

Abstract
We examined whether behavioral sensitization to amphetamine is associated with redistribution of glutamate receptors (GluR) in the rat nucleus accumbens (NAc) or dorsolateral striatum (DLSTR). Following repeated amphetamine treatment and 21 days of withdrawal, surface and intracellular levels of α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) or NMDA receptor subunits were determined using a protein cross-linking assay. In contrast to our previous results in cocaine-sensitized rats, we did not observe redistribution of GluR1 or GluR2 to the cell surface in the NAc after amphetamine withdrawal, although a small increase in total GluR1 was found in the shell subregion. Nor did we observe activation of signaling pathways associated with cocaine-induced AMPA receptor trafficking or changes in NMDA receptor subunits. No significant changes were observed in the DLSTR. We also investigated the effect of administering a challenge injection of amphetamine to amphetamine-sensitized rats 24 h prior to biochemical analysis based on prior studies showing that cocaine challenge decreases AMPA receptor surface expression in the NAc of cocaine-sensitized rats. GluR1 and GluR2 were not significantly altered in either NAc or DLSTR, although a modest effect on GluR3 cannot be ruled out. Our results suggest that glutamate transmission in the NAc is dramatically different in rats sensitized to amphetamine versus cocaine.
Keywords: α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor, amphetamine, drug abuse, nucleus accumbens, sensitization
Behavioral sensitization refers to the progressive enhancement of behavioral responses to drugs following their repeated intermittent administration, and has been well characterized for psychomotor stimulants (Robinson and Becker 1986; Post et al. 1992; Kalivas and Stewart 1991). Although most studies have measured locomotor sensitization, this is of interest mainly because it is believed to be related to incentive sensitization or enhanced ‘drug wanting’ (Robinson and Berridge 1993, 2008). Consistent with this idea, rats that develop locomotor sensitization subsequently show enhanced self-administration of psychomotor stimulants (Vezina 2004).
Although dopamine (DA) transmission plays a critical role in sensitization, it is now well accepted that glutamate transmission in the ventral tegmental area and nucleus accumbens (NAc) is important for the development and expression of behavioral sensitization, respectively (Wolf 1998; Vanderschuren and Kalivas 2000). More recent evidence suggests that glutamate transmission in the NAc is also a key link between locomotor sensitization and drug seeking. Reinstatement of cocaine seeking is blocked by intra-NAc infusion of α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) receptor (AMPAR) antagonists and can be elicited by AMPA infusion into the NAc (Cornish et al. 1999; Cornish and Kalivas 2000; Di Ciano and Everitt 2001; Bäckström and Hyytiä 2006). In rats previously sensitized to amphetamine, a lower concentration of AMPA can elicit reinstatement of cocaine seeking (Suto et al. 2004). This suggests that sensitization increases the responsiveness of NAc neurons to excitatory inputs, promoting drug seeking.
One of the most parsimonious explanations for these results is that AMPARs are up-regulated in the NAc following sensitization to psychomotor stimulants. Supporting this possibility, both biochemical and electrophysiological studies have found increased surface and synaptic AMPAR levels after withdrawal from sensitizing regimens of cocaine (Boudreau and Wolf 2005; Boudreau et al. 2007; Kourrich et al. 2007). These and other results (Wolf et al. 2004; Kauer and Malenka 2007) suggest that behavioral sensitization to cocaine shares common mechanisms with long-term potentiation (LTP) and other forms of synaptic plasticity in which the trafficking of AMPARs to and from synapses critically regulates synaptic strength (Malinow and Malenka 2002; Bredt and Nicoll 2003; Turrigiano and Nelson 2004). The purpose of the present study was to determine whether AMPAR surface expression in the NAc increases after amphetamine sensitization, as it does after cocaine sensitization (Boudreau and Wolf 2005; Boudreau et al. 2007; Kourrich et al. 2007). We also examined the effect of acute amphetamine treatment on AMPAR surface expression.
Recent research on behavioral sensitization has focused on the ventral striatum (particularly the NAc) because it serves as an interface between brain regions critically involved in motivation and behavioral output (Groenewegen et al. 1999; Kelley 2004). However, in amphetamine-sensitized rats, certain neuroadaptations are common to the dorsal striatum and NAc (Patrick et al. 1991; Robinson and Kolb 2004; Jedynak et al. 2007). Thus, we investigated both regions in the present study, focusing on a region of dorsolateral striatum (DLSTR) that exhibits increased c-fos expression after repeated cocaine (Willuhn et al. 2003) and increased spine density after repeated methamphetamine (Jedynak et al. 2007). We used a protein cross-linking assay to distinguish between surface (S) and intracellular (I) pools of AMPAR subunit proteins, and extended our investigation to assess NMDA receptor subunits and signaling cascades relevant to AMPAR trafficking. In stark contrast to results obtained by our laboratory and others for cocaine sensitization, withdrawal from amphetamine sensitization was not accompanied by increased AMPAR surface expression. Furthermore, changes in signaling pathway activation associated with AMPAR up-regulation after cocaine (Boudreau and Wolf 2006) were not observed in the NAc of amphetamine-sensitized rats. NMDAR distribution was also unaltered in the NAc. Likewise, no significant changes in AMPA or NMDA receptor surface expression were observed in the DLSTR.
Materials and methods
Animals
Male Sprague–Dawley rats weighing between 275 and 300 g at the beginning of the study (Harlan Laboratories, Indianapolis, IN, USA) were used for all experiments. Animals were group-housed with food and water available ad libitum. Animals had a 12 h day/night cycle with lights on at 6:00 am and off at 6:00 pm. All procedures were approved by the Institutional Animal Care and Use Committee of Rosalind Franklin University of Medicine and Science. All animals were handled extensively and housed in the colony for at least 7 days prior to use.
Drug treatment and behavioral analysis
Acute amphetamine experiment
Rats were injected on the test day (saline or 2.5 mg/kg amphetamine, s.c.; dose refers to free base), returned to home cages, and killed after 30 min, 2 h or 24 h.
General procedures for repeated amphetamine experiments
Rats were habituated to the testing environment 24 h prior to the first amphetamine or saline injection. To accomplish this, animals were placed in photobeam cages in the test room (one animal per cage; San Diego Instruments, San Diego, CA, USA), left undisturbed for 20 min, and then administered a mock injection (abdomen touched by a syringe with no needle). Rats remained in the photobeam cages for 4 h, the total time of behavioral analysis on subsequent test days. On the following day (treatment day 1), animals were weighed, placed in the photobeam cages in the testing room, and habituated for 20 min. They were then injected with either saline or amphetamine and horizontal locomotor activity (ambulation counts) was measured for 4 h. Animals were then returned to their home cages. Injections on days 2–5 occurred in home cages, and the day 6 injection occurred either in the home cage or the test cage depending on the experiment, as outlined below. All withdrawal periods occurred in home cages. On drug challenge days, procedures for injection and behavioral analysis were identical to those described for treatment day 1. Drug doses, withdrawal times, and challenge procedures varied between experiments, as described below and illustrated in Fig. 1.
Fig. 1.

Schematic diagram to illustrate the time-line of repeated amphetamine experiments. In all experiments, rats received repeated injections (REP INJ) of amphetamine (AMPH) or saline (SAL) on six consecutive treatment days (TD). For Withdrawal Experiments 1–3, rats were challenged (CHAL) on withdrawal day 7 (WD7) and killed (KILL) on WD21 (indicated by labeling above horizontal line). For the Challenge Experiment, rats were challenged on WD20 and killed on WD21 (indicated by labeling below horizontal line). See Materials and Methods for details of each regimen.
Withdrawal Experiment 1
Animals were divided into two groups, drug and control, and received an injection of amphetamine (2.5 mg/kg, s.c.; all doses refer to free base) or saline on day 1 in the test cages. On days 2–6, animals received a single injection of either amphetamine (5.0 mg/kg, s.c.) or saline in their home cages. Seven days following the final injection of the regimen (withdrawal day 7), animals were given a single injection of either amphetamine (2.5 mg/kg, s.c.) or saline in the photobeam cages to test for behavioral sensitization. Fourteen days after the test for sensitization, animals were decapitated for biochemical analysis (a total of 21 days after discontinuing repeated saline or amphetamine injections). The entire NAc (core and shell) was dissected in this experiment (see Surface receptor cross-linking for description of all dissections). This regimen was designed to resemble the cocaine regimen that produced AMPAR up-regulation in the NAc, which used a lower dose on days when locomotor activity was measured and a higher dose on intervening days (Boudreau and Wolf 2005). We assessed sensitization by challenging with amphetamine on withdrawal day 7 rather than comparing the first and last treatment days because amphetamine sensitization with this regimen required several days to fully manifest (data not shown). We selected the 2.5 mg/kg dose for treatment day 1 and challenge days because this dose enabled detection of sensitization of both stereotypy and post-stereotypy locomotor hyperactivity (Segal and Kuczenski 1987; Wolf et al. 1995).
Withdrawal Experiment 2
This experiment was identical to Withdrawal Experiment 1 except that the NAc was further dissected into core-enriched and shell-enriched portions (see Surface receptor cross-linking) which were then analyzed separately.
Withdrawal Experiment 3
Animals received an injection of saline or amphetamine (2.5 mg/kg, i.p.) in the test cages on day 1. On days 2–6, animals received either an injection of amphetamine (2.5 mg/kg, i.p.) or saline in their home cages. Seven days following the completion of the treatment regimen (withdrawal day 7), animals received an injection of either amphetamine (2.5 mg/kg, i.p.) or saline in the testing cages to assess behavioral sensitization. Fourteen days after the test for sensitization, animals were decapitated for biochemical analysis (a total of 21 days after discontinuing repeated saline or amphetamine injections). We obtained core-enriched, shell-enriched, and DLSTR tissue samples (see Surface receptor cross-linking). This regimen was designed after negative results were obtained in Withdrawal Experiments 1 and 2. The route of amphetamine administration was changed to match our previous cocaine studies (Boudreau and Wolf 2005; Boudreau et al. 2007) and the same amphetamine dose was given on all days in order to examine the effects of a less aggressive regimen which like cocaine produced locomotor hyperactivity without extensive stereotypy.
Challenge experiment
Animals were divided into four groups based on both pre-treatment (drug or saline) and challenge (drug or saline). In the pre-treatment phase, animals received the same 6-day regimen of saline or amphetamine injections as in Withdrawal Experiment 3 except that the injection on day 6 was given in the photobeam cages. These data were used in part to determine the withdrawal time necessary for amphetamine sensitization to be manifest (see Withdrawal Experiment 1). Animals were then returned to their home cages and left undisturbed for 19 days. On withdrawal day 20, animals received a challenge injection of either amphetamine (2.5 mg/kg, i.p.) or saline in the photobeam cages, and they were decapitated for biochemical analysis 24 h later (thus as in all of the above experiments rats were killed a total of 21 days after discontinuing repeated injections). The 24 h time-point was selected based on our prior studies of cocaine sensitization. While AMPAR surface expression was increased following withdrawal from cocaine sensitization, a challenge injection of cocaine reversed this effect such that decreased AMPAR surface expression was observed 24 h following the challenge (Boudreau et al. 2007). The entire NAc (core and shell) and the DLSTR were collected in this experiment.
Surface receptor cross-linking
Rat brain tissue was incubated with protein cross-linking reagent bis(sulfosuccinimidyl)suberate (BS3; Pierce Biotechnology, Rockford, IL, USA) to determine S and I levels of receptor subunit proteins as described previously (Boudreau and Wolf 2005; Boudreau et al. 2007). BS3 does not cross cell membranes so it selectively cross-links surface-expressed proteins, forming high molecular weight aggregates, while intracellular proteins are not modified. This enables S and I pools of a protein to be distinguished based on molecular weight using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and western blotting. Unlike biotinylation which requires S and I fractions to be purified, cross-linked (surface) and non-crosslinked (intracellular) bands can be quantified in the same lane (see Fig. 2 for representative blots for each antigen of interest). Briefly, animals were decapitated and their brains removed rapidly. A 2 mm coronal section containing the NAc and DLSTR (extending from approximately 0.7 to 2.7 mm anterior to Bregma; Paxinos and Watson 1998) was obtained using a brain matrix. In the case of the entire NAc, bilateral tissue was dissected on an ice-cold platform and immediately chopped into 400 μm slices with a McIllwain tissue chopper (Vibratome, St. Louis, MO, USA). To obtain core- and shell-enriched samples, the NAc was rapidly dissected using a scalpel blade, according to core/shell boundaries defined in Paxinos and Watson (1998). Each subregion was then chopped manually with a scalpel blade (the subregions were too small to be compatible with the McIllwain chopper). For the DLSTR, bilateral punches (∼2 mm in diameter) were collected from dorsolateral striatum in the same 2-mm coronal brain section. Punches were chopped manually with a scalpel blade. Immediately after chopping, tissue was added to Eppendorf tubes containing 1 ml of ice-cold artificial CSF, spiked with BS3 (final concentration of 2 mM), and incubated with gentle agitation at 4°C for 30 min (Withdrawal Experiment 1), 20 min (Withdrawal Experiment 2), or 15 min (Withdrawal Experiment 3, Challenge Experiment). Extensive methodological studies (Boudreau et al. in preparation) have established that 15–30 min of cross-linking is optimal for detection of AMPAR subunit surface expression. We used shorter cross-linking times (15–20 min) for later experiments in the hope that this might help us detect very small changes in AMPAR surface expression. However, as described below, control and amphetamine-sensitized groups did not differ regardless of the duration of cross-linking. Likewise, the relationship between control and cocaine-sensitized groups was the same regardless of whether cross-linking was performed for 30 min (Boudreau and Wolf 2005) versus 15 min (Boudreau et al. 2007). Cross-linking was terminated by quenching the reaction with 100 mM glycine for 10 min at 4°C. Slices were then centrifuged for 2 min at 20 800 g and the supernatant was discarded. The pellets were resuspended in ice-cold lysis buffer containing protease and phosphatase inhibitors [25 mM HEPES, pH 7.4; 500 mM NaCl; 2 mM EDTA; 1 mM dithiothreitol; 1 mM phenylmethanesulfonyl fluoride; 20 mM NaF; 1 mM Na orthovanadate; 10 mM Na pyrophosphate; 1 μM microcystin-LF; 1 μM okadic acid; 1× protease inhibitor cocktail (EMD Biosciences, San Diego, CA, USA); 0.1% Nonidet P-40 (v/v) (Fluka, Buchs, Switzerland)] and sonicated (5 sec). Samples were then centrifuged for 2 min at 20 800 g, and the supernatant fraction was collected for western blot analysis. Each sample was aliquoted and stored at −80°C until use. Total protein concentration for each sample was determined according to the Lowry method (Lowry et al. 1951).
Western blotting
Samples (∼30 μg of total protein/lysate sample) were loaded and run on 4–15% Tris–HCl gels (Bio-Rad, Hercules, CA, USA) or 3–8% Bis-Tris gradient gels (Invitrogen, Carlsbad, CA, USA), both under reducing conditions. Proteins were transferred onto polyvinylidene fluoride membranes for immunoblotting using Bio-Rad transfer cells. All membranes were dried and stored at 4°C following transfer. Membranes were then rewetted with methanol, washed thoroughly with water and Tris-buffered saline (TBS), and then blocked with either 5% non-fat dry milk/1% goat serum in TBS with 0.05% Tween 20 (v/v) (TBS-T) or 3% bovine serum albumin in TBS-T, pH 7.4, for 1 h at room temperature (21°C), depending on the experiment. Membranes were then briefly rinsed with TBS and incubated overnight at 4°C in primary antibody (see below). The following morning, membranes were washed extensively in TBS-T and incubated for 60 min with horse radish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies (1 : 10000; Millipore; Billerica, MA, USA). Membranes were then washed with TBS-T (5 min, six rinses), TBS (5 min, one rinse), and water (15 min). Detecting substrate (enhanced chemiluminescence; GE Healthcare, Piscataway, NJ, USA) was then applied for 1 min, and images were captured with a Versa Doc system (Bio-Rad). The adjusted volume of surface and intracellular bands was calculated using Quantity One software (Bio-Rad). Total protein in each lane was measured by staining membranes with Ponceau S (Sigma-Aldrich, St. Louis, MO, USA), avoiding reliance on a single protein as a loading control. The following primary antibodies were used in our studies: GluR1, 1 : 500, Millipore; GluR2, 1 : 2000, Millipore; GluR2/3, 1 : 2000, Millipore: NR2A/B, 1 : 2000, Millipore; NR2B, 1 : 2500, Calbiochem (Gibbstown, NJ, USA); extracellular signal-regulated protein kinase (ERK1/2), 1 : 20000, Millipore; phospho-ERK1/2 (p-ERK1/2), 1 : 10000, Millipore; antibody recognizing phosphorylated substrates of protein kinase A (PKA), 1 : 5000, Cell Signaling (Danvers, MA, USA); Ca2+-calmodulin-dependent protein kinase II (CaMKII), 1 : 20000, Millipore; phospho-CaMKII (p-CaMKII), 1 : 5000, Phophosolutions (Aurora, CO, USA). For some experiments, membranes were stripped after probing for the first antigen of interest by incubating for 20 min in ReBlot Plus Strong Antibody Stripping Solution (Millipore). After stripping, membranes were washed with water for 10 min, re-blocked, and then reprobed. Stripped blots were used to assess total levels of a protein of interest after initially probing with a phospho-specific antibody (e.g., total ERK after pERK). Because multiple aliquots were generated for each rat (∼15 from the whole NAc), we were able to analyze many proteins of interest for each rat in each group. However, for some experimental groups, the n value varies slightly depending on the protein analyzed. For example, in the Amphetamine group from Withdrawal Experiment 1, n = 11 for GluR1 (Table 2) and n = 12 for NR2A/B (Table 3). Such differences reflect problems with specific lanes in some western blots.
Data analysis
For behavioral analysis, we tested for sensitization by comparing ambulation counts on the first treatment day (day 1) and the test day using two-way anova with time as a repeated measure. For this analysis, we used ambulation counts from the post-stereotypy locomotor hyperactivity portion of the test session (60–180 min; see Results). The Huynh-Feldt correction was used to reduce the possibility of Type I error. For western blots, surface (S), intracellular (I), and total S + I values for the protein of interest were normalized to total protein in the lane (determined by staining with Ponceau S; see above) to control for loading variation. S/I, a measure of protein distribution, was not normalized because both values are obtained from the same lane. Results from experimental groups were then normalized to mean values from the saline control group. Differences between groups were determined with anova followed by appropriate post hoc t-tests. All anova procedures were conducted with spss 12.0 (SPSS Inc., Chicago, IL, USA). Significance was set at p < 0.05.
Results
Acute amphetamine injection alters AMPAR expression and distribution in the NAc
AMPAR subunit surface and intracellular pools were quantified using a membrane-impermeant cross-linking reagent (BS3) to selective modify surface-expressed proteins, enabling them to be distinguished from unmodified intracellular proteins using SDS–PAGE and western blotting (see Materials and Methods for more details). Representative blots illustrating surface and intracellular bands of interest are shown in Fig. 2. To determine the effect of acute amphetamine on AMPAR distribution in the NAc, three experiments were conducted in which tissue was collected 30 min, 2 h or 24 h after injection of 2.5 mg/kg amphetamine (s.c.). After 30 min, a trend towards an increase (∼10%) was found for both surface (S) and total (S + I) GluR1 in amphetamine-treated rats (Fig. 3); this was nearly significant for S + I [F(1,31) = 1.66; p = 0.054]. Similarly, S, I, and S + I were each increased for GluR2/3 by about 10%, but those results did not achieve statistical significance (Table 1). In rats killed 2 h following amphetamine injection, surface GluR1 levels were significantly increased [F(1,15) = 5.65; p = 0.03] and there was a trend towards increased S + I [∼ 20% increase; F(1,15) = 2.02; p = 0.18] (Fig. 3). Conversely, GluR2/3 showed a slight reduction in S, I, and S + I values (∼15% decreases; not statistically significant) (Table 1). Finally, 24 h after acute amphetamine, values for S, S + I, and the S/I ratio for GluR1 were slightly but not significantly decreased compared with control levels (Fig. 3). GluR2/3 measures were also not altered 24 h after amphetamine (Table 1). Taken together, these results suggest that acute administration of amphetamine produced a modest up-regulation of GluR1 surface expression that was transiently present 2 h post-injection but returned to baseline by 24 h post-injection.
Fig. 2.
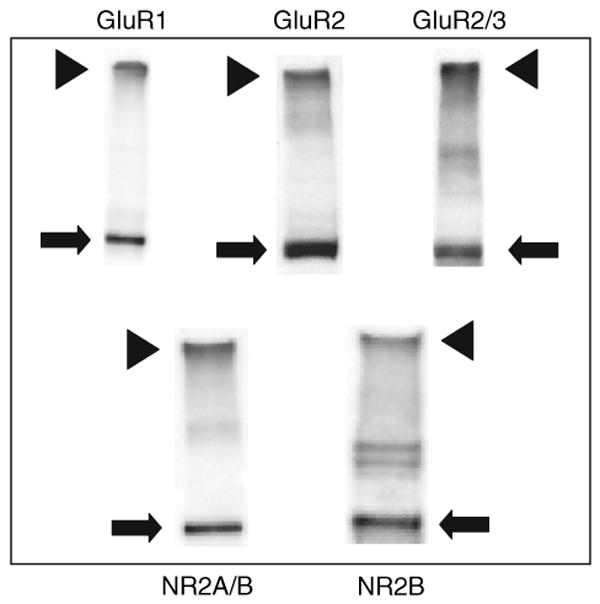
Representative immunoblots illustrating analysis of glutamate receptor subunit surface expression in nucleus accumbens tissue. Tissue was incubated for 15 min with the membrane-impermeant protein cross-linking reagent BS3, and then processed as described in the Materials and Methods section. BS3 selectively cross-links cell surface receptors, forming high molecular weight aggregates, while intracellular receptors are not modified. Thus, surface (S, arrowheads) and intracellular (I, arrows) pools can be distinguished based on molecular weight using SDS–PAGE and western blotting. Intracellular bands ran at their predicted molecular weights (in kD; GluR1, ∼101; GluR2, ∼99; GluR3, ∼99; NR2A, ∼166; NR2B, ∼166). Note that apparent differences in surface/intracellular ratios between particular subunits do not necessarily reflect differences in the relative portion of each subunit expressed on the surface because cross-linking conditions and antibody differences can affect the S/I ratio. The value of the assay lies in providing a measure of relative differences in S/I ratios between samples that are processed identically and probed with the same antibody.
Fig. 3.

Effect of acute amphetamine (AMPH) injection on GluR1 distribution in the nucleus accumbens. Rats were killed 30 min [top: n = 16, saline (SAL); n = 17, AMPH], 2 h (middle; n = 8, SAL; n = 9, AMPH), or 24 h (bottom; n = 8, SAL; n = 8, AMPH) after injection of saline or 2.5 mg/kg amphetamine. A protein cross-linking assay was used to measure surface (S), intracellular (I), and total (S + I) GluR1 levels, and determine the GluR1 S/I ratio. S, I, and S + I values are normalized to total protein in the lane determined with Ponceau S staining. All data are mean ± SEM expressed relative to SAL control values. *p < 0.05 compared with saline group (anova and post hoc t-test).
Table 1.
Effect of acute amphetamine on GluR2/3
| n | S | I | S + I | S/I | |
|---|---|---|---|---|---|
| 30 min | |||||
| Saline | 16 | 100 (8.4) | 100 (6.8) | 100 (7.3) | 100 (5.4) |
| AMPH | 17 | 107.9 (7.2) | 106.5 (4.8) | 107.2 (5.9) | 101.2 (4.8) |
| 2 h | |||||
| Saline | 8 | 100 (10.3) | 100 (17.6) | 100 (12.2) | 100 (11.6) |
| AMPH | 9 | 87.5 (7.7) | 80.6 (11.7) | 84.7 (8.5) | 108.2 (13.1) |
| 24 h | |||||
| Saline | 8 | 100 (14.6) | 100 (12.3) | 100 (12.4) | 100 (11.4) |
| AMPH | 8 | 101.6 (13.5) | 91.4 (8.6) | 97.6 (11.1) | 106.4 (8.1) |
AMPH, amphetamine; S, surface; I, intracellular.
Values are mean (SEM) expressed as percent of the saline control group.
Repeated amphetamine administration produces behavioral sensitization
We utilized two different amphetamine regimens: a more robust regimen consisting of 2.5 mg/kg on day 1 and 5 mg/kg on days 2–6 (Withdrawal Experiments 1 and 2) and a moderate regimen consisting of 2.5 mg/kg for 6 days (Withdrawal Experiment 3). In both cases, the first injection was given in the test environment and subsequent injections were given in the home cage. With both regimens, the vast majority of amphetamine-treated animals exhibited a sensitized response upon receiving a 2.5 mg/kg challenge injection on withdrawal day 7. With the more robust regimen, a reproducible biphasic pattern of activity was observed on the test day, characterized by decreased ambulation counts during the first hour followed by 2 h of locomotor hyperactivity (Fig. 4a, b). We confirmed that the decrease in ambulation counts was due to increased in-place stereotyped behaviors (data not shown). This biphasic response pattern has been described previously by our laboratory (e.g., Wolf et al. 1995) and others (Segal and Kuczenski 1987). The 2.5 mg/kg/day regimen produced a more variable response within the first hour of the test day, with animals displaying stereotypy and some hyperactivity, followed in the next 2 h by a reproducible period of locomotor hyperactivity compared with day 1 (Fig. 4c). In the Challenge Experiment, animals received a challenge injection of amphetamine (2.5 mg/kg) 20 days after completing the same regimen used for Withdrawal Experiment 3 (2.5 mg/kg for 6 days). The response pattern observed for these rats (Fig. 4d) was very similar to that observed when rats from Withdrawal Experiment 3 were tested for sensitization (Fig. 4c).
Fig. 4.

Ambulation counts (mean ± SEM) in response to amphetamine (AMPH) injection (2.5 mg/kg) on treatment day 1 and on the test (challenge) day. Panels a–d show results from Withdrawal Experiment 1 (n = 12), 2 (n = 15), 3 (n = 12) and the Challenge Experiment (n = 13). For Withdrawal Experiments 1–3, the test day was on withdrawal day 7 after discontinuing repeated amphetamine injections. For the Challenge Experiment, the test day was withdrawal day 20. The more aggressive regimen used in Withdrawal Experiments 1 and 2 (see Materials and Methods) produced a biphasic response pattern on the test day characterized by stereotypy during the first hour (reflected in decreased ambulation counts) followed by locomotor hyperactivity over the next 2 h. The more moderate regimen used for Withdrawal Experiment 3 and the Challenge Experiment did not result in consistent sensitization of stereotyped behaviors but did lead to robust post-stereotypy locomotor hyperactivity. See Results for statistical analyses.
Based on the variable response in the first hour of the test across regimens, statistical analysis of these experiments was based on activity during the locomotor hyperactivity phase (60–180 min). Two-way anova with time as the repeated measure was used to compare ambulation counts on day 1 and the challenge day in the amphetamine groups. For Withdrawal Experiment 1, there were significant effects of Day [F(1,11) = 87.71; p < 0.00] and Time [F(11,121) = 5.56; p < 0.00] as well as a significant interaction [F(11,121) = 5.20; p < 0.00]. Similar results were found for Withdrawal Experiment 2 [Day: F(1,14) = 35.76; p < 0.00); Time: F(11,154) = 5.26; p = 0.01; Interaction: F(11,154) = 2.84; p = 0.04], Withdrawal Experiment 3 [Day: F(1,11) = 19.42; p < 0.00; Time: F(11,121) = 23.80; p < 0.00; Interaction: F(11,121) = 4.09; p < 0.00], and the Challenge Experiment [Day: F(1,12) = 23.40; p < 0.00; Time: F(11,132) = 16.91; p < 0.00; Interaction: F(11,132) = 3.38; p = 0.01].
Four animals were excluded from biochemical analysis (three animals in Withdrawal Experiment 2 and one animal in the Challenge Experiment). One died before the challenge test, one had no significant locomotor response to the day 1 amphetamine injection, one had no response to the challenge injection, and one showed a very small locomotor response to amphetamine that did not differ between day 1 and the challenge day (ambulation counts plotted as a function of time were nearly superimposable).
GluR1 expression and distribution in the NAc are not significantly altered following withdrawal from repeated amphetamine administration
The major goal of these studies was to determine AMPAR distribution in the NAc following a period of withdrawal from a sensitizing amphetamine regimen (see Fig. 1 for schematic of experimental design). First, we examined GluR1 distribution 21 days after the cessation of repeated amphetamine treatment (Withdrawal Experiment 1; 2.5 mg/kg on day 1, 5 mg/kg on days 2–6, amphetamine challenge on withdrawal day 7 to establish behavioral sensitization, and tissue preparation on withdrawal day 21). We found no significant effects for any GluR1 measure (S, I, S + I, S/I) in amphetamine-treated animals when compared with controls (Table 2). We believe that these results were not influenced by the amphetamine challenge on withdrawal day 7 because we obtained the same negative results in rats killed on withdrawal day 21 without the withdrawal day 7 challenge (Nelson and Wolf, unpublished observations).
Table 2.
Effect of repeated amphetamine on GluR1
| n | S | I | S + I | S/I | |
|---|---|---|---|---|---|
| Withdrawal Experiment 1 | |||||
| NAc: Saline | 12 | 100 (11.7) | 100 (13.2) | 100 (12.2) | 100 (9.2) |
| NAc: AMPH | 11 | 95.1 (6.4) | 103.8 (10.2) | 101.5 (8.8) | 89.5 (5.7) |
| Withdrawal Experiment 2 | |||||
| Core: Saline | 18 | 100 (3.8) | 100 (3.6) | 100 (3.0) | 100 (4.6) |
| Core: AMPH | 14 | 104.9 (6.0) | 100.9 (3.2) | 103.7 (3.8) | 103.0 (6.0) |
| Shell: Saline | 18 | 100 (5.5) | 100 (4.7) | 100 (3.9) | 100 (6.4) |
| Shell: AMPH | 15 | 120.8 (11.6) | 124.6 (12.1) | 123.9 (10.0) | 105.6 (15.7) |
| Withdrawal Experiment 3 | |||||
| Core: Saline | 12 | 100 (6.8) | 100 (5.5) | 100 (4.1) | 100 (10.1) |
| Core: AMPH | 12 | 100.6 (9.5) | 103.1 (7.6) | 102.2 (5.7) | 100.2 (13.5) |
| Shell: Saline | 12 | 100 (7.1) | 100 (5.8) | 100 (4.4) | 100 (12.1) |
| Shell: AMPH | 12 | 105.6 (6.0) | 116.6 (7.2) | 113.6 (5.8) | 88.1 (6.1) |
| DLSTR: Saline | 12 | 100 (8.7) | 100 (3.1) | 100 (2.5) | 100 (10.5) |
| DLSTR: AMPH | 12 | 100.3 (8.5) | 113.9 (6.4) | 111.8 (6.3) | 86.4 (5.8) |
DLSTR, dorsolateral striatum; NAc, nucleus accumbens; AMPH, amphetamine; S, surface; I, intracellular.
Values are mean (SEM) expressed as percent of the saline control group. Shaded area indicates significance (p < 0.05).
Based on numerous studies indicating differential roles for core and shell subregions of the NAc in animal models of addiction (Everitt and Robbins 2005; Ikemoto 2007), we next treated additional rats with the same regimen and then dissected portions of the NAc enriched for either core or shell (Withdrawal Experiment 2). Similar to the initial experiment, we found no significant effects of amphetamine treatment on GluR1 measures in the core (Table 2). In the shell, however, there was a modest but significant increase in total levels (S + I) of GluR1 [F(1,31) = 5.62; p = 0.03]. This effect was reflected in both the S [F(1,31) = 2.92; p = 0.10] and I [F(1,31) = 4.11; p = 0.05] components. No change in the GluR1 S/I ratio was found.
Finally, we utilized a more moderate regimen (Withdrawal Experiment 3; 2.5 mg/kg/day for days 1–6, amphetamine challenge on withdrawal day 7 to establish sensitization, and tissue collection on withdrawal day 21) and evaluated core- and shell-enriched dissections separately. No significant alterations in GluR1 were demonstrated in the core of the NAc following repeated amphetamine treatment (Table 2). However, similar to Withdrawal Experiment 2, the shell showed a trend towards an increase in total (S + I) GluR1 levels [F(1,22) = 3.46; p = 0.08], attributable mostly to an increase in I [F(1,22) = 3.19; p = 0.09].
GluR2 and GluR3 expression and distribution in the NAc are not significantly altered following withdrawal from repeated amphetamine administration
Additional aliquots of tissue from the same cohorts of rats used to evaluate GluR1 were analyzed using an antibody recognizing both the GluR2 and GluR3 subunits (GluR2/3; Table S1). No amphetamine-treated group in any of the three withdrawal experiments demonstrated any significant alteration in GluR2/3 measures (S, I, S + I, S/I) in core, shell, or total NAc when compared with corresponding controls. NAc tissue from rats in Withdrawal Experiments 1 and 3 was also analyzed using an antibody selective for GluR2 (Table S2). Similar to results obtained with the GluR2/3 antibody, amphetamine-treated rats did not demonstrate any significant changes when compared with controls. Negative results with both the GluR2/3 and GluR2 antibodies suggest that GluR3 distribution and expression were not altered.
NR2A and NR2B expression and distribution in the NAc are not significantly altered following repeated amphetamine administration
Additional aliquots of NAc tissue from the same rats analyzed for AMPAR subunit expression (Withdrawal Experiments 1–3) were probed with an antibody recognizing both the NR2A and NR2B subunits (NR2A/B; Table 3). No significant differences were found between experimental groups for NR2A/B. However, in Withdrawal Experiment 1 (the experiment with the more robust amphetamine regimen and the full NAc dissection), there were trends towards a modest increase in I [F(1,22) = 1.82; p = 0.19] and a modest decrease in the S/I ratio [F(1,22) = 2.59; p = 0.12]. However, in tissue from Withdrawal Experiments 2 and 3, these trends were not apparent (Table 3). We also utilized an NR2B-selective antibody to probe tissue from Withdrawal Experiments 1 and 3 (Table 4). In tissue from Withdrawal Experiment 1, there was a modest decrease in the S/I ratio (∼12%), similar to that observed with the NR2A/B antibody, mostly driven by a decrease in S [F(1,21) = 2.84; p = 0.11]. However, this trend was not evident in tissue from Withdrawal Experiment 3 (Table 4). NR2A was not analyzed in this or other experiments because we have not found an NR2A-selective antibody that is compatible with this protein cross-linking assay, but negative results with NR2A/B and NR2B antibodies suggest that NR2A was not altered.
Table 3.
Effect of repeated amphetamine on NR2A/B
| n | S | I | S + I | S/I | |
|---|---|---|---|---|---|
| Withdrawal Experiment 1 | |||||
| NAc: Saline | 12 | 100 (6.6) | 100 (6.3) | 100 (5.7) | 100 (8.4) |
| NAc: AMPH | 12 | 98.9 (10.3) | 114.7 (8.9) | 101.8 (9.6) | 83.3 (6.5) |
| Withdrawal Experiment 2 | |||||
| Core: Saline | 16 | 100 (10.4) | 100 (7.8) | 100 (7.0) | 100 (13.1) |
| Core: AMPH | 15 | 105.0 (9.4) | 103.0 (7.7) | 103.5 (7.9) | 95.0 (5.9) |
| Shell: Saline | 18 | 100 (5.2) | 100 (6.1) | 100 (4.9) | 100 (6.1) |
| Shell: AMPH | 15 | 107.7 (8.7) | 104.6 (9.8) | 105.4 (7.1) | 108.5 (9.2) |
| Withdrawal Experiment 3 | |||||
| Core: Saline | 12 | 100 (16.5) | 100 (14.4) | 100 (14.3) | 100 (14.3) |
| Core: AMPH | 12 | 109.6 (8.4) | 119.2 (20.8) | 114.5 (13.2) | 116.8 (21.2) |
| Shell: Saline | 12 | 100 (6.5) | 100 (11.0) | 100 (5.2) | 100 (12.0) |
| Shell: AMPH | 12 | 94.1 (4.4) | 84.2 (13.0) | 90.5 (6.0) | 113.1 (10.9) |
| DLSTR: Saline | 12 | 100 (5.9) | 100 (10.8) | 100 (7.5) | 100 (10.6) |
| DLSTR: AMPH | 12 | 95.8 (6.3) | 103.5 (10.0) | 100.0 (7.2) | 89.4 (7.1) |
DLSTR, dorsolateral striatum; NAc, nucleus accumbens; AMPH, amphetamine; S, surface; I, intracellular.
Values are mean (SEM) expressed as percent of the saline control group.
Table 4.
Effect of repeated amphetamine on NR2B
| n | S | I | S + I | S/I | |
|---|---|---|---|---|---|
| Withdrawal Experiment 1 | |||||
| NAc: Saline | 12 | 100 (5.0) | 100 (10.1) | 100 (5.1) | 100 (10.9) |
| NAc: AMPH | 11 | 88.3 (4.8) | 96.4 (9.8) | 92.6 (6.5) | 88.4 (7.9) |
| Withdrawal Experiment 3 | |||||
| Core: Saline | 12 | 100 (7.5) | 100 (6.3) | 100 (5.9) | 100 (6.2) |
| Core: AMPH | 12 | 99.8 (7.2) | 105.7 (9.0) | 103.7 (7.2) | 97.2 (7.7) |
| Shell: Saline | 12 | 100 (12.7) | 100 (9.3) | 100 (9.9) | 100 (8.1) |
| Shell: AMPH | 12 | 83.4 (6.5) | 88.9 (10.1) | 86.9 (8.6) | 98.6 (5.2) |
| DLSTR: Saline | 12 | 100 (5.5) | 100 (9.5) | 100 (8.1) | 100 (8.5) |
| DLSTR: AMPH | 12 | 87.2 (5.5) | 90.4 (6.6) | 89.7 (5.9) | 92.9 (6.2) |
DLSTR, dorsolateral striatum; NAc, nucleus accumbens; AMPH, amphetamine; S, surface; I, intracellular
Values are mean (SEM) expressed as percent of the saline control group.
Withdrawal from repeated amphetamine does not significantly affect AMPA or NMDA receptor expression or distribution in the DLSTR
In Withdrawal Experiment 3, we collected the DLSTR in addition to the core and shell subregions of the NAc. First, we examined AMPAR distribution by utilizing the same GluR1, GluR2/3, and GluR2 antibodies used in the NAc experiments (Table 2, Tables S1 and S2). For GluR1, there was a trend towards an increase in both I [F(1,22) = 3.76; p = 0.07] and S + I [F(1,22) = 3.02; p = 0.10]. For GluR2 and GluR2/3, no significant changes were demonstrated. Similarly, no significant alterations were detected in the DLSTR of amphetamine-treated groups using the NR2A/B and NR2B antibodies (Tables 3 and 4).
Amphetamine challenge does not significantly alter AMPAR subunits in the NAc
In the Challenge Experiment, rats were treated with repeated amphetamine (2.5 mg/kg × 6 days), a 2.5 mg/kg challenge injection was administered on day 20, and 24 h later the NAc was dissected and cross-linked with BS3. There were four groups in this experiment: animals that were both pre-treated and challenged with saline (S/S), animals pre-treated with saline and challenged with amphetamine (S/A), animals pre-treated with amphetamine and challenged with saline (A/S), and animals both pre-treated and challenged with amphetamine (A/A). Cross-linked tissue from all groups was probed with antibodies to GluR1 and GluR2/3, and compared with the S/S control group. For GluR1, no significant alterations were found in S, I, S + I, or S/I (Fig. 5). For GluR2/3 (Table 5), initial analyses revealed significant pre-treatment × challenge interactions for S + I [F(1,49) = 5.04; p = 0.03] and I [F(1,49) = 5.02; p = 0.03]. Follow-up analyses revealed that the S + I interaction reflected a significant decrease in the A/A group compared with the S/A group [F(1,24) = 5.36; p = 0.03]. For I, the most pronounced difference was between the same two groups [F(1,24) = 3.33; p = 0.08; again, A/A tended to be lower than S/A]. Following the GluR2/3 analysis, the GluR2-selective antibody was tested, and no significant differences were found for any of the measures or groups tested (Table 6). Collectively, these data suggest that amphetamine-pretreated rats may exhibit small decreases in GluR3-containing receptors 24 h after amphetamine challenge injection when compared with saline-pretreated rats. We were unable to confirm this because currently available lots of GluR3 antibody do not reliably recognize the cross-linked band.
Fig. 5.

Effects of amphetamine challenge on GluR1 distribution in the nucleus accumbens (NAc; top) and dorsolateral striatum (DLSTR: bottom). Rats were treated repeatedly with saline or amphetamine for 6 days (see Materials and Methods), administered a challenge injection of saline or 2.5 mg/kg amphetamine on withdrawal day 20 and killed 24 h later. This resulted in four experimental groups: repeated saline/saline challenge, S/S (NAc, n = 14; DLSTR, n = 13); repeated saline/amphetamine challenge, S/A (NAc, n = 14; DLSTR, n = 14); repeated amphetamine/saline challenge, A/S (NAc, n = 14; DLSTR n = 13); and repeated amphetamine/amphetamine challenge, A/A (NAc, n = 13; DLSTR n = 13). A protein cross-linking assay was used to determine surface (S), intracellular (I), and total (S + I) GluR1 levels and the S/I ratio. S, I, and S + I values are normalized to total protein in the lane determined with Ponceau S staining. All data are mean ± SEM expressed relative to S control values. anova did not indicate significant group differences.
Table 5.
Effect of amphetamine challenge on GluR2/3
| n | S | I | S + I | S/I | |
|---|---|---|---|---|---|
| Nucleus accumbens | |||||
| Saline/Saline | 13 | 100 (9.7) | 100 (3.8) | 100 (4.8) | 100 (8.6) |
| Saline/Amphetamine | 14 | 97.7 (5.8) | 109.7 (6.4) | 105.1 (5.4) | 91.0 (5.9) |
| Amphetamine/Saline | 14 | 96.8 (4.8) | 120.3 (11.7) | 111.7 (7.4) | 89.3 (10.0) |
| Amphetamine/Amphetamine | 12 | 85.9 (4.9) | 95.2 (5.9) | 91.5 (3.8) | 93.8 (8.1) |
| Dorsolateral striatum | |||||
| Saline/Saline | 13 | 100 (5.2) | 100 (5.7) | 100 (4.7) | 100 (9.3) |
| Saline/Amphetamine | 14 | 90.4 (10.3) | 107.9 (11.8) | 102.6 (11.1) | 87.3 (9.2) |
| Amphetamine/Saline | 14 | 97.3 (8.5) | 116.5 (9.4) | 110.9 (7.8) | 88.1 (11.3) |
| Amphetamine/Amphetamine | 12 | 74.2 (6.8) | 101.6 (8.0) | 94.7 (6.7) | 72.4 (7.0) |
S, surface; I, intracellular.
Values are mean (SEM) expressed as percent of the saline control group. Shaded area indicates significance (p < 0.05).
Table 6.
Effect of amphetamine challenge on GluR2
| n | S | I | S + I | S/I | |
|---|---|---|---|---|---|
| Nucleus accumbens | |||||
| Saline/Saline | 13 | 100 (5.3) | 100 (5.7) | 100 (4.7) | 100 (6.8) |
| Saline/Amphetamine | 14 | 102.2 (10.3) | 92.2 (4.1) | 95.0 (4.1) | 108.7 (10.1) |
| Amphetamine/Saline | 14 | 95.4 (11.8) | 96.1 (7.1) | 92.4 (5.3) | 102.3 (13.4) |
| Amphetamine/Amphetamine | 13 | 97.5 (9.1) | 90.4 (2.3) | 93.5 (3.8) | 104.2 (9.1) |
| Dorsolateral striatum | |||||
| Saline/Saline | 14 | 100 (9.1) | 100 (5.9) | 100 (6.1) | 100 (6.4) |
| Saline/Amphetamine | 14 | 84.5 (8.4) | 107.9 (10.6) | 105.4 (10.0) | 81.4 (8.1) |
| Amphetamine/Saline | 14 | 91.7 (7.3) | 117.4 (9.7) | 114.7 (8.9) | 83.7 (8.6) |
| Amphetamine/Amphetamine | 13 | 99.4 (9.7) | 101.1 (6.1) | 101.0 (6.2) | 98.0 (6.5) |
S, surface; I, intracellular.
Values are mean (SEM) expressed as percent of the saline control group. Shaded area indicates significance (p < 0.05).
Amphetamine challenge does not significantly alter AMPAR subunits in the DLSTR
In the same animals used to obtain NAc tissue, a portion of DLSTR was also dissected and analyzed for potential changes in AMPAR distribution following amphetamine challenge (Challenge Experiment). Results for S/A, A/S, and A/A groups were compared with control levels in the S/S group (see previous section for details). No robust or consistent effects of amphetamine challenge were found. For GluR1, no significant alterations were found for S, I, S + I or S/I for any of the groups tested, though there was a trend towards a pre-treatment × challenge interaction for the S/I ratio [F(1,49) = 3.44; p = 0.07], apparently driven by a decreased S/I ratio in the S/A group (Fig. 5). There were also trends towards interactions for I [F(1,49) = 3.08; p = 0.09] and S + I [F(1,49) = 3.47; p = 0.07], driven primarily by modest increases in the S/A and A/S groups (Fig. 5). For GluR2/3, no significant differences between groups were found for any measure (Table 5), though there was a strong trend for a main effect of challenge on S [F(1,49) = 4.01; p = 0.051]. This trend was not evident using the GluR2-selective antibody (Table 6), perhaps suggesting that the GluR2/3 antibody detected modest internalization of GluR3-containing receptors in the DLSTR of amphetamine-sensitized rats after amphetamine challenge. Using the GluR2-selective antibody (Table 6), we found a significant interaction for the S/I ratio [F(1,51) = 4.80; p = 0.03] but follow-up t-tests revealed no significant differences between any two groups. However, there was a trend for S/I to be lower in the S/A group than the S/S group [F(1,26) = 3.26; p = 0.08].
Amphetamine challenge does not significantly alter NR2A or NR2B in the NAc or DLSTR
Next, tissue from the Challenge Experiment (NAc and DLSTR) was probed using NR2A/B and NR2B-selective antibodies. No robust or consistent effects of amphetamine challenge were found in either region, although modest differences between groups sometimes achieved statistical significance. In the NAc, using the NR2A/B antibody (Table 7), we found a significant pre-treatment × challenge interaction for the S/I ratio [F(1,51) = 4.92; p = 0.03]. Further analysis revealed a strong trend towards increased S/I in the S/A group when compared with the S/S group [F(1,26) = 3.77; p = 0.06], indicating a potential effect of a single dose of amphetamine on NR2A/B distribution in the NAc. Similarly, there was a significant interaction for I [F(1,51) = 4.35; p = 0.04]. Further analysis revealed that I was significantly lower in the S/A group when compared with the S/S group [F(1,26) = 4.44; p = 0.045], again indicating the potential for a single dose of amphetamine to affect NR2A/B distribution. Using the NR2B-selective antibody (Table 8), we also found a pre-treatment × challenge interaction for S/I in the NAc [F(1,51) = 5.12; p = 0.03], but the individual t-tests between groups only revealed weak trends. Similarly, trends towards changes in I found with the NR2B antibody were similar to those observed with the NR2A/B antibody (Tables 7 and 8), but were also not significant. The DLSTR did not show any significant alterations in NR2A/B or NR2B in any of the groups tested (Tables 7 and 8).
Table 7.
Effect of amphetamine challenge on NR2A/B
| n | S | I | S + I | S/I | |
|---|---|---|---|---|---|
| Nucleus accumbens | |||||
| Saline/Saline | 14 | 100 (7.0) | 100 (8.5) | 100 (5.8) | 100 (9.6) |
| Saline/Amphetamine | 14 | 96.8 (6.4) | 76.6 (7.1) | 85.3 (6.5) | 123.7 (7.4) |
| Amphetamine/Saline | 14 | 96.5 (7.3) | 79.9 (7.4) | 86.0 (6.1) | 116.9 (8.2) |
| Amphetamine/Amphetamine | 13 | 95.5 (7.7) | 86.0 (4.0) | 89.8 (4.9) | 103.9 (7.4) |
| Dorsolateral striatum | |||||
| Saline/Saline | 14 | 100 (11.0) | 100 (10.1) | 100 (9.8) | 100 (8.0) |
| Saline/Amphetamine | 13 | 89.3 (10.2) | 91.2 (12.6) | 89.8 (11.6) | 99.2 (8.6) |
| Amphetamine/Saline | 14 | 83.5 (8.7) | 91.1 (14.4) | 87.0 (11.2) | 95.8 (6.8) |
| Amphetamine/Amphetamine | 13 | 86.4 (10.7) | 85.5 (11.0) | 85.1 (10.4) | 100.9 (7.5) |
S, surface; I, intracellular.
Values are mean (SEM) expressed as percent of the saline control group. Shaded area indicates significance (p < 0.05).
Table 8.
Effect of amphetamine challenge on NR2B
| n | S | I | S + I | S/I | |
|---|---|---|---|---|---|
| Nucleus accumbens | |||||
| Saline/Saline | 14 | 100 (7.7) | 100 (8.8) | 100 (7.2) | 100 (9.5) |
| Saline/Amphetamine | 14 | 102.1 (7.1) | 85.9 (7.8) | 89.6 (5.8) | 122.9 (13.1) |
| Amphetamine/Saline | 14 | 97.3 (7.5) | 82.6 (9.6) | 85.2 (6.2) | 127.3 (15.3) |
| Amphetamine/Amphetamine | 13 | 91.6 (6.2) | 90.2 (6.7) | 90.2 (6.1) | 97.2 (6.2) |
| Dorsolateral striatum | |||||
| Saline/Saline | 13 | 100 (9.5) | 100 (6.8) | 100 (6.8) | 100 (6.9) |
| Saline/Amphetamine | 13 | 89.3 (6.6) | 105.3 (9.3) | 100.2 (7.9) | 88.4 (6.9) |
| Amphetamine/Saline | 14 | 93.9 (10.0) | 111.8 (13.1) | 104.1 (9.6) | 87.8 (8.6) |
| Amphetamine/Amphetamine | 12 | 101.1 (10.8) | 106.5 (9.4) | 103.8 (8.5) | 96.5 (8.5) |
S, surface; I, intracellular.
Values are mean (SEM) expressed as percent of the saline control group. Shaded area indicates significance (p < 0.05).
PKA, CaMKII, and ERK signaling after repeated amphetamine treatment
We showed previously that increased AMPAR surface expression in the NAc of cocaine-sensitized rats was accompanied by time-dependent changes in activation of CaMKII and ERK (measured using phospho-specific antibodies recognizing activated forms of these enzymes) as well as an increase in PKA substrate phosphorylation (measured using an antibody recognizing the phosphorylated PKA consensus sequence) (Boudreau et al. 2007; Boudreau and Wolf 2006). Therefore these measures were evaluated in NAc tissue from rats in Withdrawal Experiments 1 and 2. No changes in these measures were found in amphetamine-treated rats (data not shown).
Discussion
Amphetamine sensitization is not accompanied by AMPAR up-regulation in the NAc
We found no significant changes in AMPA or NMDA receptor surface expression in the NAc or DLSTR after withdrawal from sensitizing regimens of amphetamine. Although our biochemical approach does not permit us to rule out a very small increase in AMPARs confined to the synaptic region, our negative findings in the NAc after amphetamine contrast with previous studies indicating increased surface and synaptic AMPAR levels in the NAc of cocaine-sensitized rats on withdrawal days 14 and 21 (Boudreau and Wolf 2005; Boudreau et al. 2007; Kourrich et al. 2007). We had expected amphetamine and cocaine to cause similar AMPAR adaptations based on cross-sensitization (e.g., Kalivas and Weber 1988; Pierce and Kalivas 1995; Bonate et al. 1997; Liu et al. 2007), similar long-term changes in dendritic morphology (Robinson and Kolb 2004), and other interactions between cocaine and amphetamine suggestive of common AMPAR-related mechanisms. For example, prior exposure to a sensitizing amphetamine regimen led to a withdrawal-dependent increase in the ability of intra-NAc AMPA infusion to reinstate cocaine seeking (Suto et al. 2004).
There are several ways to conceptualize these divergent findings. First, it is possible that increased glutamate transmission in the NAc is required for expression of sensitization (see Pierce et al. 1996) and explains cross-sensitization but that this occurs through different mechanisms for cocaine and amphetamine. For cocaine, AMPAR up-regulation may be a major underlying mechanism (above). In the case of amphetamine, however, it is possible that pre-synaptic changes account for increased excitatory drive after sensitization. Supporting this, Lodge and Grace (2008) demonstrated that persistent activation of the ventral hippocampus-NAc pathway occurred in amphetamine-sensitized rats and that interference with this activation (by hippocampal tetrodotoxin injection) blocked the expression of sensitization. Interestingly, whereas LTP in this pathway was occluded in cocaine-sensitized rats, LTP was not altered in amphetamine-sensitized rats (Goto and Grace 2005). We suggest that LTP is occluded after cocaine due to a ceiling level of AMPAR up-regulation in the NAc (Boudreau and Wolf 2005), whereas it occurs normally after amphetamine because AMPAR surface expression is still at baseline level, permitting further increases.
Another possibility is that AMPAR transmission in the NAc of amphetamine-sensitized rats is enhanced, but through mechanisms other than increased AMPAR surface expression. For example, phosphorylation of GluR1 at Ser831 (by protein kinase C and CaMKII) or at Ser845 (by PKA) can enhance AMPAR currents (Song and Huganir 2002; Derkach et al. 2007). We could not assess phosphorylation of these residues in cross-linked tissue because the surface band was poorly detected by the GluR1 phospho-specific antibodies that we tested (A. C. Boudreau, C. L. Nelson, M. Milovanovic, M. E. Wolf; unpublished observations). However, phosphorylation of Ser845 is associated with increased AMPAR surface expression (e.g., Ehlers 2000; Chao et al. 2002a,b; Oh et al. 2006), which we did not detect. We also failed to obtain evidence for activation of CaMKII. Collectively, these results do not support the hypothesis that increased AMPAR channel function due to phosphorylation occurs during amphetamine withdrawal, although it cannot be ruled out.
Our findings of AMPAR up-regulation in the NAc of cocaine- but not amphetamine-sensitized rats may seem to be at odds with our prior report that NAc neurons recorded from both cocaine- and amphetamine-sensitized rats showed subsensitivity to iontophoretic glutamate (White et al. 1995). In the earlier study, recordings were performed after 3 days of withdrawal, a time when AMPAR up-regulation may not have occurred in the cocaine-treated rats (Boudreau and Wolf 2005). The subsensitive response to glutamate may have reflected decreased calcium and sodium currents in NAc neurons at this withdrawal time (Zhang et al. 1998, 2002; Hu et al. 2004). Another factor to consider is that different techniques sample different AMPAR populations. Iontophoresis delivers glutamate close to the somatic recording site, whereas protein cross-linking detects AMPARs on all parts of the cell. This is an important point because GluR1 in medium spiny neurons is primarily targeted to the dendrites and dendritic spines; perikaryal GluR1 labeling is sparse (Chen et al. 1998).
Other than the present results, which showed no change in AMPAR surface expression in the DLSTR of amphetamine-sensitized rats, we are not aware of any published data on glutamate receptor surface expression in the dorsal striatum after repeated cocaine or amphetamine treatment. However, in preliminary studies of cocaine-sensitized rats, we found that GluR1 distribution in the DLSTR was unchanged while surface and total GluR2/3 were modestly decreased (Wolf and Ferrario 2008).
Amphetamine challenge does not significantly alter AMPAR surface expression in sensitized rats
An acute injection of amphetamine administered to naïve rats produced a transient elevation in surface expression of GluR1-containing AMPARs in the NAc (evident 2 h but not 30 min or 24 h after the injection). However, when a challenge injection of amphetamine was administered to amphetamine-sensitized rats, no changes in GluR1 or GluR2 surface expression occurred in the NAc or in the DLSTR. This contrasts with evidence that surface expression of GluR1/2-containing AMPARs in the NAc is decreased 24 h after a cocaine challenge is administered to cocaine-sensitized rats (Thomas et al. 2001; Boudreau et al. 2007; Kourrich et al. 2007). Interestingly, intra-NAc injection of a peptide that prevents AMPAR internalization prevented the expression of amphetamine sensitization (Brebner et al. 2005). It is possible that a small amount of AMPAR internalization occurs soon after amphetamine challenge and is required for the behavioral response. This would not necessarily be detected 24 h after the challenge injection, although it is interesting that our results suggest a small decrease in GluR3 24 h after amphetamine challenge in both NAc and DLSTR. An alternative discussed previously (Boudreau et al. 2007) is that after AMPARs have been activated, internalization and subsequent recycling to the surface are necessary for overcoming desensitization and enabling a prolonged response sufficient for motor activation.
Why would different AMPAR adaptations occur after amphetamine versus cocaine?
Regardless of functional implications (above), it is interesting to consider what may underlie the divergent AMPAR responses to cocaine and amphetamine. Cocaine and amphetamine elevate DA levels through different mechanisms of action on the DA nerve terminal (Sulzer et al. 2005). However, it seems clear based on a great number of microdialysis studies that both drugs produce a robust increase in extracellular DA levels in the NAc on each day of treatment. Therefore, it seems unlikely that differences in DA receptor signaling in the post-synaptic NAc neuron are a major contributor to divergent AMPAR adaptations. Instead, differences in activity of glutamate afferents to NAc neurons are more likely to be responsible, in keeping with the fact that changes in the pattern of pre-synaptic activity are primarily responsible for driving changes in AMPAR synaptic content during LTP; monoamines only modulate this process (Jay 2003; Wolf et al. 2004). Thus, we hypothesize that glutamate-containing projections to NAc neurons exhibit different firing patterns during cocaine withdrawal versus amphetamine withdrawal, leading to AMPAR up-regulation only in cocaine-treated rats. Why might this occur? Glutamate projections to the NAc originate in cortical and limbic brain regions (Groenewegen et al. 1999; Kelley 2004) that express serotonin and norepinephrine transporters as well as DA transporters (e.g., Miner et al. 2000, 2003). Cocaine and amphetamines differ in their relative affinities for these transporters (Han and Gu 2006) and therefore may produce different effects on monoamine transmission, leading to different modulatory effects on glutamatergic principal neurons in these regions and ultimately to different patterns of glutamate transmission in target regions such as NAc. This may account in part for differences in the plasticity evoked in the NAc. The distinct locomotor responses to cocaine and amphetamine (compare present results for amphetamine to cocaine results in Boudreau and Wolf 2005) support the idea that they activate very different neuronal circuits despite their common ability to elevate monoamine levels.
In the case of cocaine sensitization, AMPAR up-regulation could occur through LTP-like mechanisms as a result of increased pre-synaptic glutamate activity. However, this is an overly simple hypothesis in light of temporal differences between AMPAR up-regulation after cocaine withdrawal versus hippocampal LTP. Furthermore, the pattern of signaling pathway activation in each case shows some overlap but also quite a bit of divergence (Boudreau et al. 2007; Boudreau and Wolf 2006). Alternatively, AMPAR up-regulation in cocaine-sensitized rats could result from synaptic scaling as suggested previously (Boudreau and Wolf 2005). Synaptic scaling is a form of homeostatic plasticity in which a prolonged decrease in the activity of excitatory inputs to a cell leads to a compensatory increase in synaptic transmission that is mediated in large part by increased post-synaptic AMPAR content; a prolonged increase in activity produces the opposite effect (Turrigiano and Nelson 2004; Turrigiano 2008). The possibility that AMPARs ‘scale up’ during cocaine withdrawal is supported by brain imaging studies showing decreased metabolic activity in cortical regions that send excitatory projections to the NAc after extended abstinence (1–6 weeks) from cocaine use in humans (Volkow et al. 1992; Goldstein and Volkow 2002). In primates trained to self-administer cocaine, measures of brain metabolism have been made in conjunction with the last self-administration session, not after withdrawal, but nevertheless indicate relatively lower frontal cortical and ventral striatal metabolic rates as a result of chronic cocaine exposure (Porrino et al. 2004; Beveridge et al. 2006; Porrino et al. 2007). Similar results have been obtained from brain metabolism studies in rats (Hammer and Cooke 1994; Macey et al. 2004). Furthermore, there is electrophysiological evidence for decreased basal activity in the rat prefrontal cortex (PFC) after several weeks of cocaine self-administration (Sun and Rebec 2006). Only one rat study was conducted after withdrawal; it reported decreased metabolism in some cortical areas, as well as the NAc, after 72 h of withdrawal from binge cocaine self-administration (Hammer et al. 1993).
It is possible that this is where amphetamine and cocaine diverge, that is, excitatory drive from frontal cortex to the NAc is not depressed during amphetamine withdrawal, and thus AMPARs in the NAc do not scale up. Several studies support this hypothesis by providing evidence for increases rather than decreases in the activity of frontal cortex during amphetamine withdrawal. First, results of in vivo recording studies in rats are consistent with increased activation of the PFC-NAc pathway after 21–28 days of withdrawal from repeated amphetamine treatment (Onn and Grace 2000). Second, rat PFC neurons recorded after 3 days of withdrawal from repeated amphetamine injections showed increased responsiveness to glutamate (Peterson et al. 2000), although it is interesting that methamphetamine sensitization is associated with a long-lasting decrease in stimulation-evoked transmitter release from corticostriatal terminals (Bamford et al. 2008). Finally, human methamphetamine abusers (abstinent for weeks to months) displayed a tendency towards increased frontal cortical metabolism (Volkow et al. 2001a). It should be kept in mind, however, that some of methamphetamine's effects on brain metabolism may be related to its neurotoxicity (Chang et al. 2007; Baicy and London 2007).
Although these findings support our hypothesis, several cautions must be kept in mind. First, none of the available evidence permits a ‘head-to-head’ comparison of the two drugs in identical experiments examining the level of excitatory drive to NAc neurons. Second, while evidence for cortical hypoactivity after withdrawal from cocaine self-administration exists in the brain imaging literature (above), it is not known whether excitatory synaptic transmission onto NAc neurons (originating from PFC or other regions) is depressed after withdrawal from non-contingent cocaine regimens such as the one shown to produce AMPAR up-regulation in the rat NAc (Boudreau and Wolf 2005). In fact, the synaptic scaling hypothesis is perhaps a better fit with the qualitatively different AMPAR up-regulation observed after prolonged withdrawal from cocaine self-administration (Conrad et al. 2008), because there is strong evidence for cortical hypoactivity in this situation (above). Third, it must be kept in mind that similarities between effects of cocaine and amphetamines have been found in human imaging studies focusing on task-dependent brain activation (Aron and Paulus 2007) or markers of DA transmission (Volkow et al. 2001b; Chang et al. 2007; Martinez et al. 2007).
A final consideration is the potential impact of changes in the excitability of NAc neurons themselves after withdrawal from the two drugs. Withdrawal from a sensitizing regimen of cocaine (3 days) is associated with decreased intrinsic excitability of NAc neurons due to changes in voltage-gated channels (Zhang et al. 1998, 2002; Hu et al. 2004; Dong et al. 2006). Parallel studies have not been conducted with amphetamine. However, NAc neurons recorded from methamphetamine-sensitized rats after several weeks of withdrawal showed no change in many baseline measures of excitability and no change in responsiveness to PFC stimulation relative to saline controls (Brady et al. 2003, 2005), suggesting that decreased intrinsic excitability may be unique to cocaine (although whether it occurs after long withdrawal from cocaine has yet to be established). Nevertheless, it is unclear whether decreased intrinsic excitability of NAc neurons after cocaine withdrawal can account for a homeostatic increase in AMPARs (see Turrigiano 2008 for a discussion of conditions that elicit synaptic scaling).
Unfortunately, additional information about NAc neurons during withdrawal cannot be gleaned from the literature on recordings from awake rats using microwire electrode arrays. While recordings have been performed after abstinence from cocaine self-administration, they focused on neuronal responsiveness when drug-seeking resumed rather than neuronal activity during abstinence (Hollander and Carelli 2005, 2007; Ghitza et al. 2003; Peoples et al. 2007).
Signaling pathways in the NAc after amphetamine withdrawal
We found no significant alterations in any of the signaling pathways we investigated (ERK, CaMKII, or PKA) in the NAc of amphetamine-sensitized rats, in contrast with our previous findings for cocaine sensitization. In the NAc of cocaine-sensitized rats, ERK2 activation paralleled AMPAR up-regulation on withdrawal days 7–21, PKA phosphorylation was increased on withdrawal day 21 and CaMKII was transiently activated (Boudreau and Wolf 2006; Boudreau et al. 2007). Supporting a link between activation of these signaling pathways and AMPAR up-regulation in the NAc of cocaine-sensitized rats, both are absent in the NAc of amphetamine-sensitized rats. It should be noted that CaMKII is implicated in responses of amphetamine-sensitized rats to a challenge injection of amphetamine (e.g., Pierce and Kalivas 1997; Pierce et al. 1998). This is not at odds with our failure to observe CaMKII activation during withdrawal from a sensitizing regimen of amphetamine.
We are not aware of any other study measuring ERK or CaMKII activation in the NAc after withdrawal from repeated amphetamine treatment. However, Suemaru et al. (2000) found no change in basal CaMKII activity in the NAc after 1 or 4 weeks of withdrawal from repeated methamphetamine treatment, consistent with our negative findings in amphetamine-sensitized rats. A prior study found decreased PKA activity in the NAc of amphetamine-sensitized rats after 1 day of withdrawal (Crawford et al. 2004), whereas we found no significant change in PKA substrate phosphorylation following weeks of withdrawal from repeated amphetamine. The difference could be due to different withdrawal times or to the fact that our measure of PKA substrate phosphorylation reflects the net result of changes in both PKA and protein phosphatase activity.
We did not examine signaling pathway activation in the dorsal striatum because our purpose was to compare signaling results in the NAc of amphetamine-sensitized rats with our prior results in the NAc of cocaine-sensitized rats. However, there is evidence that amphetamine sensitization and withdrawal influence CaMKII (Suemaru et al. 2000; Greenstein et al. 2007) as well as calmodulin (Gnegy 2000) in the dorsal striatum.
Conclusions
Repeated amphetamine administration reproducibly resulted in behavioral sensitization but failed to produce the significant increases in AMPAR surface expression (and accompanying alterations in signaling pathways) that were previously demonstrated in the NAc of cocaine-sensitized rats. It is puzzling that cocaine and amphetamine exert very different effects on AMPARs in the NAc yet ultimately produce similar effects on dendritic spines, the site of excitatory synapses (Robinson and Kolb 2004). Solving the puzzle will require studies of pre-synaptic glutamate inputs to the NAc during drug withdrawal and of post-synaptic mechanisms apart from AMPAR plasticity that influence the excitability of NAc neurons.
Supplementary Material
Acknowledgments
This work was supported by U.S.P.H.S. grants DA09621 and DA00453 to MEW. CLN was supported by postdoctoral National Research Service Award DA021067. We thank Dr. Carrie R. Ferrario for helpful comments on the manuscript.
Abbreviations used
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionate
- AMPAR
AMPA receptor
- BS3
bis(sulfosuccinimidyl)suberate
- CaMKII
Ca2+-calmodulin-dependent protein kinase II
- DA
dopamine
- DLSTR
dorsolateral striatum
- ERK
extracellular signal-regulated protein kinase
- GluR
glutamate receptor
- LTP
long-term potentiation
- NAc
nucleus accumbens
- PFC
prefrontal cortex
- PKA
protein kinase A
- TBS
tris-buffered saline
- TBS-T
TBS with tween
Footnotes
Supporting Information: Additional Supporting Information may be found in the online version of this article:
Table S1 Effect of repeated amphetamine on GluR2/3.
Table S2 Effect of repeated amphetamine on GluR2.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aron JL, Paulus MP. Location, location: using functional magnetic resonance imaging to pinpoint brain differences relevant to stimulant use. Addiction. 2007;102(Suppl 1):33–43. doi: 10.1111/j.1360-0443.2006.01778.x. [DOI] [PubMed] [Google Scholar]
- Bäckström P, Hyytiä P. Ionotropic and metabotropic glutamate receptor antagonism attenuates cue-induced cocaine seeking. Neuropsychopharmacology. 2006;31:778–786. doi: 10.1038/sj.npp.1300845. [DOI] [PubMed] [Google Scholar]
- Baicy K, London ED. Corticolimbic dysregulation and chronic methamphetamine abuse. Addiction. 2007;102(Suppl 1):5–15. doi: 10.1111/j.1360-0443.2006.01777.x. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Joyce JA, et al. Repeated exposure to methamphetamine causes long-lasting presynaptic corticostriatal depression that is renormalized with drug readministration. Neuron. 2008;58:89–103. doi: 10.1016/j.neuron.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beveridge TJ, Smith HR, Daunais JB, Nader MA, Porrino LJ. Chronic cocaine self-administration is associated with altered functional activity in the temporal lobes of non human primates. Eur J Neurosci. 2006;23:3109–3118. doi: 10.1111/j.1460-9568.2006.04788.x. [DOI] [PubMed] [Google Scholar]
- Bonate PL, Swann A, Silverman PB. Context-dependent cross-sensitization between cocaine and amphetamine. Life Sci. 1997;60:L1–L7. doi: 10.1016/s0024-3205(96)00591-7. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Dysregulation of signal transduction accompanies AMPA receptor trafficking in the nucleus accumbens during behavioral sensitization to cocaine. Abstr Soc Neurosci. 2006;32:394–7. [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady AM, Glick SD, O’Donnell P. Changes in electrophysiological properties of nucleus accumbens neurons depend on the extent of behavioral sensitization to chronic methamphetamine. Ann NY Acad Sci. 2003;1003:358–363. doi: 10.1196/annals.1300.026. [DOI] [PubMed] [Google Scholar]
- Brady AM, Glick SD, O’Donnell P. Selective disruption of nucleus accumbens gating mechanisms in rats behaviorally sensitized to methamphetamine. J Neurosci. 2005;25:6687–6695. doi: 10.1523/JNEUROSCI.0643-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science. 2005;310:1340–1343. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Chang L, Alicata D, Ernst T, Volkow ND. Structural and metabolic brain changes in the striatum associated with methamphetamine abuse. Addiction. 2007;102(Suppl 1):16–21. doi: 10.1111/j.1360-0443.2006.01782.x. [DOI] [PubMed] [Google Scholar]
- Chao SZ, Lu W, Lee HK, Huganir RL, Wolf ME. D1 dopamine receptor stimulation increases GluR1 phosphorylation in postnatal nucleus accumbens cultures. J Neurochem. 2002a;81:984–992. doi: 10.1046/j.1471-4159.2002.00877.x. [DOI] [PubMed] [Google Scholar]
- Chao SZ, Ariano MA, Peterson DA, Wolf ME. D1 dopamine receptor stimulation increases GluR1 surface expression in nucleus accumbens neurons. J Neurochem. 2002b;83:704–712. doi: 10.1046/j.1471-4159.2002.01164.x. [DOI] [PubMed] [Google Scholar]
- Chen Q, Veenman L, Knopp K, Yan Z, Medina L, Song WJ, Surmeier DJ, Reiner A. Evidence for the preferential localization of glutamate receptor-1 subunits of AMPA receptors to the dendritic spines of medium spiny neurons in the rat striatum. Neuroscience. 1998;83:749–761. doi: 10.1016/s0306-4522(97)00452-1. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish JL, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. 2000;20:RC89. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish JL, Duffy P, Kalivas PW. A role for nucleus accumbens glutamate transmission in the relapse to cocaine-seeking behavior. Neuroscience. 1999;93:1359–1367. doi: 10.1016/s0306-4522(99)00214-6. [DOI] [PubMed] [Google Scholar]
- Crawford CA, Choi FY, Kohutek JL, Yoshida ST, McDougall SA. Changes in PKA activity and Gs alpha and Golf alpha levels after amphetamine- and cocaine-induced behavioral sensitization. Synapse. 2004;51:241–248. doi: 10.1002/syn.10301. [DOI] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Di Ciano P, Everitt BJ. Dissociable effects of antagonism of NMDA and AMPA/KA receptors in the nucleus accumbens core and shell on cocaine-seeking behavior. Neuropsychopharmacology. 2001;25:341–360. doi: 10.1016/S0893-133X(01)00235-4. [DOI] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Ghitza UE, Fabbricatore AT, Prokopenko V, Pawlak AP, West MO. Persistent cue-evoked activity of accumbens neurons after prolonged abstinence from self-administered cocaine. J Neurosci. 2003;23:7239–7245. doi: 10.1523/JNEUROSCI.23-19-07239.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnegy ME. Ca2+/calmodulin signaling in NMDA-induced synaptic plasticity. Crit Rev Neurobiol. 2000;14:91–129. [PubMed] [Google Scholar]
- Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. 2002;159:1642–1652. doi: 10.1176/appi.ajp.159.10.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Grace AA. Dopamine-dependent interactions between limbic and prefrontal cortical plasticity in the nucleus accumbens: disruption by cocaine sensitization. Neuron. 2005;47:255–266. doi: 10.1016/j.neuron.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Greenstein R, Novak G, Seeman P. Amphetamine sensitization elevates CaMKIIbeta mRNA. Synapse. 2007;61:827–834. doi: 10.1002/syn.20429. [DOI] [PubMed] [Google Scholar]
- Groenewegen HJ, Wright CI, Beijer AV, Voorn P. Convergence and segregation of ventral striatal inputs and outputs. Ann NY Acad Sci. 1999;877:49–63. doi: 10.1111/j.1749-6632.1999.tb09260.x. [DOI] [PubMed] [Google Scholar]
- Hammer RP, Jr, Cooke ES. Gradual tolerance of metabolic activity is produced in mesolimbic regions by chronic cocaine treatment, while subsequent cocaine challenge activates extrapyramidal regions of rat brain. J Neurosci. 1994;14:4289–4296. doi: 10.1523/JNEUROSCI.14-07-04289.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer RP, Jr, Pires WS, Markou A, Koob GF. Withdrawal following cocaine self-administration decreases regional cerebral metabolic rate in critical brain reward regions. Synapse. 1993;14:73–80. doi: 10.1002/syn.890140110. [DOI] [PubMed] [Google Scholar]
- Han DD, Gu HH. Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol. 2006;6:6. doi: 10.1186/1471-2210-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander JA, Carelli RM. Abstinence from cocaine self-administration heightens neural encoding of goal-directed behaviors in the accumbens. Neuropsychopharmacology. 2005;30:1464–1474. doi: 10.1038/sj.npp.1300748. [DOI] [PubMed] [Google Scholar]
- Hollander JA, Carelli RM. Cocaine-associated stimuli increase cocaine seeking and activate accumbens core neurons after abstinence. J Neurosci. 2007;27:3535–3539. doi: 10.1523/JNEUROSCI.3667-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XT, Basu S, White FJ. Repeated cocaine administration suppresses HVA-Ca2+ potentials and enhances activity of K+ channels in rat nucleus accumbens neurons. J Neurophysiol. 2004;92:1597–1607. doi: 10.1152/jn.00217.2004. [DOI] [PubMed] [Google Scholar]
- Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev. 2007;56:27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TM. Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog Neurobiol. 2003;69:375–390. doi: 10.1016/s0301-0082(03)00085-6. [DOI] [PubMed] [Google Scholar]
- Jedynak JP, Uslaner JM, Esteban JA, Robinson TE. Methamphetamine-induced structural plasticity in the dorsal striatum. Eur J Neurosci. 2007;25:847–853. doi: 10.1111/j.1460-9568.2007.05316.x. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Weber B. Amphetamine injection into the ventral mesencephalon sensitizes rats to peripheral amphetamine and cocaine. J Pharmacol Exp Ther. 1988;245:1095–1102. [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Ventral striatal control of appetitive motivation: role in ingestive behavior and reward-related learning. Neurosci Biobehav Rev. 2004;27:765–776. doi: 10.1016/j.neubiorev.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Morgan D, Roberts DC. Cross-sensitization of the reinforcing effects of cocaine and amphetamine in rats. Psychopharmacology (Berl) 2007;195:369–375. doi: 10.1007/s00213-007-0909-6. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Amphetamine activation of hippocampal drive of mesolimbic dopamine neurons: a mechanism of behavioral sensitization. J Neurosci. 2008;28:7876–7882. doi: 10.1523/JNEUROSCI.1582-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Macey DJ, Rice WN, Freedland CS, Whitlow CT, Porrino LJ. Patterns of functional activity associated with cocaine self-administration in the rat change over time. Psychopharmacol. 2004;172:384–392. doi: 10.1007/s00213-003-1676-7. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Martinez D, Kim JH, Krystal J, Abi-Dargham A. Imaging the neurochemistry of alcohol and substance abuse. Neuroimag Clin N Am. 2007;17:539–555. doi: 10.1016/j.nic.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Miner LH, Schroeter S, Blakely RD, Sesack SR. Ul-trastructural localization of the serotonin transporter in superficial and deep layers of the rat prelimbic prefrontal cortex and its spatial relationship to dopamine terminals. J Comp Neurol. 2000;427:220–234. doi: 10.1002/1096-9861(20001113)427:2<220::aid-cne5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Miner LH, Schroeter S, Blakely RD, Sesack SR. Ul-trastructural localization of the norepinephrine transporter in superficial and deep layers of the rat prelimbic prefrontal cortex and its spatial relationship to probable dopamine terminals. J Comp Neurol. 2003;466:478–494. doi: 10.1002/cne.10898. [DOI] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Onn SP, Grace AA. Amphetamine withdrawal alters bistable states and cellular coupling in rat prefrontal cortex neurons and nucleus accumbens neurons recorded in vivo. J Neurosci. 2000;20:2332–2345. doi: 10.1523/JNEUROSCI.20-06-02332.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick SL, Thompson TL, Walker JM, Patrick RL. Concomitant sensitization of amphetamine-induced behavioral stimulation and in vivo dopamine release from rat caudate nucleus. Brain Res. 1991;538:343–346. doi: 10.1016/0006-8993(91)90453-3. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press; 1998. [Google Scholar]
- Peoples LL, Kravitz AV, Guillem K. The role of accumbal hypoactivity in cocaine addiction. ScientificWorldJournal. 2007;7:22–45. doi: 10.1100/tsw.2007.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JD, Wolf ME, White FJ. Altered responsiveness of medial prefrontal cortex neurons to glutamate and dopamine after withdrawal from repeated amphetamine treatment. Synapse. 2000;36:342–344. doi: 10.1002/(SICI)1098-2396(20000615)36:4<342::AID-SYN11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Kalivas PW. Amphetamine produces sensitized increases in locomotion and extracellular dopamine preferentially in the nucleus accumbens shell of rats administered repeated cocaine. J Pharmacol Exp Ther. 1995;275:1019–1029. [PubMed] [Google Scholar]
- Pierce RC, Kalivas PW. Repeated cocaine modifies the mechanism by which amphetamine releases dopamine. J Neurosci. 1997;17:3254–3261. doi: 10.1523/JNEUROSCI.17-09-03254.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Quick EA, Reeder DC, Morgan ZR, Kalivas PW. Calcium-mediated second messengers modulate the expression of behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1998;286:1171–1176. [PubMed] [Google Scholar]
- Porrino LJ, Lyons D, Smith HR, Daunais JB, Nader MA. Cocaine self-administration produces a progressive involvement of limbic, association, and sensorimotor striatal domains. J Neurosci. 2004;24:3554–3562. doi: 10.1523/JNEUROSCI.5578-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrino LJ, Smith HR, Nader MA, Beveridge TJ. The effects of cocaine: a shifting target over the course of addiction. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1593–1600. doi: 10.1016/j.pnpbp.2007.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post RM, Weiss SRB, Pert A. Sensitization and kindling effects of chronic cocaine administration. In: Lakoski J, Galloway MP, White FJ, editors. Cocaine: Pharmacology, Physiology and Clinical Strategies. CRC Press; Boca Raton: 1992. pp. 115–161. [Google Scholar]
- Robinson TE, Becker JB. Enduring changes in brain and behavior produced by chronic amphetamine administration: a review and evaluation of animal models of amphetamine psychosis. Brain Res. 1986;396:157–198. doi: 10.1016/s0006-8993(86)80193-7. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The incentive sensitization theory of addiction: some current issues. Phil Trans R Soc B Biol Sci. 2008;363:3137–3146. doi: 10.1098/rstb.2008.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Segal DS, Kuczenski R. Behavioral and neurochemical characteristics of stimulant-induced augmentation. Psychopharmacol Bull. 1987;23:417–424. [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Suemaru J, Akiyama K, Tanabe Y, Kuroda S. Methamphetamine decreases calcium-calmodulin dependent protein kinase II activity in discrete rat brain regions. Synapse. 2000;36:155–166. doi: 10.1002/(SICI)1098-2396(20000601)36:3<155::AID-SYN1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Sun W, Rebec GV. Repeated cocaine self-administration alters processing of cocaine-related information in rat prefrontal cortex. J Neurosci. 2006;26:8004–8008. doi: 10.1523/JNEUROSCI.1413-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto N, Tanabe LM, Austin JD, Creekmore E, Pham CT, Vezina P. Previous exposure to psychostimulants enhances the reinstatement of cocaine seeking by nucleus accumbens AMPA. Neuropsychopharmacology. 2004;29:2149–2159. doi: 10.1038/sj.npp.1300533. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural substrate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of pre-clinical studies. Psychopharmacology (Berl) 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- Vezina P. Sensitization of midbrain dopamine neuron reactivity and the self-administration of psychomotor stimulant drugs. Neurosci Biobehav Rev. 2004;27:827–839. doi: 10.1016/j.neubiorev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Hitzemann R, Wang GJ, Fowler JS, Wolf AP, Dewey SL, Handlesman L. Long-term frontal brain metabolic changes in cocaine abusers. Synapse. 1992;11:184–190. doi: 10.1002/syn.890110303. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, et al. Higher cortical and lower subcortical metabolism in detoxified methamphetamine abusers. Am J Psychiatry. 2001a;158:383–389. doi: 10.1176/appi.ajp.158.3.383. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, et al. Low level of brain dopamine D2 receptors in methamphetamine abusers: association with metabolism in the orbitofrontal cortex. Am J Psychiatry. 2001b;158:2015–2021. doi: 10.1176/appi.ajp.158.12.2015. [DOI] [PubMed] [Google Scholar]
- White FJ, Hu XT, Zhang XF, Wolf ME. Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J Pharmacol Exp Ther. 1995;273:445–454. [PubMed] [Google Scholar]
- Willuhn I, Sun W, Steiner H. Topography of cocaine-induced gene regulation in the rat striatum: relationship to cortical inputs and role of behavioural context. Eur J Neurosci. 2003;17:1053–1066. doi: 10.1046/j.1460-9568.2003.02525.x. [DOI] [PubMed] [Google Scholar]
- Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Ferrario CR. Alterations in AMPA receptor expression in the nucleus accumbens of cocaine sensitized rats after re-exposure to cocaine. Abstr Soc Neurosci. 2008;43:662–7. [Google Scholar]
- Wolf ME, Dahlin SL, Hu XT, Xue CJ, White K. Effects of lesions of prefrontal cortex, amygdala, or fornix on behavioral sensitization to amphetamine: comparison with N-methyl-d-aspartate antagonists. Neuroscience. 1995;69:417–439. doi: 10.1016/0306-4522(95)00248-h. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Sun X, Mangiavacchi S, Chao SZ. Psychomotor stimulants and neuronal plasticity. Neuropharmacology. 2004;47(Suppl 1):61–79. doi: 10.1016/j.neuropharm.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Zhang XF, Hu XT, White FJ. Whole-cell plasticity in cocaine withdrawal: reduced sodium current in nucleus accumbens neurons. J Neurosci. 1998;18:488–498. doi: 10.1523/JNEUROSCI.18-01-00488.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Cooper DC, White FJ. Repeated cocaine treatment decreases whole-cell calcium current in rat nucleus accumbens neurons. J Pharmacol Exp Ther. 2002;301:1119–1125. doi: 10.1124/jpet.301.3.1119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.