

Abstract
DNA methylation is an epigenetically imposed mark of transcriptional repression that is essential for maintenance of chromatin structure and genomic stability. Genome-wide methylation patterns are mediated by the combined action of three DNA methyltransferases: DNMT1, DNMT3A and DNMT3B. Compelling links exist between DNMT3B and chromosome stability as emphasized by the mitotic defects that are a hallmark of ICF syndrome, a disease arising from germline mutations in DNMT3B. Centromeric and pericentromeric regions are essential for chromosome condensation and the fidelity of segregation. Centromere regions contain distinct epigenetic marks, including dense DNA hypermethylation, yet the mechanisms by which DNA methylation is targeted to these regions remains largely unknown. In the present study, we used a yeast two-hybrid screen and identified a novel interaction between DNMT3B and constitutive centromere protein CENP-C. CENP-C is itself essential for mitosis. We confirm this interaction in mammalian cells and map the domains responsible. Using siRNA knock downs, bisulfite genomic sequencing and ChIP, we demonstrate for the first time that CENP-C recruits DNA methylation and DNMT3B to both centromeric and pericentromeric satellite repeats and that CENP-C and DNMT3B regulate the histone code in these regions, including marks characteristic of centromeric chromatin. Finally, we demonstrate that loss of CENP-C or DNMT3B leads to elevated chromosome misalignment and segregation defects during mitosis and increased transcription of centromeric repeats. Taken together, our data reveal a novel mechanism by which DNA methylation is targeted to discrete regions of the genome and contributes to chromosomal stability.
INTRODUCTION
DNA methylation is a heritable epigenetic mark associated with transcriptionally inactive regions of the genome. In mammals, covalent addition of methyl groups to the C-5 position of cytosine within the CpG dinucleotide is mediated by three DNA methyltransferases, DNMT1, DNMT3A and DNMT3B, and an enzymatically inactive regulatory protein, DNMT3L (1). Proper establishment of DNA methylation patterns is critical for mammalian embryonic development and maintenance of these patterns in somatic cells is essential for transcriptional regulation, chromatin structure and genome stability (2). Disruption of DNA methylation patterns during development leads to a variety of human genetic diseases, such as Angelman or Prader–Willi syndrome, and is also a direct and early contributor to cellular transformation (3). Tumor cells exhibit global hypomethylation, primarily affecting repetitive regions of the genome such as the centromeric and pericentric regions, and gene-specific hypermethylation events targeting CpG island-associated promoters (4). DNA methylation is intermingled with a complex array of epigenetic marks imparted on the core histones, also known as the histone code. Some histone marks, such as histone H3 lysine 4 (H3K4) trimethylation, are associated with transcriptional activation whereas others, such as H3K9 and H3K27 trimethylation, are associated with transcriptionally repressed regions (5). The latter two marks often overlap with DNA methylation. It remains largely unknown, however, how DNA methylation is targeted throughout the genome and how it interfaces with the histone code.
Compelling links between DNA methylation and genome stability, particularly during mitosis, have been reported. One of the most dramatic manifestations of this link comes from the study of patients with Immunodeficiency, Centromere instability, Facial anomalies (ICF) syndrome, a rare human disease caused by hypomorphic germline mutations in the DNMT3B gene. While immunodeficiency (defects in B cell function such as reduced immunoglobulin production), facial anomalies (epicanthic folds and flat nasal bridge) and mental retardation/developmental delay constitute common phenotypic abnormalities among ICF patients, one of the disease's most defining features is loss of DNA methylation from centromeric and pericentromeric repeat regions (alpha satellite and satellite 2/satellite 3 repeats, respectively) and marked loss of chromosomal condensation during mitosis (6,7). Chromosome 1 and to a lesser extent chromosomes 9 and 16 are the most affected by DNA hypomethylation in ICF patients. Pericentromere-region decondensation in ICF cells leads to formation of multiradials involving the decondensed chromosomes, translocations and telomeric associations (6). Interestingly, DNMT3B localizes to both centromeric and pericentromeric regions in mouse and human cells (8) and Dnmt3a/Dnmt3b-deficient ES cells exhibit significantly elevated rates of centromeric sister chromatid exchange (9). Taken together, these findings suggest that DNMT3B plays a critical role in chromosomal stability and maintenance of peri-/centromeric region chromatin structure. Exactly how DNMT3B accomplishes this function and is targeted to the centromeric region remains unclear.
The kinetochore, which assembles at the primary constriction and is made up of large protein complexes and centromeric heterochromatin, is critical for proper chromosome alignment by providing an attachment site for spindle microtubules needed for chromosome movement. On the basis of electron microscopy studies, the kinetochore is made up of an inner plate, comprised constitutive centromere proteins such as CENP-A and CENP-C and centromeric heterochromatin, and an outer plate composed of microtubule binding and motor proteins, separated by an electron-translucent middle domain (10). Epigenetic determinants play a dominant role in establishing centromere function and position. For example, while alpha satellite DNA is present at all human centromeres, there is no homology between centromeric repeat sequences across species (10). In contrast, the complement of proteins and epigenetic marks present at the centromere are highly conserved. Centromeric chromatin in humans contains interspersed blocks of CENP-A and H3-containing nucleosomes (11). Human centromeric chromatin is flanked by pericentromeric heterochromatin enriched in repressive H3K9 di- and trimethylation. Centromeric H3 nucleosomes are marked by H3K4 dimethylation, a mark associated with transcriptional permissiveness and by a lack of H3 and H4 acetylation, marks associated with transcriptionally active regions (12). Other studies using chromatin immunoprecipitation (ChIP) have revealed enrichment of H3K9 dimethylation at alpha satellite DNA and to a lesser extent H3K9 trimethylation and H3K27 mono- and trimethylation. H3K9 dimethylation may act as a boundary between centromeric chromatin containing CENP-A and pericentromeric heterochromatin containing H3K9 trimethylation (13). These findings suggest that a balance between repressive heterochromatin and CENP-A-containing heterochromatin is essential for proper centromere function.
CENP-C, like CENP-A, localizes to all active centromeres, neocentromeres and the active centromere of dicentric chromosomes throughout the cell cycle. While there is clear evidence that CENP-C binds alpha satellite DNA, it remains unclear whether it is truly sequence-specific (14). CENP-C also localizes to neocentromeres, which do not contain alpha satellite arrays, suggesting that DNA sequence alone is insufficient for CENP-C targeting. Cenp-c knockout mice are embryonic lethal at or before the implantation stage and cells derived from these embryos display irregular nuclear morphology and micronuclei. In addition, mitotic chromosomes are highly disorganized and do not segregate properly (15). Conditional deletion of CENP-C in chicken DT40 cells induces mitotic delay, aneuploidy and chromosome missegregation, implicating CENP-C as an important player in the metaphase-to-anaphase transition (16).
In the present study, we sought to better understand the mechanisms by which DNA methylation and DNMT3B are targeted throughout the genome by identifying new DNMT3B-interacting proteins. We therefore undertook an exhaustive yeast two-hybrid screen using full-length DNMT3B as bait and a human testis cDNA library as prey. We discovered multiple independent clones encoding the constitutive centromere protein CENP-C and confirmed that full-length CENP-C interacted with DNMT3B in yeast using a β-galactosidase assay. The CENP-C–DNMT3B interaction was further confirmed by co-immunoprecipitation (co-IP) in human cells and further supported by immunofluorescence microscopy. We then examined the function of this interaction in regulating centromeric DNA methylation, histone marks, transcription and mitotic fidelity. SiRNA knockdown of CENP-C led to a significant loss of DNA methylation, marked changes in the histone code and reduced DNMT3B binding at centromeric and pericentromeric regions. DNMT3B siRNA knockdown also led to reduced CENP-C binding. Finally, knock downs of CENP-C or DNMT3B led to enhanced centromeric transcription and elevated mitotic chromosome instability. Taken together, these data reveal a novel link between DNMT3B and centromere dynamics and suggest that CENP-C and DNMT3B mutually reinforce each other's recruitment and play a significant role in regulating epigenetic marks at the centromere.
RESULTS
Yeast two-hybrid screening reveals that DNMT3B interacts with CENP-C
Although several DNMT3B interacting proteins have been identified by us and others, there is much that we do not understand regarding how DNMT3B is targeted throughout the genome and why its loss leads to chromosomal instability in ICF syndrome (6,17,18). We therefore undertook a yeast two-hybrid screen using full-length Dnmt3b1 as bait and a human testis cDNA library as prey. We have shown previously that DNMT3B is highly expressed in human testis (19) and presumably its interacting partners are also highly expressed there. We established an AH109 yeast strain stably expressing full-length murine Dnmt3b1 fused to the GAL4 DNA binding domain (DBD, data not shown), then used this strain to transform and screen the human testis cDNA library fused to the GAL4 activation domain (AD). Approximately 1 × 107 independent clones from the library were screened under high-stringency conditions (-Ade/His/Leu/Trp plus blue/white screening). Positive clones were restreaked to confirm growth and color phenotype and then episomal DNA was isolated and sequenced. This analysis identified three clones that encoded portions of the constitutive centromere protein CENP-C (Fig. 1, the largest clone encompassed amino acid 200–943 of CENP-C). Human DNMT3B1 also interacted with CENP-C and a nearly full-length CENP-C interacted with Dnmt3b1 (Fig. 1).
Figure 1.

Identification of constitutive centromere protein CENP-C as a DNMT3B interacting protein in a yeast two-hybrid screen. (A) Summary of yeast two-hybrid results. Full-length human or murine DNMT3B1 fused to the GAL4-DBD interacts with the partial CENP-C clone (amino acids 200–943) isolated from the human testis cDNA library. Positive (p53 + TAg and DNMT3B self) and negative (p53 + Lamin and DNMT3B + empty GAL4-AD vector) controls for interaction are also shown. Results are presented as β-galactosidase activity units. (B) Mapping the CENP-C interacting region on DNMT3B. The murine Dnmt3b1 deletion constructs were fused to the GAL4-DBD and co-transfected into yeast strain Y190 with CENP-C (amino acids 23–943) fused to the GAL4-AD. Numbering refers to the amino acids of full-length murine Dnmt3b1. (C) Mapping the DNMT3B interacting region on CENP-C. As in (B), the indicated human CENP-C deletion constructs (fused to the GAL4-AD) were co-transfected into Y190 with full-length human DNMT3B1 (fused to the GAL4-DBD). Boxed regions in (B and C) indicate minimal interaction domains. ‘CBD’-centromere binding domain, roman numerals—conserved catalytic DNMT motifs.
We then set out to map the regions of DNMT3B and CENP-C that mediate the interaction. To accomplish this, the Y190 yeast strain was co-transformed with GAL4-AD–CENP-C and a series of Dnmt3b deletion constructs fused to the GAL4-DBD and interaction assessed using a β-galactosidase assay. The extreme N-terminal region of Dnmt3b from amino acids 1–140 was required for interaction with CENP-C (Fig. 1B). Using a similar strategy with full-length Dnmt3b1 and a series of CENP-C deletion mutants, we identified an interaction with the C-terminus of CENP-C encompassing amino acids 638–943 (Fig. 1C), although this region was unable to reconstitute full interaction strength with Dnmt3b1, suggesting that other regions of CENP-C may also be important (to be addressed later).
DNMT3B interacts with constitutive centromere protein CENP-C and is a component of the mammalian centromere
To further confirm and characterize the interaction between DNMT3B and CENP-C, and demonstrate its relevance in human cells, we performed co-IPs following transient co-transfection of 293T cells with FLAG-tagged DNMT3B1 and HA-tagged CENP-C. FLAG-DNMT3B1 immunoprecipitated HA-CENP-C and vice versa in reciprocal co-IP assays (Fig. 2A, top panels). In addition, antibody against the endogenous protein was also able to reciprocally co-IP the ectopically expressed DNMT3B or CENP-C (Fig. 2A, lower panel). To gain further evidence that this is indeed a physiologically relevant interaction, we prepared nuclear extract from untransfected HeLa cells for co-IP. Using two different antibodies directed against CENP-C and one directed against DNMT3B, we readily detected an interaction between DNMT3B and CENP-C (Fig. 2B). This interaction was detectable in nuclear extracts derived from unsynchronized cells and from HeLa cells synchronized by double thymidine block and harvested at the G1/S boundary (Supplementary Material, Fig. S1) and in M phase (Fig. 2B).
Figure 2.

DNMT3B co-immunoprecipitates (co-IPs) with CENP-C and CENP-A in mammalian cells. (A) Ectopically expressed DNMT3B and CENP-C interact in mammalian cells. The indicated constructs were transiently transfected into 293T cells then whole cell extract was prepared. The immunoprecipitating antibody (IP Ab) is indicated along the top of the western panel and the antibody used in western blotting (WB) is shown below it. Reciprocal co-IPs were performed. (B) Endogenous DNMT3B and CENP-C co-immunoprecipitate from HeLa nuclear extract (M phase). In the left panel, two different CENP-C antibodies (labeled 1 and 2) are used in the co-IP. In the right panel, the positive control of CENP-C interacting with CENP-A is also shown. (C) Confirmation that DNMT3B is a centromere-associated protein as shown by its ability to co-IP with the centromere-specific H3 variant CENP-A. The indicated constructs were co-transfected into 293T cells and used for reciprocal co-IP. Input—whole cell (A and C) or nuclear extract (B) prior to co-IP, IgG—negative control co-IP with species matched normal IgG.
CENP-C is known to reside in a complex with constitutive centromere protein CENP-A in a CENP-C/CENP-B/CENP-A nucleosome complex (20). To gain additional support for our findings that DNMT3B is a centromere-associated protein, we asked whether DNMT3B would co-IP with CENP-A, presumably via its interaction with CENP-C. Indeed, we detected such an interaction in cells transiently co-transfected with FLAG-CENP-A and HA-DNMT3B1 (Fig. 2C). As a control, we also showed that CENP-C co-immunoprecipitated with CENP-A from nuclear extracts (Fig. 2B, right panel). This finding lends additional support for DNMT3B as a centromere-associated protein.
Mapping the region on CENP-C that interacts with DNMT3B in yeast cells showed that amino acids 638–943 were at least partially responsible, but could not reconstitute β-galactosidase activity to the levels of full-length CENP-C. Although this may reflect expression levels of the deletion constructs in yeast, it suggested to us that other regions of CENP-C may play a role. To examine this directly, we co-transfected a series of HA-tagged CENP-C deletion constructs (Fig. 3A), and FLAG-DNMT3B1, into 293T cells and performed co-IPs with FLAG antibody. This analysis confirmed the importance of the C-terminal region of CENP-C in interacting with DNMT3B and allowed this domain to be further refined to include amino acids 638–760 (Fig. 3A and B, region 2). Interestingly, however, the central region of CENP-C (amino acids 426–537) was also capable of interacting with DNMT3B (Fig. 3A and B, region 1). These two regions of CENP-C mediate its recruitment to centromeres (amino acids 426–537) and its ability to bind alpha-satellite DNA (amino acids 638–943) (21). All CENP-C deletion constructs expressed at comparable levels in 293T cells (Fig. 3C). Taken together, these data reveal that DNMT3B interacts with two discrete functional domains of CENP-C and is present at the human centromere.
Figure 3.

DNMT3B interacts with two regions of CENP-C in mammalian cells. (A) Schematic representation of CENP-C with the deletion constructs used in the mapping studies indicated below. Numbering corresponds to amino acids of human CENP-C. Regions 1 and 2 are the minimal DNMT3B interacting regions mapped in (B). (B) Refining the DNMT3B interacting regions on CENP-C by co-immunoprecipitation. The CENP-C constructs indicated along the top of the western panel, fused to the HA tag, were co-transfected with full-length FLAG-tagged DNMT3B1 into 293T cells, whole cell extracts were prepared, and co-IP's were carried out with FLAG-agarose beads. The interaction between CENP-C and DNMT3B is lost when CENP-C regions 426–537 (region 1) and 638–760 (region 2) are deleted. (C) Inputs for the co-IP reactions in (B). The CENP-C constructs (denoted with arrows) were detected in whole cell extract of transfected 293T cells with HA antibody. The near full-length CENP-C (23–943) was run on a separate, lower percentage gel.
A fraction of DNMT3B co-localizes with CENP-C most highly during metaphase
We and others have shown that DNMT3B co-localizes with centromeric and pericentromeric regions and ‘paints’ the mitotic chromosomes using immunofluorescence microscopy (8,18). In order to examine the extent of co-localization of DNMT3B and CENP-C, we transfected HeLa cells with green fluorescent protein (GFP)-tagged DNMT3B1 and stained transfected cells with an antibody against endogenous CENP-C. Interphase cells displayed a wide nuclear distribution of DNMT3B1 with some concentration in DAPI-dense heterochromatic regions, whereas CENP-C was present in a small number of discrete foci (Fig. 4, top panels), which co-localized with anti-centromere antibody staining (data not shown). Closer examination of the DNMT3B staining revealed that there were discrete foci of DNMT3B within the more diffuse nucleoplasmic staining and these DNMT3B foci often co-localized with or were adjacent to CENP-C foci (Fig. 4, top panels). These data are consistent with our ability to co-IP DNMT3B and CENP-C throughout the cell cycle. During the early stage of mitosis (prophase), more DNMT3B foci begin to co-localize with CENP-C. Co-localization peaks during metaphase, where there is clear enrichment of DNMT3B at CENP-C-containing foci (Fig. 4, middle panel). During anaphase less DNMT3B and CENP-C foci co-localize and this declines further during telophase (Fig. 4). Taken together, these results demonstrate that although it has a wide distribution in the nucleus, discrete foci of DNMT3B exist adjacent to or coincident with CENP-C foci and human centromeres.
Figure 4.

A fraction of DNMT3B co-localizes with CENP-C particularly during metaphase. HeLa cells were transfected with GFP-tagged DNMT3B1 (green panels), synchronized with a double thymidine block, released, then fixed when cells were in mitosis. Interphase cells were also examined. Transfected cells were stained with anti-CENP-C (red panels) antibody. DNA was stained with DAPI (blue panels). An overlay of the red and green channels is shown in the right-most panels. Representative images of cells in interphase (top row) or the different phases of mitosis (lower four rows) are shown. Select regions are enlarged in the small red-boxed regions of the overlay panel to highlight closely opposed or overlapping DNMT3B and CENP-C foci. Double red arrowheads indicate closely juxtaposed foci. Scale bar—10 µm.
CENP-C influences centromere-region DNA methylation levels
To test whether CENP-C is involved in regulating DNA methylation at the centromere by recruiting DNMT3B, we transfected human HCT116 colorectal carcinoma cells with siRNA directed against CENP-C. We also separately knocked down DNMT3B expression by siRNA in HCT116 cells as a comparison. The siRNAs efficiently reduced the levels of both CENP-C and DNMT3B mRNA and protein (Supplementary Material, Fig. S2). We then used bisulfite genomic sequencing (BGS) to examine the effect of the knock downs on DNA methylation at the centromeric alpha satellite repeat and the pericentromeric satellite 2 repeat. Interestingly, BGS revealed that HCT116 cells depleted for CENP-C or DNMT3B exhibited nearly 30 and 20% reductions in DNA methylation at the alpha satellite repeat region, respectively (Fig. 5A, top panel and Supplementary Material, Fig. S3). These reductions were highly significant. Seven CpG sites were examined in the alpha satellite PCR BGS amplicon and all were moderately affected by CENP-C knock down except CpG #1, whereas all sites except CpG #4 (which was never methylated in our clones) and #7 were hypomethylated upon DNMT3B siRNA knock down (Fig. 5B). CpG sites 1 and 2 are within the CENP-B box and their DNA methylation status is known to regulate CENP-B binding (22). In comparison, siRNA knock down of CENP-C or DNMT3B both led to an ∼20–25% reduction in pericentromeric satellite 2 repeat methylation (Fig. 5A, bottom panel) and most of the 23 CpGs within the satellite 2 BGS amplicon were affected (Fig. 5C and Supplementary Material, Fig. S4). Taken together, this analysis reveals that CENP-C is at least partially responsible for targeting DNA methylation to the centromeric region. Given that our siRNA knock downs are not 100% efficient, the effect might be expected to be even more dramatic in cells completely deficient for CENP-C, which are not viable (15). Although we expected siRNA knock down of DNMT3B to influence pericentromeric DNA methylation levels, surprisingly, CENP-C also significantly affected DNA methylation within this region, suggesting CENP-C broadly affects epigenetic marks within the centromeric region or, alternatively, at pericentromeric heterochromatin immediately adjacent to centromeric heterochromatin. Given that we are examining repetitive sequences, we cannot precisely determine which repeats within the centromere or pericentromere are most affected.
Figure 5.

CENP-C modulates DNA methylation at centromeric and pericentric regions. (A) Summary of bisulfite genomic sequencing (BGS) results, as the total percent methylation at all CpG sites examined in the alpha satellite (top) or pericentric satellite 2 (bottom) regions in HCT116 cells mock transfected or transfected with siRNA directed against CENP-C or DNMT3B. Statistical significance was determined using the χ2-test. For the alpha satellite region, BGS summary data is derived from 38 mock, 42 CENP-C and 41 DNMT3B siRNA knock down clones. For satellite 2, summary data is derived from 35 mock, 35 CENP-C and 39 DNMT3B siRNA knock down clones. (B) BGS analysis of the seven CpG sites within the alpha satellite region in mock or siRNA targeted HCT116 cells. Sites 1 and 2 are within the CENP-B binding region as indicated on the graph. The total percent methylation at each CpG site is summarized and derived from data in Supplementary Material, Figure S3. (C) BGS analysis of the satellite 2 region containing 23 CpG sites. Data is summarized as in (B) and is derived from data in Supplementary Material, Figure S4.
CENP-C and DNMT3B mutually reinforce each other's binding and exert profound effects on centromere-region histone marks
Given that CENP-C is required for normal levels of DNA methylation in the centromeric region, we next asked what influence CENP-C and DNMT3B exert on the histone code at the centromere. We therefore knocked down CENP-C or DNMT3B in HCT116 cells with siRNA and prepared soluble chromatin by micrococcal nuclease digestion. We then performed ChIP examining levels of histone marks associated with transcriptional activity/permissiveness (di- and trimethylated H3K4 and H3 acetylated at K9 and K18) and transcriptionally repressed regions (di- and trimethylated H3K9 and H3K27), followed by quantitative PCR for alpha satellite or satellite 2. Consistent with other reports, we detected histone marks characteristic of both permissive and repressive chromatin at the centromere, while the pericentric region was enriched primarily for repressive histone marks (Fig. 6) (13,23). Upon siRNA knock down of CENP-C, there was a marked loss of dimethylated H3K4 and acetylated H3 at the alpha satellite repeat. In addition, alpha satellite chromatin gained di- and trimethylated H3K9 and dimethylated H3K27 (Fig. 6A, left panel). SiRNA knock down of DNMT3B, like CENP-C, resulted in increased di- and trimethylated H3K9 and trimethylated H3K4, but in contrast to CENP-C knock down, resulted in increased H3 acetylation and H3K4 dimethylation (Fig. 6A). The histone code changes we observed were not the result of alterations in total levels of histone H3 or CENP-A occupancy, as these remained constant in all knock downs (Fig. 6A). These results therefore demonstrate that CENP-C and DNMT3B regulate aspects of the histone code at the centromere and suggest that loss of either one may permit the encroachment of pericentromeric histone marks into the centromere.
Figure 6.

CENP-C and DNMT3B modulate epigenetic marks at the centromeric and pericentromeric regions. Chromatin immunoprecipitation (ChIP) and quantitative PCR were used to evaluate the effect of CENP-C (light gray bars) or DNMT3B (dark gray bars) siRNA knock down in HCT116 cells, relative to a mock transfection (black bars). (A) ChIP for the indicated histone marks, HP1α and histone H3/CENP-A (left graph) or DNMT3B and CENP-C (right panel) followed by PCR for alpha satellite DNA. (B) ChIP with the same panel of antibodies as in (A) followed PCR for satellite 2 sequences. Results are presented as the average fold-enrichment relative to the input (1% of the supernatant from the IgG ChIP reaction). All reactions were repeated at least in triplicate from two independent siRNA knock downs. The error bar denotes the standard deviation from the mean. A similar analysis of control single copy genes (WIF1 and GAPDH) is shown in Supplementary Material, Figure S6. The scale in the right panels was expanded as the ChIP signal for CENP-C and DNMT3B was generally lower than that of the histone marks. IgG—negative control ChIP with normal rabbit IgG to determine background binding, 2XMe, 3XMe—di- and trimethylated forms, Ac—acetylation.
We extended this analysis to the pericentromeric satellite 2 region. Upon CENP-C knock down, there was an increase in dimethylated H3K9 and loss of trimethylated H3K9 and K27 and acetylated H3 at satellite 2 repeats (Fig. 6B). Unlike the centromeric region, CENP-C knock down did not alter the relatively low level of H3K4 methylation we detected at satellite 2 repeats. When we knocked down DNMT3B, levels of trimethylated H3K9 and H3K27 decreased and levels of di- and trimethylated H3K4, H3 acetylation and dimethylated H3K27 increased at satellite 2 repeats (Fig. 6B, left panel). There was no change in the total level of histone H3 or CENP-A, and CENP-A enrichment was much lower at the pericentric compared with the centromeric region, consistent with what is known about CENP-A's localization (Fig. 6B). DNMT3B therefore appears to help maintain primarily repressive histone marks in pericentromeric heterochromatin, since its loss resulted in an increase in H3K4 methylation and the ‘intermediate’ marks H3K9/K27 dimethylation. DNMT3B and CENP-C may have a role in regulating the transition between centromeric and pericentromeric chromatin, since both proteins influenced H3K9 dimethylation, a mark proposed to demarcate this boundary (13).
Lastly, we performed ChIP to assay binding of HP1α, DNMT3B and CENP-C. HP1α binds to and interacts with components of both centromeric and pericentromeric chromatin and is essential for cohesin recruitment and mitotic chromosome segregation (24,25). SiRNA knock down of both CENP-C and DNMT3B led to a precipitous reduction in HP1α binding (Fig. 6A–B). Significantly, upon knock down of CENP-C there was marked loss of DNMT3B binding at both the alpha satellite and the satellite 2 regions (Fig. 6A–B, right panels). CENP-C binding was also reduced consistent with the siRNA knock down efficiency we observed (Supplementary Material, Fig. S2). Knock down of DNMT3B in HCT116 cells also resulted in a small but reproducible decrease in CENP-C binding. Given that the siRNA knock down was not 100% efficient, we confirmed this result using HCT116 cells engineered to contain a genetic knockout of DNMT3B (3BKO). ChIP from parental versus 3BKO HCT116 cells revealed a consistent 2–3-fold decrease in the binding of CENP-C at both regions analyzed (Supplementary Material, Fig. S5). These results therefore suggest that CENP-C and DNMT3B, or the epigenetic marks they regulate, are involved in mutually reinforcing each other's binding to the centromeric region. Finally, we show that the effect of CENP-C knock down on the histone code and factor binding was restricted to the centromere because none of the examined modifications were altered at a transcriptionally repressed tumor suppressor gene (WIF1) (26) or a constitutively active housekeeping gene (GAPDH), and CENP-C was not enriched above the IgG background at either locus (Supplementary Material, Fig. S6).
Functional consequences of disrupting the CENP-C–DNMT3B interaction on centromere function
Links between DNMT3B and CENP-C have separately and in other systems been linked to chromosomal stability and proper mitotic progression (16,27). In order to directly assess the function of the CENP-C–DNMT3B interaction in relation to chromosomal stability using our systems, we used siRNA to knock down CENP-C or DNMT3B in HCT116 cells (which have a relatively normal karyotype) and then monitor for chromosomal alignment or segregation defects during mitosis. Following the siRNA transfection, cells were synchronized with a double thymidine block, released and then fixed 8–9 h later (when cells entered M phase). Cells were then stained with Hoescht 33352 (for DNA), anti-centromere antibody and anti-tubulin antibody (mitotic spindle) and then visualized using immunofluorescence microscopy. The major mitotic defects we noted in knock down cells were chromosomes that had not properly aligned at the metaphase plate (misaligned chromosomes) and cells in anaphase with lagging chromosomes or bridges (anaphase bridges). Representative examples of these defects are shown in Figure 7A–B and were rare in mock transfected cells. Quantitation revealed that both misaligned chromosomes and anaphase bridges were significantly elevated in the DNMT3B and CENP-C knock down cells relative to the mock (Fig. 7C–D). Anaphase bridges were somewhat more common in CENP-C knock down cells, whereas misaligned chromosomes were more frequent in DNMT3B knock down cells. Thus, CENP-C and DNMT3B both contribute to proper mitotic progression and chromosomal segregation.
Figure 7.

Disrupting the CENP-C–DNMT3B interaction results in enhanced mitotic defects. HCT116 cells were transfected with siRNA against CENP-C or DNMT3B or were mock transfected. Following transfection, cells were synchronized, released and then fixed during M phase. Cells were then stained for DNA (blue panels), the centromere (anti-centromere antibody, green) and the mitotic spindle with anti-tubulin antibody (red). Mock transfected cells showed few mitotic defects, whereas cells with reduced levels of CENP-C or DNMT3B demonstrated elevated mitotic defects (misaligned chromosomes and anaphase bridges). (A and B) Representative images of cells undergoing normal or defective mitoses. (C and D) Quantification of mitotic defects in mock or siRNA knock down cells. At least 50 mitotic cells were counted from three independent siRNA knock downs. Statistical significance was evaluated using the Students t-test. Scale bar—10 µm.
Results from other laboratories have shown that centromeric and pericentric repeats are transcribed in both murine and human cells in a cell cycle- or stress-dependent manner (24). Maintenance of centromeric transcripts at proper levels is important for mitosis and chromosomal segregation; with forced over-expression leading to defects in segregation and altered centromeric epigenetic marks (24,28,29). Given the known role for DNA methylation and the histone code in regulating transcription, and the elevated mitotic defects we observed upon CENP-C and DNMT3B knock down, we asked whether centromeric transcription was altered in our knock down cells. We therefore isolated RNA from CENP-C and DNMT3B siRNA transfected HCT116 cells for quantitative or semi-quantitative reverse transcriptase (RT)–PCR reactions to detect levels of alpha satellite and satellite 2 transcripts, respectively. We were unable to develop quantitative RT–PCR primers for the satellite 2 repeats. Levels of alpha satellite transcripts were markedly elevated in both knock downs relative to the mock transfection control (Fig. 8A). Following siRNA knock down of CENP-C or DNMT3B, levels of satellite 2-derived transcripts were also elevated, with the CENP-C knock down having a greater effect than DNMT3B knock down (Fig. 8B). We also confirmed the elevated centromeric and pericentromeric transcription in HCT116 3BKO cells, although the effect was less dramatic possibly due to the acquisition of compensatory epigenetic marks during long-term selection of the knockout lines (Fig. 8A–B). Taken together, these data demonstrate that loss of CENP-C results in DNA hypomethylation, altered epigenetic marks and elevated centromeric transcription in human cells. Increased transcript levels from these regions may, in turn, contribute to the enhanced genomic instability we observed.
Figure 8.

Loss of CENP-C and DNMT3B-mediated epigenetic marks at the centromere region result in elevated levels of repeat transcription. (A) Quantitative RT–PCR analysis of alpha satellite repeat transcripts in HCT116 cells either mock transfected or transfected with siRNA targeting CENP-C or DNMT3B. HCT116 cells with a genetic knockout of DNMT3B (3BKO) are also shown. Values are the average of triplicate RT–PCR reactions relative to GAPDH as a loading control. The error bar is the standard deviation from the mean. (B) Semi-quantitative RT–PCR analysis of satellite 2 repeat transcription from the same knock down/knockout panel as in (A). RT–PCR reactions were repeated three times and a representative ethidium bromide stained agarose gel photo is shown. Bands were quantified using BioRad Quantity One software and set relative to an independent amplification for GAPDH (lower panel). Although samples were DNase treated, we included a no RT (-RT) control to ensure that we were not amplifying contaminating satellite DNA in the RNA preparation.
DISCUSSION
In the present study, we identified a novel interaction between de novo methyltransferase DNMT3B and constitutive centromere protein CENP-C using a yeast two-hybrid screen. We confirmed this interaction in multiple ways in mammalian cells and mapped the domains on each protein responsible for the interaction. Using siRNA-mediated knock downs of DNMT3B and CENP-C, we showed that CENP-C recruited DNA methylation and DNMT3B to the centromere and, to a lesser extent, the pericentromeric region of human chromosomes. In addition, both proteins played an important role in regulating the histone code and DNA methylation at the centromere, with knock down of either one leading to alterations in marks which typify this region. Loss of either CENP-C or DNMT3B led to enhanced chromosome alignment and segregation defects during mitosis. This effect may be mediated, at least in part, by elevated centromeric transcription in knock down cells, which is known to regulate mitotic chromosome dynamics (28). These studies therefore reveal a novel mechanism for recruiting DNMT3B and DNA methylation to a discrete region of the genome—the centromere—and they demonstrate new roles for DNMT3B and CENP-C in regulating mitotic fidelity and centromere-region epigenetic marks, further underscoring the link between DNA methylation and the histone code. Our results also suggest that aberrations in DNMT3B in cancer cells, such as altered expression, alternative splicing or DNMT3B mutations in ICF syndrome cells, may contribute to genomic instability by reducing CENP-C function and/or recruitment.
Little is known about how DNA methylation is targeted throughout the genome and how it interfaces with the histone code. Interactions between DNMT3B and the H3K9 trimethylating enzyme SUV39H1, for example, play a role in targeting DNA methylation to the murine pericentromeric region. Interestingly, however, methylation targeting to the centromere was independent of SUV39HI (17). Chen et al. (30) showed that the PWWP domain, within the N-terminal regulatory region, mediates DNMT3B's recruitment to pericentromeric regions. Deletion of the PWWP domain resulted in loss of DNA methylation at murine major satellite repeats. Re-expression of WT Dnmt3b in highly demethylated Dnmt3a/Dnmt3b double knockout ES cells resulted in remethylation of major and minor satellite repeats, demonstrating that both regions are bona fide Dnmt3b targets, consistent with our data. Interestingly, however, re-expression of PWWP-deleted Dnmt3b in double knockout ES cells destroyed its ability to remethylate the major, but not the minor satellite repeat showing that distinct regions of Dnmt3b are responsible for targeting it to centromeric and pericentric regions (30). The region of interaction between DNMT3B and CENP-C that we mapped is N-terminal to the PWWP domain (amino acids 1–140 for CENP-C interaction) and suggests that DNMT3B's interaction with CENP-C independently targets it to the centromere. This interaction could also account for the SUV39H1-independent mechanism of DNA methylation targeting to the centromere reported by Lehnertz et al. (17). Cancer cells are known to not only express elevated levels of DNMT3B, but also alternatively spliced DNMT3B isoforms (19,31,32). Some of these transcripts are predicted to lack part, or all, of the CENP-C interaction domain. On the basis of our results, expression of these variants may uncouple the DNMT3B–CENP-C interaction and lead to altered epigenetic marks within the centromeric region and reduced CENP-C binding. This, in turn, may enhance genomic instability, a hallmark of cancer cells. It will be of interest to test such ideas in future studies.
Our findings presented here tie in well with a recent paper showing that CENP-B regulates DNA methylation and the histone code at centromeric regions (33). CENP-B resides in a chromatin complex with CENP-A and CENP-C (20) and binds alpha-satellite DNA in a sequence-specific manner at the CENP-B box. Okada et al. (33) demonstrated that CENP-B promotes de novo DNA methylation and assembly of repressive H3K9 trimethylation-containing chromatin, mediated by SUV39H1, on alpha satellite DNA when integrated into a chromosome with an already functional centromere, but promotes de novo centromere formation of extrachromosomal alphoid DNA arrays. Since binding of CENP-B itself is methylation sensitive, it appears to promote DNA methylation at adjacent CpG sites. CENP-B's interaction with CENP-C may therefore provide a means to recruit DNA methylation to centromeric sites, although CENP-C is also capable of binding to centromeres independent of CENP-B (16). A direct interaction between DNA methyltransferases and CENP-B was not demonstrated in the prior study (33). When we knocked down CENP-C and DNMT3B in HCT116 cells, the alpha satellite and satellite 2 regions lost ∼20–30% of their DNA methylation. Losses of this magnitude are comparable to those reported in a recent bisulfite-based analysis of satellite 2 hypomethylation in glioma (34). Although we are not aware of other studies examining alpha satellite methylation in cancers using bisulfite sequencing, this region is well known to become hypomethylated in tumors (35). These studies, coupled with our data, suggest that disruption of the CENP-C–DNMT3B interaction could contribute to hypomethylation and genomic instability, predisposing to cancer.
Given the tight relationship between CENP-C and centromere function, we were somewhat surprised to find that CENP-C bound to the pericentromeric satellite 2 repeat by ChIP. This may be due to CENP-C complexes binding at interface regions between alpha satellite and satellite 2 chromatin as the exact size of these domains has not been defined and they may be rather fluid since they are epigenetically determined. Interestingly, loss of CENP-C or DNMT3B led to elevated H3K9 dimethylation at the alpha satellite, a mark thought to reside at this boundary region (13). Knock down of CENP-C and DNMT3B led to an increase in the repressive pericentromeric heterochromatin mark H3K9 trimethylation at the centromere and a reduction in this mark in pericentric regions, suggesting that one function of the DNMT3B–CENP-C interaction is to regulate the size of each region. That this boundary is of functional significance is underscored by a recent report showing that when a potent transcriptional repressor is targeted to a human artificial centromere, it resulted in marked centromere dysfunction and loss of CENP-A and H3K4 dimethylation, likely through nucleating the formation of more classic heterochromatin (36).
HP1 binding to trimethylated H3K9 at pericentromeric heterochromatin is required for proper chromosome cohesion and segregation. Transcription of pericentromeric heterochromatin is upregulated during mitosis then abruptly repressed at the metaphase to anaphase transition. Condensin, which interacts with DNMT3B (18), binds to chromatin during mitosis and represses pericentromeric transcription during M phase in yeast (37). In mammals, DNMT3B may be independently recruited to both centromeric (via CENP-C) and pericentromeric regions (possibly via condensin) to repress satellite repeat transcription by DNA methylation and/or recruitment of repressive histone marks. HP1, which is displaced from chromosomes at the beginning of mitosis, rebinds at the metaphase to anaphase transition, which also coincides with elevated condensin binding and repression of pericentromeric transcription (38–40). Interestingly, in yeast transcription of pericentromeric heterochromatin and the subsequent incorporation of these transcripts into the RNAi machinery is required for initial establishment of CENH3 (homologous to mammalian CENP-A) at centromeres (41), suggesting that heterochromatin plays an essential role in centromere formation and/or maintenance. In addition, neocentromeres, which form at euchromatic sites but are functionally similar to normal human centromeres and bind CENP-A and CENP-C, have significantly elevated levels of DNA methylation compared with the same region on the chromosome with the centromere at its normal position. Consistent with this, inhibition of DNA methylation with 5-azadC treatment led to neocentromere hypomethylation and elevated mitotic instability (42). It remains unknown to what extent pericentromeric heterochromatin contributes to CENP-A recruitment in mammals, however, given the importance of DNA methylation to mammalian cells, it is not unreasonable to hypothesize that the recruitment of DNMT3B, along with a myriad of other epigenetic modifiers, has been co-opted to help recruit CENP-A to centromeric regions.
Several other connections between DNA methylation, centromere function and condensin are worthy of note. In addition to repressive histone modifications, pericentric regions are marked by H3S10 phosphorylation during G2 mediated by aurora B kinase. Aurora B is recruited preferentially to large pericentromeric domains (e.g. chromosomes 1, 9 and 16), which, as we have shown here, are targets of DNMT3B and are preferentially susceptible to mitotic defects in ICF syndrome cells (6). Interestingly, aurora B co-localizes with densely staining 5-methylcytosine foci during mitosis. Disruption of DNA methylation by 5-azadC treatment inhibits aurora B recruitment to pericentromeric regions leading to loss of H3S10 phosphorylation (43). Aurora B is also a critical player in recruiting other chromatin complexes essential for mitotic chromosome segregation. For example, phosphorylation of CENP-A by aurora A and B kinases is essential for kinetochore function (44). Aurora B also regulates the association of condensin I and condensin II with mitotic chromosomes. Inhibition of aurora B, in turn, leads to reduced condensin recruitment and subsequent defects in mitosis (45,46). Although we do not know, at present, the sequence of events leading to recruitment of these various factors or if DNMT3B interfaces with aurora B directly, it is tempting to speculate that DNMT3B itself bound at the centromere, or DNA methylation of alpha satellite and satellite 2 repeats mediated by DNMT3B, is involved in recruiting other factors such as aurora B, condensin or HP1 proteins. In support of this notion, the localization of all HP1 isoforms is markedly altered in ICF syndrome cells (47). Alternatively, DNMT3B may help stabilize the binding of these proteins at the centromere once recruited by other factors. It will be of significant interest to test these models as part of future studies as they have important implications for defining the mechanisms responsible for the chromosomal instability thought to underlie many cancers.
MATERIALS AND METHODS
Yeast two-hybrid screening
The Matchmaker Two-Hybrid System 3 (Clontech) was used to screen a human adult testis cDNA library (Clontech) with full-length Dnmt3b1 as bait according to the manufacturer's instructions. Expression of the Dnmt3b-bait fusion protein in yeast was confirmed by western blotting with anti-GAL4-DBD antibody (Santa Cruz Biotechnology, data not shown). The library screen was performed using yeast strain AH109 stably expressing full-length Dnmt3b1 under high-stringency conditions (Ade−, His−, Leu−, Trp− media) using sequential transformation. Approximately 1 × 107 clones were screened and positives from the primary screen were re-streaked on the same media and confirmed with a liquid β-galactosidase assay. Plasmid DNA from positive colonies was isolated, transformed into E. coli, and sequenced to identify clones. Plasmids for the bait and the library clone identified by sequencing were co-transformed into yeast strain Y190 (using Trp−, Leu− media), and the interaction was verified with a β-galactosidase assay. The β-galactosidase activity was calculated using the formula: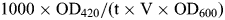 , where t =incubation time, V = 0.1 ml × dilution factor and OD600 = A600 of 1 ml of culture and is represented in Miller units where 1 U of β-galactosidase is defined as the amount which hydrolyzes 1 µmol of o-nitrophenyl-β-galactoside to o-nitrophenol and d-galactose per minute per cell.
, where t =incubation time, V = 0.1 ml × dilution factor and OD600 = A600 of 1 ml of culture and is represented in Miller units where 1 U of β-galactosidase is defined as the amount which hydrolyzes 1 µmol of o-nitrophenyl-β-galactoside to o-nitrophenol and d-galactose per minute per cell.
Plasmids
Creation of the full-length Dnmt3b1 GAL4-DBD fusion protein for yeast expression was described previously (48). The full-length DNMT3B1 cDNA was cloned into the EcoRI and XhoI sites of pCMV-Tag2A (Stratagene) and pEGFP-C2 mammalian expression vectors (Clontech) to create FLAG- and GFP-tagged fusion constructs, respectively. Creation of the HA-tagged full-length and deletion mutant CENP-C constructs are described by Trazzi et al. (21). HA-Dnmt3a and HA-DNMT1 plasmids were described previously (48,49).
Cell lines
HeLa and parental HCT116 cells were purchased from the American Type Culture Collection. Isogenic HCT116 cells with a knockout of the DNMT3B gene (3BKO) were provided by Dr Bert Vogelstein (Johns Hopkins University). Cells were cultured in McCoy's-5A medium supplemented with 10% heat-inactivated fetal bovine serum and 2 mm l-glutamine (Invitrogen).
ChIP and qPCR
ChIP was performed as described by Lefevre and Bonifer (50). Briefly, formaldehyde cross-linked cells were lysed in buffer containing 10 mm Tris–HCl, pH 7.4, 10 mm NaCl, 5 mm MgCl2 and 0.2% NP-40 for 1 h at 4°C. The supernatant was discarded and the nuclei were resuspended in glycerol buffer (10 mm Tris–HCl, pH 7.4, 0.1 mm EDTA, 5 mm MgAc2 and 25% glycerol). To generate nucleosomal material, nuclei were resuspended in an equal volume of 2× MNase buffer containing 50 mm KCl, 8 mm MgCl2, 2 mm CaCl2, 100 mm Tris–HCl, pH 7.4 and treated with 100 U/ml for micrococcal nuclease for 15 min at 37°C. The reaction was stopped by adding EDTA to a final concentration of 10 mm. The digested nuclei were resuspended in immunoprecipitation buffer containing 25 mm Tris–HCl, pH 8.0, 2 mm EDTA, 150 mm NaCl, 1% Triton X-100 and 0.1% SDS. The sample was cleared of debris by centrifugation at 1500 g for 5 min and the supernatant was used as the chromatin template for ChIP–PCR reactions. After MNase treatment, the ChIP experiment was performed similarly to conventional ChIP (51). Purified, immunoprecipitated DNA was analyzed by quantitative PCR. The change in binding was represented as fold-enrichment compared with the input signals where the supernatant from the irrelevant antibody served as a positive control (‘input’, 1% of the ChIP material). Antibodies used in ChIP experiments are listed in Supplementary Material, Table S1.
Quantitative PCR was performed to evaluate the binding of DNMT3B and CENP-C as well as the level of histone modifications at the alpha satellite and satellite 2 repeat regions. The human WNT inhibitory factor-1 (WIF1) and human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene promoter regions were used as inactive (26) and active single copy gene comparisons, respectively. The enrichment was calculated with respect to the input DNA (2(Ct(Input)−Ct(sample)). Primers used for these regions are listed in Supplementary Material, Table S2.
Immunofluorescence
HeLa cells were grown on 22 mm2 glass coverslips in six-well plates. Cells were transfected with GFP-DNMT3B expression constructs using TransIT-LT1 transfection reagent according to the manufacturer's protocol (Mirus). The cells were fixed with 2% paraformaldehyde in 1× PBS, pH 7.0 48 h post-transfection. Cells were permeabilized with 0.5% Triton X-100 and incubated with anti-CENP-C antibody (Abcam) diluted 1:50 in 1× PBS with 0.1% Tween-20 for 1 h at room temperature. The cells were washed three times with PBST and subsequently incubated with anti-mouse-TRITC secondary antibody. Cells were washed and stained with Hoechst 33342 for DNA before being mounted onto a glass slide using fluoromount G (Southern Biotech). For mitosis studies, HeLa cells were synchronized as described previously (18). Briefly, HeLa cells were transfected (as described above) for 24 h, after which they were subjected to a single thymidine block (2.5 mm thymidine) for 17 h and subsequently washed and allowed to recover in complete medium. After 8–9 h, cells were fixed with 2% paraformaldehyde and subjected to immunostaining. The single thymidine block resulted in an enrichment of mitotic cells (∼40% of the cells plated were in M-phase). Images were captured using a Nikon TE-2000 inverted microscope and deconvolved using Nikon Elements advanced software.
Mitotic defect analysis
Immunofluorescence analysis of HCT116 cells for the presence of mitotic defects was performed after transfecting cells with CENP-C or DNMT3B siRNA in comparison to the control (mock) treatment. Cells were stained with antibodies to the centromere (anti-centromere antibody—Antibodies, Inc.) and α-tubulin (Calbiochem) and counterstained with Hoechst 33352 for DNA. Mitotic cells with misaligned centromeres or chromosome bridges from three independent HCT116 CENP-C, DNMT3B or mock transfections were counted and the statistical significance was evaluated using the Students t-test.
Immunoprecipitation and western blotting
Whole cell extracts from transfected HCT116 cells for immunoprecipitation assays were prepared by lysing cells in 250 µl of lysis buffer containing 50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% NP-40, 10% glycerol and 1.0 µg/ml of protease inhibitors (aprotinin, leupeptin and pepstatin A). Lysates were clarified by centrifugation and the supernatant was subject to immunoprecipitation. For immunoprecipitations, 250 µl of whole cell extract was diluted to 800 µl in lysis buffer then pre-cleared with 20 µl of protein A/G agarose beads (Santa Cruz Biotechnology) for 1 h at 4°C. Samples were centrifuged and the supernatant was incubated with 10 µl of anti-FLAG M2 affinity agarose gel (Sigma) or anti-HA agarose beads (Sigma) overnight at 4°C. Samples were washed four times with wash buffer containing 20 mm Tris–HCl pH 7.4, 150 mm NaCl, 0.1% Tween, 0.2 mm EDTA and 10% glycerol. Proteins bound to the beads were denatured with sample buffer and analyzed by western blotting following SDS–PAGE and electrophoretic transfer to PVDF membrane. Immunoprecipitation of endogenous proteins was performed using nuclear extracts from HeLa cells in G1/S or M phase by subjecting them to a double thymidine block. Briefly, cells were trypsinized and freshly plated in medium containing 2.5 mm thymidine for 18 h. Cells were washed and recovered for 8–9 h in complete medium and again treated with 2.5 mm thymidine for 17 h. The cells were harvested (G1) or washed and recovered in complete medium for 8–9 h and harvested for nuclear extract (M phase). The nuclear extracts were prepared as described by Tsai and Carstens (52) and were subject to immunoprecipitation as described previously (49) using antibodies against endogenous CENP-C or DNMT3B. Antibodies are listed in Supplementary Material, Table S1.
For testing siRNA knock down efficiency, whole cell extract from transfected cells was resolved on 10% SDS–PAGE gels and transferred overnight onto PVDF membrane. The membrane was blocked in 5% milk in TBST. Expression of the relevant proteins after 72 h of siRNA KD was determined by probing for DNMT3B and CENP-C using the antibodies listed in Supplementary Material, Table S1. GAPDH antibody was used to determine the total protein loading for these samples.
siRNA knock downs
Endogenous DNMT3B and CENP-C were knocked down using the pre-designed siRNA Smart Pool from Dharmacon. The siRNA transfection was performed twice over a period of 72 h with Dharmafect transfection reagent according to the manufacturer's instructions. The siRNA treated cells were processed for DNA, RNA, protein or chromatin 72 h post-transfection.
Quantitative reverse transcriptase–PCR
Quantitative PCR was performed as described previously (49). Total mRNA expression was analyzed by qRT–PCR for human DNMT3B and CENP-C. GAPDH expression was measured as an internal control. The mRNA expression was normalized to GAPDH expression levels using the (2(Ct(GAPDH)−Ct(DNMT3B)) for DNMT3B expression and (2(Ct(GAPDH)−Ct(CENPC1)) for CENP-C. Sequences of primers used for the qRT–PCR are listed in Supplementary Material, Table S2.
Satellite repeat transcription analysis
Total RNA was prepared from cells transfected with CENP-C or DNMT3B siRNAs as well as a control (mock) sample using the RNAeasy kit with DNase treatment according to the manufacturer's instructions (Qiagen). Transcript expression was analyzed by qRT–PCR with human alpha satellite-specific primers (Supplementary Material, Table S2). Human GAPDH expression was measured as an internal control. The transcript expression was normalized to the GAPDH expression levels using the formula (2(Ct(GAPDH)−Ct(satellite alpha)). Satellite 2 transcript analysis was performed using semi-quantitative RT–PCR with primers 5′-TTT CCG TTT GGT GTT GAT AC-3′ and 5′-ACA GAA TTG AAT GGA ATG GTC-3′. The quantification of SAT2 expression was performed using BioRad Quantity One Image analysis software and normalized to GAPDH expression.
Bisulfite genomic sequencing
Genomic DNA extracted from cell lines was bisulfite treated as described previously (51). Bisulfite treated DNA was used as a template to amplify satellite regions of interest using primers listed in Table S2. The PCR product amplified from bisulfite treated DNA was gel purified using the Qiaex II gel extraction kit (Qiagen) and TOPO cloned using the TA Cloning Kit (Invitrogen). Cloned products were sequenced in a 96-well plate format using the M13 reverse primer at the University of Florida Interdisciplinary Center for Biotechnology Research and methylation levels analyzed. Statistical significance was determined using the χ2-test.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by National Institutes of Health grant R01CA114229 (KDR).
REFERENCES
- 1.Goll M.G., Bestor T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 2.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nature Rev. Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 3.Robertson K.D. DNA methylation and human disease. Nature Rev. Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 4.Jones P.A., Baylin S.B. The fundamental role of epigenetic events in cancer. Nature Rev. Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 5.Berger S.L. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 6.Ehrlich M., Jackson K., Weemaes C. Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF) Orphanet J. Rare Dis. 2006;1:2. doi: 10.1186/1750-1172-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miniou P., Jeanpierre M., Bourc'his D., Barbosa A.C.C., Blanquet V., Viegas-Pequignot E. α-Satellite DNA methylation in normal individuals and in ICF patients: heterogeneous methylation of constitutive heterochromatin in adult and fetal tissues. Hum. Genet. 1997;99:738–745. doi: 10.1007/s004390050441. [DOI] [PubMed] [Google Scholar]
- 8.Craig J.M., Earle E., Canham P., Wong L.H., Anderson M., Choo K.H.A. Analysis of mammalian proteins involved in chromatin modification reveals new metaphase centromeric proteins and distinct chromosomal distribution patterns. Hum. Mol. Genet. 2003;12:3109–3121. doi: 10.1093/hmg/ddg330. [DOI] [PubMed] [Google Scholar]
- 9.Jaco I., Canela A., Vera E., Blasco M.A. Centromere mitotic recombination in mammalian cells. J. Cell Biol. 2008;181:885–892. doi: 10.1083/jcb.200803042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choo K.H.A. Centromere DNA dynamics: latent centromeres and neocentromere formation. Am. J. Hum. Genet. 1997;61:1225–1233. doi: 10.1086/301657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schueler M.G., Sullivan B.A. Structural and functional dynamics of human centromeric chromatin. Annu. Rev. Genomics Hum. Genet. 2006;7:301–313. doi: 10.1146/annurev.genom.7.080505.115613. [DOI] [PubMed] [Google Scholar]
- 12.Sullivan B.A., Karpen G.H. Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nature Struct. Mol. Biol. 2004;11:1076–1083. doi: 10.1038/nsmb845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lam A.L., Boivin C.D., Bonney C.F., Rudd M.K., Sullivan B.A. Human centromeric chromatin is a dynamic chromosomal domain that can spread over noncentromeric DNA. Proc. Natl Acad. Sci. USA. 2006;103:4186–4191. doi: 10.1073/pnas.0507947103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Politi V., Perini G., Trazzi S., Pliss A., Raska I., Earnshaw W.C., Della Valle G. CENP-C binds alpha-satellite DNA in vivo at specific centromere domains. J. Cell Sci. 2002;115:2317–2327. doi: 10.1242/jcs.115.11.2317. [DOI] [PubMed] [Google Scholar]
- 15.Kalitsis P., Fowler K.J., Earle E., Hill J., Choo K.H.A. Targeted disruption of mouse centromere protein C gene leads to mitotic disarray and early embryo death. Proc. Natl Acad. Sci. USA. 1998;95:1136–1141. doi: 10.1073/pnas.95.3.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon M.-S., Hori T., Okada M., Fukagawa T. CENP-C is involved in chromosome segregation, mitotic checkpoint function, and kinetochore assembly. Mol. Biol. Cell. 2007;18:2155–2168. doi: 10.1091/mbc.E07-01-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehnertz B., Ueda Y., Derijck A.A.H.A., Braunschweig U., Perez-Burgos L., Kubicek S., Chen T., Li E., Jenuwein T., Peters A.H.F.M. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003;13:1192–1200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 18.Geiman T.M., Sankpal U.T., Robertson A.K., Chen Y., Mazumdar M., Heale J.T., Schmiesing J.A., Kim W., Yokomori K., Zhao Y., et al. Isolation and characterization of a novel DNA methyltransferase complex linking DNMT3B with components of the mitotic chromosome condensation machinery. Nucleic Acids Res. 2004;32:2716–2729. doi: 10.1093/nar/gkh589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robertson K.D., Uzvolgyi E., Liang G., Talmadge C., Sumegi J., Gonzales F.A., Jones P.A. The human DNA methyltransferases (DNMTs) 1, 3a, and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291–2298. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ando A., Yang H., Nozaki N., Okazaki T., Yoda K. CENP-A, -B, and -C chromatin complex that contains the I-type α-satellite array constitutes the prekinetochore in HeLa cells. Mol. Cell. Biol. 2002;22:2229–2241. doi: 10.1128/MCB.22.7.2229-2241.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trazzi S., Bernardoni R., Diolaiti D., Politi V., Earnshaw W.C., Perini G., Della Valle G. In vivo functional dissection of human inner kinetochore protein CENP-C. J. Struct. Biol. 2002;140:39–48. doi: 10.1016/s1047-8477(02)00506-3. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka Y., Kurumizaka H., Yokoyama S. CpG methylation of the CENP-B box reduces human CENP-B binding. FEBS J. 2005;272:282–289. doi: 10.1111/j.1432-1033.2004.04406.x. [DOI] [PubMed] [Google Scholar]
- 23.Peters A.H.F.M., Kubicek S., Mechtler K., O'Sullivan R.J., Derijck A.A.H.A., Perez-Burgos L., Kohlmaier A., Opravil S., Tachibana M., Shinkai Y., et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell. 2003;12:1577–1589. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- 24.Lu J., Gilbert D.M. Cell cycle regulated transcription of heterochromatin in mammals vs. fission yeast: functional conservation or coincidence? Cell Cycle. 2008;7:1907–1910. doi: 10.4161/cc.7.13.6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Obuse C., Iwasaki O., Kiyomitsu T., Goshima G., Toyoda Y., Yanagida M. A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nature Cell Biol. 2004;6:1135–1141. doi: 10.1038/ncb1187. [DOI] [PubMed] [Google Scholar]
- 26.Ai L., Tao Q., Zhong S., Fields C.R., Kim W.-J., Lee M.W., Cui Y., Brown K.D., Robertson K.D. Inactivation of Wnt inhibitory factor-1 (WIF1) expression by epigenetic silencing is a common event in breast cancer. Carcinogenesis. 2006;27:1341–1348. doi: 10.1093/carcin/bgi379. [DOI] [PubMed] [Google Scholar]
- 27.Dodge J.E., Okano M., Dick F., Tsujimoto N., Chen T., Wang S., Ueda Y., Dyson N., Li E. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 2005;280:17986–17991. doi: 10.1074/jbc.M413246200. [DOI] [PubMed] [Google Scholar]
- 28.Bouzinba-Segard H., Guais A., Francastel C. Accumulation of small murine minor satellite transcripts leads to impaired centromeric architecture and function. Proc. Natl Acad. Sci. USA. 2006;103:8709–8714. doi: 10.1073/pnas.0508006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frescas D., Guardavaccaro D., Kuchay S.M., Kato H., Poleshko A., Basrur V., Elenitoba-Johnson K.S., Katz R.A., Pagano M. KDM2A represses transcription of centromeric satellite repeats and maintains the heterochromatic state. Cell Cycle. 2008;7:3539–3547. doi: 10.4161/cc.7.22.7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen T., Tsujimoto N., Li E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell. Biol. 2004;24:9048–9058. doi: 10.1128/MCB.24.20.9048-9058.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L., Wang J., Sun S., Rodriguez M., Yue P., Jang S.J., Mao L. A novel DNMT3B subfamily, ΔDNMT3B, is the predominant form of DNMT3B in non-small cell lung cancer. Int. J. Oncol. 2006;29:201–207. [PubMed] [Google Scholar]
- 32.Ostler K.R., Davis E.M., Payne S.L., Gosalia B.B., Exposito-Cespedes J., Le Beau M.M., Godley L.A. Cancer cells express aberrant DNMT3B transcripts encoding truncated proteins. Oncogene. 2007;26:5553–5563. doi: 10.1038/sj.onc.1210351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okada T., Ohzeki J.-i., Nakano M., Yoda K., Brinkley W.R., Larionov V., Masumoto H. CENP-B controls centromere formation depending on the chromatin context. Cell. 2007;131:1287–1300. doi: 10.1016/j.cell.2007.10.045. [DOI] [PubMed] [Google Scholar]
- 34.Cadieux B., Ching T.-T., Van den Berg S.R., Costello J.F. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006;66:8469–8476. doi: 10.1158/0008-5472.CAN-06-1547. [DOI] [PubMed] [Google Scholar]
- 35.Narayan A., Ji W., Zhang X.-Y., Marrogi A., Graff J.R., Baylin S.B., Ehrlich M. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int. J. Cancer. 1998;77:833–838. doi: 10.1002/(sici)1097-0215(19980911)77:6<833::aid-ijc6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 36.Nakano M., Cardinale S., Noskov V.N., Gassmann R., Vagnarelli P., Kandels-Lewis S., Larionov V., Earnshaw W.C., Masumoto H. Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev. Cell. 2008;14:507–522. doi: 10.1016/j.devcel.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen E.S., Zhang K., Nicolas E., Cam H.P., Zofall M., Grewal S.I.S. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature. 2008;451:734–737. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- 38.Lu J., Gilbert D.M. Proliferation-dependent and cell cycle regulated transcription of mouse pericentric heterochromatin. J. Cell Biol. 2007;179:411–421. doi: 10.1083/jcb.200706176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu R., Singh P.B., Gilbert D.M. Uncoupling global and fine-tuning replication timing determinants for mouse pericentric heterochromatin. J. Cell Biol. 2006;174:185–194. doi: 10.1083/jcb.200601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerlich D., Hirota T., Koch B., Peters J.M., Ellenberg D.J. Condensin I stabilizes chromosomes mechanically through a dynamic interaction in live cells. Curr. Biol. 2006;16:333–344. doi: 10.1016/j.cub.2005.12.040. [DOI] [PubMed] [Google Scholar]
- 41.Folco H.D., Pidoux A.L., Urano T., Allshire R.C. Heterochromatin and RNAi are required to establish CENP-A chromatin at centromeres. Science. 2008;319:94–97. doi: 10.1126/science.1150944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong N.C., Wong L.H., Quach J.M., Canham P., Craig J.M., Song J.Z., Clark S.J., Choo K.H.A. Permissive transcriptional activity at the centromere through pockets of DNA hypomethylation. PLoS Genet. 2006;2:e17. doi: 10.1371/journal.pgen.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monier K., Mouradian S., Sullivan K.F. DNA methylation promotes aurora-B-driven phosphorylation of histone H3 in chromosomal subdomains. J. Cell Sci. 2007;120:101–114. doi: 10.1242/jcs.03326. [DOI] [PubMed] [Google Scholar]
- 44.Kunitoku N., Sasayama T., Marumoto T., Zhang D., Honda S., Kobayashi O., Hatakeyama K., Ushio Y., Saya H., Hirota T. CENP-A phosphorylation by aurora-A in prophase is required for enrichment of aurora-B at inner centromeres and for kinetochore function. Dev. Cell. 2003;5:853–864. doi: 10.1016/s1534-5807(03)00364-2. [DOI] [PubMed] [Google Scholar]
- 45.Ono T., Fang Y., Spector D.L., Hirano T. Spatial and temporal regulation of condensins I and II in mitotic chromosome assembly in human cells. Mol. Biol. Cell. 2004;15:3296–3308. doi: 10.1091/mbc.E04-03-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lipp J.J., Hirota T., Poser I., Peters J.M. Aurora B controls the association of condensin I but not condensin II with mitotic chromosomes. J. Cell Sci. 2007;120:1245–1255. doi: 10.1242/jcs.03425. [DOI] [PubMed] [Google Scholar]
- 47.Luciani J.J., Depetris D., Missirian C., Mignon-Ravix C., Metzler-Guillemain C., Megarbane A., Moncla A., Mattei G. Subcellular distribution of HP1 proteins is altered in ICF syndrome. Eur. J. Hum. Genet. 2004;13:41–51. doi: 10.1038/sj.ejhg.5201293. [DOI] [PubMed] [Google Scholar]
- 48.Ling Y., Sankpal U.T., Robertson A.K., McNally J.G., Karpova T., Robertson K.D. Modification of de novo DNA methyltransferase 3a (Dnmt3a) by SUMO-1 modulates its interaction with histone deacetylases (HDACs) and its capacity to repress transcription. Nucleic Acids Res. 2004;32:598–610. doi: 10.1093/nar/gkh195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palii S.S., Van Emburgh B.O., Sankpal U.T., Brown K.D., Robertson K.D. DNA methylation inhibitor 5-aza-2′-deoxycytidine (5-azadC) induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell. Biol. 2008;28:752–771. doi: 10.1128/MCB.01799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lefevre P., Bonifer C. Analyzing histone modification using crosslinked chromatin treated with micrococcal nuclease. Methods Mol. Biol. 2006;325:315–325. doi: 10.1385/1-59745-005-7:315. [DOI] [PubMed] [Google Scholar]
- 51.Jin B., Tao Q., Peng J., Soo H.M., Wu W., Ying J., Fields C.R., Delmas A.L., Liu X., Qiu J., et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum. Mol. Genet. 2008;17:690–709. doi: 10.1093/hmg/ddm341. [DOI] [PubMed] [Google Scholar]
- 52.Tsai A., Carstens R.P. An optimized protocol for protein purification in cultured mammalian cells using a tandem affinity purification approach. Nat. Protoc. 2006;1:2820–2827. doi: 10.1038/nprot.2006.371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.