

Summary
Generating the extended endoplasmic reticulum (ER) network depends on microtubules, which act as tracks for motor-driven ER tubule movement, generate the force to extend ER tubules by means of attachment to growing microtubule plus-ends and provide static attachment points. We have analysed ER dynamics in living VERO cells and find that most ER tubule extension is driven by microtubule motors. Surprisingly, we observe that ∼50% of rapid ER tubule movements occur in the direction of the centre of the cell, driven by cytoplasmic dynein. Inhibition of this movement leads to an accumulation of lamellar ER in the cell periphery. By expressing dominant-negative kinesin-1 constructs, we show that kinesin-1 drives ER tubule extension towards the cell periphery and that this motility is dependent on the KLC1B kinesin light chain splice form but not on KLC1D. Inhibition of kinesin-1 promotes a shift from tubular to lamellar morphology and slows down the recovery of the ER network after microtubule depolymerisation and regrowth. These observations reconcile previous conflicting studies of kinesin-1 function in ER motility in vivo. Furthermore, our data reveal that cytoplasmic dynein plays a role in ER motility in a mammalian cultured cell, demonstrating that ER motility is more complex than previously thought.
Keywords: ER, Dynein, Kinesin, Microtubule
Introduction
The endoplasmic reticulum (ER) is a large, dynamic organelle that fulfils crucial roles within the cell, including the synthesis and translocation of membrane and secretory proteins, calcium homeostasis and lipid synthesis (Baumann and Walz, 2001). Its morphology and organisation is not uniform, as it has regions with and without ribosomes (rough ER and smooth ER, respectively), and can contain a mixture of lamellar and tubular regions. The transition between tubular and lamellar membrane is thought to be controlled by reticulon proteins that localise and shape the peripheral ER (Voeltz et al., 2006).
The complex morphology of the ER network is generated and maintained by a combination of extension of membrane tubules and membrane fusion. Fusion is dependent on p97-p47-VCIP135 proteins (Kano et al., 2005; Vedrenne and Hauri, 2006). Even though ER tubule formation in vitro can be driven solely by the activity of reticulon proteins (Voeltz et al., 2006), in vivo the cytoskeleton plays a major role in the dynamics of ER tubules and the maintenance of ER morphology. For example, the depolymerisation of microtubules (MTs) in a variety of cultured cells leads to slow retraction of the ER and clustering around the nucleus (Lee et al., 1989; Terasaki et al., 1986; Terasaki and Reese, 1994; Waterman-Storer and Salmon, 1998). It is now clear that there are multiple ways in which the ER tubules can interact with MTs. First, they can bind to MTs statically along their length by means of MT-binding proteins such as CLIMP-63 (Klopfenstein et al., 2001). Second, they can bind to the growing tips of MTs through tip attachment complexes (TACs) (Waterman-Storer and Salmon, 1988), which are generated by an interaction between the ER transmembrane protein STIM1 and the MT plus-end-binding protein EB1 (Grigoriev et al., 2008). Third, ER tubules are actively moved along MTs by MT motors, as described below. Actin-based motors can also move ER tubules, although in animal cells this motility is likely to contribute more to short-range movement such as that seen in dendritic spines (Dekker-Ohno et al., 1996; Takagishi et al., 1996; Bridgman, 1999). Myosins might have a more important role in ER transport during mitosis when MT-based motility is inactivated (Allan and Vale, 1991; Wollert et al., 2002).
Observation of ER dynamics in living newt lung cells has shown that the majority of the newly formed ER tubules are extended towards the cell periphery by plus-end-directed MT-based motors (Waterman-Storer and Salmon, 1998). In vitro motility assays indicate that kinesin-1 is the motor that drives extension of rat liver rough ER tubules (Wozniak and Allan, 2006). However, the involvement of kinesin-1 in ER motility in intact cells is controversial. Positive support comes from antisense studies in astrocytes that showed ER clustering around the nucleus when kinesin-1 was downregulated, similar to the changes seen upon MT depolymerisation (Feiguin et al., 1994). Also, kinesin-1 seems to be responsible for movements of a specialised ER subcompartment in hippocampal dendrites (Bannai et al., 2004). By contrast, neither the knockout of the gene encoding the KIF5B kinesin-1 motor subunit in mouse nor the inhibition of kinesin-1 activity by function-blocking antibodies in sea urchin eggs had any effect on ER morphology (Tanaka et al., 1998; Wright et al., 1993). Interestingly, the minus-end-directed MT motor cytoplasmic dynein transports ER tubules in mammalian and amphibian eggs (FitzHarris et al., 2007; Allan, 1995; Niclas et al., 1996; Reinsch and Karsenti, 1997) and in the fungus Ustilago maydis (Wedlich-Söldner et al., 2002). Such dynein-driven ER movement in embryos might play a part in pro-nuclear migration (Payne et al., 2003; Reinsch and Karsenti, 1997), as well as in the general organisation of the ER (FitzHarris et al., 2007). The current view in cultured cells, however, is that dynein does not contribute to ER movement. This conclusion is based on the observations that ER tubules do not move actively towards the cell centre in newt lung cells (Waterman-Storer and Salmon, 1998), and that inhibiting dynein function by overexpressing the dynactin p50-dynamitin subunit had no effect on ER morphology in HeLa cells (Burkhardt et al., 1997).
Here, we re-examine ER motility in living mammalian cultured cells to determine the relative contributions of TACs and motors in ER dynamics. ER organisation is dependent on both MTs and motor activity as loss of motility leads to a shift from a reticular to a more lamellar morphology. We demonstrate for the first time that ER membranes in cultured mammalian cells are actively transported towards MT minus-ends at the cell centre by the MT minus-end-directed motor cytoplasmic dynein. Furthermore, we confirm that kinesin-1 drives outward movement of ER tubules in vivo.
Results
ER motility is based on MTs
Although previous evidence obtained from in vitro motility assays suggests that kinesin-1 is most likely the only MT plus-end-directed motor protein that drives ER motility (Wozniak and Allan, 2006), conclusive in vivo evidence for the motor involved is lacking. In previous studies DiOC6 was used to label the ER (Lee et al., 1989; Lee and Chen, 1988; Terasaki et al., 1984; Terasaki et al., 1986; Terasaki and Reese, 1994; Waterman-Storer and Salmon, 1998). However, this compound is not selective and stains all membranous organelles. In addition, we noticed that the ER in cells labelled with DiOC6 was much less motile than the ER in cells expressing GFP-tagged ER markers (data not shown). Although GFP tagged with an HDEL ER-retention sequence and Sec61α-GFP enabled us to follow ER movement, a proportion of cells demonstrated a high cytosolic background of mislocalised GFP (data not shown). Therefore, we prepared an ER-targeted GFP construct that associates with the ER by inserting into the cytoplasmic face of the ER membrane by means of a C-terminal tail anchor sequence (Fig. 1A). The GFP-ER construct was transfected into cells and expressed for 16–24 hours. Expression of the construct in VERO (Fig. 1B), COS7 or NRK cells (data not shown) resulted in labelling of a tubular reticular network that colocalised with the ER lumenal protein calreticulin (Fig. 1B). Expression had no effect on the MT network (Fig. 1C).
Fig. 1.
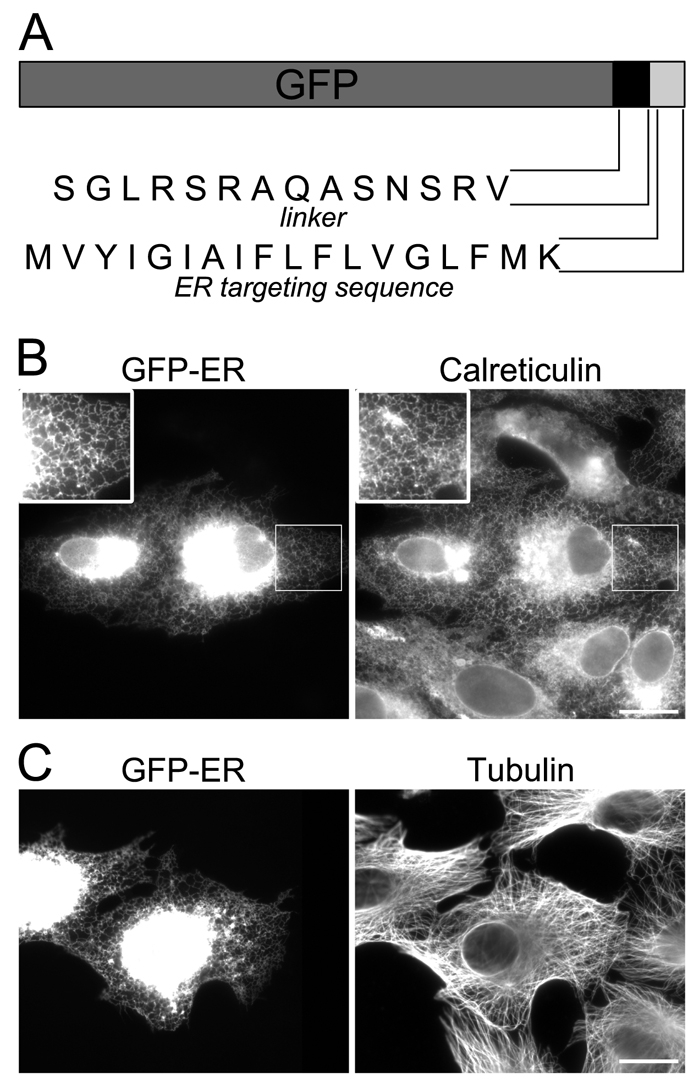
ER-GFP localises to the ER and does not affect cell morphology. (A) A C-terminal tail anchor ER-targeting sequence was attached at the C-terminus of GFP through a 14-residue linker peptide. (B,C) VERO cells were transfected with vector encoding ER-GFP and were labelled with antibodies against either calreticulin (B) or tubulin (C). Insets in B are 1.7× magnifications of the selected region. Scale bars: 20 μm. In this and all subsequent figures the brightness and contrast have been adjusted so that the peripheral ER can be seen, which results in signal saturation in the cell centre.
We then used the construct to image ER movement in living cells at high frame rates (ten frames/second). Although there was some variability between cells, we noticed that the ER network was much more motile in VERO than in COS7 cells, and the network was less dense, making imaging of moving tubules easier (data not shown). All subsequent experiments were therefore performed in VERO cells. ER motility was complex and included the extension of tubules along linear trajectories, as well as a continual stretching and remodelling of the ER network (supplementary material Movie 1), as has been described previously (Lee and Chen, 1988). ER dynamics were similar in the cell centre and in the periphery (data not shown), although individual movements were much easier to image away from the nucleus. Overlapping the first and last frames of Movie 1 revealed that the ER network had changed its organisation substantially during 30 seconds (Fig. 2A).
Fig. 2.

ER motility depends on microtubules (MTs). VERO cells were transfected with vector encoding GFP-ER: MTs were depolymerised with nocodazole (B) and the actin cytoskeleton was depolymerised with cytochalasin D (C); untreated cells were used as a control (A). Cells were imaged for 30 seconds at 10 frames/second, and the first (first row) and last (second row) frames were overlapped using ImageJ (bottom row). White areas indicate complete overlap of the first frame (magenta) with the last frame (green). Green or magenta areas show regions of the ER network that moved during imaging. Merged images in the bottom row are 2.8× magnifications of the selected boxed regions. Scale bars: 20 μm.
Previous studies have demonstrated that the motility of the ER in cultured cells strongly depends on MTs (Terasaki et al., 1986; Lee et al., 1989; Terasaki and Reese, 1994). This was also true in VERO cells as depolymerisation of MTs completely inhibited ER tubule extension and network rearrangements (supplementary material Movie 1). The first image of nocodazole-treated cells nearly completely overlapped with the final frame, indicating that there was hardly any movement during the 30 second image sequence in the absence of MTs (Fig. 2B). This lack of movement suggested that myosin-driven movement plays only a minor role in ER motility in VERO cells. In support of this conclusion, ER motility in cells treated with cytochalasin D was not obviously affected (Fig. 2C; supplementary material Movie 1), although the ER tubules appeared slightly thickened. Therefore, ER motility in VERO cells mainly depends on the MT cytoskeleton.
Analysis of ER tubule extension in living VERO cells
We could distinguish two types of ER tubule extension events in living VERO cells expressing the GFP-ER construct. First, a proportion of ER tubules extended slowly towards the cell periphery, resembling the MT plus-tip attachment complexes (TACs) (Waterman-Storer and Salmon, 1998) that have been described previously. To determine what proportion of ER tubules moved as TACs, we microinjected VERO cells with the GFP-ER construct along with a Tomato-EB3 construct, which encodes a neuronal homologue of EB1 (Nakagawa et al., 2000) fused to a red fluorescent protein (RFP) variant, dtTomato (see Materials and Methods) that localises to the growing plus-ends of MTs. An example of a TAC is shown in Fig. 3A and supplementary material Movie 2. On average, only 7% of ER tubules extended by means of TACs (n=19 cells), and, in some cells, we did not observe them at all. Instead, most moving ER tubules extended rapidly either towards and away from the cell periphery (Fig. 3B; supplementary material Movies 1 and 3), and these movements were very easy to distinguish from the relatively slow rate of TAC extension (0.14–0.25 μm/second, n=40). We tracked the movement of individual ER tubules using MetaMorph and determined the displacement between frames, allowing the maximal frame-to-frame speed of movement for each tubule to be calculated. This was comparable for both directions of movement, being 4.3–4.6 μm/second (Table 1).
Fig. 3.

ER tubules are actively transported towards and away from the plasma membrane. (A) Frames at selected time points from a movie of a cell transfected with vectors encoding GFP-ER and Tomato-EB3. Arrowheads indicate an ER tubule extended by a growing microtubule (TAC). Scale bar: 5 μm. (B) The large image shows areas that were selected to demonstrate inward (frame I) and outward (frame II) movements of ER tubules (arrowheads). Scale bars: 10 μm (large image) and 5 μm (small images). (C,D) Frames from movies of cells expressing Tomato-EB3 are shown to demonstrate (arrowheads) bending (C) and nucleation of MTs at the plasma membrane (D). Scale bars: 10 μm. (E) A minus-end-directed ER tubule was visualised in a cell expressing both dTomato-EB3 and GFP-ER (imaged at 2 frames/second, 100 millisecond exposure in each colour). The full image sequence is shown in supplementary material Movie 4. Scale bar: 10 μm.
Table 1.
Maximum rates of ER tubule extension
|
Maximum rate (μm/second) ± s.e.m.
|
||
|---|---|---|
| Construct expressed | Outward | Inward |
| RFP or mCherry | 4.58±0.1 (n=105) | 4.35±0.13 (n=61) |
| mCherry-p50 | 5.64±0.28 (n=31)** | ND |
| RFP-KHCct | 4.49±0.11 (n=101) | 4.99±0.2 (n=52)* |
| RFP | ND | 4.24±0.22 (n=26) |
| RFP-KHCtail | ND | 4.43±0.16 (n=40) |
ER tubules that moved inward or outward in cells expressing the indicated construct were tracked using MetaMorph, and the maximum frame-to-frame displacement was used to generate the maximum rate of translocation. The Student's t-test was used to determine whether differences between controls (RFP- or mCherry-expressing cells) and experimental samples were significant (*P≤0.01; **P≤0.0001). ND, not determined.
As these rapid inward movements could not be mistaken for the slow actin-dependent retrograde ER flow reported previously (Terasaki and Reese, 1994; Waterman-Storer and Salmon, 1998), we tested whether they were driven by a MT motor protein. As dynein-driven ER motility has not been reported before in cultured cells, we considered an alternative explanation, which is that some MTs might be organised with their plus-ends oriented towards the cell centre, allowing kinesin-1 to transport ER tubules inwards. We transfected VERO cells with Tomato-EB3 and counted EB3-labelled MT tips that grew inward and outward. There was considerable variability between cells: in some, MTs were organised with nearly all their plus-tips pointing towards the cell periphery, whereas, in others, ∼20% of MT tips extended inward. The average percentage of inward-growing MTs, labelled with Tomato-EB3, was 10.3±1.14% (s.e.m.), as estimated from 24 cells in three independent experiments. The inwardly pointing MTs were either a result of bending of centrally nucleated MTs (Fig. 3C) or of nucleation at the cell periphery (Fig. 3D). However, as the number of ER tubules moving rapidly inward equals those moving outward (Fig. 4G; Fig. 5F,G), it is unlikely that the 10% of MTs oriented towards the cell centre provide tracks for all these inward movements. The direct involvement of a minus-end-directed MT motor was confirmed by imaging cells expressing both GFP-ER and dTomato-EB3 (at levels where MTs were sufficiently coated along their length to be visualised) (Fig. 3E; supplementary material Movie 4). A growing MT can be seen making contact with an ER tubule, which then moves towards the MT minus-end. Taken together, these data reveal that there are two types of ER movements in VERO cells: slow extension by means of TACs and rapid motor-protein-driven movements that can be divided into plus-end- and minus-end-directed movements.
Fig. 4.

Dynein inhibition affects ER morphology and motility. VERO cells were transfected with vector encoding GFP-ER and either mCherry (A) as a control or Cherry-p50 (B-D). Images in the right-hand column are 3.7× magnifications of the selected areas. Scale bar: 20 μm. (E) Cells were transfected with vector encoding mCherry-p50 or mCherry, fixed and labelled with antibody against calreticulin. The proportion of cells with mainly reticular against predominantly lamellar ER morphology (see the Materials and Methods) was scored in mCherry- and mCherry-p50-expressing cells and in non-transfected cells (>1000 cells per condition, from three separate experiments). (F) The distribution of GFP-ER fluorescence was determined along a line from the nucleus to the cell periphery in 154 cells expressing mCherry and 184 cells expressing mCherry-p50 (from three independent experiments), as described in the Materials and Methods. An average of all traces is shown. (G) Graph showing the number of ER tubules moving either inward or outward in cells expressing GFP-ER and either mCherry or mCherry-p50. ER tubules were counted in 43 (mCherry-p50) and 35 (mCherry) cells in four independent experiments and are expressed as the number of tubules per cell counted in a box of 20×20 μm during a 30 second movie. Error bars indicate s.e.m., and P values were calculated using the Student's t-test (*P≤0.01; **P≤0.001; ***P≤0.0001).
Fig. 5.

Inhibition of kinesin-1 affects the motility of the ER network. VERO cells expressing GFP-ER and either RFP (A), RFP-DTC (B), RFP-BTC (C), RFP-KHCct (D) or RFP-KHCtail (E) were imaged for 30 seconds (10 frames/second). Cells expressing RFP proteins are identified in the top row. Acquired image stacks were collapsed (third row) using ImageJ and overlapped with the first frame of each acquisition (second row). In the merged images (bottom row, 3.4× magnifications of the boxed area), the white areas indicate complete overlap of the first frame (magenta) with the image of the collapsed stack (green). Scale bars: 20 μm. (F,G) Graphs showing the number of tubules moving inward or outward in cells microinjected with plasmids encoding GFP-ER and various RFP-tagged plasmids. Tubules were counted in >30 cells in 3-4 independent experiments and are expressed as the number counted per cell in a box of 10×10 μm (panel F) or 15×15 μm (panel G) per 30-second movie. Error bars indicate s.e.m., and P values were calculated using the Student's t-test (*P≤0.05; ** P≤0.01; *** P ≤0.001).
A role for dynein-dynactin in ER motility
We suspected that the minus-end-directed movement of ER tubules was driven by cytoplasmic dynein. As the function of this motor can be disrupted by the overexpression of the p50 (dynamitin) subunit of dynactin (Echeverri et al., 1996; Burkhardt et al., 1997), we coexpressed GFP-ER together with mCherry-p50 (or mCherry as a control) and observed ER organisation after fixation. ER morphology varied between cells in control transfections, with ∼70% of cells having a mainly tubular-reticular morphology (Fig. 4A,E) (defined as having less than ∼40% of the ER area as lamellae). As has been described previously, lamellar regions were concentrated in the cell centre (Puhka et al., 2007). By contrast, ∼30% of cells contained more extensive lamellar patches of ER. Lamellar ER was not an artefact of GFP-ER expression as a similar proportion of non-transfected cells labelled with antibody against calreticulin exhibited each phenotype (Fig. 4E). In cells transfected with GFP-ER and mCherry-p50, the situation was reversed, with ∼70% of cells having lamellar ER morphology (Fig. 4E). In the most severely affected cells, the ER did not form a reticular network at all but instead formed large lamellar patches (Fig. 4B). In other cells, the patches were smaller (Fig. 4C) or were only observed in peripheral regions (Fig. 4D). We also noticed that cells expressing p50 often had more ER in the cell periphery than control cells, as might be expected if plus-end-directed movement continued when dynein was inhibited. This impression was confirmed by measuring the fluorescence intensity along a line drawn from the nucleus to the cell periphery and plotting the average from 120 cells per condition. In p50-expressing cells, the central fluorescence intensity was reduced compared with that in RFP-expressing cells, whereas the peripheral intensity was increased (Fig. 4F).
We also counted moving ER tubules in cells cotransfected with GFP-ER and either mCherry-p50 or mCherry (Fig. 4G). The number of inward-moving ER tubules was reduced significantly in cells expressing mCherry-p50 (P<0.001), whereas outward movement was not affected significantly (P=0.45). We also determined the maximal rates of outward-moving tubules in cells expressing mCherry-p50. Interestingly, the average maximal rate was ∼1.0 μm/second higher than in control cells (P<0.01, Table 1). Taken together, our experiments indicate that dynein-dynactin is involved in ER motility and drives the movement of ER tubules towards the cell centre.
Kinesin-1 drives ER motility in VERO cells
We have previously shown that kinesin-1 is the major MT plus-end-directed motor protein for rat liver RER motility in vitro by using a range of inhibitory GST fusion proteins (Wozniak and Allan, 2006). We made similar constructs for expression in mammalian cells by swapping GST for RFP. An RFP-tagged C-terminal fragment of kinesin-1 heavy chain (RFP-KHCct) should inhibit kinesin activity by binding to the kinesin motor domain (Wozniak and Allan, 2006; Coy et al., 1999). When expressed at high levels, some MT decoration by RFP-KHCct was seen (data not shown) as this construct contains the previously described cryptic MT-binding site (Navone et al., 1992). However, cells with strong MT labelling were never selected for imaging. We also generated a shorter KHC C-terminal construct (RFP-KHCtail, containing amino acids 925-963), which displayed little or no MT binding (data not shown). In addition, to test whether specific kinesin light chains (KLCs) are involved in ER motility in vivo, we made RFP-tagged KLC1B and KLC1D constructs (RFP-BTC, RFP-DTC) that contained the tetratricopeptide repeat domain and isoform-specific C-terminal sequences (Wozniak and Allan, 2006). RFP-DTC served as a negative control as we have shown previously that a GST-DTC fusion protein has no effect on ER motility in vitro, whereas GST-BTC was inhibitory (Wozniak and Allan, 2006).
To determine whether kinesin-1 inhibition affected ER tubule movement, we microinjected cells with DNA encoding GFP-ER mixed with either RFP vector as a control or one of the kinesin constructs (Fig. 5). Outward movement of the ER was inhibited in RFP-KHCct- and RFP-KHCtail-expressing cells (Fig. 5D,E; supplementary material Movie 5). Reduced motility was also observed in RFP-BTC-expressing cells (Fig. 5C; supplementary material Movie 5). We scored moving ER tubules in at least 30 cells expressing RFP-tagged kinesin-1 fragments from three independent experiments. RFP-KHCct, RFP-KHCtail and RFP-BTC all inhibited outward motility of the ER, whereas RFP-DTC and RFP had no effect (Fig. 5F,G). The remaining motile tubules moved at a rate similar to that of controls (Table 1). Inward ER tubule movement was unaffected by RFP-BTC or RFP-DTC. The two heavy chain constructs had different effects on inward movement. RFP-KHCct caused a slight inhibition, whereas RFP-KHCtail led to a minor increase in motility (see supplementary material Movie 5, RFP-KHCtail, for four clear examples of inward movement): however, neither effect was statistically significant upon quantification (Fig. 5F,G).
We wondered whether expression of RFP-KHCct had an analogous effect on the rates of inward tubule extension as the expression of mCherry-p50 had on outward rates (Table 1). While the maximal rate of outward tubule movement was similar in RFP- and RFP-KHCct-expressing cells, the maximal rate of inward tubule movement was significantly faster upon expression of RFP-KHCct (4.35 μm/second in control cells against 4.99 μm/second in RFP-KHCct-expressing cells, P<0.01; Table 1). By contrast, expression of RFP-KHCtail had no effect on the inward movement rate (Table 1).
Role of MTs and motors in determining the ER network morphology
Previous studies have shown that the ER in some cell types collapses inwards when MTs are depolymerised (Lee et al., 1989; Terasaki et al., 1986; Terasaki and Reese, 1994; Waterman-Storer and Salmon, 1998). We were therefore surprised that ER retraction was a relatively rare consequence of inhibiting kinesin-1 (Fig. 5), even though dynein-driven ER movement persisted. However, cells expressing either RFP-KHCtail or RFP-KHCct often had a sparser peripheral ER network with larger polygons. To determine whether the observed lack of ER retraction was a consequence of incomplete kinesin-1 inhibition, we imaged the ER at slow frame rates (one frame per 6 seconds) during the process of MT depolymerisation induced by nocodazole (see Materials and Methods). Very little bulk inward movement of the ER was seen (supplementary material Movie 6; Fig. S1), suggesting that the actin-based inward flow of ER noted in some cell types (Terasaki and Reese, 1994; Waterman-Storer and Salmon, 1998) might not be occurring. This was confirmed by imaging cells expressing both GFP-ER and mCherry-actin before and during nocodazole treatment. Few VERO cells showed any evidence of inward movement of actin filaments (five out of 15 cells over three experiments), and only two cells showed any evidence of corresponding actin-based retraction of the ER network. By contrast, when the same markers were imaged in Xenopus A6 cells, actin filaments and ER moved inwards together in all cells (n=22) (supplementary material Movie 6; Fig. S1), consistent with previous observations in amphibian cell lines (Terasaki and Reese, 1994; Waterman-Storer and Salmon, 1998).
A striking feature of many nocodazole-treated VERO cells (20 out of 29 cells, from five independent experiments) was that the ER underwent a shift from reticular to lamellar morphology, with peripheral lamellar patches appearing (supplementary material Movie 6). Interestingly, disruption of kinesin function led to a significant shift of ER morphology from reticular to lamellar, with 63% of KHCct-expressing cells having a lamellar ER against 30% of control cells (Fig. 4E). Likewise, inhibition of dynein function also promoted lamellar morphology (Fig. 4E). A second phenotype was seen in nocodazole-treated cells, where the remaining ER tubules often became thicker, and the reticular network became much simpler (16 out of 29 cells). Interestingly, these thickened tubules often remained anchored at the cell periphery (supplementary material Movies 1 and 6; Figs 2, 5 and 6; supplementary material Fig. S1). The net result of these changes is that the morphology of the peripheral ER reticulum in VERO cells is profoundly altered when MTs are depolymerised, but the ER is not necessarily retracted from the cell periphery.
Fig. 6.

Re-extension of the ER after MT regrowth is strongly delayed in KHCct-expressing cells. VERO cells microinjected with plasmids encoding GFP-ER and either RFP or RFP-KHCct were treated with nocodazole to depolymerise MTs. Cells expressing RFP and RFP-KHCct are identified in the left-hand image in A and B, respectively. Nocodazole was washed off and cells were imaged 2-4 minutes later at one frame every 6 seconds, for 1 hour. Still images of cells expressing either RFP (C) or RFP-KHCct (D) are shown at selected time points. Scale bar: 20 μm.
When nocodazole was removed, the fine ER reticulum in control cells was rapidly re-established throughout the cell, with an extended, reticular ER organisation observed within ∼8 minutes (Fig. 6A). While the expression of RFP-KHCct (or RFP-BTC; data not shown) had no effect on nocodazole-induced changes (supplementary material Movie 6), their presence delayed the reappearance of a normal peripheral ER reticulum after nocodazole wash-out, with clustered or lamellar ER still present 30 minutes after nocodazole removal (Fig. 6B). However, if cells were left longer without nocodazole (∼1 hour), the ER extended to the cell periphery even in cells strongly expressing RFP-KHCct (Fig. 6B). Similar results were seen by immunofluorescence of fixed cells (data not shown). Taken together, our data indicate that kinesin-1 and dynein are motor proteins that drive ER dynamics in living cells and also contribute to the overall morphology of the ER network.
Discussion
We have examined ER motility in VERO cells and find that ER tubules move in both directions along MTs by using kinesin-1 and cytoplasmic dynein. When either motor is inhibited, or MTs are depolymerised, we observe that the ER morphology shifts from tubular to lamellar but that many ER tubules still extend right to the cell periphery.
Cytoplasmic dynein drives inward ER movement
We were surprised to find that about half of the rapid ER tubule movements in VERO cells were directed towards the cell centre. Fast bidirectional motility of vesicular ER structures has been seen in dendrites, but that movement was ascribed to kinesin-1 based on the fact that dendrites contain MTs of mixed polarity and that all of the movement was blocked when kinesin-1 was inhibited (Bannai et al., 2004). In VERO cells, only a small proportion of MTs were oriented with their plus-ends inwards, and blocking kinesin-1 function had no effect on inward tubule translocation. Using tdTomato-EB3, we confirmed that ER tubules can indeed move towards the minus-ends of MTs. Overexpressing the p50 subunit of dynactin, which disassembles the dynactin complex and so blocks dynein activity (Echeverri et al., 1996; Eckley at al., 1999), inhibited this inward movement, confirming that it is driven by dynein.
This use of dynein to drive ER motility is clearly a widespread phenomenon as it occurs in Xenopus laevis and mouse eggs and in the fungus U. maydis (Niclas et al., 1996; Allan, 1995; FitzHarris et al., 2007; Wedlich-Söldner et al., 2002) as well as in VERO cells. However, not all cultured cell types appear to exhibit this behaviour as imaging of the ER in newt lung epithelial cells revealed only slow inward movement of ER tubules in association with actin arcs (Waterman-Storer and Salmon, 1998). Furthermore, expressing p50 in HeLa cells had no effect on ER morphology (Burkhardt et al., 1997) even though in VERO cells it causes clear accumulation of lamellar ER that is often enriched in the cell periphery (Fig. 4A-D). This might be due in part to the different degrees of ER motility in these cell lines, as we have found that HeLa ER is much less motile than that in VERO cells (M.J.W., unpublished data).
Kinesin-1 and TACs drive outward ER movement
Our imaging of ER motility in living cells confirms previous observations (Waterman-Storer and Salmon, 1998; Grigoriev et al., 2008) that ER tubules move towards the cell periphery using a combination of motor-driven translocation and attachment to growing MT tips (by means of TACs). Interestingly, the balance between these two mechanisms differs between cell types (this work) (Waterman-Storer and Salmon, 1998; Grigoriev et al., 2008). Why this should be so is not clear, but one possibility worth investigating is whether TAC activity is proportional to the levels of STIM1 expression as STIM1 binds to the +TIPs EB1 and EB3 to link ER tubules to growing MT ends (Grigoriev et al., 2008).
Kinesin-1 was proposed two decades ago to be the motor that drives ER tubule extension (Dabora and Sheetz, 1988), and this was confirmed by using in vitro assays for ER motility (Lane and Allan, 1999; Wozniak and Allan, 2006). The situation in vivo has been less clear, however. For example, disrupting kinesin expression using an antisense approach led to aggregation of ER membranes around the nucleus in astrocytes (Feiguin et al., 1994), whereas cells from Kif5B-knockout mice had a normal ER distribution (Tanaka et al., 1998). In keeping with the latter situation, overexpression of our kinesin-1-inhibitory fragments had only subtle effects on ER distribution, causing a slight decrease in the density of the reticulum in peripheral regions, and an increase in lamellar morphology. Importantly, however, the same reagents strongly inhibited both ER re-extension after MT depolymerisation and outward ER tubule movement in living cells. Similar results have been obtained in dendrites, where overall ER organisation was not affected by Kif5B antisense treatment or by expressing a headless KIF5B protein, whereas the movement of a vesicular ER subdomain was inhibited (Bannai et al., 2004).
One explanation for these apparently contradictory results is that, although kinesin-1 or TACs extend ER tubules towards the cell periphery, where they often fuse with other tubules to generate the characteristic reticular tubular network, the ER is also stabilised by static interactions with the cytoskeleton. These could involve ER-localised MT-binding proteins, including CLIMP-63 and others (Klopfenstein et al., 2001; Vedrenne et al., 2005; Vedrenne and Hauri, 2006; Borgese et al., 2006). However, the ER network is very stable in semi-intact cells in the absence of MTs (Kano et al., 2005), suggesting that interactions between the ER and other cytoskeletal elements might be important. For example, ER-localised myosin V could connect ER tubules to actin filaments in the cell cortex, as well as driving short-range motility (Tabb et al., 1998; Wollert et al., 2002). Additionally, interactions between the ER and plasma membrane might contribute to stabilising the extended ER reticulum in the cell periphery, and this could explain the retention of some peripheral ER tubules over long periods in nocodazole-treated cells (Figs 2, 5 and 6). Whether STIM1 contributes to these interactions remains to be tested.
Is there competition or cooperation between kinesin-1 and dynein on the ER?
An idea that has gained widespread acceptance recently is that, if two motors of opposite polarity are present on one cargo, they will work together, rather than in competition. This is based first on data from a number of systems in which inhibiting one motor leads to simultaneous inhibition of the other motor, and second on biophysical measurements of bidirectional organelle movement (Welte, 2004). Bidirectional ER movement seems not to fit this pattern, however, as inhibition of dynein had no effect on the number of kinesin-1-driven tubule extensions (Fig. 4G). Interestingly, however, inhibition of dynein led to an increase in the rate of outward ER tubule movement (Table 1), again suggesting that the motors are in a `tug-of-war' rather than working cooperatively. Further support for this conclusion is provided by the effects of one of the inhibitory kinesin-1 constructs, RFP-KHCct, which led to a slight reduction in inward tubule movement (Fig. 5), while at the same time generating a statistically significant increase in the rate of these movements (Table 1). By contrast, however, RFP-KHCtail did not alter inward rates and led to a small but statistically insignificant increase in the number of inward tubule movements. This might reflect the fact that the interaction of KHCct with native kinesin-1 can lead to the motor binding tightly to MTs even in the presence of ATP (Wozniak and Allan, 2006), which could effectively `lock' the ER on to MTs, reducing its ability to move using dynein. Whether KHCct has the same effect on the interaction between kinesin-1 and MTs has not been tested. Alternatively, the partial coating of MTs by KHCct (even though cells exhibiting clear MT labelling were not analysed) could hinder the initial binding of dynein to MTs while not affecting, or even stimulating, the rate of translocation of dynein along the MT.
An interesting feature of the ER motility described here is the very rapid maximum rates of movement that the tubules exhibit in both directions along MTs, which far outstrip rates seen for purified kinesin-1 (∼1.4 μm/second at 35°C) (Kawaguchi and Ishiwata, 2000), although they are similar to those of Dictyostelium cytoplasmic dynein (∼3.5 μm/second at 25°C) (Shima et al., 2006). Similar maximum rates have recently been observed for outward ER tubule movement in HeLa cells (Grigoriev et al., 2008) and for dynein-driven ER motility in U. maydis (Wedlich-Söldner et al., 2002). In neurons, kinesin-1-driven cargoes also move at rates of up to 4 μm/second (Araki et al., 2007). Interestingly, in this latter case, different rates were observed for distinct cargoes, suggesting that the interaction between the motor and cargo-specific molecules can have a profound effect on motor function. This might explain the discrepancy between the rates of kinesin-1 translocation in vitro and in vivo. It has also been suggested that the rates of movement in vivo increase in an additive fashion for each additional motor engaged (1.2 μm/second per kinesin-1 or dynein on peroxisomes, up to a maximum of 12 μm/second) (Kural et al., 2005), raising the possibility that ER tubules move rapidly because several kinesin or dynein molecules are working together. However, more recent work has shown that the number of kinesin-1 molecules engaged in moving lipid droplets has no effect on their rate or distance of travel (Shubeita et al., 2008). The rapid rates of peroxisome movement seen by Kural and co-workers were probably partially attributable to sliding of the MTs themselves in the Drosophila S2 cells used (Kulic et al., 2008). We have not observed such MT sliding in VERO cells.
Relationship between motility, morphology and ER function
Our movies clearly show that the ER can be highly dynamic and that there is a continuous movement of tubules both towards and away from the cell centre. This raises the question as to what this movement contributes to ER function as the level of ER motility varies between cell types and can also differ between cells in a population (Lee and Chen, 1988) (and M.J.W., unpublished data).
Motility is obviously linked to ER morphology as the amount of lamellar ER increases while the reticular/tubular domains decrease both when MTs are depolymerised (Lee and Chen, 1988; Terasaki et al., 1986) (Figs 2 and 6) and when motors are inhibited (Figs 4 and 5). The simplest explanation for this is that the force generated by motors pulling on ER membranes plays a significant part in tubule formation by directly promoting alterations in membrane curvature. Alternatively, motor activity could promote the recruitment of members of the reticulon family of proteins whose presence triggers the formation of ER tubules and which are absent from lamellar ER regions (Voelz et al., 2006; Shibata et al., 2006). However, in living cells, reticulon proteins are particularly important for maintaining the ER network at the cell periphery (Voelz et al., 2006), while other observations suggest that tubules in this region are far less motile in VERO and COS7 cells (data not shown).
It has been suggested that lamellar regions are where protein synthesis is taking place, whereas tubules correspond to smooth ER (Shibata et al., 2006). Indeed, it has been shown that triggering enhanced protein synthesis leads to a shift from tubular to lamellar ER in a pancreatic acinar cell line (Rajasekaran et al., 1993), whereas removing ribosomes with puromycin in CHO-K1 cells has the opposite effect (Puhka et al., 2007). Although it might seem unlikely that MT depolymerisation or motor inhibition triggers an increase in rough ER in cultured cells, to our knowledge this idea has not been tested. It is certainly possible that kinesin-1 and dynein contribute to the efficiency of protein synthesis and/or export of protein from the ER, however. For example, kinesin-1 has been localised to ER exit sites (Aridor et al., 2001), and inhibition of kinesin-1 function causes a delay in the exit of newly synthesised protein from the ER (Gupta at al., 2008). Furthermore, the p150 subunit of the dynactin complex has been shown to be important for ER exit site turnover and accumulation of secretory cargo (Watson et al., 2005). Interestingly, the lateral mobility of translocon complexes is restricted by the presence of MTs (Nikonov et al., 2007), although this is most likely due to the activity of CLIMP63 rather than motors.
One benefit of having an ER network that is spread throughout the cell could be to aid the regulation of calcium levels. Although this ER distribution could be achieved by TACs and static interactions with the cytoskeleton, active translocation might facilitate the generation of contacts between the ER and plasma membrane for influx of extracellular calcium (Isoc) following depletion of intracellular stores. Indeed, the level of Isoc current has been reported to be sensitive to depolymerisation of MTs (Smyth et al., 2007; Wu et al., 2007) and to inhibition of kinesin or dynein (Wu et al., 2007). Interestingly, Isoc is not affected by disrupting the ability of STIM1 to generate TACs (Grigoriev et al., 2008), further suggesting that kinesin-1-driven ER movement plays a crucial role in the cellular response to calcium depletion. It will be interesting to monitor ER tubule movement in VERO cells following activation of Isoc to see whether the balance between inward and outward movement is affected, particularly as increased cytosolic calcium has been shown to reduce ER dynamics (Brough et al., 2005).
In summary, the data presented here support results obtained using in vitro motility assays (Wozniak and Allan, 2006) and establishes kinesin-1 as the plus-end-directed MT motor protein for the ER in living VERO cells, explaining contradictory data in the literature. In addition, we demonstrate for the first time that dynein-dynactin is actively involved in ER tubule sliding along MTs. Establishing why this motility is important for ER function is a fascinating challenge for the future.
Materials and Methods
Constructs and antibodies
The GFP-ER construct was prepared as described previously (Csordas et al., 2006). The EGFP open reading frame (ORF) from pEGFP-N1 (Clontech, Laboratories, Mountain View, CA) was amplified with specific primers that added a linker sequence SGLRSRAQASNSRV at the C-terminus of EGFP. A second PCR added the ER targeting sequence of the yeast UBC6 protein (X73234, residues 233-250: MVYIGIAIFLFVGLFMK) to the N-terminus. Finally, the EGFP ORF in the pEGFP-N1 vector was replaced with the amplified GFP-ER ORF. The Sec61α-GFP was a generous gift from Stephen High (University of Manchester, UK) (Greenfield and High, 1999). mRFP-actin was provided by Martin Humphries (University of Manchester, UK). tdTomato-EB3 encoded full-length mouse EB3, which was amplified from a C2C12 cell cDNA library and tagged with tdTomato (Shaner et al., 2004), and was a generous gift from Anne Straube (Marie Curie Research Institute, Oxted, UK). Rabbit anti-calreticulin was purchased from Affinity Bioreagents (Golden, CO). All secondary antibodies were purchased from Jackson (West Grove, PA).
The C-terminus of human kinesin heavy chain fragments (KHCct, residues 771-963 and KHCtail, residues 925-963) and hamster kinesin light chain 1 isoforms B and D (residues 228-543 and 228-605, respectively) were amplified using specific primers and cloned into the pcDNA 3.1-mRFP vector (a gift from Martin Humphries, University of Manchester, UK). The chicken p50 (dynamitin) ORF (provided by Trina Schroer (Johns Hopkins University, Baltimore, MA) was amplified with specific primers and cloned into a pcDNA3.1-mCherry vector (mCherry ORF was provided by Roger Tsien, University of California at San Diego, La Jolla, CA) (Shu et al., 2006). mRFP and mCherry proteins were fused at the N-terminus of p50 and the kinesin-1 fragments.
Immunofluorescence analysis
For immunofluorescence analysis, cells were grown on 13 mm coverslips, transfected using JetPei transfection reagent (Polyplus-transfection, New York, NY) according to the manufacturer's recommendations, then fixed 16-24 hours later in 3.6% formaldehyde, 0.2% glutaraldehyde in PBS for 25 minutes. Cells were quenched with NaBH4, permeabilised with 0.1% Triton X-100, 0.05% SDS in PBS and labelled with primary and fluorescently conjugated secondary antibodies. Cells were imaged using a 100× 1.35 NA or 60× 1.4 NA objectives on an Olympus BX60 microscope equipped with a CoolSnap ES camera (Roper Scientific) driven by MetaMorph software (Universal Imaging Corp.).
To estimate the effect of dynein inhibition on ER distribution, VERO cells were transfected with GFP-ER mixed with mCherry-p50 or mCherry (as a control) and expressed for ∼18 hours. Images of RFP- and p50-expressing cells (n=154 and n=184, respectively, over three independent experiments) were taken and the fluorescence brightness was measured along a line between the nuclear membrane (close to the microtubule-organising center, where the ER is thickest) and the plasma membrane using ImageJ (Rasband, W.S., ImageJ, US NIH, Bethesda, MD, http://rsb.info.nih.gov/ij/, 1997-2008). The values obtained per pixel were averaged and displayed as a function of distance from the nucleus.
To determine the percentage of cells with lamellar against reticular ER distribution in control (mCherry or RFP) and inhibitory conditions (RFP-KHCct and mCherry-p50 expressing cells), transfected cells were scored in more than 60 fields per condition in at least three independent experiments (>1000 cells in total per condition). Cells were classified as lamellar if more than ∼40% of the visible ER surface formed ER patches as opposed to a tubular, reticular network.
Microinjection and transfection
VERO cells grown on glass-bottomed dishes (MatTek Corp., Ashland, MA) were transferred into L15 medium containing 10% FCS and microinjected using an Eppendorf FemtoJet II and Micromanipulator 5171 on an Olympus IX50 at 37°C in a temperature-controlled box. DNA was used at 0.1 mg/ml in 10 mM Tris, pH 8.0. Microinjected cells were imaged 3-5 hours later. Cells on MatTek dishes were transfected with a total 3 μg of plasmid DNA per MatTek dish using JetPei transfection reagent and were imaged 16–24 hours later.
Live imaging and image analysis
Cells were imaged with a 100× 1.35 NA phase-contrast objective, with or without an additional 2× lens element mounted on an Olympus IX81 microscope fitted with a Prior Proscan II H1P7 motorised stage (Prior Scientific Instruments, Cambridge, UK). The stage and nose-piece were enclosed in a heated perspex chamber (Solent Scientific, Segensworth, UK) set at 37°C. Illumination at 475 nm or 575 nm (10 nm bandwidth) was provided by an Optoscan high-speed dynamic bandpass control monochromator (1800 g/mm holographic grating; Cairn Research, Faversham, UK) fitted with a 100 W mercury lamp, in combination with an EGFP/DsRed dichroic and emission filter (set 51019, Chroma Technology Corp., Rockingham, VT). Images were collected using a Photometrics Cascade 512 back-illuminated camera (Photometrics, Tucson, AZ) at ten frames/second, 100 msecond exposure, unless stated otherwise. For dual-colour imaging of GFP-ER and dTomato-EB3, sequential images were collected at 2 frames/second for each colour for a duration of 150 seconds.
Images were analysed using MetaMorph software and ImageJ. To estimate the amount of inward and outward motility in GFP-expressing cells, ER tubules were counted in a boxed area that fitted within cell boundaries. Depending on the experiment, the box dimensions were 10×10, 15×15 or 20×20 μm. Tubules that moved laterally rather than outward or inward were ignored. Rates of ER tubule movement were obtained using the point tracker function in MetaMorph. The percentage of ER tubules moving as TACs against being motor-driven was determined by scoring tubules in 19 cells expressing dTomato-EB3 and GFP-ER, using individual channels and an overlay. QuickTime movies were made from 8-bit MetaMorph stacks in ImageJ. Supplementary material Movie 4 was left uncompressed, whereas the remaining movies were made using MPEG or Sorensen3 image compression set at normal or high, depending on the image sequence.
Drug treatments
To image the behaviour of the ER alone, or together with actin, during drug treatments, cells were first microinjected with DNA encoding GFP-ER ± mRFP-actin and incubated at 37°C, 7% CO2 in DMEM plus 10% FCS for 3-5 hours. To depolymerise actin filaments, cells were incubated for 1 hour at 37°C, 7% CO2, in DMEM containing 10% FCS plus 1 μg/ml cytochalasin D. The medium was then replaced with prewarmed L15 plus 10% FCS and 1 μg/ml cytochalasin D and cells expressing GFP-ER were imaged at 37°C at ten frames per second, as described above.
To visualise the ER and actin behaviour during MT depolymerisation, cells were transferred into prewarmed L15 medium containing 10% FCS and imaged to locate 4-5 microinjected cells, marking their position using the motorised stage. The medium was then aspirated and replaced with L15 plus 10% FCS containing 3 μg/ml nocodazole. Following adjustment of the focus at each position, cells were imaged for up to 40 minutes at one frame per 6 seconds (in one or two colours, depending on the experiment) starting at 2-4 minutes after nocodazole addition. To image ER recovery after nocodazole treatment, cells were first incubated in ice-cold L15 medium plus 10% FCS plus 1 μg/ml nocodazole for 5 minutes on ice, then transferred to the 37°C microscope chamber for 1 hour. Nine or ten cells expressing GFP-ER were located and their positions marked, as described above. The nocodazole-containing medium was aspirated and changed four times with prewarmed L15 plus 10% FCS. After adjusting the focus, imaging commenced 2-4 minutes later and continued for 30-60 minutes at one frame per 6 seconds.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/12/1979/DC1
We would like to thank the following people for generous gifts of reagents: Stephen High, Martin Humphries (both at The University of Manchester, Manchester, UK), Trina Schroer (The Johns Hopkins University, Baltimore, MD), Anna Straube (Marie Curie Research Institute, Oxted, UK) and Roger Tsien (University of California at San Diego, La Jolla, CA). We are particularly grateful to Lisa Swanton and Philip Woodman for commenting on the manuscript, and to Philip Woodman for stimulating discussions throughout this project. This work was supported by The Wellcome Trust (project grant 078825), the EU (TMR Network grant HPRN-CT-2000-00081) and the Medical Research Council (Co-operative Group Award G9722026). B.B. was supported by a Wellcome Trust PhD studentship (075373), and K.B. is supported by a BBSRC PhD studentship. Deposited in PMC for release after 6 months.
References
- Allan, V. (1995). Protein phosphatase 1 regulates the cytoplasmic dynein-driven formation of endoplasmic reticulum networks in vitro. J. Cell Biol. 128, 879-891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan, V. J. and Vale, R. D. (1991). Cell cycle control of microtubule-based membrane transport and tubule formation in vitro. J. Cell Biol. 113, 347-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki, Y., Kawano, T., Taru, H., Saito, Y., Wada, S., Miyamoto, K., Kobayashi, H., Ishikawa, H. O., Ohsugi, Y., Yamamoto, T. et al. (2007). The novel cargo Alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J. 26, 1475-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor, M., Fish, K. N., Bannykh, S., Weissman, J., Roberts, T. H., Lippincott-Schwartz, J. and Balch, W. E. (2001). The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J. Cell Biol. 152, 213-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai, H., Inoue, T., Nakayama, T., Hattori, M. and Mikoshiba, K. (2004). Kinesin dependent, rapid, bi-directional transport of ER sub-compartment in dendrites of hippocampal neurons. J. Cell Sci. 117, 163-175. [DOI] [PubMed] [Google Scholar]
- Baumann, O. and Walz, B. (2001). Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 205, 149-214. [DOI] [PubMed] [Google Scholar]
- Borgese, N., Francolini, M. and Snapp, E. (2006). Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgman, P. C. (1999). Myosin Va movements in normal and dilute-lethal axons provide support for a dual filament motor complex. J. Cell Biol. 146, 1045-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brough, D., Schell, M. J. and Irvine, R. F. (2005). Agonist-induced regulation of mitochondrial and endoplasmic reticulum motility. Biochem. J. 392, 291-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt, J. K., Echeverri, C. J., Nilsson, T. and Vallee, R. B. (1997). Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J. Cell Biol. 139, 469-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coy, D. L., Hancock, W. O., Wagenbach, M. and Howard, J. (1999). Kinesin's tail domain is an inhibitory regulator of the motot domain. Nat. Cell Biol. 1, 288-292. [DOI] [PubMed] [Google Scholar]
- Csordas, G., Renken, C., Varnai, P., Walter, L., Weaver, D., Buttle, K. F., Balla, T., Mannella, C. A. and Hajnoczky, G. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabora, S. L. and Sheetz, M. P. (1988). Microtubule dependent formation of a tubularvesicular network with characteristics of the endoplasmic reticulum from cultured cell extracts. Cell 54, 27-35. [DOI] [PubMed] [Google Scholar]
- Dekker-Ohno, K., Hayasaka, S., Takagishi, Y., Oda, S., Wakasugi, N., Mikoshiba, K., Inouye, M. and Yamamura, H. (1996). Endoplasmic reticulum is missing in dendritic spines of Purkinje cells of the ataxic mutant rat. Brain Res. 714, 226-230. [DOI] [PubMed] [Google Scholar]
- Echeverri, C. J., Paschal, B. M., Vaughan, K. T. and Vallee, R. B. (1996). Molecular characterization of the 50-kD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. J. Cell Biol. 132, 617-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckley, D. M., Gill, S. R., Melkonian, K. A., Bingham, J. B., Goodson, H. V., Heuser, J. E. and Schroer, T. A. (1999). Analysis of dynactin subcomplexes reveals a novel actin-related protein associated with the arp1 minifilament pointed end. J. Cell Biol. 147, 307-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiguin, F., Ferreira, A., Kosik, K. S. and Caceres, A. (1994). Kinesin-mediated organelle translocation revealed by specific cellular manipulations. J. Cell Biol. 127, 1021-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzHarris, G., Marangos, P. and Carroll, J. (2007). Changes in endoplasmic reticulum structure during mouse oocyte maturation are controlled by the cytoskeleton and cytoplasmic dynein. Dev. Biol. 305, 133-144. [DOI] [PubMed] [Google Scholar]
- Greenfield, J. J. and High, S. (1999). The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J. Cell Sci. 112, 1477-1486. [DOI] [PubMed] [Google Scholar]
- Grigoriev, I., Gouveia, S. M., van der Vaart, B., Demmers, J., Smyth, J. T., Honnappa, S., Splinter, D., Steinmetz, M. O., Putney, J. W. J., Hoogenraad, C. C. et al. (2008). STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr. Biol. 18, 177-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, V., Palmer, K. J., Spence, P., Hudson, A. and Stephens, D. J. (2008). Kinesin-1 (uKHC/KIF5B) is required for bidirectional motility of ER exit sites and efficient ER-to-Golgi transport. Traffic 9, 1850-1866. [DOI] [PubMed] [Google Scholar]
- Kano, F., Kondo, H., Yamamoto, A., Kaneko, Y., Uchiyama, K., Hosokawa, N., Nagata, K. and Murata, M. (2005). NSF/SNAPs and p97/p47/VCIP135 are sequentially required for cell cycle-dependent reformation of the ER network. Genes Cells 10, 989-999. [DOI] [PubMed] [Google Scholar]
- Kawagishi, K. and Ishiwata, S. (2000). Temperature dependence of force, velocity and processivity of single kinesin molecules. Biochem. Biophys. Res. Commun. 272, 895-899. [DOI] [PubMed] [Google Scholar]
- Klopfenstein, D. R., Klumperman, J., Lustig, A., Kammerer, R. A., Oorschot, V. and Hauri, H. P. (2001). Subdomain-specific localization of CLIMP-63 (p63) in the endoplasmic reticulum is mediated by its luminal alpha-helical segment. J. Cell Biol. 153, 1287-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulic, I. M., Brown, A. E. X., Kim, H., Kural, C., Blehm, B., Selvin, P. R., Nelson, P. C. and Gelfand, V. I. (2008). The role of microtubule movement in bidirectional organelle transport. Proc. Natl. Acad. Sci. USA 105, 10011-10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kural, C., Kim, H., Syed, S., Goshima, G., Gelfand, V. I. and Selvin, P. R. (2005). Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordingated movement? Science 308, 1469-1472. [DOI] [PubMed] [Google Scholar]
- Lane, J. D. and Allan, V. J. (1999). Microtubule-based endoplasmic reticulum motility in Xenopus laevis: activation of membrane-associated kinesin during development. Mol. Biol. Cell 10, 1909-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, C. and Chen, L. B. (1988). Dynamic behavior of endoplasmic reticulum in living cells. Cell 54, 37-46. [DOI] [PubMed] [Google Scholar]
- Lee, C., Ferguson, M. and Chen, L. B. (1989). Construction of the endoplasmic reticulum. J. Cell Biol. 109, 2045-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, H., Koyama, K., Murata, Y., Morito, M., Akiyama, T. and Nakamura, Y. (2000). EB3, a novel member of the EB1 family preferentially expressed in the central nervous system, binds to a CNS-specific APC homologue. Oncogene 19, 210-216. [DOI] [PubMed] [Google Scholar]
- Navone, F., Niclas, J., Hom-Booher, N., Sparks, L., Bernstein, H. D., McCaffrey, G. and Vale, R. D. (1992). Expression of the human kinesin heavy chain gene in CV-1 cells: interaction of the C-terminal domain with cytoplasmic microtubules. J. Cell Biol. 117, 1263-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niclas, J., Allan, V. J. and Vale, R. D. (1996). Cell cycle regulation of dynein association with membranes modulates microtubule-based organelle transport. J. Cell Biol. 133, 585-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonov, A. V., Hauri, H., Lauring, B. and Kreibich, G. (2007). Climp-63-mediated binding of microtubules to the ER affects the lateral mobility of translocon complexes. J. Cell Sci. 120, 2248-2258. [DOI] [PubMed] [Google Scholar]
- Payne, C., St., John, J., CRamalho-Santos, J. and Schatten, G. (2003). LIS1 association with dynactin is required for nuclear motility and genomic union in the fertilized mammalian oocyte. Cell Motil. Cytoskeleton 56, 245-251. [DOI] [PubMed] [Google Scholar]
- Puhka, M., Vihinen, H., Joensuu, M. and Jokitalo, E. (2007). Endoplasmic reticulum remains continuous and undergoes sheet-to-tubule transformation during cell division in mammalian cells. J. Cell Biol. 179, 895-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran, A. K., Morimoto, T., Hanzel, D. K., Rodriguez-Boulan, E. and Kreibich, G. (1993). Structural reorganization of the rough endoplasmic reticulum without size expansion accounts for dexamethasone-induced secretory activity in AR42J cells. J. Cell Sci. 105, 333-345. [DOI] [PubMed] [Google Scholar]
- Reinsch, S. and Karsenti, E. (1997). Movement of nuclei along microtubules in Xenopus egg extracts. Curr. Biol. 7, 211-214. [DOI] [PubMed] [Google Scholar]
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N., Palmer, A. E. and Tsien, R. Y. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572. [DOI] [PubMed] [Google Scholar]
- Shibata, Y., Voeltz, G. K. and Rapoport, T. A. (2006). Rough sheets and smooth tubules. Cell 126, 435-439. [DOI] [PubMed] [Google Scholar]
- Shima, T., Kon, T., Imamula, K., Ohkura, R. and Sutoh, K. (2006). Two modes of microtubule sliding driven by cytoplasmic dynein. Proc. Natl. Acad. Sci. USA 103, 17736-17740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, X., Shaner, N. C., Yarbrough, C. A., Tsien, R. Y. and Remington, S. J. (2006). Novel chromophores and buried charges control color in mFruits. Biochemistry 45, 9639-9647. [DOI] [PubMed] [Google Scholar]
- Shubeita, G. T., Tran, S. L., Xu, J., Cermelli, S., Cotton, S. L., Welte, M. A. and Gross, S. P. (2008). Consequences of motor copy number on the intracellular transport of kinesin-1-driven lipid droplet movement. Cell 135, 1098-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth, J. T., DeHaven, W. I., Bird, G. S. and Putney, J. W. J. (2007). Role of the microtubule cytoskeleton in the function of the store-operated Ca2+ channel activator STIM1. J. Cell Sci. 120, 3762-3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb, J. S., Molyneaux, B. J., Cohen, D. L., Kuznetsov, S. A. and Langford, G. M. (1998). Transport of ER vesicles on actin filaments in neurons by myosin V. J. Cell Sci. 111, 3221-3234. [DOI] [PubMed] [Google Scholar]
- Takagishi, Y., Oda, S., Hayasaka, S., Dekker-Ohno, K., Shikata, T., Inouye, M. and Yamamura, H. (1996). The dilute-lethal (dl) gene attacks a Ca2+ store in the dendritic spine of Purkinje cells in mice. Neurosci. Lett. 215, 169-172. [DOI] [PubMed] [Google Scholar]
- Tanaka, Y., Kanai, Y., Okada, Y., Nonaka, S., Takeda, S., Harada, A. and Hirokawa, N. (1998). Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 93, 1147-1158. [DOI] [PubMed] [Google Scholar]
- Terasaki, M. and Reese, T. S. (1994). Interactions among endoplasmic reticulum, microtubules, and retrograde movements of the cell surface. Cell Motil. Cytoskeleton 29, 291-300. [DOI] [PubMed] [Google Scholar]
- Terasaki, M., Song, J., Wong, J. R., Weiss, M. J. and Chen, L. B. (1984). Localization of endoplasmic reticulum in living and glutaraldehyde-fixed cells with fluorescent dyes. Cell 38, 101-108. [DOI] [PubMed] [Google Scholar]
- Terasaki, M., Chen, L. B. and Fujiwara, K. (1986). Microtubules and the endoplasmic reticulum are highly interdependent structures. J. Cell Biol. 103, 1557-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedrenne, C. and Hauri, H. (2006). Morphogenesis of the endoplasmic reticulum: beyond active membrane expansion. Traffic 7, 639-646. [DOI] [PubMed] [Google Scholar]
- Vedrenne, C., Klopfenstein, D. R. and Hauri, H. (2005). Phosphorylation controls CLIMP-63-mediated anchoring of the endoplasmic reticulum to microtubules. Mol. Biol. Cell 16, 1928-1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz, G. K., Prinz, W. A., Shibata, Y., Rist, J. M. and Rapoport, T. A. (2006). A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124, 573-586. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer, C. M. and Salmon, E. D. (1998). Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol. 8, 798-806. [DOI] [PubMed] [Google Scholar]
- Watson, P., Forster, R., Palmer, K. J., Pepperkok, R. and Stephens, D. J. (2005). Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nat. Cell Biol. 7, 48-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedlich-Söldner, R., Schulz, I., Straube, A. and Steinberg, G. (2002). Dynein supports motility of endoplasmic reticulum in the fungus Ustilago maydis. Mol. Biol. Cell 13, 965-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte, M. A. (2004). Bidirectional transport along microtubules. Curr. Biol. 14, R525-R537. [DOI] [PubMed] [Google Scholar]
- Wollert, T., Weiss, D. G., Gerdes, H. and Kuznetsov, S. A. (2002). Activation of myosin V-based motility and F-actin-dependent network formation of endoplasmic reticulum during mitosis. J. Cell Biol. 159, 571-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak, M. J. and Allan, V. J. (2006). Cargo selection by specific kinesin light chain 1 isoforms. EMBO J. 25, 5457-5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, B. D., Terasaki, M. and Scholey, J. M. (1993). Roles of kinesin and kinesin-like proteins in sea urchin embryonic cell division: evaluation using antibody microinjection. J. Cell Biol. 123, 681-689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, S., Chen, H., Alexeyev, M. F., King, J. A. C., Moore, T. M., Stevens, T. and Balczon, R. D. (2007). Microtubule motors regulate ISOC activation necessary to increase endothelial cell permeability. J. Biol. Chem. 282, 34801-34808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.