

Abstract
Motivation: Quantitative mass spectrometry-based proteomics requires protein-level estimates and associated confidence measures. Challenges include the presence of low quality or incorrectly identified peptides and informative missingness. Furthermore, models are required for rolling peptide-level information up to the protein level.
Results: We present a statistical model that carefully accounts for informative missingness in peak intensities and allows unbiased, model-based, protein-level estimation and inference. The model is applicable to both label-based and label-free quantitation experiments. We also provide automated, model-based, algorithms for filtering of proteins and peptides as well as imputation of missing values. Two LC/MS datasets are used to illustrate the methods. In simulation studies, our methods are shown to achieve substantially more discoveries than standard alternatives.
Availability: The software has been made available in the open-source proteomics platform DAnTE (http://omics.pnl.gov/software/).
Contact: adabney@stat.tamu.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
In mass spectrometry-based, bottom-up, proteomics, protein abundance measurements must be translated from MS peak heights for constituent peptides (Nesvizhskii et al., 2007). However, this translation is complicated by many factors. MS intensities are derived from peak heights or areas but do not represent absolute abundance levels. Intensities can vary greatly across peptides from the same protein (Fig. 1), due to, for example, differing ionization efficiencies or other chemical characteristics. For ‘shotgun’ proteomics measurements (Aebersold and Mann, 2003), many peptides that are observed in some samples are not observed in others, resulting in widespread missing values (Fig. 2). Furthermore, the fact that a peak was not observed for a peptide is often due to that peptide's presence at a lower abundance than the instrument can detect. Because of this informative missingness, care must be taken when handling the missing values to avoid biasing abundance estimates. There will inevitably be some highly variable peptides, as well as incorrect identifications in the list of peptides used in an analysis, and these may greatly diminish the quality of the data for the proteins to which they are assigned. Diagnostic tools for finding and excluding such outlier peptides are needed.
Fig. 1.

Peptide profiles for two proteins in the diabetes data that were found to be overexpressed in diabetics relative to controls: (A) lumican (IPI:00020986.2) and (B) an antithrombonin III variant (IPI:00032179.2). Note the peptide-specific baseline intensities, the apparent censoring of low intensities and the consistent group differences across peptides. The data have been log2-transformed. Peptides that have been shaded out vertically were filtered from the analysis due to poor data quality by our model-based filtering routine. The horizontal lines and shaded regions show our protein-level estimates for each group, as well as approximate 95% confidence intervals for those estimates.
Fig. 2.

Missingness proportions by observed mean for peptides in the first sample from the diabetes experiment. Each point represents a peptide. The x-axis indicates the average of the observed intensities, and the y-axis indicates the proportion of missing peaks. There are generally more peaks missing for peptides with lower observed intensities, suggesting that many missing peaks have been censored. The solid curve is a natural cubic spline with 5 degrees of freedom, and the dashed line is the estimated random missing proportion for this sample.
Ideally, MS intensity would be a linear function of absolute abundance. At the least, we would hope that relative intensities for a single peptide under different conditions change monotonically with actual abundance. There are several ways in which these ideals might not be realized, including ion suppression effects, ionization inefficiencies, peptide misidentification, etc. (Tang et al., 2004). However, there is evidence that linear relationships between intensity and abundance can be attained using microcapillary liquid chromatography (μLC) separation coupled with electrospray ionization (ESI) MS/MS, based on either stable isotope labeling or label-free approaches (Gygi et al., 1999; Wang et al., 2003). In this article, we focus on the label-free approach, basing abundance estimates on peak areas or heights, although the methodology applies equally well to labeled experiments.
Existing approaches to protein quantitation do not generally address the information present in censored peak intensities. Instead, analysis is either carried out on complete data, excluding the missing values [Hill et al. (2008); Oberg et al. (2008); Wang et al. (2003); also, commercial software like Agilent's Spectrum Mill], or based on imputed data using standard imputation routines like k-nearest neighbors (KNN) (Troyanskaya et al., 2001) for filling in the missing values. Both of these strategies are designed for the scenario in which missingness is statistically independent of both its intensity, had it been observed, and the intensities of other peaks (Little and Rubin, 2002). In the presence of censoring, missingness is not independent of intensity, and these strategies can lead to extremely biased estimates, especially in proteomics data where there is frequently very high (≈50%) missingness (Fig. 2). Wang et al. (2006) discuss this issue and propose a probability model for censoring that can be used to impute censored values. However, their model is specific to a single experimental design, in which replicate measurements are taken over a sequence of days and protein abundances are expected to decrease systematically with time. Protein estimation and inference is typically done by the ANOVA analysis of either peptide- or protein-level peak intensities. Protein rollup is often accomplished by averaging the intensities for a protein's peptides, after suitable scaling or normalization of the sibling peptides. The DAnTE software (Polpitiya et al., 2008), for example, constructs a median protein abundance profile across samples and scales all other proteins to this profile.
We present here a comprehensive statistical model, applicable to a wide range of experimental designs, for protein-level abundance in mass spectrometry experiments that carefully accounts for expected missingness mechanisms. A likelihood model is formulated that expresses protein abundance in terms of peptide-level intensities. The model accounts for the fact that many peptide measurements will be unobserved. Two missingness mechanisms are modeled, one completely random and the other abundance-dependent. Completely random missingness occurs when the fact that a peptide was unobserved in a sample has nothing to do with its abundance or the abundance of any other peptides. This is expected to affect a relatively small proportion of the peptides considered in an analysis. Abundance-dependent missingness boils down to censoring, where a peptide is either not present or is present at too low an abundance to be detected by the instrument. In this case, we have partial information for the peptide intensity, in that we know it must be less than the detection limit of the instrument.
Model parameters are estimated by empirically maximizing the likelihood function. We also use our model to derive an automated filtering routine, where formal concepts of information content from maximum likelihood theory guide the selection and exclusion of proteins and peptides in an analysis (Fig. 1). Furthermore, we report an imputation routine that uses our model to generate random values for the missing intensities. We provide the evidence that using the imputation routine followed by standard ANOVA or regression analyses results in estimates and inferential decisions that are very similar to those obtained by empirical maximization (Fig. 3). The model and tools apply to a wide range of experimental designs and allow inference on any protein or peptide contrast of interest. Finally, in simulations, the model is shown to achieve substantially more discoveries than standard alternatives (Fig. 3 and Table 1).
Fig. 3.

ROC curve comparing the performance of (a) numerical estimation of our model, (b) estimation of our model after model-based imputation, (c) ANOVA on just the complete data, throwing out all missing values and (d) ANOVA after simple row-mean imputation. Our model achieves substantially higher sensitivity for each specificity, as compared with the standard alternatives.
Table 1.
Summary of all simulations
| Method | Simulation |
|||||
|---|---|---|---|---|---|---|
| S1 |
S2 |
S3 |
||||
| (i) | (ii) | (i) | (ii) | (i) | (ii) | |
| (a) | 0.85 | 0.90 | 0.79 | 0.82 | 0.77 | 0.83 |
| (b) | 0.84 | 0.89 | 0.79 | 0.78 | 0.79 | 0.83 |
| (c) | 0.74 | 0.89 | 0.75 | 0.67 | 0.76 | 0.77 |
| (d) | 0.60 | 0.85 | 0.51 | 0.56 | 0.70 | 0.72 |
Shown are the sensitivities achieved at a specificity of 0.90. Simulations S1 have 50% of the proteins differentially expressed and π=0.05, with (i) 40% and (ii) 20% overall missingness, respectively. Simulations S2 have 40% overall missingness with π=0.05, with (i) 30% and (ii) 15% of the proteins differentially expressed. Simulations S3 have 50% differential expression, 40% overall missingness, with (i) π=0.15 and (ii) π=0.10. Both implementations of our model outperform simple alternatives in the presence of >20% missing data. Note that Simulation S1(i) is shown in more detail in Figure 3.
2 METHODS
2.1 Experiments
2.1.1 Diabetes
Our samples consisted of frozen human serum samples from the DASP (years 2000–2005), with 10 healthy control individuals and 10 patients recently diagnosed with T1DM. Six high−abundant plasma proteins that constitute ∼85% of the total protein mass of human plasma were removed and the serum extracted. The samples were then analyzed following the accurate mass and time tag (AMT) strategy (Pasa-Tolic et al., 2004; Zimmer et al., 2006). The final LC-FTICR MS datasets were processed using the PRISM Data Analysis system (Kiebel et al., 2006), a series of software tools developed in-house.
2.1.2 Simulation
We also created six synthetic datasets by computer simulation, based on our model for peak intensities described in Section 2.2. The structure of the data mimicked that of the diabetes data described above, with the same number of proteins and the same numbers of peptides per protein, similar effect sizes and similar residual errors. The simulations differed in terms of (i) the proportion of missing data, (ii) the proportion of proteins that are differentially expressed and (iii) the proportion of missing values that are missing due to ‘completely random’ mechanisms (Section 2.2). These simulated data allow us to evaluate the proposed method in terms of precise performance measures such as sensitivity and specificity. For full details of the simulations, see the Supplementary Material.
2.1.3 Salmonella virulence
The goal of this experiment was to assess the effects of deleting 13 transcriptional regulators essential for S.typhimurium virulence (spvR, fruR, himD, phoP/Q, ssrA/B, slyA, hnr, rpoE, csrA, rpoS, STM3120, crp and ompR/envZ) in mice by global proteomics profiling. Bacteria were grown in a low-pH, low-Mg2+ minimal media (MgM) designed to mimic the intracellular environment of the macrophage and shown to induce the virulence program in S.typhimurium. Three biological replicates for each mutant were grown, then pooled and partitioned into a soluble and an insoluble fraction. Proteins were isolated and subjected to LC/MS analysis with three technical replicates per sample analyzed using the AMT tag approach; six technical replicates were also obtained for the wild-type. This resulted in 90 individual datasets overall, representing different regulator mutant/culture condition combinations. For the purposes of this analysis, we only considered the 45 samples from the soluble fraction.
2.2 A model for protein-level abundance
We begin the model-building process by examining Figure 1, which shows the observed log2-intensity profiles for two proteins from the diabetes data. The first is a lumican protein (IPI:00020986.2), for which six peptides were observed. Lumican has been found to be associated with diabetes in previous studies (Lehti et al., 2006). It is apparent from the figure that average intensity differs across peptides. Also, while many control intensities were apparently censored on the low end, it appears reasonable to assume that group differences remain roughly constant across peptides. In other words, whereas the average intensity of one peptide may differ from other peptides of the same protein, the difference between control and diabetic intensities in the same protein should remain approximately constant. The second protein is an antithrombonin III variant (IPI:00032179.2). In both proteins, peptides that have been shaded out vertically were filtered from the analysis due to poor data quality, as determined by a model-based filtering routine described in the later section.
The observations above suggest a protein-specific additive model involving main effects for peptide and group. In particular, let yijkl be the log2-transformed intensity for protein i and peptide j in comparison group k and sample l; we consider a model of the form
| (1) |
Here, Proti represents the overall average intensity for protein i, Pepij represents the effect of peptide j in protein i and Grpik represents the effect of group k in protein i. For a given protein, the peptide effects are constrained to sum to zero; that is, ∑j Pepij=0. Similarly, for a given protein, the group effects are constrained to sum to zero; ∑k Grpik=0. The errorijkl term represents random error, assumed to follow the Normal distribution with mean zero and variance σij2. Note that we assume a separate error variance for each peptide but a common variance between comparison groups in the same peptide.
For the purposes of comparing protein abundance levels, the parameters of interest are the Grpik. In the diabetes study, for example, peptide j of protein i has overall mean intensity Proti+Pepij. This is an average of the intensities for this peptide in the control and diabetic groups. If k=1 indicates the diabetic group and k=2 the control group, then Grpi2−Grpi1 is the expected difference in intensity between controls and diabetics in protein i. To test for differential expression, we can test the null hypothesis that Grpi2=Grpi1=0; note that, since ∑kGrpik=0, this is equivalent to testing whether Grpi2=0. Furthermore, the model naturally handles more than two comparison groups (K, say), in which case we test a null hypothesis that Grpi1=Grpi2=···=GrpiK=0.
2.2.1 Missing and censored values
Figure 2 compares the proportion of missing peaks for a peptide to the average of the peptide's observed peaks in the first sample from the diabetes experiment. Overall, 32% of the attempted recordings were unsuccessful. The systematic pattern reflects the fact that low-abundance peptides are more likely to have missing peaks. Many unobserved peaks therefore correspond to peptides that are either absent in a sample or present at levels below the detection limit of the mass spectrometer. In statistical parlance, such peaks have been censored to the left. While we do not know the actual peak height, we do know that the peak height is below the instrument's detection threshold.
Consider, for example, the second lumican peptide in Figure 1. This peptide was observed in only 2 of 10 controls, but in 9 of 10 diabetics. Suppose that the detection limit of the instrument was 16.9 (the smallest observed intensity in the entire experiment was 16.91). A reasonable explanation for the observed profile for the second peptide would then be that the abundance level in controls is lower than that in diabetics, with most control samples giving rise to peaks below the instrument's detection limit. To quantify peptide abundance in the two groups, we might average the observed peaks. While this would be reasonable for the diabetics, we would overestimate the abundance of the controls. A similar problem arises from replacing the missing control values with the average of the two observed intensities. This underestimation of the control group would lead to an estimate of the group difference that is attenuated toward zero; such attenuation has been reported in validation studies (Old et al., 2005). In other words, failure to account for censored peaks reduces our ability to detect differences. Note that replacing the missing values with a small number will underestimate the variability of intensities, resulting in underestimated standard errors and hence overestimated significance levels. As discussed below, standard statistical techniques can be employed to carefully account for censoring.
On the other hand, other unobserved peaks are likely unobserved for completely random reasons, regardless of the peptide abundance levels. This can happen due to the ionization inefficiencies, ion-suppression effects and other technical factors (Tang et al., 2004). These peaks are said to be ‘missing completely at random’ and can be safely ignored or imputed (Little and Rubin, 2002). Incorrectly treating randomly missing peaks as censored or vice versa will result in biased abundance estimates. We cannot know whether any one unobserved peak is randomly missing or censored. However, we can estimate probabilities of the two events from the entire collection of data and use these to construct unbiased abundance estimates.
2.2.2 Probability model
We use maximum likelihood estimates (MLEs) of protein abundance under the above model assumptions. These are obtained by first translating our model assumptions into probability statements, combining these probabilities into the likelihood, then choosing the values of the unknown model parameters that maximize the likelihood. The likelihood gauges how likely it would be to observe our data, given a particular set of values for the model parameters. Maximum likelihood estimation therefore seeks parameter values for the model that best explain the data (Lehmann and Casella, 2003). We describe the model and estimation approach in detail in what follows.
Consider the j-th peptide of the i-th protein in the k-th comparison group and the l-th sample. The expected intensity is μijk=Proti+Pepij+Grpik. Further, based on the random error distributions, we assume that sampled intensities follow the normal distribution with mean μijk and variance σ2ij. We assume that there are two independent mechanisms by which an intensity will not be observed. The first is due to censoring at the unknown detection threshold ci for protein i. The probability that censoring occurs is the left-hand tail probability of the N(μijk, σ2ij) distribution, evaluated at a peptide-specific censoring threshold cij. We denote this by Φ ((cij−μijk)/σij)), where Φ is the cumulative distribution function (cdf) of the N(0, 1) distribution. The second mechanism is random missingness, and any peak in sample l is assumed to be affected with probability πl.
Let Wijkl be an indicator of whether yijkl is unobserved (0 if observed and 1 if unobserved). Note that Wijkl=0, and we observe yijkl, if and only if the peak is neither randomly missing nor censored. By the independence of these two mechanisms, we therefore have that the probability of a missing peak is
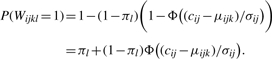 |
This is the contribution to the likelihood of an unobserved peak. Similarly, the likelihood contribution of an observed peak yijkl>cij is (1−πl)ϕ((yijkl−μijk)/σij)/σij, where ϕ is the probability distribution function (pdf) of the N(0, 1) distribution. Putting these pieces together and combining all peaks, the likelihood for protein i is of the form
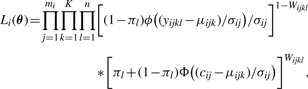 |
(2) |
with the restriction that all observed yijkl exceed the relevant detection thresholds cij. Actually, the full likelihood is the product of the protein−specific pieces, L(θ)=∏i=1MLi(θ). However, as discussed below, heuristic estimates for the πl allow us to consider each protein-specific likelihood separately, and this greatly reduces the computational burden.
2.2.3 Maximum likelihood estimation
The likelihood is viewed as a function of the unknown parameters πl, cij, μijk and σij, i=1, 2,…, M, j=1, 2,…, mi, k=1, 2,…, K and l=1, 2,…, n, which we have collectively denoted as θ. Our estimation goal is to maximize expression (2) for each protein with respect to the unknown parameters, resulting in the MLEs 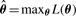 .
.
Since the likelihood is increasing in the cij, and any observed yijkl had to exceed the thresholds, the MLE of cij equals the minimum observed intensity for peptide j of protein i. Analytical solutions for the other MLEs are not available, necessitating an iterative solution. However, the random missingness probabilities πl have heuristic estimates, based on a simple observation. Recall that Figure 2 shows missingness proportions versus observed average intensities in the diabetes example. At the far right side of the x-axis, there are many complete peptides. Assuming that the intensity distributions of these peptides are shifted enough to avoid censoring, we can interpret any systematic vertical deviation from zero as being due to random missingness. To estimate the systematic part to Figure 2, we fit a natural cubic spline with 5 degrees of freedom, shown by the solid curve in the figure. We then estimate πl as the fitted value of the curve at the maximum average intensity, shown by the dashed line in the figure; see the Supplementary Material for details. This results in the estimate 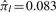 , meaning that we expect 8.3% of all peaks to be missing randomly in this sample.
, meaning that we expect 8.3% of all peaks to be missing randomly in this sample.
While the remaining mean and SD parameters do not have analytical solutions, numerical optimization algorithms can be used to iteratively search for their MLEs. Searching the parameter space of all proteins and peptides simultaneously is not practical, since finding the best direction to search requires a prohibitive amount of computation. Instead, since we initially estimate the πl parameters, we can break the likelihood into protein-specific pieces and maximize each separately. Thus, estimation proceeds by first forming the MLEs of the censoring thresholds cij and the random missingness probabilities πl, then treating these as known in protein-specific algorithms for the mean and SD parameters. We use a generic Newton–Raphson algorithm and provide the requisite first and second derivatives of the likelihood in the Supplementary Material.
2.2.4 Hypothesis testing and the false discovery rate
Recall that, in terms of the model statement in Equation (1), the relevant null hypothesis for testing differential expression in protein i is H0i: Grpi1=Grpi2=···=GrpiK=0. We use likelihood ratio statistics to test this null hypothesis for each protein. A likelihood ratio statistic is a ratio of the maximized likelihood computed under the null hypothesis to the maximized likelihood computed under the alternative hypothesis. In practice, we estimate the null likelihood by plugging in MLEs 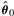 computed under the null and the alternative likelihood by plugging in unconstrained MLEs
computed under the null and the alternative likelihood by plugging in unconstrained MLEs  . Thus, the likelihood ratio for protein i is
. Thus, the likelihood ratio for protein i is
The standard likelihood ratio test statistic is actually −2logΛi, as this has a χ2-distribution with K−1 degrees of freedom under the null hypothesis (Lehmann, 1997). A P-value for protein i can therefore be computed as the right-hand tail probability of the χ2K−1-distribution, evaluated at −2 logΛi. By relying on a parametric form, we are assuming that our model is approximately correct or that the sample size is large. We point out, however, that standard alternatives like ANOVA require parametric assumptions in small samples and thus are susceptible to similar small-sample difficulties. While it would be preferable to assign significance levels by non-parametric resampling techniques like the bootstrap, the bootstrap is not easily applied to the complex data structure considered here.
In a typical comparative quantitative proteomics experiment, hundreds to thousands of hypothesis tests will be performed. When using P-values to assign statistical significance to proteins, we are focusing on the probability of a single false positive out of all tests. In order to keep this probability small when conducting a large number of hypothesis tests, we must choose a P-value cutoff that is much smaller than the traditional cutoffs of 0.01 or 0.05. While this will stringently control false positives, such a small cutoff will mean that many interesting proteins are missed. A more practical error measure in the context of many hypothesis tests is the false discovery rate (FDR) (Benjamini and Hochberg, 1995) and its associated Q-value (Storey and Tibshirani, 2003). For example, selecting all proteins with a Q-value of 0.05 leads to a FDR of 5% among all significant proteins. In contrast, a P-value threshold of 5% leads to a 5% false positive rate among all null proteins. The Q-value is therefore a relevant significance error measure for the selected proteins.
2.3 Preprocessing
2.3.1 Model-based filtering
Peptides and proteins of poor quality are typically filtered out prior to analysis. The definition of ‘poor quality’ is usually subjective, however, having to do with (i) the number of times a peptide or protein was observed, (ii) the variability of a peptide relative to other peptides from the same protein, (iii) the agreement of a peptide's behavior across samples with other peptides from the same protein, etc. Peptides or proteins with too few observations contribute little information to the analysis. Peptides that differ wildly from sibling peptides from the same proteins may be false identifications.
One of the benefits of having a probability model is that we can use it to quantify the information content of a protein or peptide. In likelihood theory, the information matrix is the negative expected value of the second derivative matrix of the log-likelihood function. In large samples, the inverse of this matrix contains the variances of the maximum likelihood estimators (Ferguson, 1996). Hence, large information matrix entries correspond to parameters whose estimates we can be highly confident in. In the context of our model (1), the parameters of interest are the protein-level group differences. For any protein and set of peptides within that protein, we can estimate the information matrix and quantify the information content for the protein-level group difference parameters by taking the scaled determinant of the corresponding matrix block. We filter out proteins for which no collection of peptides can produce an identifiable model, with non-zero information matrix determinant. We then use a greedy search algorithm to select peptide sets for each remaining protein that produce an optimal information content and filter out the rest. For the two proteins in Figure 1, peptides that are shaded out vertically were filtered from the analysis due to insufficient information content. As an example, the two filtered peptides for the lumican protein had too many censored values and did not contribute any further information over the other, more complete, peptides. In the examples considered here, our filtering routine resulted in ∼30% of all peptides being removed from the analysis. For details of the filtering algorithm, see the Supplementary Material.
2.3.2 Model-based imputation
Our model can also be used to impute missing values. With estimates of the model parameters in hand, we can simply replace the missing values with random numbers drawn from the estimated likelihood model. Standard visualization, estimation and inference can then be performed without the use of any special methodology. Furthermore, inference on the resulting complete data will be unbiased under the model assumptions. We obtain preliminary estimates of our model and use these to simulate random numbers from the estimated intensity distributions to replace missing values. To avoid overfitting issues associated with single imputation as well as the awkward data management required with multiple imputation (Little and Rubin, 2002), we implement a hybrid approach. A single imputation is carried out, but then we adjust the P-value distribution to minimize the effect of overfitting. For details of the imputation algorithm, see the Supplementary Material.
As we discuss with the examples below, the results from standard regression on complete data from our model-based imputation routine are very similar to the full likelihood model results obtained by numerical optimization. This is particularly relevant in the analysis of complex experiments, like the Salmonella mutant virulence example considered below. In this case, there are many comparison groups, and the nature of the scientific question of interest requires non-standard hypothesis testing procedures and an implementation of the bootstrap, neither of which are easy in the context of the full censored likelihood model (1). Because of the similarity between the results based on the censored likelihood model and imputation, together with the flexibility gained by working with complete data, the model-based imputation routine may be preferred in complex experimental designs and/or very large datasets.
3 RESULTS
3.1 Simulated data
We carried out four analyses: (a) filtering, then numerical estimation; (b) filtering and model-based imputation, then ANOVA; (c) ANOVA on just the complete data, throwing out all missing values; and (d) ANOVA after imputation by simple row-means. As mentioned above (full details in the Supplementary Material), the simulations were constructed to mimic the diabetes data as closely as possible. Thus, these results describe the performance of our method under the assumptions of our model, in a scenario that closely resembles a real-world proteomics dataset. Figure 3 shows a receiver operating characteristic (ROC) curve for the simulation with 40% missingness. As can be seen in the ROC curve, our method achieves substantially higher sensitivity at any given specificity, as compared with both simple alternatives. This is true for the numerical estimates of our model as well as ANOVA on the data after applying our model-based imputation routine. Both versions of our method resulted in null P-values that were approximately uniformly distributed between 0 and 1, as expected (Dabney and Storey, 2006; Storey, 2002). As compared with the estimates from our method, methods (c) and (d) resulted in attenuated group effects and underestimated standard errors. Table 1 summarizes the results of all six simulations, in which it is apparent that both implementations of our method outperform standard alternatives under various model scenarios, and particularly when there is >20% missingness. When there are few missing values, our method performs very similarly to the ANOVA on the complete data. This is as expected since, by inspection of our likelihood model (2), our method reduces to standard ANOVA when there are no missing values. Taken together, these results indicate that the proposed method produces valid estimates and inference in general, and can achieve substantially more discoveries than standard approaches in typical MS-based proteomics datasets for which there are widespread missing measurements.
3.2 Diabetes data
We found substantial differential expression overall in the diabetes experiment. After filtering, we were left with 151 proteins, of which 90 were called significant at q=0.05. Nearly all of the selected proteins were overexpressed in diabetics relative to controls. Figure 1 shows two examples of differentially expressed proteins: lumican and antithrombonin III. For lumican, the estimated group difference (control minus diabetic) is −1.28 on the log2 scale, meaning that control raw abundance is estimated to be 2−1.28≈40% that of diabetics on average. The estimated standard error of this estimate is 0.17, giving an approximate 95% confidence interval of [−1.62, −0.94]. Model-based standard errors and confidence intervals are also easily attained for peptides and any model contrast of interest. For example, the horizontal lines and shaded regions in Figure 1 show our protein-level estimates for each group, together with approximate 95% confidence intervals. We note that the lumican protein, as well as alpha-2-glycoprotein (zinc), were selected by our analysis and agree with a previously published biomarker study using these data (Metz et al., 2008). Also, using our model-based imputation routine resulted in a similar number of differentially expressed proteins at a 5% FDR (104 versus 90), while the standard alternative methods considered in the simulation studies, ANOVA on the complete data only and ANOVA after simple row-mean imputation, both found only 42 proteins at 5% FDR. This is a more substantial discrepancy than was observed in the simulations, perhaps due to a greater impact of our model-based filtering routine. We did not specifically include cases in the simulations that would highlight the filtering routine, but to the extent that it catches falsely identified or otherwise poor quality peptides, the filtering routine can substantially improve the quality of the data and hence significance analysis performance.
3.3 Salmonella mutant virulence data
The goal of this experiment was to find proteins that differ from wild-type Salmonella in the majority of the 13 mutant strains, as such proteins are thought to be best targets for disrupting virulence. After filtering, we used standard least-squares regression to fit the model
where WTi is the mean protein-level abundance for the wild-type, the PEPij are peptide effects and the MUTik are mean differences between the mutant and wild-type groups. We therefore wish to test the null hypothesis that all MUTik terms equal zero, for each protein.
For the i-th protein, we first compute mutant-specific test statistics
where 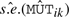 is the model-based standard error estimate for
is the model-based standard error estimate for 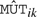 , k=1, 2,…, 13. A large absolute value for the k-th of these test statistics is indicative of a difference between mean abundance of the k-th mutant and wild-type. We use Ti=#{|Tik|≥2 : k=1, 2,…, 13} as the protein-level test statistic, where a large value of Ti is the evidence that many mutants differ from wild-type. To compute a P-value, we perform a bootstrap analysis, simulating many realizations of Ti under the null hypothesis and computing the proportion of these that exceed the observed statistic; see the Supplementary Material for more details. Using a Q-value threshold of 0.05, we select 55 of the 112 proteins. Included in this list of selected proteins are STM0831 and STM1044, both of which are known to contribute to Salmonella virulence in mice. A third protein known to be relevant to virulence, STM0448, was not selected by our method. This indicates that our analysis has reasonable statistical power to detect interesting proteins in this context.
, k=1, 2,…, 13. A large absolute value for the k-th of these test statistics is indicative of a difference between mean abundance of the k-th mutant and wild-type. We use Ti=#{|Tik|≥2 : k=1, 2,…, 13} as the protein-level test statistic, where a large value of Ti is the evidence that many mutants differ from wild-type. To compute a P-value, we perform a bootstrap analysis, simulating many realizations of Ti under the null hypothesis and computing the proportion of these that exceed the observed statistic; see the Supplementary Material for more details. Using a Q-value threshold of 0.05, we select 55 of the 112 proteins. Included in this list of selected proteins are STM0831 and STM1044, both of which are known to contribute to Salmonella virulence in mice. A third protein known to be relevant to virulence, STM0448, was not selected by our method. This indicates that our analysis has reasonable statistical power to detect interesting proteins in this context.
4 DISCUSSION
Observations for peptides from the same protein are correlated within a single sample, although preliminary investigation indicates that this correlation is negligible relative to other sources of variation in a typical MS-based proteomics experiment. Protein-level inference could also be carried out by first averaging the post-imputation complete peptide-level data, similar to what is done for rolling probesets up to the probe level in expression arrays. To the extent that within-sample correlation between peptides is indeed negligible, employing a peptide-level model like that in Equation (1) is to be preferred, as adjustment for peptide shifts will lower the standard error of the protein-level group effects.
We have avoided the issue of degenerate peptides, or peptides that could come from more than one protein. Instead, we simply randomly assign a single protein identity. Degenerate peptides constitute a small fraction of the total collection of peptides, ∼5% for the datasets considered here. We have also avoided the issue of peptide identification confidence levels. Since all peptide intensities are dependent on correct peptide identification, it is clear that protein quantitation is closely tied to peptide identification. In the datasets considered here, we have applied stringent identification criteria, typically using PeptideProphet posterior probabilities (Keller et al., 2002) of 0.95 or more for MS/MS-based identifications and in-house confidence measures for LC/MS-based identification. We intend to incorporate more systematic treatments of these issues in future work.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Navdeep Jaitly, Nathan Manes, Vlad Petyuk and Ashoka Polpitiya for helpful discussions. Therese Clauss and Marina Gritsenko assisted with sample preparation and instrument operation in the Salmonella mutant virulence experiment.
Funding: This work was sponsored by a subcontract from PNNL and by the NIH R25-CA-90301 training grant at TAMU. Additional support was provided by NIH grant DK070146 and by the National Institute of Allergy and Infectious Diseases (NIH/DHHS through interagency agreement Y1-AI-4894-01).
Conflict of Interest: none declared.
REFERENCES
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochbergv Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Dabney AR, Storey JD. A reanalysis of a published affymetrix genechip control dataset. Genome Biol. 2006;7:401. doi: 10.1186/gb-2006-7-3-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson TS. A Course in Large Sample Theory. London: Chapman and Hall; 1996. [Google Scholar]
- Gygi SP, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- Hill EG, et al. A statistical model for iTRAQ data analysis. J. Proteome Res. 2008;7:3091–3101. doi: 10.1021/pr070520u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, et al. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Kiebel G, et al. PRISM: a data management system for high-throughput proteomics. Proteomics. 2006;6:1783–1790. doi: 10.1002/pmic.200500500. [DOI] [PubMed] [Google Scholar]
- Lehmann EL. Testing Statistical Hypotheses. New York, NY: Springer; 1997. [Google Scholar]
- Lehmann EL, Casella G. Theory of Point Estimation. New York, NY: Springer; 2003. [Google Scholar]
- Lehti TM, et al. Effects of streptozotocin-induced diabetes and physical training on gene expression of extracellular matrix proteins in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2006;290:E900–E907. doi: 10.1152/ajpendo.00444.2005. [DOI] [PubMed] [Google Scholar]
- Little RJA, Rubin DB. Statistical Analysis with Missing Data. Hoboken, New Jersey: Wiley-Interscience; 2002. [Google Scholar]
- Metz TO, et al. Application of proteomics in the discovery of candidate protein biomarkers in a diabetes autoantibody standardization program sample subset. J. Proteome Res. 2008;7:698–707. doi: 10.1021/pr700606w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, et al. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods. 2007;4:787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- Oberg AL, et al. Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J. Proteome Res. 2008;7:225–233. doi: 10.1021/pr700734f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Old WM, et al. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics. 2005;4:1487–1502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- Pasa-Tolic L, et al. Proteomic analyses using an accurate mass and time tag strategy. BioTechniques. 2004;37:621–636. doi: 10.2144/04374RV01. [DOI] [PubMed] [Google Scholar]
- Polpitiya AD, et al. DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics. 2008;24:1556–1558. doi: 10.1093/bioinformatics/btn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. J. R. Stat. Soc. B. 2002;64:479–498. [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, et al. Charge competition and the linear dynamic range of detection in electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2004;15:1416–1423. doi: 10.1016/j.jasms.2004.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanskaya O, et al. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- Wang P, et al. Normalization regarding non-random missing values in high-throughput mass spectrometry data. Pacific Symposium Biocomputing. 2006:315–326. [PubMed] [Google Scholar]
- Wang W, et al. Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal. Chem. 2003;75:4818–4826. doi: 10.1021/ac026468x. [DOI] [PubMed] [Google Scholar]
- Zimmer JS, et al. Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom. Rev. 2006;23:450–482. doi: 10.1002/mas.20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.