

Abstract
In the past decade, palladium-catalyzed C–H activation/C–C bond forming reactions have emerged as promising new catalytic transformations; however, development in this field is still at an early stage compared to the state of the art in cross-coupling reactions using aryl and alkyl halides. This Review begins with a brief introduction of four extensively investigated modes of catalysis for forming C–C bonds from C–H bonds: Pd(II)/Pd(0), Pd(II)/Pd(IV), Pd(0)/Pd(II)/Pd(IV) and Pd(0)/Pd(II) catalysis. More detailed discussion is then directed towards the recent development of Pd(II)-catalyzed coupling of C–H bonds with organometallic reagents through a Pd(II)/Pd(0) catalytic cycle. Despite much progress made to date, improving the versatility and practicality of this new reaction remains a tremendous challenge.
Keywords: C-H activation, palladium, C-C coupling, organometallic reagent
1. Introduction
Among the myriad important transition metal-catalyzed synthetic transformations, palladium-catalyzed Heck coupling, cross-coupling (Kumada, Stille, Negishi, Suzuki–Miyaura, Hiyama), Tsuji–Trost allylation and Buchwald–Hartwig amination reactions using organohalides and other surrogates are particularly valuable tools in synthetic chemistry.[1, 2] A common and critical feature of these catalytic processes is the formation of aryl or alkyl Pd(II) intermediates which can be subsequently functionalized to form carbon–carbon and carbon–heteroatom bonds (Scheme 1).
Scheme 1.

Pd(0)-catalyzed reactions of aryl(alkyl) halides
The impressive versatility of these C–C and C–heteroatom bond forming processes ultimately stems from the reactivity of the corresponding aryl and alkyl Pd(II) species. Thus the development of the most straightforward and economical sequences to prepare such intermediates would certainly further improve these reactions. In particular, there exist virtually unlimited opportunities for using unactivated carbon–hydrogen (C–H) bonds,[3] which can readily be cleaved by Pd(II) catalysts, as reaction partners (Scheme 2). From the viewpoint of synthetic analysis, such reactions offer not only complementary reactivity, but also represent novel synthetic disconnections when the regioselective introduction of halides into molecules is not straightforward in a given synthetic plan.
Scheme 2.

Pd(II)-catalyzed functionalization of C–H bonds
Cyclopalladation with C–H bond-containing molecules has been extensively documented[4,5,6] and has been found to proceed along a variety of pathways (Scheme 3).[7] These studies were a major part of the impetus for us to launch our efforts in developing catalytic transformations that are based on a sequence of C–H activation followed by cross-coupling with organometallic reagents. In hindsight, the fact that a strongly coordinating nitrogen-containing directing group is typically needed to promote facile cyclopalladation severely limits the substrate scope; nevertheless, such a class of substrates has served as a pivotal platform for our discovery and optimization of this unprecedented mode of catalysis.
Scheme 3.
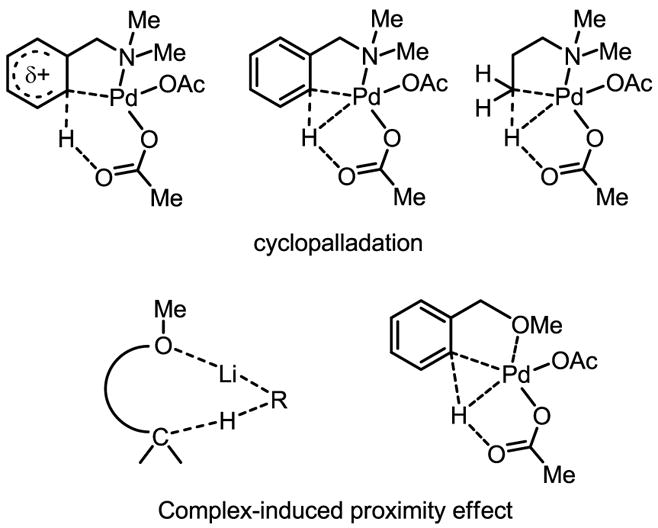
C–H activation through cyclopalladation or the CIPE
We envisioned ultimately expanding the scope of these reactions to include more synthetically useful oxygen-containing directing groups (e.g. carboxylic acids, ketones, esters, alcohols, etc.). We were encouraged on this front by studies using less coordinative oxy-functional groups such as Boc and OMe to direct lithiation through the complex-induced proximity effect (CIPE), a term originally coined by Beak and Snieckus (Scheme 3).[8] The distinction between the CIPE and directed cyclopalladation is that the thermodynamic stability of the resulting intermediates in the case of the CIPE is generally much lower. As such, these complexes are, in general, not isolable, even though they are incredibly intriguing from a synthetic perspective. Indeed a pioneering example of ortho-C–H functionalization of acetophenone by a Ru(0) catalyst has been reported.[9] Inspired by these precedents, we focused on developing new conditions and reagents that would promote C–H insertion driven by not only by traditional cyclopalladation, but also by the CIPE.
Following a brief survey of various approaches in Pd-catalyzed C–H activation/C–C bond formation, this review describes the early adventures and recent developments in Pd-catalyzed coupling of C–H bonds with organometallic reagents to form sp2–sp2, sp2–sp3 and sp3–sp3 carbon–carbon bonds. The versatility and practicality of these types of reactions in their current forms are evaluated with respect to the efficiency of catalysis, substrate scope, and operational costs. Key problems and potential solutions in this field are also discussed.
2. Olefination of sp2 C–H Bonds: Pd(II)/Pd(0) Catalysis
The past five decades have witnessed noticeable progress in the development of Pd-catalyzed C–H activation/C–C bond forming processes. Research in this field has largely focused on the discovery of new modes of catalysis and the expansion of substrate scope. One of the earliest examples concerns C–H activation of benzene by Pd(OAc)2 and subsequent carbopalladation and β-hydride elimination to afford olefinated arenes (Scheme 4).[10]
Scheme 4.

Pd(II)-catalyzed olefination of arenes: Pd(II)/Pd(0) catalysis
This early report by Fujiwara demonstrated the impressive reactivity of Pd(II) in activating aryl C–H bonds; however, two major drawbacks largely hampered the application of this catalytic reaction.[11] First, a large excess of the arene was required (often used as the solvent). Second, there was a lack of control of the regioselectivity when mono-substituted benzene was used as the substrate. Addressing this latter shortcoming, an early attempt of using benzoic acid to achieve ortho-selectivity represented an encouraging step forward (Scheme 5).[12]
Scheme 5.

Directed ortho-olefination of benzoic acid
In response to this regioselectivity problem, an instrumental development using a directing group was reported by de Vries (Scheme 6).[13] The use of an anilide substrate afforded high ortho-selectivity and allowed the arene to be used as the limiting reagent. In this reaction, benzoquinone is believed to be crucial for the C–C bond forming step, and the use of TsOH was also found to be beneficial.
Scheme 6.

ortho-Selective olefination of arenes
It must be noted that the mechanism of the C–H cleavage step for this electron-rich arene may be distinct from that of the reaction with benzene. Among the three known reaction mechanisms,[7] the cleavage of the C–H bonds in the anilide substrate is likely to proceed via electrophilic palladation of the electron-rich arene followed by loss of the ortho-proton (SArE). This mechanism is consistent with the relatively electron-rich nature of this substrate and is further supported by kinetic data collected for a series of substituted anilides.[13,14]
Importantly, this study together with Fujiwara’s early work has spurred recent studies on C–H activation/Heck coupling reactions using arenes possessing either high electron density or directing groups.[15] Notably, two elegant synthetic applications using an indole olefination have further inspired efforts towards improving this reaction (Scheme 7 and Scheme 8).[16] In the synthesis of ibogamine the carbon–palladium bond was reduced by NaBH4 to give the desired product. An unexpected ring expansion of the alkylpalladium intermediate served extraordinarily well in the synthesis of (+)-austamide.
Scheme 7.

Synthesis of ibogamine
Scheme 8.

Synthesis of (+)-austamide
Although catalytic olefination of indoles using Pd(OAc)2 and Ag(I) and Cu(II) salts as the reoxidants was reported as early as 1983 by Itahara (Scheme 9),[17] several recent studies have greatly advanced this chemistry. Notably, Stoltz’s work using molecular oxygen as the reoxidant in the intramolecular olefination of indoles was a significant development (Scheme 10).[18] On the other hand, by using allylic acetates as the olefin partner, Ma cleverly avoided the need for an oxidant (Scheme 11).[19] In this latter study, the authors put forth a mechanism whereby Pd(II)-catalyzed C–H activation takes place as the first step, followed by intermolecular carbopalladation of the allylic acetate. β-acetate elimination then regenerates the active catalyst, Pd(OAc)2, without formal reduction to Pd(0) during the catalytic cycle.
Scheme 9.

Catalytic olefination of indoles via electrophilic palladation
Scheme 10.

Intramolecular olefination of indoles using O2 as the oxidant
Scheme 11.

Oxidant-free olefination of indoles
Achieving regioselective functionalization at either the 2- or 3-position of pyrroles through the use of different protecting groups is also synthetically useful (Scheme 12).[20] In this case, the agreement of the observed regioselectivity with that of the electrophilic bromination reaction of protected pyrroles[21] lends valuable evidence to the hypothesis that an electrophilic palladation process is involved in these olefination reactions.
Scheme 12.

Regioselective olefination of pyrroles
To expand the synthetic utility of directed C–H activation/olefination, a concise and general route for the preparation of heterocyclic compounds from triflate-protected arylalkylamines has recently been developed using highly acidic triflamide groups to direct C–H activation (Scheme 13).[22]
Scheme 13.

Heterocycle synthesis via olefination of arenes
A recent report by Chang describes a very useful olefination of pyridine N-oxides (Scheme 14).[23] The reactivity of Pd catalysts with pyridine N-oxides was also documented previously by Fagnou in arylation reactions. [24]
Scheme 14.

Olefination of pyridine N-oxides
Finally, efforts in establishing a meta-C–H activation/olefination process have yielded unprecedented reactivity (Scheme 15).[25] The use of a rationally designed mutually repulsive ligand was crucial for C–H activation of electron-deficient arenes, which were previously found to be unreactive. Owing to the electron-poor nature of the substrates and the observed distribution of products (roughly 4:1 meta:para isomers), an electrophilic palladation mechanism[15] in this case is unlikely. Rather, a combination of C–H acidity and steric hinderance seem to govern the reactivity of the different sites, suggesting a concerted mechanism whereby acetate serves as an internal base (Scheme 3). This novel ligand also allowed 1 atm O2 to be used as the sole oxidant, which represents a potentially important step forward in developing highly practical C–H functionalization reactions.
Scheme 15.

meta-Selective olefination of electronic-deficient arenes
3. Arylation of sp2 and sp3 C–H Bonds: Pd(II)/Pd(IV) Catalysis
Owing to their versatility, Pd(0)/Pd(II) catalysis and Pd(II)/Pd(0) catalysis have both been extensively exploited for the development of catalytic reactions. On the other hand, redox chemistry involving Pd(IV) is studied far less often, despite the early proposal of the existence of this oxidation state[26,27] and the unambiguous supporting evidence obtained subsequently.[28] Tremont reported the first intriguing methylation of ortho-C–H bonds in anilide (Scheme 16). In this work, the reactivity of the cyclopalladated intermediate with MeI was established, and a plausible Pd(II)/Pd(IV) mechanism was presented (Scheme 17).[29]
Scheme 16.

ortho-Methylation of anilides
Scheme 17.

Proposed Pd(II)/Pd(IV) catalytic cycle
The proposed oxidation of Pd(II) to Pd(IV) by MeI was conclusively supported by X-ray crystallography, the first crystal structure being obtained by Canty (Scheme 18).[28] Recently, additional corroborative physical evidence has been obtained by Sanford in X-ray crystallographic studies of Pd(IV) intermediates generated in her acetoxylation reaction.[30] Furthermore, the isolation of quantitative amounts of PdI2 after the completion the asymmetric catalytic iodination reaction developed in our laboratory[31] and an earlier example of azo-directed iodination[32] are also important pieces of evidence in support of Pd(II)/Pd(IV) redox chemistry.
Scheme 18.

X-ray structures of Pd(IV) complexes
This early alkylation reaction via a Pd(II)/Pd(IV) cycle was further exploited to develop catalytic arylation reactions. In 2000, an intriguing report from Chen described a Pd-catalyzed arylation of aldehydic C–H bonds using a hypervalent iodine reagent, [Ph2I]Br (Scheme 19).[33] In seeking to explain the observed reactivity, the authors invoke a Pd(0)/Pd(II) catalytic cycle; however, given the strength of [Ph2I]Br as an oxidant, a Pd(II)/Pd(IV) catalytic cycle cannot conclusively be ruled out, particularly when considering more recent literature in C–H activation using hypervalent iodine reagents.
Scheme 19.

Pd-catalyzed arylation of aldehydic C–H bonds
Sanford and Daugulis independently developed a more general approach using directed C–H activation and [Ph2I]PF6 and [Ph2I]BF4 for arylation of C–H bonds (Scheme 20).[34] It is believed that this reaction follows a Pd(II)/Pd(IV) mechanism, whereby [Ph2I]PF6 and [Ph2I]BF4 play a similar role to that of MeI in earlier studies mentioned above. Building on this work, Sanford has also successfully extended this chemistry to the arylation of indoles at room temperature.[35]
Scheme 20.


Arylation of C–H bonds via Pd(II)/Pd(IV) catalysis
Especially noteworthy is the discovery by Daugulis that the arylation of C–H bonds can be performed using cheap and practical ArI under neat conditions or using CF3COOH (TFA) as the solvent (Scheme 21).[34b] This protocol represents the most efficient arylation reaction via Pd(II)/Pd(IV) catalysis to date. These conditions have also been applied to the arylation of sp3 C–H bonds by linking a pyridyl group to carboxylic acids via an amide bond (Scheme 21).[36]
Scheme 21.

Arylation of C–H bonds using ArI
4. Sequential Ortho-alkylation and Olefination of Aryl Iodides: Pd(0)//Pd(II)/Pd(IV) Catalysis
A highly complex yet efficient catalytic reaction involving Pd(0), Pd(II) and Pd(IV) in the reaction pathway was invented by Catellani in 1997 following her early studies on Pd(IV) complexes in 1988 (Scheme 22).[37] The key feature of this reaction is the dialkylation of both ortho-C–H bonds of the aryl iodide substrate. The final Heck coupling of the aryl Pd(II) intermediate with an olefin serves as a critical step for closing the catalytic cycle. The biggest advantage of this catalytic cycle is that no external oxidant is needed. As a demonstration of the flexibility and power of Pd redox chemistry, this complex and elegant catalytic cycle is unparalleled.
Scheme 22.


ortho-Alkylation of C–H bonds via Pd(0)/Pd(II)/Pd(IV) catalysis
Despite the many merits of this transformation, the lack of simplicity (due to the formation of multiple bonds, some of which may not be desired in a synthetic application) constitutes a noticeable limitation. Spectacular efforts from Lautens and others to overcome this drawback and make this transformation more amenable for synthesis have yielded substantial improvements.[38] For example, the interception of aryl palladium intermediates by a cyanation event is a significant departure from the early alkylation/olefination sequence, affording valuable versatility for synthetic applications (Scheme 23).[39]
Scheme 23.

ortho-Alkylation and cyanation of arenes
The use of an alkyl halide containing a tethered acetylene has also led to a spectacular route to tetrasubstituted helical alkenes (Scheme 24).[40]
Scheme 24.

Synthesis of tetrasubstituted helical alkenes
Interestingly, an analogous form of “cascade catalysis” for arylation was also achieved by Carretero without using norbornene as the mediator (Scheme 25).[41]
Scheme 25.

Early arylation of C–H bonds involving Pd(IV) intermediates
5. Arylation and Alkylation of sp2 and sp3 C–H Bonds: Pd(0)/Pd(II) Catalysis
Oxidative addition of aryl halides to Pd(0) is one of the most important modes of reactivity in modern palladium chemistry, because it serves as the first step in Heck coupling, cross-coupling and Buchwald–Hartwig amination reactions. This reactivity has also been drawn upon extensively in the development of C–H activation/arylation reactions in the past three decades. The initial proof of concept was established using electron-rich (hence, more reactive) heterocycles as substrates (Scheme 26).[42]
Scheme 26.

Arylation of electron-rich heterocycles via Pd(0)/Pd(II) catalysis
Among the numerous examples showcasing the arylation of various heterocycles,[43] subtle effects of the choice of the catalyst, aryl halide and N-protecting groups on both reactivity and selectivity have been observed.[44] Understanding these trends is important for synthetic applications (Scheme 27).
Scheme 27.

Regioselective arylation of heterocycles
The arylation and alkylation of non-heterocyclic arenes was initially limited to intramolecular reactions.[45] An elegant and useful development of this reaction is the synthesis of oxindoles initiated by the oxidative addition of alkyl halides to Pd(0) (Scheme 28).[46]
Scheme 28.

Alkylation of arenes by alkyl halides
An important early observation was made by Rawal that phenolic hydroxyl groups promote ortho-arylation from an ether-tethered aryl bromide (Scheme 29).[47] This appears to be the first example of arylation of non-heterocyclic arenes since the discovery by Sakai and Ohta in 1982. Within the same year, intermolecular arylation of 2-phenylphenol was demonstrated.[48] Together, these two results constitute a highly significant contribution towards the development of intermolecular arylation reactions with non-heterocyclic arenes using Pd(0)/Pd(II) catalysis.
Scheme 29.

Development of intermolecular arylation reactions with arenes
Impressive ortho-coupling of broadly useful substrates, including benzanilides, benzaldehydes and benzoic acids, has since been reported (Scheme 30).[49]
Scheme 30.

ortho-Arylation of benzanilides, benzadehydes and benzoic acids
Exploiting the reactivity of Pd(II) with excess benzene,[10] a major advance was made by adding pivalic acid to the reaction system, allowing for the coupling of benzene with aryl bromides (Scheme 31).7i However, the long-standing problems associated with non-directed arene C–H activation (see Section 2) still persist in this case: the arene is used as a co-solvent, and there is a lack of regioselectivity with mono-substituted arenes.
Scheme 31.

Arylation of benzene with aryl bromides
Finally, research towards the intramolecular arylation of sp3 C–H bonds has also progressed during the last fifteen years, albeit with far fewer examples. The first example reported by Dyker impressively encompasses both Pd(0)/Pd(II) and Pd(II)/Pd(IV) redox chemistry (Scheme 32).[50]
Scheme 32.

Intramolecular Arylation of sp3 C–H bonds
An intriguing and useful carbocyclization reaction was developed by Baudoin using this reactivity to give a strained benzocyclobutene (Scheme 33). This chemistry has also recently been applied to the synthesis of complex natural products by the same group.[51]
Scheme 33.

Carbocyclization through arylation of C–H bonds
In another case, this Pd(0)/Pd(II) chemistry was beautifully combined with a Suzuki–Miyaura coupling reaction to perform the arylation of sp3 C–H bonds with external phenylboronic acids (Scheme 34).[52] In fact, this finding inspired for our own recent studies concerning the direct coupling of C–H bonds with organoboron reagents, the main subject of this review (See Section 6).
Scheme 34.

Arylation of sp3 C–H bonds with external ArB(OH)2
In an insightful mechanistic study of a related process, intramolecular sp3 C–H bonds were pivalated in the presence of CsO2CtBu (Scheme 35).[53] The use of a bulky carboxylate group such as pivalate was believed to be beneficial for the reaction. This observation supports a “through space” migration of Pd from an aryl to an allylic carbon atom. However, one could argue that the activation of the allylic sp3 C–H bond by the ArPdI species can not necessarily be ruled out in the absence of further evidence.
Scheme 35.

Reaction involving the migration of aryl Pd to allylic Pd
Recently, this reactivity was elegantly utilized by Fagnou to develop a general method for the preparation of dihydrobenzofurans (Scheme 36).[54] In this case, the presence of bulky carboxylate anions was also found to dramatically improve the yield.
Scheme 36.

Synthesis of dihydrobenzofurans
With respect to the simplicity and cost of the catalytic system, intermolecular arylation using Pd(0)/ArI/ligand is the closest to conventional Heck coupling and cross-coupling reactions. A major challenge that remains for this mode of catalysis is still the relatively limited scope and versatility.
6. Arylation and Alkylation of sp2 and sp3 C–H Bonds with Organometallic Reagents: Pd(II)/Pd(0) catalysis
6.1 Rh(I) and Ru(II)-Catalyzed Arylation of sp2 C–H Bonds
Although the development of C–H activation/C–C bond forming reactions using other metals is beyond the scope of this review,[55,56,57] we wish to illustrate two coupling reactions with organometallic reagents catalyzed by Rh(I) and Ru(II) catalyst to highlight the significant advancements in the field (Scheme 37).[58]
Scheme 37.

Rh(I) and Ru(II)-catalyzed ortho-C–H coupling
Conceptually, it is important to distinguish the Pd(II)-catalyzed C–H activation/C–C coupling reactions developed in our laboratory from this chemistry. In using Pd rather than other transition metals, we sought to build our chemistry upon the established reactivity of aryl(alkyl) halides with Pd(0). In doing so, we endeavored to access the reactivity of known catalytic cycles of Pd from new entry points, rather than using the various redox manifolds of other transition metals.
6.2 Establishment of the First Catalytic Cycle for a Pd(II)-Catalyzed C–H Activation/C–C Coupling Reaction
Following our initial development of diastereoselective iodination and acetoxylation of C–H bonds of oxazoline substrates via Pd(II)/Pd(IV) catalysis,[31] we attempted to harness the excellent reactivity of the oxazoline directing group to establish the proof of concept for an unprecedented coupling process via Pd(II)/Pd(0) catalysis. The choice of organotin reagents as the coupling partners was inspired by Hartwig’s early observation of a transmetalation process between a cyclopalladated complex and Me3SnPh.[59]
A brief comparison of the proposed C–H activation/C–C coupling process to the Pd(0)-catalyzed cross-coupling reactions[60] of aryl halide and alkyl halides was helpful in identifying potential problems in our early attempts (Scheme 38).
Scheme 38.

Comparison of conventional cross-coupling with C–H activation/C–C coupling
This proposed catalytic cycle has two obvious differences from that of the cross-coupling reactions: a) an oxidation system is required for the reoxidation of Pd(0); b) the ligands commonly used to promote the desired transmetalation and reductive elimination steps are not compatible with the C–H activation step.
The most challenging obstacle for establishing this new catalytic cycle, however, was that Pd(II) species tend to react preferentially with organometallic reagents rather than the more inert C–H bonds, resulting in rapid precipitation of Pd(0). Indeed, reactions of oxazoline substrates with Pd(OAc)2 and organotin reagents under various conditions consistently resulted in full recovery of Pd(0) precipitates, despite the fact that each individual step in the potential catalytic cycle had precedent (Scheme 39).
Scheme 39.

Problematic homocoupling of organometallic reagents in the presence of Pd(II)
These results were frustrating during our exploratory studies because no meaningful information could be extracted for guidance from our extensive screening experiments. To facilitate a progressive screening process, we made a decision to compromise by adding the organotin reagents batchwise, which we expected would slow down the reaction of Pd(OAc)2 with the organotin reagents. This simple operational change was vital for us to begin to observe the desired coupling products and to establish a meaningful assay. The resulting data allowed for a more rational screening to be performed. It was ultimately established that the combination of Cu(OAc)2, benzoquinone and CH3CN gave the best results for this new coupling reaction (Scheme 40).[61]
Scheme 40.

C–H coupling with organotin reagents
Although benzoquinone is a well established oxidant for Pd(0) and promoter for C–C bond formation in a wide range of Pd-catalyzed reactions,[62] our studies on the formation of cyclopalladated intermediates and their subsequent reaction with organotin reagents revealed an additional role for benzoquinone: promoting C–H activation. In particular, the previously reported use of benzoquinone to promote C–C bond formation[13] in an arene C–H activation/olefination reaction was most relevant to our study (Scheme 6).
6.3 Expanding Coupling Partner Scope: Versatility
Having established the proof of concept, we moved forward to test if organoboronic acids, the most widely used coupling partners,[1] could be used for this reaction. As described in Scheme 34, the Pd(0)/ArI initiated C–H coupling with phenyl boronic acid reported by Buchwald was encouraging in the early stages.[52] A single example of stoichiometric coupling of a cyclopalladated complex with vinyl boronic acid has also previously been reported by Sames.[63] Our preliminary results showed that coupling of oxazoline substrates with organoboronic acids gave approximately 10% yield. Exploring other directing groups, we were pleased to find that coupling of pyridine substrates with alkylboronic acids was successful (Scheme 41).[64] The use of Ag(I) oxidants was critical both for transmetalation and for catalytic turnover in this case. Additionally, we found that arylboronic acids could also be used in this reaction (20–30% yields). However, rather than optimizing this coupling protocol with 2-phenylpyridine, we sought applications with more synthetically applicable substrates and thus focused our efforts on developing this reaction using a carboxylic acid directing group (see Section 6.4).
Scheme 41.

C–H coupling with organoboron reagents
The potential generality of Pd(II)-catalyzed C–H activation/C–C coupling with organometallic reagents has been further demonstrated by Shi in the coupling of anilides with arylsilanes (Scheme 42).[65] These highly reactive anilide substrates can also be ortho-coupled with arylboronic acids.[66] As discussed in Section 2, de Vries’s early study[13] suggested that reaction of this anilide with Pd(OAc)2 proceeds by electrophilic palladation.
Scheme 42.

C–H coupling with organosilane reagents
6.4 Expanding Substrate Scope: A Significant Challenge
As discussed in Section 6.2, one of the major problems in these coupling reactions is the undesired reaction between the Pd(II) catalysts and the organometallic reagents. This side reaction becomes predominant if C–H activation of the substrates is not rapid. The aforementioned coupling reactions benefited greatly from the use of electron-rich aryl rings or from the presence of strong coordinating groups to ensure rapid binding of the substrate with the Pd(II) catalysts. Typically, nitrogen-containing directing groups are used to aid coordination; however, the presence of such groups severely restricts the substrate scope, preventing potential broad synthetic applications. Thus, expanding the scope to include simple substrates such as carboxylic acids and alcohols was major hurdle to clear on the way to general applicability. Compared to the classical nitrogen atom-directed cyclopalladation reactions, Pd(II) insertion into C–H bonds promoted by oxygen atom coordination (via CIPE)[8] is rather rare.
Considerable difficulties have been met during our effort to promote Pd(II) insertion into inert C–H bonds in both aliphatic and aryl carboxylic acids. We hypothesized that the observed lack of reactivity of carboxylic acids with Pd(II) catalysts was due to the presence of several possible known coordination modes (Scheme 43). In these complexes, the CIPE is absent because the Pd center is locked away from the β -C–H bonds by κ2-coordination.
Scheme 43.

Coordination modes of Pd(II) with carboxylic acids
We then made a critical discovery that the presence of wide range of cationic counter ions, including Na+, promoted Pd(II) insertion into C–H bonds in carboxylic acid substrates. In our working model, the sodium cation coordinates with the carboxylate group in a κ2 fashion, thereby forcing Pd(II) to coordinate with the unhindered oxygen lone pair (Scheme 44). The assembly of this pre-transition state is believed to trigger C–H insertion through the CIPE. Subsequent structural studies using X-ray crystallography and 1H NMR spectroscopy have also provided evidence for the formation of such a structure from toluic acid.[67] The dramatic influence of Na+ or K+ on the reactivity of carboxylic acids was later also observed in other reactions of these substrates. Strikingly, the mere use of table salt was sufficient for the promotion of C–H insertion.
Scheme 44.

A working model for table salt-promoted C–H insertion
It is commonly believed that a shift from a κ2 to a κ1 metal carboxylate,[68] as has been observed for stoichiometric Rh(I) and Ir(I) insertions[69] into the ortho positions of benzoic acids, is the operative mechanism for late transition metals in general. However, in the case of Pd, the energetic preference is for Pd to remain in a κ2 acetate-bound configuration, which seems to disfavor a shift to a κ1 Pd(II) carboxylate (Scheme 45).
Scheme 45.

Comparison of iridium carboxylates and palladium carboxylates
This newly discovered mode of reactivity made possible the application of our coupling protocol to substrates without strong directing groups (Scheme 46).[70] In this preliminary report, yields were generally poor, and the substrate scope was limited to only a few benzoic acids. The use of sp3-boronic acids was limited to MeB(OH)2, most likely due to β-hydride elimination, which could occur with other alkylboronic acids after the transmetalation step. The use of Ag2CO3 as a stoichiometric oxidant was another major practical drawback. Nonetheless, the ability to use a simple functional group to promote C–H insertion by Pd(II) was encouraging. Moreover, the C–H insertion intermediates are different from the commonly obtained cyclopalladated complexes in that the latter have unusually high thermodynamic stability. The stability of such C–H insertion intermediates is beneficial for the C–H activation step but may also cause difficulties for further functionalization. This cation-promoted reactivity was further demonstrated in the arylation of sp3 C–H bonds using Pd(II)/Pd(IV) catalysis. Previous conditions developed by Daugulis[36] were modified by adding excess NaOAc to improve the yields.
Scheme 46.

Coupling of C–H bonds with substrates without a nitrogen-containing directing group
The versatility and practicality of this coupling reaction was then substantially improved by using potassium aryltrifluoroborates as the coupling partners (Table 1).[71,72] Under these new conditions, the use of air or O2 as the oxidant instead of Ag2CO3 was made possible. Although the use of 20 atm of air or O2 is needed to shorten the reaction time, this coupling reaction could also be performed under 1 atm of air or O2 with prolonged reaction time. Most importantly, a wide range of functional groups was tolerated. The compatibility with electron-withdrawing groups, such as nitro and acetyl, which are usually deactivating, is valuable in synthesis. Mechanistically, the reactivity observed with arenes containing both a carboxyl and nitro group renders an electrophilic palladation pathway unlikely.
Table 1.
Versatile biaryl synthesis via C–H activation/C–C coupling
 | ||
|---|---|---|
 83% 65% (1 atm O2, 72h) 52% (1 atm air, 72h) |
 91% 91%62% (1 atm O2, 72h) 55% (1 atm air, 72h) |
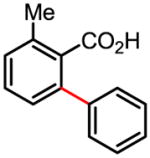 87% 70% (1 atm O2, 72h) 65% (1 atm air, 72h) |
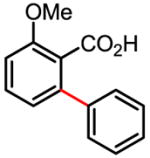 45% |
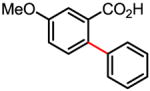 82% 58% (1 atm O2, 72h) |
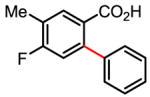 91% |
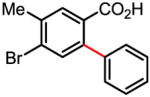 85% |
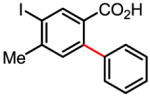 36% |
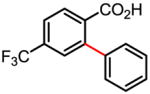 83% |
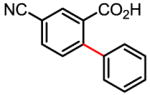 68% |
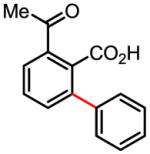 69% |
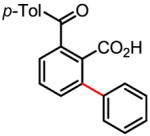 88% 57% (1 atm O2, 72h) |
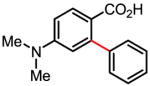 43% |
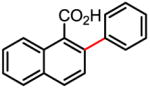 98% 67% (1 atm O2, 72h) |
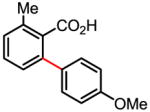 79% |
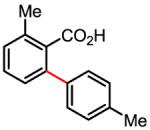 78% 69% (1 atm O2, 72h) |
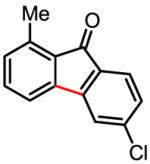 89%a |
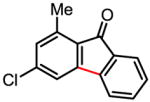 87%a |
Treatment of the coupling product with oxalyl chloride gave the product shown
The excellent results obtained with benzoic acid substrates prompted us to test whether this coupling protocol could be applied to phenyl acetic acid substrates as well. It is worth noting that the broadly useful lithiation/iodination/arylation sequence (Scheme 47) is incompatible with this type of substrate due to the presence of the acidic α-hydrogen atom.[73] A direct ortho-arylation would therefore provide an unprecedented disconnection for biaryl synthesis.
Scheme 47.

ortho-Lithiation
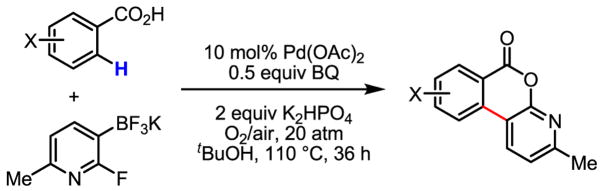
We were very pleased that a wide range of phenyl acetic acid substrates was reactive under our newly developed C–H activation/C–C coupling conditions (Table 2). Intriguingly, the removal of the Ag(I) oxidant was crucial for this reaction to occur. Common functional groups on the aryl boronic acids, including methoxyl, carbonyl, and halo groups, were also tolerated. Currently, the scope of heterocyclic boronic acids is still limited (Table 3). As shown for pyridyl boronic acid substrates, 2,6-disubstitution is required to obtain the corresponding product in good yields.
Table 2.
Versatile biaryl synthesis via C–H activation/C–C coupling
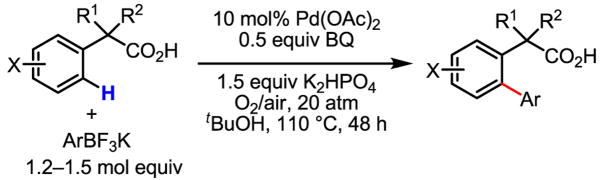
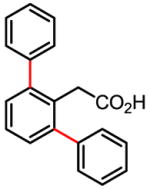
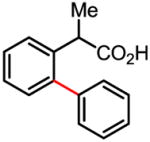
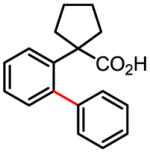
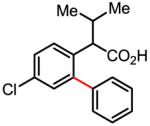
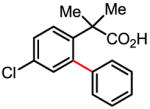
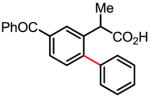
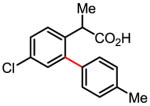
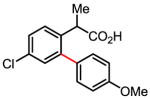
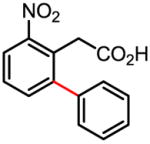
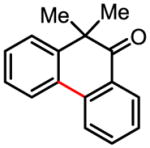
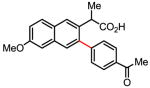
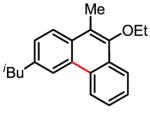
Treatment of the coupling product with oxalyl chloride gave the product shown
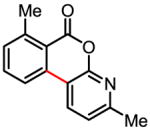
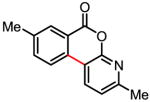
Table 3.
Coupling with heterocyclic trifluoroborates
 | ||
|---|---|---|
 74% |
 82% |
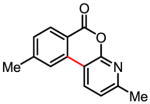 81% |
The compatibility with both benzoic acid and phenyl acetic acid substrates makes this aryl–aryl coupling reaction a versatile way to construct biaryl molecules with different carbon skeletons. In addition, benzoic acids and phenyl acetic acids are among the most abundant starting materials in synthesis. The only drawback is the requirement for the presence of a carboxyl group; however, the rich chemical reactivity of carboxyl groups also offers opportunity for a wide range of chemical manipulations to meet synthetic needs. Furthermore, our ongoing work suggests that other broadly useful substrates are also compatible with this coupling protocol. For instance, triflate-protected phenylalkyl amines, which have recently been found to be reactive substrates for ortho-C–H activation,[22] similarly undergo successful ortho-coupling under identical conditions. Further inclusion of a broader range of synthetically useful directing groups will minimize the inherent limitation of directed C–H coupling to a great extent because a particular directing group can be chosen to meet the needs of a desired synthetic application.
Intrigued by the drastically enhanced reactivity in C–H activation, we are currently investigating whether a different mechanism is operative in this coupling reaction. For instance, transmetalation between Pd(II) and ArBF3K could take place as the first step in the catalytic cycle (Scheme 48). If this pathway were to hold, in theory the electron-rich Ph–Pd–OAc species could be oxidized to a Pd(IV) intermediate, which could then cleave C–H bonds more efficiently. However, these hypotheses remain pure speculation in the absence of comprehensive mechanistic studies and further structural characterization.
Scheme 48.
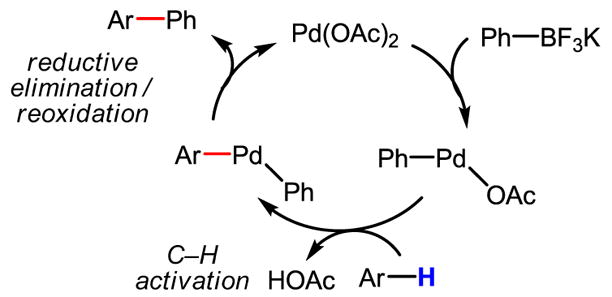
An alternative catalytic cycle
The next major task is to test the applicability of this newly developed C–H activation/C–C coupling reaction for a wide range of substrates containing no proximate chelating functional groups. While it is encouraging to see that coupling of electron-rich and hence highly activated olefins, arenes and indoles with organoboron[74] and organotin[75] reagents is feasible (Scheme 49), key challenges in this endeavor remain to be addressed. For example, the reaction of Pd(II) with benzene still requires a large excess of benzene. Additionally, Pd(II) generally reacts with mono-substituted benzene at the ortho-, meta- and para-positions in an unselective fashion, limiting the potential for synthetic applications. The solution to both of these problems most likely hinges on an innovative design of a new ligand that will impart an appropriate steric and electronic bias on Pd(II) so that selective C–H coupling of mono-substituted arenes can be accomplished. In this respect, our initial report on meta-C–H activation/Heck coupling represents a promising step forward (Scheme 15),[25] but a significant amount of work remains to be done to attain higher selectivity and efficiency and to expand this chemistry to C–C cross-coupling. Continued efforts to confront the fundamental challenges of coupling unactivated arenes with organometallic reagents regioselectively are expected to yield both novel ligands and improved catalytic systems (Scheme 50).
Scheme 49.

Coupling of electron-rich arenes with organometallic reagents
Scheme 50.

meta- and para-Selective coupling of mono-substituted arenes with organometallic reagents: a lingering challenge
6.5 Coupling of C–H Bonds with Arenes: a Related Reaction
Recently, a closely related coupling reaction involving two different arene substrates as the coupling partners has attracted significant attention. Since the early discovery of Pd(II)-catalyzed arene–arene coupling,[76] a great deal of effort has been devoted to eliminating the less desirable arene–arene homocoupling pathway. Substantial progress towards this goal has been made by Lu using Pd(II)/Pd(0) catalysis, although the obtained selectivity of heterocoupling product versus homocoupling product is not yet high enough for widespread synthetic application (Scheme 51).[77]
Scheme 51.

An early example of arene–arene coupling
The replacement of one of the arene partners by an electron-rich heterocycle substantially improves the selectivity for the heterocoupling reaction (Scheme 52).[78] Notably, sub-stoichiometric coupling of N-acetylindole with benzene was previously reported by Itahara.[79] The highly efficient Pd(II)-catalyzed intramolecular homocoupling of pyrroles was also successfully exploited by Boger to achieve the total synthesis of prodigiosin.[76c] Recently, Buchwald reported a drastically improved protocol that allowed the coupling of anilide substrates with 4–11 equivalents of benzene.[80]
Scheme 52.

Coupling of heterocycles with benzene
The use of a directing group has also been successfully employed to suppress homocoupling (Scheme 53).[81] A highly efficient heterocoupling process was developed by Sanford using a pyridyl moiety as the directing group. [81a] Similar to the coupling protocol developed in our group,[64] in this case, the use of benzoquinone as a C–H activation promoter and Ag2CO3 as the oxidant was also found to be critical. Additionally, concurrent to these studies, You found that oxazoline was a suitable directing group for this coupling reaction.[81b] Diastereoselective coupling using chiral oxazolines was also found to be possible by the same group. Notably, our group has also used oxazoline groups as auxiliaries to achieve C–H coupling with organotin reagents using Cu(OAc)2 as the oxidant.[61]
Scheme 53.

Heterocoupling using directing groups
6.6 Coupling of sp3 C–H Bonds with Organometallic Reagents
Despite recent impressive progress made in sp3–sp3 cross coupling using alkyl halides,[82] efforts to couple sp3 C–H bonds with organometallic reagents have been met with numerous problems, likely due to the lack of assistance from an appropriate ligand. Nevertheless, the feasibility of such a process was first demonstrated in pyridyl-directed C–H activation/C–C coupling (Scheme 54).[64] Although generally, the C–H bonds involved in this process are primary, secondary C–H bonds are also reactive under these conditions, albeit in much lower yields.
Scheme 54.
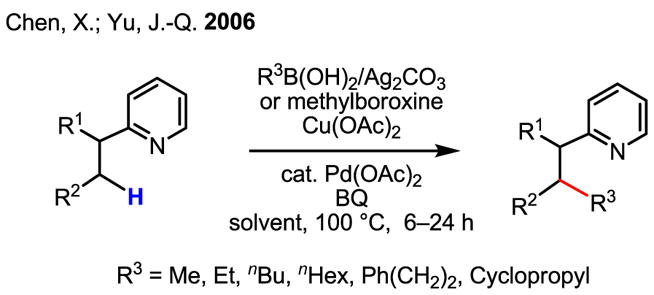
An early example of sp3–sp3 C–H activation/C–C coupling
Expansion of this C–C coupling protocol to aliphatic carboxylic acid substrates afforded sp3–sp3 coupling products in poor yields (10–20%). The carboxyl-directed C–H activation inspired us to test structurally analogous hydroxamic acids as substrates (Scheme 55). We envisioned that the CONH moiety could exhibit behavior similar to that of the CO2H group. We also hypothesized that the methoxy group could provide steric hindrance, which is believed to be important for preventing β-hydride elimination in sp3–sp3 cross-coupling reactions.[82] Extensive screening established conditions for an unprecedented reaction to accomplish β-C–C bond formation with aliphatic acid derivatives.[83] Considering the significance of the classical α-lithiation/alkylation of carboxylic acids in chemical synthesis, this newly developed β-C–C bond forming reaction is likely to find broad utility as a novel synthetic disconnection.
Scheme 55.

sp3 C–H activation/C–C coupling
The utility of this coupling reaction was further demonstrated in the alkylation of the hydroxamic acid derived from dehydroabietic acid, a natural product identified as an efficient BK channel opener (Scheme 56).[84] Due to their bioactivity, molecules of this type could ultimately be used for treatments of diseases such as acute stroke, epilepsy and asthma. Generally, however, diversification of such core structures is difficult because of the lack of reactive chemical functional groups, aside from the carboxylic acid moiety, which is essential for biological activity. Masking the carboxylic acid as the hydroxamic acid allows for functionalization at the methyl C–H bond, affording a novel class of analogues that could ultimately display improved pharmacokinetic properties.
Scheme 56.

Derivatives of a biologically active natural product
6.7 Enantioselective C–H Activation/C–C Coupling
To date, asymmetric catalysis is largely based on chiral recognition of a π-face (re versus si) possessed by olefins or carbonyl compounds.[85] Only a few examples involve chiral recognition of sp3 carbon centers,[86–88] including asymmetric Heck-cyclization, asymmetric metathesis and kinetic resolution of alcohols.[89] Nonetheless, research concerning enantioselective carbene insertion into sp3 C–H bonds has made impressive progress during the past few decades.[90] Recently, an enantioselective nitrene insertion process has also emerged as promising asymmetric C–H amination method.[91]
Despite the remarkable success in developing Pd-catalyzed asymmetric catalysis more generally, studies towards the enantioselective functionalization of C–H bonds via Pd insertion have been largely unsuccessful.[92–95] Based on our experience, it seems that two pervasive problems have historically plagued research in this field. Firstly, the relatively high reaction temperatures required in C–H activation reactions make chiral recognition of sp3 carbon centers challenging. Secondly, most commonly used chiral ligands are problematic. Typically, effective chiral induction in asymmetric catalysis occurs as a result of the chiral ligands promoting the favored reaction pathway. However, in the case of C–H insertion processes, these ligands either outcompete the substrate for binding to the Pd(II) center or deactivate Pd(II) for cleavage of the desired C–H bond, even if the required L(substrate)PdX2 complex is formed.
Encouraged by the recent progress in Pd(II)-catalyzed C–H activation/C–C coupling, we sought to develop enantioselective variants of these reactions (Scheme 57). Our initial efforts focused on desymmetrization of prochiral C–H bonds on geminal aryl or methyl groups. In choosing such systems for early investigation, we envisioned that any insights into the stereoselection model for these substrates would be directly applicable to the desymmetrization of other C–H bonds. In particular, we looked towards desymmetrization of geminal C–H bonds of methylene groups as a long-term goal (Scheme 58), though the reactivity of methylene C–H bonds is usually markedly lower.[64]
Scheme 57.
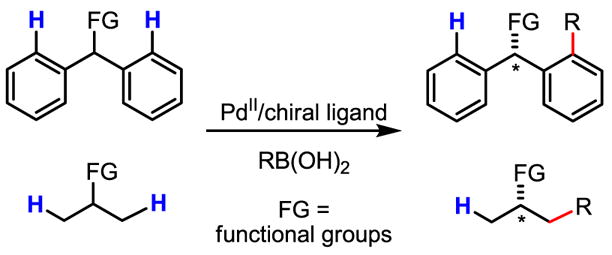
Desymmetrization of germinal aryl and methyl groups
Scheme 58.
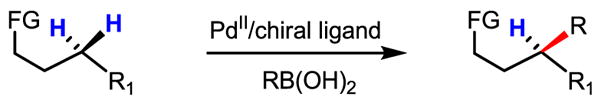
Desymmetrization of methylene C–H bonds
Using a highly efficient C–H activation/C–C coupling reaction at a relatively mild temperature (60 °C) as an assay, we were able to establish a proof of concept for Pd(II)-catalyzed enantioselective C–H activation using chiral carboxylic acids with a constrained conformation (Scheme 59).[96] Analysis of these data revealed that only the α-chiral center is important for chiral recognition.
Scheme 59.

Proof of concept
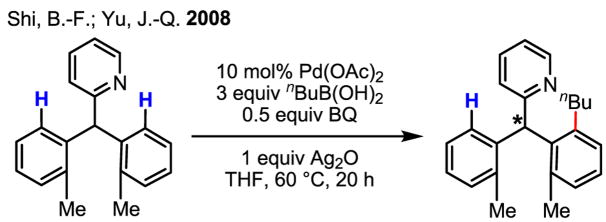
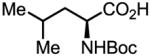
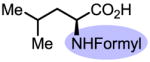
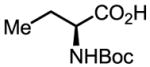
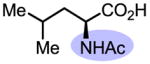
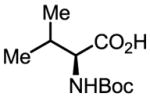
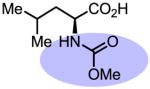
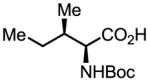
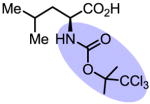
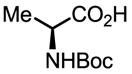
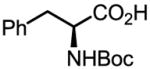
Upon further investigation, it was found that a wide range of mono-protected amino acids were effective chiral ligands for this enantioselective coupling reaction (Table 4). Of particular importance was the finding that mono-N-protection of the amino acid ligands is crucial for chiral recognition.
Table 4.
Pd-Catalyzed enantioselective C–H coupling
 | |||||
|---|---|---|---|---|---|
| Ligand | Yield (%) | ee | Ligand | Yield (%) | ee |
|
63 | 90 |
|
53 | 6 |
|
47 | 85 |
|
74 | 80 |
|
69 | 70 |
|
88 | 79 |
|
85 | 72 |
|
89 | 85 |
|
60 | 80 | |||
|
66 | 81 |
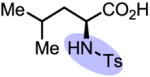
|
89 | 60 |
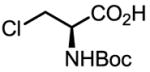
|
83 | 83 |
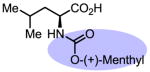
|
87 | 85 |
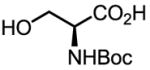
|
65 | 88 |
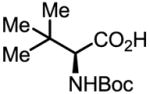
|
91 | 87 |
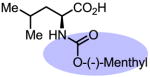
|
60 | 52 | |||
Analysis by 1H NMR spectroscopy and X-ray crystallography led us to propose the involvement of a key reactive intermediate B (Scheme 60). The interaction between bound substrate and the mono-protected chiral amino acid ligand on the Pd center results in the assembly of an intermediate complex B in which C–H cleavage is not retarded to a noticeable degree. Contrasting this favorable pre-transition state with the unfavorable pre-transition state formed from A, it is clear that the unfavorable steric interactions in A decrease the efficiency of this pathway, leading to the high enantioselectivity of this process (Scheme 60).
Scheme 60.

A simplified stereomodel for asymmetric C–H insertion
In hindsight, mono-protection of the nitrogen atom in the amino acid ligands offers a tremendous advantage for chiral control in metal-mediated reactions. With this modification, the chirality on the α-carbon (which is spatially remote) is relayed to the nitrogen atom attached to the metal center, a process that hinges upon the bisdentate coordination of the ligand. This chiral relay can be thought of as a “gearing effect.” Notably, this concept is closely related to the Evans’s pioneering work on the rational design of a chiral mixed phosphorus/sulfur ligand for asymmetric hydrogenation in which the chiral sulfur is assembled through a “gearing effect” as well.[97]
In an effort to expand the scope of this reaction, we explored coupling of prochiral sp3 C–H bonds. Use of the ligands listed in Table 4 gave poor enantioselectivity (10–15% ee). A significant improvement was made by using a more rigid ligand to give 37% ee (Scheme 61). The sensitive response of enantioselectivity to the ligand structure suggests that there is vast opportunity for additional tuning of existing ligand structures, as well as for the design of entirely new ligand architectures to achieve enantioselective C–H activation reactions with more general substrate scope. To this end, we are currently synthesizing a wide range of chiral amino acid ligands[98] with the aim of applying this new asymmetric C–C bond forming reaction to broader classes of substrates.
Scheme 61.

Enantioselective coupling of sp3 C–H bonds
7. Conclusions and Outlook
Recently, palladium-catalyzed C–H activation/C–C bond forming reactions have emerged as a promising set of synthetic transformation for the assembly of carbon–carbon bonds. Various catalytic cycles have been developed to accomplish the olefination, arylation and alkylation of unactivated C–H bonds, including Pd(II)/Pd(0), Pd(II)/Pd(IV), Pd(0)/Pd(II)/Pd(IV) and Pd(0)/Pd(II) catalytic cycles. Our group developed the first protocol for successful C–H activation/C–C coupling with organometallic reagents using Pd(II)/Pd(0) catalysis. Since its initial discovery, this mode of catalysis has been expanded to include a broad range of coupling partners, including organotin, organoboron and organosilicon reagents. Importantly, sp2–sp2, sp2–sp3 and sp3–sp3 coupling reactions have all been demonstrated. One major goal, the use of simple substrates with this Pd(II)/Pd(0) coupling such as carboxylic acids and amines, has been achieved. Due to the ubiquity of these functional groups, this catalytic reaction will likely find immediate synthetic applications, especially in the early stages of a synthesis and in medicinal chemistry.
Despite these advancements, C–H activation/C–C coupling still falls short of the remarkably high standards for efficiency and practicality set by palladium-catalyzed cross-coupling reactions of aryl and alkyl halides. In this context, a number of major challenges must still be overcome before these reactions will find broad applicability.
*Air as the oxidant. Development of an efficient catalytic system that uses 1 atm of air as the sole oxidant, rather than co-oxidants such as Cu(II) and Ag(I) salts, or benzoquinone would make this new process more comparable to conventional cross-coupling reactions in terms of costs and practicality.
*Reduced Catalyst Loading. In many cases, C–H activation reactions with Pd require 5–10 mol% catalyst. Thus, from the standpoint of atom economy and overall cost, discovering more efficient catalytic systems with improved turnover is paramount.
*Regioselective arene C–H activation. The design of novel ligands to promote C–H activation of mono-substituted benzene regioselectively at the meta-[25] or para-positions would represent a new paradigm in reactivity and would greatly expand the scope of this new C–C bond forming reaction.
*Enantioselective C–H activation of sp3 C–H bonds. Although, this goal may seem elusive based the results of our laboratory to date, continued efforts will eventually lead to a general asymmetric C–H activation/C–C coupling protocol, a reaction that would deliver a completely new disconnection for asymmetric C–C bond formation. These reactions will greatly simplify target synthesis by allowing for overarching strategies that start from simpler and more abundant starting materials. Furthermore, the understanding gleaned from the development of chiral ligands will greatly facilitate the design of new ligands to promote catalysis and to control the regioselectivity of C–H activation.
Acknowledgments
We wish to thank the U.S. National Science Foundation (NSF CHE-0615716), the U.S. National Institute of Health (NIGMS, 1 R01 GM084019-01A1), the A. P. Sloan Foundation, Pfizer, Eli Lilly and Amgen for financial support. Additionally, we would like to express our gratitude to the Royal Society for financial support during the early stages of this work (oxazoline-directed C–H activation) in 2002–2003 (RG36873). K.M.E. gratefully acknowledges the U.S. Department of Defense, the U.S. National Science Foundation, the Skaggs Oxford Scholarship program and The Scripps Research Institute for predoctoral fellowships.
References
- 1.For reviews, see: Stille JK. Angew Chem Int Ed. 1986;25:508.Heck RF. In: Comprehensive Organic Synthesis. 4. Trost BM, Fleming I, editors. Pergamon; Oxford: 1991. p. 833.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457.Diederich F, Stang PJ, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; New York: 1998. Luh TY, Leung MK, Wong KT. Chem Rev. 2000;100:3187. doi: 10.1021/cr990272o.Hiyama T. J Organomet Chem. 2002;653:58.E-i Negishi, Hu Q, Huang Z, Qian M, Wang G. Aldrichimica Acta. 2005;38:71.Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4442. doi: 10.1002/anie.200500368.Surry DS, Buchwald SL. Angew Chem Int Ed. 2008;47:6338. doi: 10.1002/anie.200800497.Hartwig JF. Nature. 2008;455:314. doi: 10.1038/nature07369.Denmark SE, Regens CS. Acc Chem Res. 2008;41:1486. doi: 10.1021/ar800037p.
- 2.For ligand developments, see: Old DW, Wolfe JP, Buchwald SL. J Am Chem Soc. 1998;120:9722.Littke AF, Fu GC. Angew Chem Int Ed. 1998;37:3387. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3387::AID-ANIE3387>3.0.CO;2-P.Herrmann WA, Reisinger CP, Spiegler M. J Organomet Chem. 1998;557:93.Hamann BC, Hartwig JF. J Am Chem Soc. 1998;120:7369.Zhang C, Huang J, Trudell ML, Nolan SP. J Org Chem. 1999;64:3804.Rataboul F, Zapf A, Jackstell R, Harkal S, Riermeier T, Monsees A, Dingerdissen U, Beller M. Chem Eur J. 2004;10:2983. doi: 10.1002/chem.200306026.
- 3.For reviews on C–H activation chemistry, see: Crabtree RH. Chem Rev. 1985;85:245.Shilov AE, Shul’pin GB. Chem Rev. 1997;97:2879. doi: 10.1021/cr9411886.Stahl SS, Labinger JA, Bercaw JE. Angew Chem Int Ed. 1998;37:2181. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2180::AID-ANIE2180>3.0.CO;2-A.Bergman RG. Nature. 2007;446:391. doi: 10.1038/446391a.Brookhart M, Green MLH, Parkin G. Proc Natl Acad Sci. 2007;104:6908. doi: 10.1073/pnas.0610747104.
- 4.For comprehensive reviews on cyclopalladation reactions, see: Ryabov AD. Synthesis. 1985:233.Canty AJ. In: Comprehensive organometallic chemistry II: a review of the literature 1982–1994. Abel EW, Stone FGA, Wilkinson G, editors. Pergamon; 1995. pp. 225–255.
- 5.a) For an early example of cyclopalladation of sp2 C-H bonds directed by azo groups, see: Cope AC, Siekman RW. J Am Chem Soc. 1965;87:3272.b) For an early example of cyclopalladation of sp3 C–H bonds directed by oximes see: Constable AG, McDonald WS, Sawkins LC, Shaw BL. J Chem Soc Chem Commun. 1978:1061.
- 6.For early examples of exploiting cyclopalladation reactions in synthesis, see: Carr K, Sutherland JK. J Chem Soc Chem Commun. 1984:1227.Baldwin JE, Jones RH, Najera C, Yus M. Tetrahedron. 1985;41:699.Bore L, Honda T, Gribble GW. J Org Chem. 2000;65:6278. doi: 10.1021/jo0008688.
- 7.For development of mechanistic models of cyclopalladation reactions see: Ryabov AD, Sakodinskaya IK, Yatsimirsky AK. J Chem Soc Dalton Trans. 1985:2629.Burger P, Bergman RG. J Am Chem Soc. 1993;115:10462.Canty AJ, van Koten G. Acc Chem Res. 1995;28:406.Gómez M, Granell J, Martinez M. Organometallics. 1997;16:2539.Gómez M, Granell J, Martinez M. J Chem Soc, Dalton Trans. 1998:37.Vigalok A, Uzan O, Shimon LJW, Ben-David Y, Martin JML, Milstein D. J Am Chem Soc. 1998;120:12539.Webster CE, Fan Y, Hall MB, Kunz D, Hartwig JF. J Am Chem Soc. 2003;125:858. doi: 10.1021/ja028394c.Davies DL, Donald SMA, Macgregor SA. J Am Chem Soc. 2005;127:13754. doi: 10.1021/ja052047w.Lafrance M, Fagnou K. J Am Chem Soc. 2006;128:16496. doi: 10.1021/ja067144j.García-Cuadrado D, de Mendoza P, Braga AAC, Maseras F, Echavarren AM. J Am Chem Soc. 2007;129:6880. doi: 10.1021/ja071034a.
- 8.a) Beak P, Snieckus V. Acc Chem Res. 1982;15:306. [Google Scholar]; b) Snieckus V. Chem Rev. 1990;90:879. [Google Scholar]; c) Mortier J, Moyroud J, Bennetau B, Cain PA. J Org Chem. 1994;59:4042. [Google Scholar]; d) Whisler MC, MacNeil S, Snieckus V, Beak P. Angew Chem Int Ed. 2004;43:2206. doi: 10.1002/anie.200300590. [DOI] [PubMed] [Google Scholar]; e) Schlosser M, Mongin F. Chem Soc Rev. 2007;36:1161. doi: 10.1039/b706241a. [DOI] [PubMed] [Google Scholar]
- 9.Murai S, Kakiuchi F, Sekine S, Tanaka Y, Kamatani A, Sonoda M, Chatani N. Nature. 1993;366:529. [Google Scholar]
- 10.Moritani I, Fujiwara Y. Tetrahedron Lett. 1967;8:1119. [Google Scholar]
- 11.(a) Jia C, Piao D, Oyamada J, Lu W, Kitamura T, Fujiwara Y. Science. 2000;287:1992. doi: 10.1126/science.287.5460.1992. [DOI] [PubMed] [Google Scholar]; b) Yokota T, Tani M, Sakaguchi S, Ishii Y. J Am Chem Soc. 2003;125:1476. doi: 10.1021/ja028903a. [DOI] [PubMed] [Google Scholar]; c) Dams M, De Vos DE, Celen S, Jacobs PA. Angew Chem Int Ed. 2003;42:3512. doi: 10.1002/anie.200351524. [DOI] [PubMed] [Google Scholar]
- 12.Miura M, Tsuda T, Satoh T, Pivsa-Art S, Nomura M. J Org Chem. 1998;63:5211. [Google Scholar]
- 13.Boele MDK, van Strijdonck GPF, de Vries AHM, Kamer PCJ, de Vries JG, van Leeuwen PWNM. J Am Chem Soc. 2002;124:1586. doi: 10.1021/ja0176907. [DOI] [PubMed] [Google Scholar]
- 14.a) Li JJ, Giri R, Yu JQ. Tetrahedron. 2008;64:6979. [Google Scholar]; b) Desai LV, Stowers KJ, Sanford MS. J Am Chem Soc. 2008;130:13285. doi: 10.1021/ja8045519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Trost BM, Toste FD. J Am Chem Soc. 1996;118:6305. [Google Scholar]; b) Liu C, Widenhoefer RA. J Am Chem Soc. 2004;126:10250. doi: 10.1021/ja046810i. [DOI] [PubMed] [Google Scholar]; c) Zaitsev VG, Daugulis O. J Am Chem Soc. 2005;127:4156. doi: 10.1021/ja050366h. [DOI] [PubMed] [Google Scholar]; d) Capito E, Brown JM, Ricci A. Chem Commun. 2005:1854. doi: 10.1039/b417035k. [DOI] [PubMed] [Google Scholar]; e) Cai G, Fu Y, Li Y, Wan X, Shi Z. J Am Chem Soc. 2007;129:7666. doi: 10.1021/ja070588a. [DOI] [PubMed] [Google Scholar]; f) Wang JR, Yang CT, Liu L, Guo QX. Tetrahedron Lett. 2007;48:5449. [Google Scholar]; g) Houlden CE, Bailey CD, Ford JG, Gagné MR, Lloyd-Jones GC, Booker-Milburn KI. J Am Chem Soc. 2008;130:10066. doi: 10.1021/ja803397y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Trost BM, Godleski SA, Genêt JP. J Am Chem Soc. 1978;100:3930. [Google Scholar]; b) Baran PS, Corey EJ. J Am Chem Soc. 2002;124:7904. doi: 10.1021/ja026663t. [DOI] [PubMed] [Google Scholar]
- 17.a) Itahara T. Synthesis. 1979:151. [Google Scholar]; b) Itahara T, Ikeda M, Sakakibara T. J Chem Soc Perkin Trans I. 1983:1361. [Google Scholar]
- 18.Ferreira EM, Stoltz BM. J Am Chem Soc. 2003;125:9578. doi: 10.1021/ja035054y. [DOI] [PubMed] [Google Scholar]
- 19.Ma S, Yu S. Tetrahedron Lett. 2004;45:8419. [Google Scholar]
- 20.a) Grimster NP, Gauntlett C, Godfrey CRA, Gaunt MJ. Angew Chem Int Ed. 2005;44:3125. doi: 10.1002/anie.200500468. [DOI] [PubMed] [Google Scholar]; b) Beck EM, Grimster NP, Hatley R, Gaunt MJ. J Am Chem Soc. 2006;128:2528. doi: 10.1021/ja058141u. [DOI] [PubMed] [Google Scholar]; c) Beck EM, Hatley R, Gaunt MJ. Angew Chem Int Ed. 2008;47:3004. doi: 10.1002/anie.200705005. [DOI] [PubMed] [Google Scholar]
- 21.Muchowski JM, Solas DR. Tetrahedron Lett. 1983;24:3455. [Google Scholar]
- 22.Li JJ, Mei TS, Yu JQ. Angew Chem Int Ed. 2008;47:6452. doi: 10.1002/anie.200802187. [DOI] [PubMed] [Google Scholar]
- 23.Cho SH, Hwang SJ, Chang S. J Am Chem Soc. 2008;130:9254. doi: 10.1021/ja8026295. [DOI] [PubMed] [Google Scholar]
- 24.Campeau LC, Rousseaux S, Fagnou K. J Am Chem Soc. 2005;127:18020. doi: 10.1021/ja056800x. [DOI] [PubMed] [Google Scholar]
- 25.Zhang YH, Shi BF, Yu JQ. J Am Chem Soc. 2009 doi: 10.1021/ja900327e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) For early proposal of formation of Pt(IV) complexes, see: Pope WJ, Peachy SJ. Proc Chem Soc London. 1907;23:86.b) For oxidation of Pt(II) to Pt(IV) by O2 see: Rostovtsev VV, Henling LM, Labinger JA, Bercaw JE. Inorg Chem. 2002;41:3608. doi: 10.1021/ic0112903.
- 27.a) Uson R, Fornies J, Navarro R. J Organomet Chem. 1975;96:307. [Google Scholar]; b) Trost BM, Tanoury GJ. J Am Chem Soc. 1987;109:4753. [Google Scholar]; c) Canty AJ. Acc Chem Res. 1992;25:83. [Google Scholar]
- 28.a) Byers PK, Canty AJ, Skelton BW, White AH. J Chem Soc Chem Commun. 1986:1722. [Google Scholar]; b) Canty AJ, Denney MC, van Koten G, Skelton BW, White AH. Organometallics. 2004;23:5432. [Google Scholar]
- 29.Tremont SJ, Rahman HU. J Am Chem Soc. 1984;106:5759. [Google Scholar]
- 30.a) Dick AR, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:2300. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; b) Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; c) Dick AR, Kampf JW, Sanford MS. J Am Chem Soc. 2005;127:12790. doi: 10.1021/ja0541940. [DOI] [PubMed] [Google Scholar]
- 31.a) Giri R, Chen X, Yu JQ. Angew Chem Int Ed. 2005;44:2112. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]; b) Giri R, Liang J, Lei JG, Li JJ, Wang DH, Chen X, Naggar IC, Guo C, Foxman BM, Yu JQ. Angew Chem Int Ed. 2005;44:7420. doi: 10.1002/anie.200502767. [DOI] [PubMed] [Google Scholar]
- 32.For early Pd(II)-catalyzed iodination reactions involving Pd(II)/Pd(IV) catalysis, see: Fahey DR. J Organometal Chem. 1971;27:283.Andrienko OS, Goncharov VS, Raida VS. Russ J Org Chem. 1996;32:89.
- 33.Xia M, Chen ZC. Syn Commun. 2000;30:531. [Google Scholar]
- 34.a) Kalyani D, Deprez NR, Desai LV, Sanford MS. J Am Chem Soc. 2005;127:7330. doi: 10.1021/ja051402f. [DOI] [PubMed] [Google Scholar]; b) Daugulis O, Zaitsev VG. Angew Chem Int Ed. 2005;44:4046. doi: 10.1002/anie.200500589. [DOI] [PubMed] [Google Scholar]
- 35.Deprez NR, Kalyani D, Krause A, Sanford MS. J Am Chem Soc. 2006;128:4972. doi: 10.1021/ja060809x. [DOI] [PubMed] [Google Scholar]
- 36.a) Zaitsev VG, Shabashov D, Daugulis O. J Am Chem Soc. 2005;127:13154. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]; b) Reddy BVS, Reddy LR, Corey EJ. Org Lett. 2006;8:3391. doi: 10.1021/ol061389j. [DOI] [PubMed] [Google Scholar]
- 37.a) Catellani M, Chiusoli GP. J Organomet Chem. 1988;346:C27. [Google Scholar]; b) Catellani M, Chiusoli GP. J Organomet Chem. 1992;425:151. [Google Scholar]; c) Catellani M, Frignani F, Rangoni A. Angew Chem Int Ed. 1997;36:119. [Google Scholar]
- 38.Alberico D, Scott ME, Lautens M. Chem Rev. 2007;107:174. doi: 10.1021/cr0509760.For a mechanistic study see: Crdenas DJ, Martn-Matute B, Echavarren AM. J Am Chem Soc. 2006;128:5033. doi: 10.1021/ja056661j.
- 39.Mariampillai B, Alliot J, Li M, Lautens M. J Am Chem Soc. 2007;129:15372. doi: 10.1021/ja075599i. [DOI] [PubMed] [Google Scholar]
- 40.Gericke KM, Chai DI, Bieler N, Lautens M. Angew Chem Int Ed. 2009;48:1447. doi: 10.1002/anie.200805512. [DOI] [PubMed] [Google Scholar]
- 41.Mauleon P, Alonso I, Carretero JC. Angew Chem Int Ed. 2001;40:1291. doi: 10.1002/1521-3773(20010401)40:7<1291::aid-anie1291>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 42.a) Nakamura N, Tajima Y, Sakai K. Heterocycles. 1982;17:235. [Google Scholar]; b) Akita Y, Ohta A. Heterocycles. 1982;19:329. [Google Scholar]; c) Pivsa-Art S, Satoh T, Kawamura Y, Miura M, Nomura M. Bull Chem Soc Jpn. 1998;71:467. [Google Scholar]
- 43.For recent arylation of heterocycles with aryltosylates and aryltriflates see: Ackermann L, Althammer A, Fenner S. Angew Chem Int Ed. 2009;48:201. doi: 10.1002/anie.200804517.Roger J, Doucet H. Org Biomol Chem. 2008;6:169. doi: 10.1039/b715235c.Mousseau JJ, Larivée A, Charette AB. Org Lett. 2008;10:1641. doi: 10.1021/ol800396v.
- 44.a) Park CH, Ryabova V, Seregin IV, Sromek AW, Gevorgyan V. Org Lett. 2004;6:1159. doi: 10.1021/ol049866q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Touré BB, Lane BS, Sames D. Org Lett. 2006;8:1979. doi: 10.1021/ol053021c. [DOI] [PubMed] [Google Scholar]
- 45.a) Roesch KR, Larock RC. J Org Chem. 1998;63:5306. [Google Scholar]; b) Dong CG, Hu QS. Angew Chem Int Ed. 2006;45:2289. doi: 10.1002/anie.200504310. [DOI] [PubMed] [Google Scholar]; c) Ren H, Knochel P. Angew Chem Int Ed. 2006;45:3462. doi: 10.1002/anie.200600111. [DOI] [PubMed] [Google Scholar]; d) Cruz ACF, Miller ND, Willis MC. Org Lett. 2007;9:4391. doi: 10.1021/ol702044z. [DOI] [PubMed] [Google Scholar]; e) Watanabe T, Oishi S, Fujii N, Ohno H. Org Lett. 2008;10:1759. doi: 10.1021/ol800425z. [DOI] [PubMed] [Google Scholar]
- 46.Hennessy EJ, Buchwald SL. J Am Chem Soc. 2003;125:12084. doi: 10.1021/ja037546g. [DOI] [PubMed] [Google Scholar]
- 47.Hennings DD, Iwasa S, Rawal VH. J Org Chem. 1997;62:2. doi: 10.1021/jo961876k. [DOI] [PubMed] [Google Scholar]
- 48.Satoh T, Kawamura Y, Miura M, Nomura M. Angew Chem Int Ed. 1997;36:1740. [Google Scholar]
- 49.a) Kametani Y, Satoh T, Miura M, Nomura M. Tetrahedron Lett. 2000;41:2655. [Google Scholar]; b) Gürbüz N, Özdemir I, Çetinkaya B. Tetrahedron Lett. 2005;46:2273. [Google Scholar]; c) Chiong HA, Pham QN, Daugulis O. J Am Chem Soc. 2007;129:9879. doi: 10.1021/ja071845e. [DOI] [PubMed] [Google Scholar]
- 50.a) Dyker G. Angew Chem Int Ed. 1992;31:1023. [Google Scholar]; b) Dyker G. J Org Chem. 1993;58:6426. [Google Scholar]; c) Dyker G. Chem Ber. 1994;127:739. [Google Scholar]; d) Dyker G. Angew Chem Int Ed. 1994;33:103. [Google Scholar]
- 51.a) Baudoin O, Herrbach A, Guéritte F. Angew Chem Int Ed. 2003;42:5736. doi: 10.1002/anie.200352461. [DOI] [PubMed] [Google Scholar]; b) Hitce J, Retailleau P, Baudoin O. Chem Eur J. 2007;13:792. doi: 10.1002/chem.200600811. [DOI] [PubMed] [Google Scholar]; c) Chaumontet M, Piccardi R, Audic N, Hitce J, Peglion JL, Clot E, Baudoin O. J Am Chem Soc. 2008;130:15157. doi: 10.1021/ja805598s. [DOI] [PubMed] [Google Scholar]; d) Chaumontet M, Piccardi R, Baudoin O. Angew Chem Int Ed. 2009;48:179. doi: 10.1002/anie.200804444. [DOI] [PubMed] [Google Scholar]
- 52.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 53.Zhao J, Campo M, Larock RC. Angew Chem Int Ed. 2005;44:1873. doi: 10.1002/anie.200462327. [DOI] [PubMed] [Google Scholar]
- 54.Lafrance M, Gorelsky SI, Fagnou K. J Am Chem Soc. 2007;129:14570. doi: 10.1021/ja076588s. [DOI] [PubMed] [Google Scholar]
- 55.a) Lenges CP, Brookhart M. J Am Chem Soc. 1999;121:6616. [Google Scholar]; b) Chen H, Schlecht S, Semple TC, Hartwig JF. Science. 2000;287:1995. doi: 10.1126/science.287.5460.1995. [DOI] [PubMed] [Google Scholar]; c) Weissman H, Song X, Milstein D. J Am Chem Soc. 2001;123:337. doi: 10.1021/ja003361n. [DOI] [PubMed] [Google Scholar]; d) Cho J-Y, Tse MK, Holmes D, Maleczka RE, Jr, Smith MR., III Science. 2002;295:305. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]; e) Periana RA, Mironov O, Taube D, Bhalla G, Jones CJ. Science. 2003;301:814. doi: 10.1126/science.1086466. [DOI] [PubMed] [Google Scholar]; f) Sadow AD, Tilley TD. J Am Chem Soc. 2003;125:7971. doi: 10.1021/ja021341a. [DOI] [PubMed] [Google Scholar]; g) Lim SG, Ahn JA, Jun CH. Org Lett. 2004;6:4687. doi: 10.1021/ol048095n. [DOI] [PubMed] [Google Scholar]; h) Vetter AJ, Flaschenriem C, Jones WD. J Am Chem Soc. 2005;127:12315. doi: 10.1021/ja042152q. [DOI] [PubMed] [Google Scholar]; i) Das S, Incarvito CD, Crabtree RH, Brudvig GW. Science. 2006;312:1941. doi: 10.1126/science.1127899. [DOI] [PubMed] [Google Scholar]; j) Yanagisawa S, Sudo T, Noyori R, Itami K. J Am Chem Soc. 2006;128:11748. doi: 10.1021/ja064500p. [DOI] [PubMed] [Google Scholar]; k) Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; l) Li Z, Capretto DA, Rahaman R, He C. Angew Chem Int Ed. 2007;46:5184. doi: 10.1002/anie.200700760. [DOI] [PubMed] [Google Scholar]; m) Stokes BJ, Dong H, Leslie BE, Pumphrey AL, Driver TG. J Am Chem Soc. 2007;129:7500. doi: 10.1021/ja072219k. [DOI] [PubMed] [Google Scholar]; n) Luedtke AT, Goldberg KI. Angew Chem Int Ed. 2008;47:7694. doi: 10.1002/anie.200800524. [DOI] [PubMed] [Google Scholar]
- 56.For recent development of copper-catalyzed C–H functionalizations, see: Chen X, Hao XS, Goodhue CE, Yu JQ. J Am Chem Soc. 2006;128:6790. doi: 10.1021/ja061715q.Uemura T, Imoto S, Chatani N. Chem Lett. 2006;35:842.Li Z, Cao L, Li CJ. Angew Chem Int Ed. 2007;46:6505. doi: 10.1002/anie.200701782.Do HQ, Daugulis O. J Am Chem Soc. 2008;130:1128. doi: 10.1021/ja077862l.Brasche G, Buchwald SL. Angew Chem Int Ed. 2008;47:1932. doi: 10.1002/anie.200705420.Phipps RJ, Grimster NP, Gaunt MJ. J Am Chem Soc. 2008;130:8172. doi: 10.1021/ja801767s.Do HQ, Khan RMK, Daugulis O. J Am Chem Soc. 2008;130:15185. doi: 10.1021/ja805688p.Ackermann L, Potukuchi HK, Landsberg D, Vicente R. Org Lett. 2008;10:3081. doi: 10.1021/ol801078r.
- 57.For recent development of Iron-catalyzed C–H functionalizations, see: Zhang Y, Li CJ. Eur J Org Chem. 2007:4654.Li Z, Yu R, Li H. Angew Chem Int Ed. 2008;47:7497. doi: 10.1002/anie.200802215.Norinder J, Matsumoto A, Yoshikai N, Nakamura E. J Am Chem Soc. 2008;130:5858. doi: 10.1021/ja800818b.
- 58.a) Oi S, Fukita S, Inoue Y. Chem Commun. 1998:2439. [Google Scholar]; b) Kakiuchi F, Kan S, Igi K, Chatani N, Murai S. J Am Chem Soc. 2003;125:1698. doi: 10.1021/ja029273f. [DOI] [PubMed] [Google Scholar]; c) Pastine SJ, Gribkov DV, Sames D. J Am Chem Soc. 2006;128:14220. doi: 10.1021/ja064481j. [DOI] [PubMed] [Google Scholar]; d) Vogler T, Studer A. Org Lett. 2008;10:129. doi: 10.1021/ol702659a. [DOI] [PubMed] [Google Scholar]
- 59.Louie J, Hartwig JF. Angew Chem Int Ed. 1996;35:2359. [Google Scholar]
- 60.For a review on the mechanisms of the Stille reaction see: Espinet P, Echavarren AM. Angew Chem Int Ed. 2004;43:4704. doi: 10.1002/anie.200300638.
- 61.Chen X, Li JJ, Hao XS, Goodhue CE, Yu JQ. J Am Chem Soc. 2006;128:78. doi: 10.1021/ja0570943. [DOI] [PubMed] [Google Scholar]
- 62.a) Albéniz AC, Espinet P, Martín-Ruiz B. Chem Eur J. 2001;7:2481. doi: 10.1002/1521-3765(20010601)7:11<2481::aid-chem24810>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]; b) Verboom RC, Plietker BJ, Bäckvall JE. J Organomet Chem. 2003;687:508. [Google Scholar]; c) Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970. doi: 10.1021/ja0500198. [DOI] [PubMed] [Google Scholar]
- 63.Dangel BD, Godula K, Youn SW, Sezen B, Sames D. J Am Chem Soc. 2002;124:11856. doi: 10.1021/ja027311p. [DOI] [PubMed] [Google Scholar]
- 64.Chen X, Goodhue CE, Yu JQ. J Am Chem Soc. 2006;128:12634. doi: 10.1021/ja0646747. [DOI] [PubMed] [Google Scholar]
- 65.Yang S, Li B, Wan X, Shi Z. J Am Chem Soc. 2007;129:6066. doi: 10.1021/ja070767s. [DOI] [PubMed] [Google Scholar]
- 66.Shi Z, Li B, Wan X, Cheng J, Fang Z, Cao B, Qin C, Wang Y. Angew Chem Int Ed. 2007;46:5554. doi: 10.1002/anie.200700590. [DOI] [PubMed] [Google Scholar]
- 67.For detailed discussion and evidence for cation-promoted Pd insertion into C–H bonds, see: Giri R, Yu JQ. J Am Chem Soc. 2008;130:14082. doi: 10.1021/ja8063827.
- 68.Sanford MS, Valdez MR, Grubbs RH. Organometallics. 2001;20:5455. [Google Scholar]
- 69.Kisenyi JM, Sunley GJ, Cabeza JA, Smith AJ, Adams H, Salt NJ, Maitlis PM. J Chem Soc Dalton Trans. 1987:2459. [Google Scholar]
- 70.Giri R, Maugel N, Li JJ, Wang DH, Breazzano SP, Saunders LB, Yu JQ. J Am Chem Soc. 2007;129:3510. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]
- 71.Wang DH, Mei TS, Yu JQ. J Am Chem Soc. 2008;130 doi: 10.1021/ja806681z. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.For the use of ArBF3K see: Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J Org Chem. 1995;60:3020.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q.Darses S, Genet JP. Chem Rev. 2008;108:288. doi: 10.1021/cr0509758.
- 73.Beak P, Snieckus V. Acc Chem Res. 1982;15:306. [Google Scholar]
- 74.a) Ge H, Niphakis MJ, Georg GI. J Am Chem Soc. 2008;130:3708. doi: 10.1021/ja710221c. [DOI] [PubMed] [Google Scholar]; b) Yang SD, Sun CL, Fang Z, Li BJ, Li YZ, Shi ZJ. Angew Chem Int Ed. 2008;47:1473. doi: 10.1002/anie.200704619. [DOI] [PubMed] [Google Scholar]; c) Zhao J, Zhang Y, Cheng K. J Org Chem. 2008;73:7428. doi: 10.1021/jo801371w. [DOI] [PubMed] [Google Scholar]
- 75.Kawai H, Kobayashi Y, Oi S, Inoue Y. Chem Commun. 2008:1464. doi: 10.1039/b717251f. [DOI] [PubMed] [Google Scholar]
- 76.a) van Helden R, Verberg G. Recl Trav Chim Pays-Bas. 1965;84:1263. [Google Scholar]; b) Fujiwara Y, Moritani I, Ikegami K, Tanaka R, Teranishi S. Bull Chem Soc Jpn. 1970;43:863. [Google Scholar]; c) Boger DL, Patel M. Tetrahedron Lett. 1987;28:2499. [Google Scholar]; d) Li X, Yang J, Kozlowski MC. Org Lett. 2001;3:1137. doi: 10.1021/ol015595x. [DOI] [PubMed] [Google Scholar]
- 77.Li R, Jiang L, Lu W. Organometallics. 2006;25:5973. [Google Scholar]
- 78.a) Stuart DR, Fagnou K. Science. 2007;316:1172. doi: 10.1126/science.1141956. [DOI] [PubMed] [Google Scholar]; b) Dwight TA, Rue NR, Charyk D, Josselyn R, DeBoef B. Org Lett. 2007;9:3137. doi: 10.1021/ol071308z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.a) Itahara T. J Chem Soc Chem Comm. 1981:254. [Google Scholar]; b) Itahara T, Kawasaki K, Ouseto F. Bull Chem Soc Jpn. 1984;57:3488. [Google Scholar]; c) Itahara T. J Org Chem. 1985;50:5272. [Google Scholar]; d) Itahara T. J Org Chem. 1985;50:5546. [Google Scholar]; e) Itahara T. Heterocycles. 1986;24:2557. [Google Scholar]
- 80.Brasche G, García-Fortanet J, Buchwald SL. Org Lett. 2008;10:2207. doi: 10.1021/ol800619c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.a) Hull KL, Sanford MS. J Am Chem Soc. 2007;129:11904. doi: 10.1021/ja074395z. [DOI] [PubMed] [Google Scholar]; b) Xia JB, You SL. Organometallics. 2007;26:4869. [Google Scholar]; c) Li BJ, Tian SL, Fang Z, Shi ZJ. Angew Chem Int Ed. 2008;47:1115. doi: 10.1002/anie.200704092. [DOI] [PubMed] [Google Scholar]
- 82.a) Netherton MR, Dai C, Neuschütz K, Fu GC. J Am Chem Soc. 2001;123:10099. doi: 10.1021/ja011306o. [DOI] [PubMed] [Google Scholar]; b) Saito B, Fu GC. J Am Chem Soc. 2007;129:9602. doi: 10.1021/ja074008l. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Saito B, Fu GC. J Am Chem Soc. 2008;130:6694. doi: 10.1021/ja8013677. [DOI] [PubMed] [Google Scholar]
- 83.Wang DH, Wasa M, Giri R, Yu JQ. J Am Chem Soc. 2008;130:7190. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]
- 84.Ohwada T, Nonomura T, Maki K, Sakamoto K, Ohya S, Muraki K, Imaizumi Y. Bioorg Med Chem Lett. 2003;13:3971. doi: 10.1016/j.bmcl.2003.08.072. [DOI] [PubMed] [Google Scholar]
- 85.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. Springer; New York: 1999. [Google Scholar]
- 86.For enantioselective lithiation of sp3 C-H bonds see: Whisler MC, MacNeil S, Snieckus V, Beak P. Angew Chem. 2004;116:2256. doi: 10.1002/anie.200300590.Angew Chem Int Ed. 2004;43:2206.
- 87.Groves JT, Viski P. J Am Chem Soc. 1989;111:8537. [Google Scholar]
- 88.a) Zhou XG, Yu XQ, Huang JS, Che CM. Chem Commun. 1999:2377. [Google Scholar]; b) Larrow JF, Jacobsen EN. J Am Chem Soc. 1994;116:12129. [Google Scholar]; c) Punniyamurthy T, Miyafuji A, Katsuki T. Tetrahedron Lett. 1998;39:8295. [Google Scholar]; d) Murahashi SI, Noji S, Hirabayashi T, Komiya N. Tetrahedron: Asymmetry. 2005;16:3527. [Google Scholar]
- 89.a) Sato Y, Sodeoka M, Shibasaki M. J Org Chem. 1989;54:4738. [Google Scholar]; b) Carpenter NE, Kucera DJ, Overman LE. J Org Chem. 1989;54:5846. [Google Scholar]; c) Hoveyda AH, Schrock RR. Chem Eur J. 2001;7:945. doi: 10.1002/1521-3765(20010302)7:5<945::aid-chem945>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]; d) Jensen DR, Pugsley JS, Sigman MS. J Am Chem Soc. 2001;123:7475. doi: 10.1021/ja015827n. [DOI] [PubMed] [Google Scholar]; e) Ferreira EM, Stoltz BM. J Am Chem Soc. 2001;123:7725. doi: 10.1021/ja015791z. [DOI] [PubMed] [Google Scholar]
- 90.For reviews on enantioselective C–H activation via carbene and nitrene insertions, see: Müller P, Fruit C. Chem Rev. 2003;103:2905. doi: 10.1021/cr020043t.Doyle MP. J Org Chem. 2006;71:9253. doi: 10.1021/jo061411m.Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485.
- 91.a) Zalatan DN, Du Bois J. J Am Chem Soc. 2008;130:9220. doi: 10.1021/ja8031955. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Milczek E, Boudet N, Blakey S. Angew Chem Int Ed. 2008;47:6825. doi: 10.1002/anie.200801445. [DOI] [PubMed] [Google Scholar]
- 92.For elegant approaches of combining C–H activation and asymmetric hydrorhodation or carbopalladation, see: Mikami K, Hatano M, Terada M. Chem Lett. 1999:55.Thalji RK, Ellman JA, Bergman RG. J Am Chem Soc. 2004;126:7192. doi: 10.1021/ja0394986.
- 93.For Pd(II)-catalyzed asymmetric allylic C–H oxidation, see: Yang H, Khan MA, Nicholas KM. J Mol Catal. 1994;91:319.El-Qisiari AK, Qaseer HA, Henry PM. Tetrahedron Lett. 2002;43:4229.Covell DJ, White MC. Angew Chem Int Ed. 2008;47:6448. doi: 10.1002/anie.200802106.
- 94.For Ru- and Rh-catalyzed atropselective alkylation of biaryls see: Kakiuchi F, Gendre PL, Yamada yA, Ohtaki H, Murai S. Tetrahedron: Asymmetry. 2000;11:2647.
- 95.For a review on Cu(I)-catalyzed asymmetric Kharasch-Sosnovsky reaction, see: Eames J, Watkinson M. Angew Chem Int Ed. 2001;40:3567. doi: 10.1002/1521-3773(20011001)40:19<3567::AID-ANIE3567>3.0.CO;2-C.
- 96.Shi BF, Maugel N, Zhang YH, Yu JQ. Angew Chem Int Ed. 2008;47:4882. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]
- 97.Evans DA, Michael FE, Tedrow JS, Campos KR. J Am Chem Soc. 2003;125:3534. doi: 10.1021/ja012639o. [DOI] [PubMed] [Google Scholar]
- 98.a) Peris G, Jakobsche CE, Miller SJ. J Am Chem Soc. 2007;129:8710. doi: 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao Y, Rodrigo J, Hoveyda AH, Snapper ML. Nature. 2006;443:67. doi: 10.1038/nature05102. [DOI] [PubMed] [Google Scholar]; c) Lewis CA, Miller SJ. Angew Chem Int Ed. 2006;45:5616. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]