

Abstract
Motivation: In some applications, prior biological knowledge can be used to define a specific pattern of association of multiple endpoint variables with a genomic variable that is biologically most interesting. However, to our knowledge, there is no statistical procedure designed to detect specific patterns of association with multiple endpoint variables.
Results: Projection onto the most interesting statistical evidence (PROMISE) is proposed as a general procedure to identify genomic variables that exhibit a specific biologically interesting pattern of association with multiple endpoint variables. Biological knowledge of the endpoint variables is used to define a vector that represents the biologically most interesting values for statistics that characterize the associations of the endpoint variables with a genomic variable. A test statistic is defined as the dot-product of the vector of the observed association statistics and the vector of the most interesting values of the association statistics. By definition, this test statistic is proportional to the length of the projection of the observed vector of correlations onto the vector of most interesting associations. Statistical significance is determined via permutation. In simulation studies and an example application, PROMISE shows greater statistical power to identify genes with the interesting pattern of associations than classical multivariate procedures, individual endpoint analyses or listing genes that have the pattern of interest and are significant in more than one individual endpoint analysis.
Availability: Documented R routines are freely available from www.stjuderesearch.org/depts/biostats and will soon be available as a Bioconductor package from www.bioconductor.org.
Contact: stanley.pounds@stjude.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Microarrays have opened exciting new possibilities for biological research by enabling investigators to simultaneously measure thousands or even millions of genomic features in a biological specimen. Statistical analysis is used to identify features that are associated with an endpoint of interest. The statistical analysis often includes one or more hypothesis tests for each genomic feature to explore the association with the endpoint of interest. Gene-set enrichment analysis (GSEA; Jiang and Gentleman, 2007) can be a useful complement to the feature-by-feature analysis. Given a collection of predefined sets of genes that share a common biological function or jointly participate in a specific biological process, GSEA performs a statistical test for each gene set to determine whether the member genes are ‘enriched’ among the most statistically significant results. The feature-by-feature and GSEA each lead to a multiple-testing problem, so that several false discoveries may result even if very stringent P-value thresholds are applied. Pounds (2006) reviewed several methods that address this multiple-testing problem by estimating or controlling the false discovery rate (FDR; Benjamini and Hochberg, 1995; Storey and Tibshirani 2003). Nevertheless, after the statistical analyses are complete, the biologist is still left with the problem of using the results to select the most promising candidates for follow-up research.
One strategy to select the most promising leads from a list of genes that show a statistically significant association with one endpoint is to identify genes that show a biologically plausible pattern of association with related endpoints. For example, Yang et al. (2009) explored the association of the genotype of 600K single nucleotide polymorphisms (SNPs) with the level of minimal residual disease of acute lymphoblastic leukemia after initial treatment with chemotherapy. To refine the list of SNPs identified in the initial genome-wide analysis, they explored the association of the genotypes of the significant SNPs with pharmacokinetics (PK) endpoints that are associated with response to therapy. The SNPs with the most biologically plausible pattern of association with the clinical and pharmacologic endpoints are now considered promising candidates for future pharmacogenetic research in this field. In this study, prior biological knowledge regarding the relationships of endpoints to one another helps to define patterns of association for genomic variables that are biologically most plausible.
However, to our knowledge, there is not a published statistical procedure that is designed to identify genomic features that show a specific pattern of association with multiple endpoint variables. In general, it may be difficult to incorporate prior biological knowledge regarding the relationships among multiple endpoint variables when trying to interpret the results of several analyses that each explores the association of genomic variables with an individual endpoint. For example, how does one characterize the statistical significance of a gene that has P=0.0001 for association with one endpoint, but P=0.04 for association with another endpoint in the context of multiple testing? If each individual endpoint analysis is corrected for multiple testing, then there may not be any gene that meets the stringent criteria for statistical significance in more than one individual endpoint analysis. Thus, to identify genes with interesting patterns of association, the P-value threshold must be made less stringent, opening the possibility for a large number of false discoveries.
Here, we propose projection onto the most interesting statistical evidence (PROMISE) as a statistical procedure to identify genomic features that show a specific pattern of association with multiple endpoints that is biologically most plausible or biologically most interesting. PROMISE performs one hypothesis test for the specified pattern for each genomic variable, thus avoiding the inferential problems described above. Additionally, PROMISE is a flexible procedure that can accommodate various types of endpoints, which classical multivariate procedures are not designed to manage. In Section 2, we describe the PROMISE procedure. Section 3 describes how PROMISE differs from other procedures. Section 4 presents simulation studies, and Section 5 presents the results from an example application. Finally, Section 6 provides the discussion and concluding remarks.
2 THE PROMISE METHOD
Suppose that g=1,…, m genomic features are measured for i=1,…, n subjects. Also, suppose that data on j=1,…, k endpoint variables are available for these subjects. For i=1,…, n and g=1,…, m, let yig represent the value of genomic feature g for subject i. Additionally, let xij represent the value of endpoint variable j for subject i. For j=1,…, k, let xj represent the vector (x1j, x2j,…, xnj) of values of endpoint variable j for all subjects. Similarly, for g=1,…, m, let yg represent the vector (y1g, y2g,…, yng) of values for genomic variable g for all subjects. Let Y represent the set of all yig and let X represent the set of all xij.
PROMISE is a general procedure to identify genomic variables with the strongest statistical evidence for the biologically most interesting pattern of associations with the endpoint variables. For j=1,…, k, let Tjg(xj, yg) be a statistic measuring the association of genomic feature yg (for any g) with endpoint variable xj. Now, define
| (1) |
as the vector of the statistics measuring the association of genomic feature g with the endpoint variables j=1,…, k.
In many applications, biological knowledge can be used to define a vector d=(d1,…, dk) that corresponds to the biologically most interesting values for Tg. For example, suppose that a set of subjects is treated with a drug that inhibits DNA synthesis. Drug levels and DNA synthesis rates after the treatment are measured as endpoint variables on these subjects. Genomic data are also collected on the same set of subjects. In this example, there are k=2 endpoints. Also, for each genomic variable g we have Tg=(T1g, T2g) where T1g and T2g are the correlations of genomic variable g with drug level and DNA synthesis rate, respectively. Conceptually, a biologically most interesting result would be T1g=+1 and T2g=−1. Thus, the vector d=(+1, −1) defines a biologically most interesting statistical result for T. Additionally, the result −d would be another biologically most interesting statistical result. Thus, d defines a vector in the space of Tg that corresponds to the biologically most interesting result.
The dot-product of d and Tg,
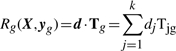 |
(2) |
is a statistic that measures the similarity of the vector Tg of observed associations to the vector d of the biologically most interesting statistical results. The sign of Rg indicates the direction of Tg relative to d and the magnitude of Rg is proportional to the length of the projection of Tg onto d. Thus, (2) defines the PROMISE statistic.
The significance (i.e. P-value) of the PROMISE statistic can be determined via permutation. Let rg represent the value of Rg computed from the observed data. Also, let X⋆l, represent an endpoint dataset obtained by l=1,…, b permutations of the subject indices of X. Let R⋆gl=R(X⋆l, yg) and let ρ0g be a specified ‘null value’ of Rg. Then, for g=1,…, m,
| (3) |
where I(·) is the indicator function that equals 1 if the enclosed statement is true and equals 0 otherwise. Note that (3) defines a two-sided permutation P−value giving the probability that 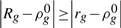 under the complete (permutation) null.
under the complete (permutation) null.
Here, the null hypothesis is exchangeability of the assignments of genomic data to endpoint data. Also, we note that the endpoint data are permuted jointly; each endpoint variable is not permuted individually. Permuting endpoint variables individually breaks the correlation of the endpoints with one another and thus tests a different null hypothesis than permuting the endpoint data jointly. As a consequence, permuting endpoints individually is likely to yield results that are very statistically significant, but not biologically meaningful because the small P-values may indicate that the endpoints are strongly correlated with one another instead of indicating that the genomic variable has the interesting pattern of association with the endpoint variables.
3 OTHER APPROACHES
There are other approaches that could be taken to identify genomic variables that exhibit a specific pattern of association with multiple endpoints. A seemingly straightforward approach would be to screen the association of the genomic variables with each endpoint individually and then identify genes that are significant in each analysis and have the desired pattern of association. This approach is problematic because it lacks statistical power, and the results are difficult to interpret statistically. The analysis for each endpoint involves multiple testing. After adjusting each endpoint's results for multiple testing, it is quite likely that no gene will meet the criteria for inclusion as a ‘significant’ result because it is unlikely that a genomic variable could meet the stringent P-value threshold for each endpoint. Additionally, with this approach, if any genes are identified, it would be difficult to assign a meaningful FDR estimate to the result. For example, what FDR estimate should be ascribed to a set of genes that are inferred to have the association pattern of interest if these genes are selected because they meet a certain FDR or P-value threshold in several single-endpoint analyses? PROMISE avoids these problems by performing a single test for the pattern of association for each gene. For each genomic variable, PROMISE performs one test that directly addresses the question of whether a gene shows the association pattern of interest. This improves the statistical power and simplifies the interpretation, as seen in the example application of Section 5.
Classical multivariate methods, such as principal components (PC) or canonical correlation (CC), are other potential approaches to the problem. For example, one could determine the first PC (PC1) for the endpoint data and then test for the association of each genomic variable with the PC. However, in general, there is no reason to expect that the association of the genomic variable with the PC is a measure of evidence for a specific pattern of association with the endpoints. Clearly, a PC differs markedly from the definition of the PROMISE statistic in (2). The first PC is the linear combination of the endpoint variables that explains the greatest variation in the set of endpoint variables. Unlike PCs, PROMISE does not define a new endpoint variable by a linear combination of the individual endpoint variables. Instead, the PROMISE statistic in (2) is a linear combination of the statistics characterizing the associations of the individual endpoints with the genomic variable.
Also, one could compute the CC of each gene with the endpoint variables and test whether the CC is non-zero. However, CC also differs from the definition of the PROMISE statistic in (2). The CC of two sets of variables is the maximum possible correlation of a linear combination of the first set of variables with a linear combination of the second set of variables. The index selection method of quantitative genetics (Falconer and Mackay, 1996, pp. 240–245) is similar to CC. These approaches do not measure a specific pattern of association of one variable with a set of other variables as in (2).
Furthermore, in many applications, classical multivariate methods such as PCs and CC are not well suited to handle the endpoints of interest. For example, in clinical studies, the endpoint of greatest interest may be a censored time-to-event variable such as relapse-free survival. A censored time-to-event variable is one that is only partially observed in some subjects. For a subject of a clinical trial, relapse-free survival is the duration of time between study enrollment and death or relapse. Many subjects are living and remain relapse-free at the conclusion of the study. For these subjects, it is known that the relapse-free period is greater than the length of time they were followed, but the full duration of the relapse-free period is unknown. The relapse-free period for these subjects is considered censored. This type of endpoint does not fit into the classical multivariate normal framework. As seen in the example application of Section 5, the definition of the PROMISE statistic in (2) is flexible enough to accommodate censored time-to-event variables and ordered categorical variables.
GSEA and other gene-set analysis approaches [such as that of Nettleton et al. (2008)] could be used to determine whether gene sets identified from the analysis of association with one endpoint are associated with another endpoint. For example, one could identify genes that are associated with one endpoint and then explore whether the set of identified genes is associated with another endpoint. While this exercise may provide useful biological insights, it does not give results with the same interpretation as PROMISE. PROMISE provides a P-value for each gene, whereas gene-set methods give a P-value for each gene set. Additionally, the interpretation of gene-set results may be difficult. For instance, what if the list of genes associated with endpoint A are associated with endpoint B, but the list of genes associated with endpoint B are not associated with endpoint A? Such questions could easily become quite frustrating when more than two endpoints are involved. Nevertheless, permutation-based gene-set analyses can be performed in conjunction with PROMISE to identify gene sets that are enriched among genes that show the association pattern of interest. Integrating gene-set methods with PROMISE may prove to be a synergistic combination in terms of improving statistical power to reveal important biological insights.
4 SIMULATIONS
Simulations were performed to compare the statistical power of PROMISE to that of other approaches in a collection of simple settings. Let (Y, X1, X2) be a random vector corresponding to a genomic variable and two endpoint variables. Suppose that (Y, X1, X2) follows a multivariate normal distribution with mean (0, 0, 0) and that Y, X1 and X2 each have unit variance. Let ρx=cor(X1, X2), ρ1=cor(Y, X1) and ρ2=cor(Y, X2). Suppose that the interesting pattern of association is for Y to show the same direction of association with X1 and X2. In terms of the framework above, the test is performed using d=(+1, +1) in Equation (2). For the PROMISE procedure, the statistic R is defined as the average of Pearson's correlation of Y with X1 and Pearson's correlation of Y with X2.
Several alternative approaches were considered in the simulation study. The classical t-test for Pearson's correlation coefficient was used to test the association of Y with X1 and the association of Y with X2. Also, Pearson's correlation of Y with the PC1 of (X1, X2) was computed. Additionally, the CC of Y with (X1, X2) was computed. In each simulation, two-sided P-values for the PROMISE statistic, the correlation of Y with PC1 and the CC statistic were determined using the same set of 1000 permutations of Y. An overlap (OV) analysis identified genes that were significant in each of the single−endpoint analyses. A total of 1000 independent replications were performed for each simulation setting defined by a unique set of values for the sample size n, ρ1, ρ2 and ρx. Simulations were performed for all combinations of ρ1={−0.4, −0.3,…, 0.3, 0.4}, ρ2={−0.4, −0.3,…, 0.3, 0.4}, ρx={−0.1, 0.0, 0.1, 0.2} and n=10, 20, 50, 100. The Supplementary Materials include tables and contour plots with the results of all simulations.
PROMISE had the greatest probability to give a P-value less than α=0.01 for values of (ρ1, ρ2) along the line ρ1=ρ2 (Fig. 1A). Recall that the interesting pattern of association is for Y to have the same direction of association with X1 and X2. The values of (ρ1, ρ2) that match this pattern of interest are the bottom-left and upper-right quadrants of the plots in the Supplementary Materials. The line ρ1=ρ2 passes through the center of this region of interesting patterns of association. Simulations using other combinations of n and ρx give qualitatively similar results regarding the performance of PROMISE relative to that of other procedures (Supplementary Materials).
Fig. 1.

Simulation results. Each plot gives simulation results for ρx=0 and n=50. (A) The power of PROMISE, PC, CC, OV and individual endpoint analyses (X1 only and X2 only) at the α=0.01 level along the line ρ1=ρ2. Note that the line ρ1=ρ2 is perpendicular to the null line ρ1+ρ2=0 of the PROMISE procedure. (B) The probability that the procedures reject their respective null hypotheses along the null line |ρ1+ρ2|=0 of the PROMISE procedure. The Supplementary Materials provides the results of all simulations.
Additionally, PROMISE maintains its level along the line H0: ρ1+ρ2=0 (Fig. 1B). For values of ρ1 and ρ2 along the line |ρ1+ρ2|=0, other procedures test a different null hypothesis and therefore have a greater than α=0.01 probability of giving a P-value less than α=0.01 (Fig. 1B). For example, the X1-only analysis tests H0: ρ1=0, and the CC analysis tests the null hypothesis that the CC of Y with (X1, X2) is zero. However, the null hypothesis H0: ρ1+ρ2=0 is the null hypothesis of interest, and thus the high probabilities of small P-values for values of (ρ1, ρ2) along this line are undesirable in this setting.
It is quite interesting that PROMISE outperforms classical multivariate procedures in this setting. The classical multivariate procedures were developed for this type of setting and are known to perform very well in this setting. The key observation here is that the classical multivariate procedures were designed to detect any non-zero correlations whereas PROMISE is designed to detect a particular pre-specified pattern of correlations. The simulations clearly show that PROMISE performs better for the latter purpose than other methods.
5 EXAMPLE APPLICATION
The example application uses data from the St. Jude AML97 clinical trial (Crews et al., 2002; Rubnitz et al. 2009). Ross et al. (2004) used Affymetrix U133A microarrays to measure gene expression in the leukemic cells of diagnostic bone marrow samples of 42 subjects in this trial. Additionally, PK, pharmacodynamic (PD) and clinical endpoint data were collected for these same subjects. Concentrations of ara-CTP (the active form of the chemotherapy drug cytarabine; ara-C) in leukemic cells of the bone marrow were obtained on the first day (CTP1; after ara-C alone) and the second day (CTP2; after combining cladribine with ara-C) of therapy. The rate of DNA synthesis in leukemic cells from the marrow was measured at diagnosis (baseline) and on days 1 and 2 of the therapy. From these measurements of DNA synthesis rates, we computed the log-ratio of the day 1 rate to the baseline rate (DNA1) and the log-ratio of the day 2 rate to the baseline rate (DNA2). The white blood count (WBC) in peripheral blood is a measure of tumor burden. The log-ratio of the WBC after 48 h of therapy to the WBC at the initiation of therapy was determined. Initial response (RESP) was determined by morphologic examination of a bone marrow aspirate collected after completion of the first course of chemotherapy and categorized as no response (RESP = 0), partial response (RESP = 1) or complete response (RESP = 2). Also, event-free survival (EFS) was defined as zero and considered uncensored for patients who did not achieve complete remission after two courses of therapy. For the remaining patients, EFS was defined as the time elapsed from study enrollment to relapse, development of a second malignancy or death, with patients having experienced none of those events censored at the date of last follow-up. For the purposes of PROMISE analysis in this application, the gene expression data were considered as genomic features and the other variables as endpoint variables. All 22 215 probe sets represented on the microarray were included in the analysis. We have previously noted that excluding probe sets on the basis of present–absent calls may be of limited value (Pounds and Cheng, 2005).
The correlation of gene expression with each endpoint can be measured using published statistical methods. The association of expression with CTP1, CTP2, DNA1, DNA2, WBC and RESP is measured with Spearman's correlation coefficient. For this application, these statistics are denoted Tctp1, Tctp2, Tdna1, Tdna2, Twbc and Tresp. The association of expression with the risk of relapse and death (EFS) is measured using the rank-based statistic developed by Jung et al. (2005) that accounts for follow-up and censoring. For this application, this statistic is denoted Tefs. The statistic Tefs has the form of a dot product and was scaled so that −1≤Tefs≤1. For each endpoint, the association statistics were computed using all subjects with pairwise complete data (i.e. having gene expression data and data for the specific endpoint). This technique for managing missing data allows us to use all available data. The same approach was used for computing permutation statistics.
All AML97 subjects were randomly assigned to receive one of two infusion schedules for ara-C during the initial course of therapy. An amendment to the study protocol added one dose of intrathecally delivered ara-C before the first course of intravenous chemotherapy. Thus, each patient received one of four distinct therapies (short infusion or continuous infusion of ara-C with or without intrathecal ara-C). The statistical analyses of the association of gene expression with the PK endpoints (CTP1 and CTP2), PD endpoints (DNA1, DNA2 and WBC) and clinical endpoints (RESP and EFS) must account for the different therapeutic strategies. Therefore, for each endpoint and expression probe set, the correlation with expression was computed separately for each of four therapy-defined groups of subjects, and then the final correlation statistic was the sample-size weighted average of the group-specific correlations. This type of adjustment for therapy is called a stratified analysis, and the therapy-defined groups are called strata (or one group is called a stratum).
In this application, prior biological and clinical knowledge was used to define the most interesting result for the association of gene expression with the seven endpoints. First, it is most interesting if the correlation of expression with CTP1 and CTP2 are both equal to ±1. For purposes of constructing the vector d, let dctp1=dctp2=1. Given this selection for dctp1 and dctp2, the most interesting correlation of expression with DNA1 and DNA2 is ddna1=ddna2=−1, because the PD effect of ara-CTP is to interfere with DNA synthesis. Interference with DNA synthesis results in cell death and therefore leads to a reduction in WBC. Thus, dwbc=−1. Increased levels of ara−C in leukemic blasts should reduce the amount of tumor in the marrow (a better tumor response), and therefore dresp=1. Effective therapy should reduce the risk of relapse and death, thus defs=−1. Therefore, by (2), the PROMISE statistic for this application was defined as
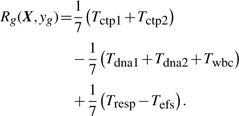 |
(4) |
The subscript g is omitted from the right-hand side of (4) for simplicity of notation. The PROMISE statistic is scaled by 1/7 so that −1≤Rg≤1. A positive Rg indicates that the expression of probe set g shows a therapeutically beneficial pattern of correlation with the endpoint variables, i.e. higher expression associates with therapeutically desirable values of the endpoint variables. Similarly, negative Rg indicates that the expression of probe set g shows a therapeutically detrimental pattern of association with the endpoint variables.
The statistical significance of each individual endpoint's association statistic and the PROMISE statistic were determined using the same set of 10 000 permutations of the assignment of expression data to endpoint data. The permutations were restricted so that data reassignments were performed separately within each therapy-defined stratum because the differences in therapy are important factors for ara-C pharmacology and clinical outcome, as previously described (Rubnitz et al., 2009). For each gene g, the P-value was computed by letting ρg0=0 in Equation (3). Several interesting biological findings will be reported in detail elsewhere. Here, we describe a few results that illustrate the advantages of PROMISE.
The results for the human equilibrative nucleoside transporter 1 (hENT1) gene (probe set 201802_at) clearly illustrate the interpretative advantage of PROMISE. hENT1 is a solute carrier that brings ara-C into the cell (Hubeek et al., 2005). Given this role in ara-C metabolism, one would expect hENT1 to show a therapeutically beneficial pattern of association. This was indeed the case (Table 1 and Fig. 2). The expression of hENT1 was positively associated with CTP1, positively associated with CTP2, negatively associated with DNA1, negatively associated with DNA2, negatively associated with WBC, positively associated with RESP and negatively associated with the risk of an EFS event (Table 1). However, it is difficult to interpret the statistical significance of the association pattern with the seven individual P-values. The PROMISE analysis indicated that the beneficial pattern of association was very significant (Rg=0.23, P=0.0033, rank = 225). Thus, PROMISE identified this gene of known relevance to ara-C metabolism and provided a straightforward interpretation of statistical significance for the interesting pattern of associations with the endpoint variables. The individual endpoint analyses also provided insights that may be helpful for biological interpretation of the results. The associations with DNA1, DNA2 and RESP were strong contributors to the final value of the PROMISE statistic (correlations from 0.30 to 0.40). The associations with CTP1 and CTP2 were moderate contributors, and the associations with WBC and EFS were relatively weak contributors. Other genomic features had similar PROMISE statistics as hENT1, but for some of those features the associations with individual endpoints were substantially different.
Table 1.
The association statistic, P-value and rank among 22 215 probe sets (by P-value) for hENT1 from each individual endpoint analysis and the PROMISE analysis
| Analysis | Corr | P-value | Rank |
|---|---|---|---|
| CTP1 (day 1 ara-CTP level) | +0.16 | 0.2579 | 6723 |
| CTP2 (day 2 ara-CTP level) | +0.17 | 0.1890 | 4374 |
| DNA1 (day 1 DNA synth.) | −0.30 | 0.0082 | 1084 |
| DNA2 (day 2 DNA synth.) | −0.39 | 0.0214 | 726 |
| WBC | − 0.09 | 0.5662 | 13 485 |
| RESP | +0.40 | 0.0091 | 343 |
| EFS | −0.07 | 0.2765 | 6874 |
| PROMISE | +0.23 | 0.0033 | 225 |
Fig. 2.

Beneficial pattern of association for hENT1. (A) The log-CTP1 value versus the log-expression of hENT1. (B) The log-DNA1 value versus the log-expression of hENT1. (C) A boxplot of hENT log-expression for subjects with no response (NR), partial response (PR) and complete response (CR) to the initial course of chemotherapy. (D) The Kaplan–Meier estimates of EFS for subjects with hENT1 expression values greater than the median (black line) and those with hENT1 expression less than the median (gray line). Scatter plots illustrating the association of hENT1 expression with CTP2, DNA2 and WBC are not shown. Unlike the statistical analysis reported in Table 1, the above figures do not distinguish between subjects from different therapy groups.
In our application, PROMISE clearly had greater power to identify genes with interesting patterns of association. First, PROMISE identified a substantially greater proportion of all genes as significant than did any pairwise overlap analysis (Fig. 3A). In the PROMISE analysis, 498 probe sets were significant at P=0.01 level. In contrast, only 92 probe sets were significant at P=0.01 for DNA1 and DNA2, the greatest number of overlapping probe sets for any pair of individual endpoint analyses performed at the P=0.01 level. By definition, overlap of three or more endpoints detected fewer probe sets at the P=0.01 level. No probe set was significant in all seven individual endpoint analyses at a P<0.15. Second, PROMISE identified a greater proportion of genes with an interesting pattern of association than did any individual endpoint analysis (Fig. 3B). For 719 probe sets, the statistics measuring the association of expression with the individual endpoints all had signs matching (or all had signs mismatching) those of the interesting result vector d. PROMISE identified 154 of these probe sets as significant at P=0.01. The individual endpoint analysis for DNA2 detected 34 of the probe sets with an interesting pattern of association at the P=0.01 level, the best among any individual endpoint analysis.
Fig. 3.

Proportion of probe sets called significant as a function of P-value threshold. In (A and B), the results for PROMISE are shown by the thick black curve and the reference line y=x is shown by the thick gray line. In (A), the results for pairwise overlap analyses are shown by the thin black lines. In (B), the results for individual endpoint analyses are shown by the thin black lines.
A permutation-based GSEA was also performed for each individual endpoint analysis and the PROMISE analysis. The pathway column of the Affymetrix annotation dataset was used to define 233 gene sets. For each gene set, the gene-set enrichment statistic was defined as the average of the absolute value of the member genes' correlation statistics. PROMISE identified 42 of these gene sets as significant at the P=0.05 level. Biologically interesting gene sets with significant PROMISE results included the DNA replication reactome gene set (P=0.0248) and the gene set for the G1-to-S phase of the cell cycle (P=0.0113). These gene sets are very interesting in this application because ara-C interferes with DNA synthesis (or replication), and clearly the cell cycle is very important in cancer. These two gene sets were significant at the P=0.05 level in the individual endpoint analysis for DNA1, but not in any other individual endpoint analysis. No gene set was significant in all seven individual endpoint analyses at P<0.15. Thus, GSEA based on PROMISE identified more gene sets than did searching for overlap among the results of gene-set analyses for individual endpoints.
6 DISCUSSION
PROMISE is a general procedure designed specifically to increase statistical power to identify genomic features that show a biologically most interesting pattern of association with multiple endpoint variables. PROMISE defines a test statistic that measures the evidence for the association pattern of interest by projecting the observed vector of association statistics onto the vector of conceptually most interesting values for those statistics. Permutation is used to compute P-values. Unlike classical multivariate statistical methods such as PC or CC, which are designed for data with a multivariate normal distribution, PROMISE can manage ordinal and censored time-to-event endpoints (Fig. 2). Furthermore, as observed in the simulation study in Section 4, CC and PC are not designed to detect a specific pattern of association and therefore do not have as much statistical power to detect the association pattern of interest as does PROMISE. PROMISE showed better power to identify genes with an interesting pattern of association in our example application than searching for such genes within lists of significant genes identified by individual endpoint analyses. Finally, GSEA can be incorporated into PROMISE so that the advantages of both approaches may be simultaneously realized. In our example, the PROMISE-based GSEA gave biologically interesting results and showed much greater statistical power than identifying overlap among the results of the individual-endpoint GSEAs.
Certainly, PROMISE is a very general procedure that must be customized to specific applications. The general concept presented here can be easily extended to accomodate stratified analyses by incorporating a stratum variable into the statistics Tj(·) and Rg(·) and restricting the permutations appropriately. As with other permutation−based methods, defining too many strata for stratified analysis may severely limit the statistical power of the analysis by reducing the number of available permutations. Additionally, the statistics Tj(·) and the most interesting results dj must be defined in an application-appropriate manner. Also, PROMISE can be adapted so that SNP genotypes can be used as the genomic variables.
Future research should explore how to modify or generalize the correlation statistics and the way they are combined to form the PROMISE statistic. In this work, we used a geometric interpretation of the correlation vector to motivate the dot product as an objective way to uniquely define the PROMISE statistic given sufficient prior biological knowledge about the endpoint variables. Other ways to define the PROMISE statistic may prove useful in practice as well. Another approach to define the PROMISE statistic would be to define a vector d that subjectively weighs the correlations according to their practical relevance. For instance, in our example application, one may wish to give more weight to EFS due to its obvious importance for the patients. Additionally, it would be interesting to develop methods to define interesting result vectors and test statistics for applications with thousands of endpoint variables and thousands of genomic variables.
However, great caution should be exercised when using subjectively defined d because the statistical significance may be exaggerated if d is not selected a priori or if PROMISE is used as an exploratory procedure to perform many analyses with different d vectors. Users should definitely avoid using the observed correlations for specific genes to define d. If d is defined to maximize the dot-product in (2) for a specific gene, the procedure will give a very small P-value for that gene. In this case, the small P-value will greatly exaggerate statistical significance. In such a situation, the small P-value reflects the fact that the coefficients d were selected to maximize the statistic Rg instead of indicating that the observed correlations did not arise by chance. If a search is performed for d, the P-value should adjust for that search in some manner. Nesting the search within each permutation round would be one way to perform such an adjustment and give meaningful P-values.
We do not recommend that the PROMISE procedure be used for applications in which there is not adequate prior biological knowledge about the endpoint variables to objectively define the vector d. Our simulations suggest that canonical correlation is a good method for such a setting if the variables are appropriate for CC analysis.
Supplementary Material
ACKNOWLEDGEMENTS
We also thank Mr David Galloway for expert editorial assistance.
Funding: American Lebanese Syrian Associated Charities (ALSAC); National Institutes of Health (R01CA132946-01).
Conflict of Interest: none declared.
REFERENCES
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Crews KR, et al. Interim comparison of a continuous infusion versus a short daily infusion of cytarabine given in combination with cladribine for pediatric acute myeloid leukemia. J. Clin. Oncol. 2002;20:4217–4224. doi: 10.1200/JCO.2002.10.006. [DOI] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. 4. Essex: Addison Wesley Longman; 1996. [Google Scholar]
- Hubeek I, et al. The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br. J. Cancer. 2005;93:1388–1394. doi: 10.1038/sj.bjc.6602881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Gentleman R. Extensions to gene set enrichment. Bioinformatics. 2007;23:306–313. doi: 10.1093/bioinformatics/btl599. [DOI] [PubMed] [Google Scholar]
- Jung SH, et al. A multiple testing procedure to associate gene expression levels with survival. Stat. Med. 2005;24:3077–3088. doi: 10.1002/sim.2179. [DOI] [PubMed] [Google Scholar]
- Nettleton D, et al. Identification of differentially expressed gene categories in microarray studies using nonparametric multivariate analysis. Bioinformatics. 2008;24:192–201. doi: 10.1093/bioinformatics/btm583. [DOI] [PubMed] [Google Scholar]
- Pounds SB. Estimation and control of multiple testing error rates for the analysis of microarray data. Brief. Bioinform. 2006;7:25–36. doi: 10.1093/bib/bbk002. [DOI] [PubMed] [Google Scholar]
- Pounds S, Cheng C. Statistical development and evaluation of gene expression data filters. J. Comput. Biol. 2005;12:482–495. doi: 10.1089/cmb.2005.12.482. [DOI] [PubMed] [Google Scholar]
- Ross MB, et al. Gene expression profiling of pediatric acute myleogenous leukemia. Blood. 2004;104:3679–3687. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- Rubnitz JE, et al. Combination of cladribine and cytarabine is effective for childhood acute myeloid leukemia: results of the St. Jude AML97 trial. Leukemia. 2009 doi: 10.1038/leu.2009.30. to appear. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, et al. Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. J. Am. Med. Assoc. 2009;301:393–403. doi: 10.1001/jama.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.