10.1158/1078-0432.ccr-0497-03. [DOI] [PubMed] [Google Scholar]
- 17.Therasse P, Arbuck SG, Eisenhauer EA, et al. New Guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 18.Look KY, Blessing JA, Nelson BE, et al. A phase II trial of isotretinoin and alpha interferon in patients with recurrent squamous cell carcinoma of the cervix: a Gynecologic Oncology Group study. Am J Clin Oncol. 1998;21:591–4. doi: 10.1097/00000421-199812000-00012. [DOI] [PubMed] [Google Scholar]
- 19.Mannel RS, Blessing JA, Boike G. Cisplatin and pentoxifylline in advanced or recurrent squamous cell carcinoma of the cervix: a phase II trial of the Gynecologic Oncology Group. Gynecol Oncol. 2000;79:64–6. doi: 10.1006/gyno.2000.5874. [DOI] [PubMed] [Google Scholar]
- 20.Rose PG, Blessing JA, Arseneau J. Phase II evaluation of altretamine for advanced and recurrent squamous cell carcinoma of the cervix: a Gynecologic Oncology Group Study. Gynecol Oncol. 1996;62:100–2. doi: 10.1006/gyno.1996.0196. [DOI] [PubMed] [Google Scholar]
- 21.Bookman MA, Blessing JA, Hanjani P, et al. Topotecan in squamous cell carcinoma of the cervix: A phase II study of the Gynecologic Oncology Group. Gynecol Oncol. 2000;77:446–9. doi: 10.1006/gyno.2000.5807. [DOI] [PubMed] [Google Scholar]
- 22.Rose PG, Blessing JA, Van Le L, et al. Prolonged oral etoposide in recurrent or advanced squamous cell carcinoma of the cervix: a Gynecologic Oncology Group study. Gynecol Oncol. 1998;70:263–6. doi: 10.1006/gyno.1998.5097. [DOI] [PubMed] [Google Scholar]
- 23.Schilder RJ, Blessing JA, Morgan M, et al. Evaluation of gemcitabine in patients with squamous cell carcinoma of the cervix: a phase II study of the Gynecologic Oncology Group. Gynecol Oncol. 2000;76:204–7. doi: 10.1006/gyno.1999.5671. [DOI] [PubMed] [Google Scholar]
- 24.Monk BJ, Sill MW, Burger RA, et al. Phase II trial of bevacizumab in the treatment of persistent or recurrent squamous cell carcinoma of the cervix: a Gynecologic Oncology Group study. J Clin Oncol. doi: 10.1200/JCO.2008.18.9043. IN PRESS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen TT, Ng TH. Optimal flexible designs in phase II clinical trials. Stat Med. 1998;17:2301–12. doi: 10.1002/(sici)1097-0258(19981030)17:20<2301::aid-sim927>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 26.Kawamoto T, Sato JD, Le A, et al. Growth stimulation of A431 cells by epidermal growth factor: identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proc Natl Acad Sci USA. 1983;80:1337–41. doi: 10.1073/pnas.80.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan Z, Masui H, Altas I, et al. Blockade of epidermal growth factor receptor function by bivalent and monovalent fragments of 225 anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res. 1993;53:4322–8. [PubMed] [Google Scholar]
- 28.Goncalves A, Fabbro M, Lhomme C, et al. A phase II trial to evaluate gefitinib as second- or third-line treatment in patients with recurring locoregionally advanced or metastatic cervical cancer. Gynecol Oncol. 2008;108:42–6. doi: 10.1016/j.ygyno.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 29.Arias-Pulido H, Joste N, Chavez A, et al. Absence of epidermal growth receptor mutations in cervical cancer. Int J Gynecol Cancer. 2008;18:749–54. doi: 10.1111/j.1525-1438.2007.01111.x. [DOI] [PubMed] [Google Scholar]
- 30.Marzano R, Corrado G, Merola R, et al. Analysis of chromosomes 3, 7, X and the EGFR gene in uterine cervical cancer progression. Eur J of Cancer. 2004;40:1624–9. doi: 10.1016/j.ejca.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 31.Rocha-Lima CM, Soares HP, Raez LE, et al. EGFR targeting of solid tumors. Cancer Control. 2007;14:295–304. doi: 10.1177/107327480701400313. [DOI] [PubMed] [Google Scholar]
- 32.Yamasaki F, Johansen MJ, Zhang D, et al. Acquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res. 2007;67:5779–88. doi: 10.1158/0008-5472.CAN-06-3020. [DOI] [PubMed] [Google Scholar]
- 33.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 34.Burtness B, Goldwasser MA, Flood WB, et al. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: an Eastern Cooperative Oncology Group study. J Clin Oncol. 2005;23:8646–54. doi: 10.1200/JCO.2005.02.4646. [DOI] [PubMed] [Google Scholar]
- 35.Vermorken JB, Trigo J, Hitt R, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol. 2007;25:2171–7. doi: 10.1200/JCO.2006.06.7447. [DOI] [PubMed] [Google Scholar]
- 36.Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 37.Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 38.Van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–64. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 39.Knecht R, Peters S, Adunka O, et al. Carcinomas unresponsive to either cisplatinum or anti-EGFR therapy can be growth inhibited by combination therapy of both agents. Anticancer Res. 2003;23:2577–83. [PubMed] [Google Scholar]