

Abstract
The Drosophila Swiss Cheese (SWS) protein and its vertebrate ortholog Neuropathy Target Esterase (NTE) are required for neuronal survival and glial integrity. In humans, NTE is the target of organophosphorous compounds which cause a paralyzing axonal degeneration and recently mutations in NTE have been shown to cause a Hereditary Spastic Paraplegia called NTE-related Motor-Neuron Disorder. SWS and NTE are concentrated in the endoplasmic reticulum and both have been shown to have an esterase function against an artificial substrate. However, the functional mechanisms and the pathways in which SWS/NTE are involved in are still widely unknown. Here, we show that SWS interacts specifically with the C3 catalytic subunit of cAMP activated protein kinase (PKA-C3), which together with orthologs in mouse (Pkare) and human (PrKX) forms a novel class of catalytic subunits of unknown function. This interaction requires a domain of SWS which shows homology to regulatory subunits of PKA and, like conventional regulatory subunits, the binding of SWS to the PKA-C3 inhibits its function. Consistent with this result, expression of additional PKA-C3 induces degeneration and enhances the neurodegenerative phenotype in sws mutants. We also show that the complex formation with the membrane-bound SWS tethers PKA-C3 to membranes. We therefore propose a model in which SWS acts as a noncanonical subunit for PKA-C3, whereby the complex formation regulates the localization and kinase activity of PKA-C3, and that disruption of this regulation can induce neurodegeneration.
Keywords: Drosophila, SWS, NTE, PKA, catalytic subunit, neurodegeneration
Introduction
Swiss-cheese (sws) mutant flies show age-dependent neurodegeneration, glial hyperwrapping, and neuronal apoptosis (Kretzschmar et al., 1997). SWS is the ortholog of vertebrate Neuropathy Target Esterase (NTE) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Lush et al., 1998; Moser et al., 2000), which plays an important role in organophosphate (OP)-induced delayed neuropathy (OPIDN), occurring after intoxication with organophosphorous compounds (Glynn, 2000; Moretto, 2000) found in pesticides and nerve agents (Lotti and Moretto, 2005). OPIDN, which has first been described after a poisoning epidemic in the southern United States (Smith et al., 1930), is characterized by degeneration of long axons in the central and peripheral nervous systems (Ehrich and Jortner, 2001). Recently, mutations in human NTE have also been shown to cause a Hereditary Spastic Paraplegia called NTE-Related Motor-Neuron Disorder (Rainier et al., 2008). Mice lacking NTE show severe growth retardation and die approximately on day 9 of embryonic development (Moser et al., 2004), whereas neuronal specific NTE knock-out mice show a strikingly similar phenotype to sws mutants, including vacuolization, abnormal myelin figures, and neuronal death (Akassoglou et al., 2004). We have also shown that these proteins are functionally conserved because mouse NTE can completely replace SWS in Drosophila (Mühlig-Versen et al., 2005).
Both, SWS and mouse NTE are widely expressed in the nervous system with a more restricted pattern to large neurons in older animals (Moser et al., 2000; Mühlig-Versen et al., 2005). Both have been localized to the endoplasmic reticulum (ER) (Akassoglou et al., 2004; Mühlig-Versen et al., 2005) and Li et al. (2003) described that NTE transfected into COS cells is inserted into ER membranes with most of it exposed on the cytoplasmic face.
NTE and SWS exhibit esterase activity against the artificial substrate phenyl-valerate (Johnson, 1977; Mühlig-Versen et al., 2005), which requires a serine residue within a highly conserved domain. A point mutation in this serine in SWS abolishes esterase activity and interferes with the function of SWS in vivo (Mühlig-Versen et al., 2005). In addition, SWS contains several regions that show homology to the regulatory subunit of cAMP-dependent protein kinase (PKA). One of these regions contains a tandem cyclic nucleotide binding site found in canonical regulatory subunits, whereas a third consists of a single cyclic nucleotide binding site (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material). The forth region shows homology to the motif required for the interaction of the regulatory subunit with the catalytic subunit of PKA, including the pseudosubstrate site (Kretzschmar et al., 1997). PKA holoenzymes are tetramers consisting of two catalytic and two regulatory subunits, which are activated by dissociation of the regulatory subunits allowing the catalytic subunits to unfold their kinase activity (Francis et al., 2002; Taylor et al., 2005). The binding between the subunits is mediated by the pseudosubstrate site which resembles the R-R-X-S-X consensus site found in PKA substrates (Poteet-Smith et al., 1997). In this study, we show that SWS acts similar to regulatory subunits specifically binding PKA-C3, and we show that this interaction plays a role in the neurodegenerative phenotype of sws.
Materials and Methods
Drosophila stocks and UAS-lines.
The sws1 allele has been described by Kretzschmar et al. (1997). If not stated differently, yw, the genetic background of all the transgenic lines, and sws1 was used as control. Actin-GAL4, GMR-GAL4, elav-GAL4, and Df(3L)brm11 were provided by the Bloomington Stock Center (Indiana University, Bloomington, IN) and Appl-GAL4 was provided by L. Torroja (Universidad Autonoma de Madrid, Spain). Stocks were maintained and raised under standard conditions. To create the UAS-SWSR133A construct, we used the pUAST-sws construct described in (Mühlig-Versen et al., 2005) and replaced the arginine133 in the PKA pseudosubstrate site by alanine, using primers that substituted the CGG triplet by GAA. The SWSS985D construct has been described in the study by Mühlig-Versen et al. (2005). The UAS-PKA-C3 line was created by inserting the PKA-C3 coding region (the PKA-C3 cDNA was kindly provided by D. Kalderon, Columbia University, NY) into the pUAST vector (Brand and Perrimon, 1993).
Tissue sections and whole-mount preparations for immunohistochemistry.
Paraffin sections were performed as described in the study by Bettencourt da Cruz et al. (2005). Adult brains were dissected on ice and fixed in 4% parformaldehyde overnight. Immunohistochemistry was performed following the protocol of Buchner et al. (1989). The anti-SWS rabbit antisera was used 1:100, anti-PKA-C3 rat antisera was used 1:1000, and mouse anti-GRP78 (Stressgen) 1:1000 and applied overnight at 4°C. Cy2 and Cy3 secondary antibodies were obtained from Jackson ImmunoResearch. Preparations were observed with an Olympus Fluoview confocal 300 microscope, and optical sections taken with a thickness of 0.1 μm.
Determination of vacuole size.
To analyze the neurodegenerative phenotype of different genotypes, we photographed sections at the level of the great commissure. For a double blind analysis, pictures were taken and numbered, and the area of the vacuoles in the central brain was then calculated in Photoshop as total pixel number, which was subsequently converted into square micrometers (Bettencourt da Cruz et al., 2005). As controls, we used flies from the same cross, which did not carry the UAS construct, and flies that had a balancer chromosome instead of the deletion chromosome. Both controls showed similar values and were therefore pooled. Statistics were done using the SPSS program and independent sample t tests. The Levene's test was used to determine variance and p values.
Yeast two-hybrid screens.
The two-hybrid screens were performed using the CytoTrap Vector kit and the CytoTrap XR Adult Drosophila cDNA Library from Stratagene following the instruction manual. As bait, we used the full-length SWS protein and two fragments that did not contain any of the predicted transmembrane domains. Both of these shortened fragments contained the pseudosubstrate site as predicted from sequence similarity with the regulatory subunit of PKA. The longer fragment consisted of amino acids 60–544, the smaller one of amino acids 60–241. cDNAs for the PKA-C1 and PKA-C2 catalytic subunit were kindly provided by D. Kalderon (Columbia University, NY) and inserted into the pMyr vector.
Western blots.
Membrane and cytosolic fractions were prepared from the different genotypes following the protocol of Orgad et al. (1987). Approximately 300 heads were used for each preparation, protein amounts determined by Bradford assays (Bradford, 1976), and 12 μg was loaded per lane. Gels and blots were performed as described by Tschäpe et al. (2002). Anti-SWS was used at 1:500 and anti-PKA at 1:4000 (generated by D. Kalderon and kindly provided by B. Biteau, University of Rochester, NY). When head lysates were used directly in Western blots, five heads were loaded per lane, and a loading control was performed using anti-actin 1:500 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA).
PKA activity measurements.
The PepTag Assay for Non-Radioactive Detection of Protein Kinase C or cAMP-Dependent Protein Kinase Kit from Promega was used for PKA activity measurement. Five fly heads were homogenized in 50 μl of extraction buffer and centrifuged at 14,000 × g for 5 min. Supernatant (2 μl) was immediately used for activity measurement assays for each sample, whereas 20 μl were used to perform Bradford assays (Bradford, 1976). Activity measurements were performed using 0.04 μg/μl PepTag A1 peptide, and 250 mm IBMX (isobutylmethylxanthine) either with or without cAMP (0.4 mm) added to the reaction. The reaction was performed according to the kit protocol and evaluated by luminosity measurement of the gel picture by determining the p/q quotient of the luminosity value of phosphorylated to unphosphorylated kemptide peptide. Values were normalized per ng amount of protein in the lysate, as determined by the Bradford assays. As positive control, we used 2 μg/ml PKA provided by the kit, negative controls contained water instead of fly head lysate. Incubation time was 30 min.
Esterase assays.
Frozen flies were homogenized (5% w/w) in 50 mm Tris-HCl, 1 mm EDTA. After brief centrifugation (500 × g; 5 min) the homogenates were assayed as described by Johnson (1977). NTE-like activity is operationally defined as that portion of phenyl valerate hydrolyzing activity which is resistant to paraoxon (40 μm; a nonneuropathic OP) but sensitive to mipafox (50 μm; a neuropathic OP). The homogenates were preincubated (37°C; 20 min) with paraoxon in the presence or absence of mipafox before substrate, phenyl valerate, was added and reaction allowed to proceed for another 20 min. Activity was determined as the amount of phenol liberated and expressed as the NTE-like activity (i.e., the difference in the activity measured in the presence and absence of mipafox) per mg homogenate protein. Homogenates were prepared from ∼200 flies (2–5 d old) and measurements done in duplicates.
Results
Mutations in the interaction domain of SWS interfere with its biological function
SWS is a 1425aa long transmembrane protein with a highly conserved C-terminal domain that is involved in its function as serine esterase, and a N-terminal region that shows homology to the regulatory subunit of PKA (Kretzschmar et al., 1997), including cyclic nucleotide binding sites and a domain which in PKA is involved in binding the catalytic subunit (see Fig. 2A,B). This region, which from here on will be referred to as interaction domain, contains a site resembling the pseudosubstrate site of the regulatory subunit of PKA (PKA-R). As described above, this site is similar to the R-R-X-S-X site found in substrates of PKA and has been shown to mediate binding between the catalytic and regulatory subunits of the PKA complex. Whereas the serine residue in this motif, which is the target for phosphorylation, is maintained in the type RII subunits, it is replaced by alanine in vertebrate RI subunits and by glycine in the fly R1 subunit. The SWS pseudosubstrate site contains an asparagine at this position (see Fig. 2B). The two arginines, which are shared by all types of regulatory subunits (RIα, RIβ, RIIα, RIIβ) and by the PKA inhibitor PKI, are also conserved in SWS. Mutations in these arginines have been shown to decrease the interaction and inhibitory potency of R1α (Buechler et al., 1993; Poteet-Smith et al., 1997). To investigate the functional importance of the interaction domain, and specifically the pseudosubstrate site in SWS, we mutated the second arginine residue (R133) to an alanine. To determine whether this mutant construct (SWSR133A) is functional, we expressed SWSR133A pan-neuronally using the Appl-GAL4 driver (Torroja et al., 1999) in sws mutant flies. Determining the degree of neurodegeneration by measuring the area of vacuoles in brain sections from 14-d-old flies, we observed a reduced rescue capability of SWSR133A compared with the wild-type SWS construct expressed under the same conditions (equal expression levels of the SWS constructs were confirmed by Western blot) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Control sws1 flies, carrying the Appl-GAL4 driver but no SWS construct, showed a mean vacuole area of 52 ± 6.8 μm2 (Fig. 1A,D). Compared with these flies, sws1 mutant flies expressing SWSR133A pan-neuronally showed significantly less vacuolization (Fig. 1B) (30 ± 2.4 μm2; p < 0.01). However, the rescue ability of the SWSR133A construct was significantly less than seen after expression of the wild-type construct. Because of the cell-autonomous requirement of SWS in neurons and glia, neuronal expression even of the wild-type SWS in sws1 mutants can only prevent neuronal degeneration whereas the glial phenotype still persists (Mühlig-Versen et al., 2005). Therefore a few vacuoles are still detectable after expression of the wild-type construct, but these are much less than with SWSR133A (Fig. 1C,D) (16 ± 3.7 μm2; p < 0.05 to SWSR133A). These results show that the R to A mutation in the pseudosubstrate site does interfere with the function of the SWS protein in vivo.
Figure 2.

SWS binds specifically to PKA-C3. A, The different fragments used in the yeast two-hybrid experiments are shown as dark gray lines below a schematic of the SWS protein. The predicted transmembrane (TM) domains are shown as vertical black bars and the interaction domain (ID), cyclic nucleotide binding sites (cNMP1–3), and esterase domain are indicated as gray boxes. The active site serine which is localized in the putative third transmembrane domain within the esterase domain is indicated by an S. B, Sequence comparison of the interaction domain of SWS with the human and Drosophila R1 regulatory subunits. Identical amino acids are indicated by asterisks, highly conserved amino acids by colons, and conserved amino acids by dots. C, Using the smaller SWS (SWS60–241) fragment, we obtained colonies at the restrictive temperature when cotransfected with PKA-C3 (first row) but not after cotransfection with PKA-C2 or PKA-C1 (second and third row). Using the same fragment with a mutation in the conserved arginine133 still produced colonies after cotransfection with PKA-C3 at the restrictive temperature but these colonies grew less well (fourth row). D, The same results were obtained in cotransfection studies using the larger SWS60–544 fragment.
Figure 1.

Expression of SWSR133A in the nervous system of sws1 mutant flies does only partially rescue the degenerative phenotype and affects esterase activity. A, A sws1 fly shows the characteristic vacuolization in the neuropil (arrows). B, Expressing SWSR133A pan-neuronally in these flies leads to a significant reduction in vacuole formation. C, Expressing wild-type SWS under the same conditions almost completely reverts the mutant phenotype. D, Mean area of vacuoles in μm2 in the three genotypes shown in A, B, and C. Number of measured flies was as follows: n = 24 for Appl-GAL4, sws1 (A), n = 28 for Appl-GAL4, sws1; UAS-SWSR133A (B), and n = 34 for Appl-GAL4, sws1; UAS-SWS (C). SEMs are indicated. All sections are horizontal paraffin sections through the heads of 14-d-old flies. E, Esterase activity in fly head homogenates revealed that expressing the SWSR133A construct pan-neuronally did not restore the esterase function in sws1. In contrast, this construct showed esterase function in a wild-type background and heterozygous sws1/+ flies, approximately doubling the activity. All values are expressed relative to wild type (100%). SEMs are indicated. Scale bar: (in A) A–C, 50 μm. la, Lamina; me, medulla; lo, lobula; lb, lobula plate; cb, central brain.
In addition to its rescue ability, as measured by vacuolization, we also determined whether the R to A mutation affects the esterase activity of SWS. For these experiments, we used head homogenates from flies expressing SWSR133A in neurons (using Appl-GAL4) and measured the hydrolyzing activity against the artificial substrate phenyl valerate, a standard method to detect NTE activity. Expression of SWSR133A in the sws1 mutant background appeared not restore the esterase activity (Fig. 1E), although it has an intact esterase domain. Interestingly, the mutated construct did show esterase activity in a wild-type or heterozygote sws1 mutant background, about doubling the endogenous esterase activity. In comparison, expression of the wild-type construct restored the activity in sws mutants (116%) and also doubled the activity (197%) in wild type (Mühlig-Versen et al., 2005). This shows that the mutation in the pseudosubstrate site does not affect the esterase activity per se, however, SWS appears to require an interaction mediated via the pseudosubstrate binding site to exhibit esterase activity.
SWS interacts specifically with the C3 subunit of PKA
The requirement of the pseudosubstrate site for wild-type SWS function suggested that this domain is important for an interaction with a partner. We therefore used the two-hybrid system to isolate direct binding partners of SWS. In addition to the full-length SWS protein, we also used two smaller fragments (SWS60–544 and SWS60–241) (Fig. 2A) that both contained the interaction domain but no transmembrane domain to screen an expression library from adult Drosophila. With all three baits we isolated cDNAs for the C3 catalytic subunit of PKA (PKA-C3). The longest isolated PKA-C3 clone encoded aa138–583 of the longer (583aa) isoform (corresponding to aa55–500 of the smaller isoform), containing the entire protein kinase domain (aa274–528, http://flybase.bio.indiana.edu). The smallest one encoded aa246–482. Interestingly, we isolated only one positive clone with the full-length SWS construct which also encoded PKA-C3.
To determine whether the binding of SWS to PKA-C3 is indeed mediated by the pseudosubstrate site, we created bait fragments with the R133 to A mutation. Cotransfection of SWS60–241R133A with the PKA-C3 clone resulted in colonies that did not grow as well as colonies formed by PKA-C3 and SWS without the R to A mutation (Fig. 2C, compare the first and last row). A similar result was obtained with the SWS60–544R133A construct (Fig. 2D, first and last row). This shows that as with the vertebrate regulatory PKA subunit (Poteet-Smith et al., 1997), a point mutation in the conserved arginine in SWS significantly reduces the binding of SWS to the C3 catalytic PKA subunit.
In addition to C3, two other catalytic PKA subunits have been identified in Drosophila (Meléndez et al., 1995). Although we only isolated clones for the C3 subunit in our two-hybrid screens, we verified that SWS does not interact with the other two catalytic subunits. For this purpose, we created pMyr transformation constructs for all three catalytic PKA subunits using full-length cDNAs (kindly provided by D. Kalderon, Columbia University). As shown in Figure 2, C and D, only SWS and PKA-C3 produced colonies under the restrictive conditions, whereas cotransfection with PKA-C1 or PKA-C2 (second and third row) did not result in colonies under the same conditions. This strongly suggests that SWS specifically interacts with the C3 subunit.
SWS tethers PKA-C3 to membranes
The interaction of SWS and PKA-C3 in the yeast two-hybrid system strongly suggested that SWS and PKA-C3 are also found in a complex in vivo. We therefore performed immunohistochemistry on adult brains using our anti-SWS rabbit antiserum and an anti-PKA-C3 rat antiserum for colocalization studies (Meléndez et al., 1995). We have recently shown that SWS is expressed in most or all neurons (Mühlig-Versen et al., 2005) and PKA-C3 can also be detected at low levels in most or all neurons (Fig. 3A). In addition to this weak pan-neuronal expression, we found stronger expression in some neurons, which also appeared to contain higher levels of SWS (Fig. 3A–C, arrowheads). Interestingly, PKA-C3 was highly expressed in a few very large neurons (Fig. 3A–C, asterisks). In addition, we detected that PKA-C3 and SWS colocalize on a subcellular level because both can be found in the same vesicles (Fig. 3A–C, arrows). As mentioned above, SWS and NTE both have been shown to be enriched in the ER (Akassoglou et al., 2004; Mühlig-Versen et al., 2005), suggesting that the PKA-C3 immunopositive vesicles might be part of the ER. To test this hypothesis, we performed coimmunostainings with anti-PKA-C3 (Fig. 3D) and GRP78 (Fig. 3E), a marker for the ER. As shown in Figure 3F, some vesicles indeed contained both protein (white arrows) but others were only positive for PKA-C3 (green arrowheads) or GRP78 (red arrowheads). These results show that some PKA-C3 is localized to the ER, but like SWS it is also found in other parts of the cell, and indeed SWS and PKA-C3 seem to colocalize to a much higher degree than PKA-C3 and GRP78.
Figure 3.
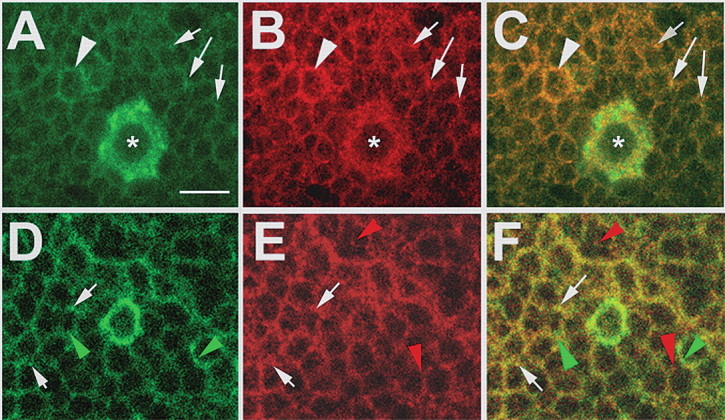
SWS and PKA-C3 colocalize in neurons. A–C, Brain whole-mounts stained with anti-PKA-C3 (green) and anti-SWS (red) show expression in most or all neurons with stronger expression of both proteins in some neurons (arrowheads), including a few large neurons, which highly express PKA-C3 (asterisks). In addition, both proteins can be colocalized to the same vesicles (arrows). D–F, In some vesicles PKA-C3 (green) colocalizes with the ER marker GRP78 (red), although both proteins can also be found separately (green arrowheads and red arrowheads). Thickness of the optical sections was 0.1 μm. Scale bar, 5 μm.
Both SWS and NTE contain several predicted transmembrane domains and SWS could therefore tether PKA-C3, which by itself is not a transmembrane protein, to membranes. To determine whether SWS does affect the subcellular location of PKA-C3, we used the PKA-C3 antiserum on brain whole mounts from sws1 mutant flies. Whereas a mostly vesicular pattern of PKA-C3 is detected in the wild-type background (Fig. 4A), its subcellular localization becomes less concentrated to vesicles in sws1 mutant brains (Fig. 4B). To verify that SWS does indeed recruit PKA-C3 to membranes, we performed Western blots from membrane and cytosolic fractions prepared from yw control and sws1 mutant heads. First, we confirmed that SWS is confined to membranes and as shown in Figure 4C, the 160 kDa SWS protein is exclusively detected in membrane fractions from control flies. We could also detect a putative PKA-C3 band of 66 kDa (the predicted size for PKA-C3) in control membrane fractions, which was missing in membrane fractions prepared from sws1 flies (Fig. 4D, asterisks). To verify that this band does correspond to PKA-C3, we created flies that contained a PKA-C3 construct under the control of the UAS sequence and expressed it in neurons using Appl-GAL4. Membrane fractions derived from these flies (PKA m) did indeed show increased levels of this band. Although PKA-C3 was depleted from the membranes of sws1 flies, we could not detect an accumulation of the 66 kDa band in the cytosolic fraction of sws1 (nor did we see it in control flies). However, a smaller band of ∼50 kDa was increased in sws1 cytosolic fractions compared with controls, which could be a degradation product of PKA-C3. To determine whether SWS affects the levels of PKA-C3, we performed Western blot with head lysates which showed that the lack of SWS does not affect PKA-C3 levels of PKA-C3 in general (supplemental Fig. 3, available at www.jneurosci.org as supplemental material, lane 4 and 5). We also induced this construct in the eye (via GMR-GAL4) or pan-neuronally (via elav-GAL4), which again resulted in the increase of the 66 kDa band. These results show that the loss of SWS does not affect PKA-C3 levels but changes its subcellular localization, depleting it from membranes.
Figure 4.

PKA-C3 is mislocalized in the absence of SWS. A, B, Whereas PKA-C3 can be found in a mostly punctuate pattern in yw control flies, its pattern is less distinct in a sws1 mutant fly (B). C, In Western blots, the SWS protein (asterisks), which contains several transmembrane domains, is exclusively found in membrane fractions (m) from yw control flies, while it is missing in sws1 mutants. D, PKA-C3 is detected as a 66 kDa band in membrane fractions from yw control flies and, in increased amount in Appl-GAL4/UAS-PKA-C3 (mPKA) flies, whereas it is not detectable in membrane fractions from sws1 mutant flies. Cytosolic fractions (c) reveal an increase in a 50 kDa band in sws1, which could be a degradation product of PKA-C3. Scale bar: (in A) A, B, 2 μm.
SWS inhibits the catalytic activity of PKA
To investigate the functional consequences of the interaction between SWS and PKA-C3, we tested whether SWS regulates the catalytic activity of PKA-C3. For this experiment, we used a PKA activity assay based on the phosphorylation of the kemptide peptide in vitro. Measuring the overall PKA activity in fly head homogenates in the presence of cAMP, we found a significant increase in the PKA activity of sws1 mutant flies (1.15 ± 0.16 p/q) versus controls (0.82 ± 0.10 p/q; p < 0.01) (Fig. 5). In contrast, expression of additional SWS in neurons via elav-GAL4 reduced the activity to 0.55 ± 0.36 p/q although not quite statistically significant (p = 0.054). To determine the activity with the intrinsic cyclic nucleotide concentration present in the lysates, we repeated these measurements without adding cAMP and also found a significant increase in the kinase activity of sws1 mutant flies compared with control flies (0.83 ± 0.11 p/q; versus 0.52 ± 0.04 p/q, p < 0.02, yw: n = 16, sws1; n = 19). These results strongly suggest that SWS binding to PKA-C3 inhibits the catalytic function of PKA, similar to the inhibitory function described for canonical regulatory subunits of PKA.
Figure 5.

PKA activity in fly head homogenates. In the presence of cAMP (0.4 mm), PKA activity is increased by ∼30% in sws1 extracts compared with yw control flies, whereas expressing additional SWS in wild type reduces PKA activity (although not quite statistically significant; p = 0.054). Expression of additional PKA-C3 via Appl-GAL4 did not increase the overall PKA activity compared with yw controls. n = 16 for yw, n = 21 for sws1, n = 13 for elav-GAL4/UAS-SWS, and n = 15 for Appl-GAL4/UAS-PKA/C3. p/q, quotient of the luminosity value of phosphorylated to unphosphorylated kemptide peptide. Values were normalized to ng protein in the lysates. SEMs are indicated. *p < 0.01.
Somewhat surprisingly, pan-neuronal expression of PKA-C3 via Appl-GAL4 in the presence of cAMP (or elav-GAL4, data not shown) did not significantly increase the overall kinase activity (0.9 ± 0.1 vs 0.83 ± 0.1 p/q) (Fig. 5) although the expression of PKA-C3 was confirmed by Western blots (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). It is possible that the additional expression of only this isoform is not sufficient to significantly change the overall level of PKA activity. Alternatively, the additional PKA-C3 is inactive because it is bound by SWS.
PKA-C3 modulates the neurodegenerative phenotype of sws
To investigate whether PKA-C3 plays a role in the neurodegenerative phenotype observed in sws1 mutant flies, we genetically altered the levels of PKA-C3 in sws1. Comparing sws1 flies with or without additional expression of PKA-C3 (via Appl-GAL4), we found a significant increase of the degenerative phenotype in the presence of additional PKA-C3 (Fig. 6B,A; sws1 alone). As shown in Figure 6C, sws1 flies overexpressing PKA-C3 showed a mean vacuole area of 79 ± 8.2 μm2 compared with 52 ± 6.8 μm2 in sws1 (p < 0.05). Next we asked whether lower levels of PKA-C3 can suppress the sws phenotype. Whereas combining sws1 with the Df(3L)brm11 deficiency which removed one copy of the PKA-C3 gene did not reduced vacuole formation in the central brain (54 ± 9.2 μm2), measurements in another part of the brain, the lamina cortex, revealed a significant suppression. Vacuole formation in this area was reduced from 39 ± 4.1 μm2 in sws1 (Fig. 6E) to 22 ± 3.7 μm2 (p < 0.05) in sws1 flies heterozygous for the deficiency (Fig. 6D). These measurements were performed in 7-d-old flies because the severe phenotype in this area often results in a loss of contact between the eye and the lamina cortex in 14-d-old sws1 flies, preventing the measurement of vacuoles. These genetic interactions show that the interaction of PKA-C3 with SWS plays a role in the progressive neurodegeneration observed in sws1. To determine whether this effect is specific for PKA-C3, we also removed one copy of PKA-C1 using flies heterozygous for the PKA-C1C2 allele (homozygous PKA-C1C2 is lethal), which did not affect the vacuole formation in sws1 (data not shown), neither in the central brain nor in the lamina cortex.
Figure 6.

Overexpression of PKA-C3 enhances the neurodegenerative phenotype of sws. A, B, Compared with the sws1 mutant fly (A), flies expressing PKA-C3 via the APPL-GAL4 driver (B) showed a significant increase in vacuolization. C, This is confirmed by measuring the mean area of vacuoles in μm2 in sws1 (n = 24) and sws1; UAS-PKA-C3 flies (n = 33). sws1 flies carrying one copy of the deficiency Df(2)brm11 showed no significant difference in the mean area of vacuoles in the central brain (n = 22). Sections in A and B are horizontal paraffin sections through the heads of 14-d-old flies. D, Vacuoles (arrows) in the lamina cortex of a 7-d-old sws1 fly heterozygous for Df(2)brm11. In this region, Df(2)brm11 significantly reduces the area of vacuoles compared with sws1 without the deficiency (E) [n = 17 for sws1, n = 13 for sws1; Df(2)brm11]. Scale bar: (in A), A, B, 50 μm. la, Lamina; me, medulla; lo, lobula; lb, lobula plate; cb, central brain; re, retina.
Additional expression of PKA-C3 does not affect the esterase activity of SWS
As described above, we found that the SWS construct with the mutation in the interaction site (SWSR133A) showed no esterase activity when expressed in sws1 flies, suggesting that the interaction of SWS with PKA-C3 might regulate its esterase activity. We therefore tested whether increased amounts of PKA-C3 affect the esterase activity of SWS. Comparing the esterase activity levels in homogenates prepared from control flies versus flies expressing additional PKA-C3 pan-neuronally revealed that elevated levels of PKA-C3 did not change the esterase activity of SWS significantly (108 ± 10 vs 100 ± 10). This suggests that binding to PKA-C3, although required to activate the esterase activity of SWS, complex formation per se is not sufficient to activate the esterase function.
Hyperactive PKA-C3 in the sws mutant contributes to the degenerative phenotype
The hyperactive PKA in sws flies and the enhanced degenerative phenotype in the presence of additional PKA-C3, suggested that increased PKA activity could play a role in the progressive neurodegeneration observed in sws. To test this hypothesis, we investigated whether additional expression of PKA-C3 in the nervous system can by itself induce a neurodegenerative phenotype. Whereas we could not detect any signs of degeneration in 1-d-old male Appl-GAL4/Y; UAS-PKA-C3/+ flies (Fig. 7A), these flies showed a few, mostly large vacuoles after 30d of aging (Fig. 7B, arrows). In ∼10% of these flies, we also found severe vacuolation in the lamina (Fig. 7C, arrowhead). Aged females, carrying one copy of each Appl-GAL4 and UAS-PKA-C3 did not show a phenotype, probably attributable to effects of dosage compensation (Lucchesi, 1996) on the X-chromosomal Appl-GAL4. Nevertheless, this shows that expression of PKA-C3 in neurons can induce a degenerative phenotype, although it is quite weak. However, as we present in Figure 5A, these flies only exhibited a slight increase in their kinase activity (which was not significant) and therefore a weak phenotype may be expected. As noted above, the lack of a significant increase in kinase activity could be due to the fact that additionally expressed PKA-C3 is catalytically inactive because it is inhibited by binding to SWS. Therefore, we repeated these experiments with PKA-C3 overexpressing flies that lack on copy of the sws gene, thereby reducing the amount of the proposed PKA-C3 inhibitor. Although heterozygous sws1 flies do not show a degenerative phenotype because of the recessive nature of the mutation, they did enhance the degeneration caused by PKA-C3 overexpression, now resulting in vacuolation in aged females (Fig. 7D) (males could not be tested because sws is localized on the X).
Figure 7.

Neuronal expression of PKA-C3 induces vacuolization. A, A 1-d-old male fly hemizygous for Appl-GAL4 and heterozygous for UAS-PKA-C3 shows no degeneration, whereas a 30-d-old one (B) shows some, sometimes quite large vacuoles (arrowhead and arrow). C, In ∼10% of these aged flies, the lamina shows severe vacuolization (arrowhead). D, Removing one copy of sws in 30-d-old females heterozygous for Appl-GAL4 and UAS-PKA-C3 also results in the formation of vacuoles similar to the ones seen in males, whereas these heterozygous females do not show vacuoles without removing one copy of sws (data not shown). All sections are horizontal paraffin head sections. Scale bars: (in A) A, B, D, 50 μm; C, 25 μm. re, Retina, la, lamina.
To get additional support that the neurodegenerative phenotype in sws is partially due to the missing inhibition of PKA-C3, and not to the possible effects of PKA-C3 on the esterase function of SWS, we performed rescue experiments with an SWS construct that has no esterase activity (Mühlig-Versen et al., 2005). As shown in Figure 8, Appl-GAL4 induced expression of this construct (SWSS985D) resulted in a partial rescue with a vacuole area of 29.4 ± 4.4 μm2 (Fig. 8B) in contrast to control sws1 flies with 52 ± 6.8 μm2 (Fig. 8A) and sws1 flies expressing wild-type SWS with 16 ± 3.7 μm2 (Fig. 1D) (both p < 0.05). This shows that a catalytically inactive SWS construct, which however has an intact PKA-C3 interaction domain, can partially rescue the neurodegenerative phenotype. Together with the results, that overexpression of PKA-C3 induces an age-dependent degenerative phenotype, which is enhanced by reducing the level of its inhibitor SWS, this strongly suggests that the missing inhibition of PKA-C3 is partially responsible for the progressive neurodegeneration observed in sws mutant flies.
Figure 8.

Expression of a catalytically inactive SWS construct results in a partial rescue. A, sws1. B, Pan-neuronal expression of SWSS985D, which has no kinase activity (Mühlig-Versen et al., 2005), but an intact PKA-C3 interaction domain in the nervous system of sws1 mutant flies can partially rescue the degenerative phenotype. Mean area of vacuoles (in μm2) in Appl-GAL4, sws1 flies (n = 24), and Appl-GAL4, sws1; UAS-SWSS985D (n = 38). Sections are horizontal paraffin sections through the heads of 14-d-old flies. SEMs are indicated. Scale bar, 50 μm. la, Lamina; me, medulla; lo, lobula; lb, lobula plate; cb, central brain.
Discussion
Our results show that SWS binds specifically to PKA-C3, inhibiting its catalytic function, similar to the inhibitory function of canonical regulatory subunits. The binding is mediated by the N-terminally localized interaction domain of SWS which contains a site, the pseudosubstrate site, that is shared by all types of canonical regulatory subunits (Poteet-Smith et al., 1997). As we have shown, a mutation in one of the conserved arginines in the pseudosubstrate site reduces the binding capacity of SWS for PKA-C3, similar to the decreased binding observed with comparable mutations in vertebrate canonical regulatory subunits (Buechler et al., 1993; Poteet-Smith et al., 1997). In addition, we verified that the interaction between SWS and PKA-C3 is of biological relevance because expression of the construct with the mutated pseudosubstrate site (SWSR133A) only partially restores the function of SWS in sws1 mutant flies. Surprisingly, we found that SWSR133A does not exhibit significant esterase activity in the sws1 background, although it has an intact esterase domain, suggesting that the interaction between SWS and PKA-C3 has a dual function: regulating the kinase activity of PKA-C3 and the esterase activity of SWS.
In addition, the binding of PKA-C3 to the transmembrane protein SWS also regulates the subcellular localization of PKA-C3; although PKA-C3 is found in membrane fractions in wild-type flies, it is missing in membranes from sws mutants. For canonical PKA complexes it has been shown that the spatial regulation plays an important role for the function because it ensures that PKA is exposed to localized cAMP gradients within a cell, which then allows accurate substrate selection (Michel and Scott, 2002). Although the targeting of canonical PKA complexes to different cellular compartments is also regulated by the regulatory subunits, in canonical complexes it is controlled by binding of the regulatory subunit to various A kinase-anchoring proteins (Feliciello et al., 2001; Michel and Scott, 2002) and is not a direct consequence of the localization of the regulatory subunit itself.
The regulatory subunits of PKA are not only controlling the localization of the complex but also the activation of its kinase function by binding cAMP, which results in the release of the catalytic subunit (Taylor et al., 2005). As shown in supplemental Figures 2 and 3, available at www.jneurosci.org as supplemental material, SWS as well as its human and mouse orthologs, contain three cyclic nucleotide binding sites; a tandem binding site similar to the one found in canonical regulatory subunits and an additional single site. Interestingly, this single site resembles more a cGMP binding site because, both SWS and NTE contain a threonine instead of an alanine within this motif (highlighted in supplemental Fig. 3A, available at www.jneurosci.org as supplemental material), which is typical for cAMP binding sites (Shabb et al., 1990). In addition, the third binding site in SWS also has a threonine (whereas NTE has an alanine, highlighted in supplemental Fig. 3B, available at www.jneurosci.org as supplemental material), suggesting that the complex formation of SWS and PKA-C3 may be regulated by cGMP. However, it has been shown that cyclic nucleotide binding sites can also bind to other ligands, including heme in the bacterial transcription factor cooA subfamily (Lanzilotta et al., 2000) and ortho-chlorophenolic compounds in the bacterial transcriptional regulators CprK (Kannan et al., 2007). Future experiments will determine whether and which compound the three cyclic nucleotide binding sites of SWS can bind.
In addition to identifying this novel PKA complex, our results also describe for the first time a function for the unique C3 subunit. PKA-C3 has been identified as a catalytic subunit by its homology to the well characterized PKA-C1 subunit (Kalderon and Rubin, 1988). Comparing PKA-C3 with the other two catalytic subunits found in Drosophila showed that it is more closely related to the C1 subunit (7e-103) than to C2 (3e-70) (supplemental Fig. 6, available at www.jneurosci.org as supplemental material), but studies expressing PKA-C3 under the control of the PKA-C1 promoter have indicated that PKA-C3 is not functionally redundant with PKA-C1 (Meléndez et al., 1995).
PKA-C3 shares 62% identity in the catalytic core region with the recently isolated X-linked protein kinases in mouse (Pkare) and human (PrKX) (Blaschke et al., 2000; Diskar et al., 2007). Notably, the homology between the human and fly proteins is actually higher than between the different catalytic subunits from any one species (Zimmermann et al., 1999). Comparing PKA-C3 with human PrKX provided an E-value of 7e-125 and PKA-C1 has an even higher homology to human PKA-Ca (7e-174) (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). It was therefore suggested that these unique catalytic subunits represent a novel class of evolutionary conserved PKAs.
Here we show that PKA-C3 is expressed in the nervous system, with very high levels in distinct large neurons, suggesting a function in the adult nervous system. This was verified by our results that additional PKA-C3 enhances the neuronal degeneration observed in sws1, whereas removing one copy of the PKA-C3 gene suppresses degeneration, confirming a neuronal function. This neuroprotective function could be solely mediated by its effects on SWS, alternatively PKA-C3 can directly induce neurodegeneration. The latter is supported by our findings that overexpression of PKA-C3 in neurons resulted in a weak but readily detectable degenerative phenotype, whereas it did not affect the esterase function of SWS. In addition, an SWS construct without esterase activity but an intact pseudosubstrate site, allowing binding and inhibition of PKA-C3, can still partially rescue the degenerative phenotype of sws1. Finally, removing one copy of the proposed inhibitor SWS in PKA-C3 overexpressing flies, enhanced the PKA-C3 induced neurodegeneration. Therefore, we propose that SWS acts as a noncanonical regulatory subunit that inhibits the catalytic activity of PKA-C3, and that the disruption of this inhibitory mechanism results in the uncontrolled release of active PKA-C3 which damages the nervous system.
Because mouse Pkare mRNA is also expressed in many regions of the adult nervous system, including hypothalamus, hippocampus, cerebellum, and olfactory bulb (Blaschke et al., 2000) while the human Pkare protein was shown to be highly expressed in brain (Li et al., 2005). Together with our data, that PKA-C3 plays a role in progressive neurodegeneration, this suggests that this novel class of catalytic subunits may have a specific function in the maintenance of the nervous system conserved between flies and mammals.
Footnotes
This work was supported by a grant from the National Institutes of Health (NS047663-01). We thank David Morton and Paul Glynn for technical help, as well as Burkhard Poeck for critical reading of this manuscript.
References
- Akassoglou K, Malester B, Xu J, Tessarollo L, Rosenbluth J, Chao MV. Brain-specific deletion of neuropathy target esterase/swisscheese results in neurodegeneration. Proc Natl Acad Sci U S A. 2004;101:5075–5080. doi: 10.1073/pnas.0401030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt da Cruz A, Schwärzel M, Schulze S, Niyyati M, Heisenberg M, Kretzschmar D. Disruption of the MAP1B-related protein FUTSCH leads to changes in the neuronal cytoskeleton, axonal transport defects, and progressive neurodegeneration in Drosophila. Mol Biol Cell. 2005;16:2433–2442. doi: 10.1091/mbc.E04-11-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke RJ, Monaghan AP, Bock D, Rappold GA. A novel murine PKA-related protein kinase involved in neuronal differentiation. Genomics. 2000;64:187–194. doi: 10.1006/geno.2000.6116. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Buchner S, Buchner E, Hofbauer A. Immunocytochemistry of the brain. In: Ashburner M, editor. Drosophila: a laboratory manual. New York: Cold Spring Harbor Laboratory; 1989. pp. 254–259. [Google Scholar]
- Buechler YJ, Herberg FW, Taylor SS. Regulation-defective mutants of type I cAMP-dependent protein kinase. Consequences of replacing arginine 94 and arginine 95. J Biol Chem. 1993;268:16495–16503. [PubMed] [Google Scholar]
- Diskar M, Zenn HM, Kaupisch A, Prinz A, Herberg FW. Molecular basis for isoform-specific autoregulation of protein kinase A. Cell Signal. 2007;10:2024–2034. doi: 10.1016/j.cellsig.2007.05.012. [DOI] [PubMed] [Google Scholar]
- Ehrich M, Jortner BS. Organophosphorus-induced delayed neuropathy. In: Krieger R, editor. Handbook of pesticide toxicology. San Diego: Academic; 2001. pp. 987–1012. [Google Scholar]
- Feliciello A, Gottesman ME, Avvedimento EV. The biological functions of A-kinase anchor proteins. J Mol Biol. 2001;308:99–114. doi: 10.1006/jmbi.2001.4585. [DOI] [PubMed] [Google Scholar]
- Francis SH, Poteet-Smith C, Busch JL, Richie-Jannetta R, Corbin JD. Mechanisms of autoinhibition in cyclic nucleotide-dependent protein kinases. Front Biosci. 2002;7:d580–d592. doi: 10.2741/A796. [DOI] [PubMed] [Google Scholar]
- Glynn P. Neural development and neurodegeneration: two faces of neuropathy target esterase. Prog Neurobiol. 2000;61:61–74. doi: 10.1016/s0301-0082(99)00043-x. [DOI] [PubMed] [Google Scholar]
- Johnson MK. Improved assay of neurotoxic esterase for screening organophosphates for delayed neurotoxicity potential. Arch Toxicol. 1977;37:113–115. doi: 10.1007/BF00293860. [DOI] [PubMed] [Google Scholar]
- Kalderon D, Rubin GM. Isolation and characterization of Drosophilas cAMP-dependent protein kinase genes. Genes Dev. 1988;2:1539–1556. doi: 10.1101/gad.2.12a.1539. [DOI] [PubMed] [Google Scholar]
- Kannan N, Wu J, Anand GS, Yooseph S, Neuwald AF, Venter JC, Taylor SS. Evolution of allostery in the cyclic nucleotide binding module. Genome Biol. 2007;8:R264. doi: 10.1186/gb-2007-8-12-r264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar D, Hasan G, Sharma S, Heisenberg M, Benzer S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J Neurosci. 1997;17:7425–7432. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzilotta WN, Schuller DJ, Thorsteinsson MV, Kerby RL, Roberts GP, Poulos TL. Structure of the CO sensing transcription activator CooA. Nat Struct Biol. 2000;7:876–880. doi: 10.1038/82820. [DOI] [PubMed] [Google Scholar]
- Li W, Yu ZX, Kotin RM. Profiles of PrKX expression in developmental mouse embryo and human tissues. J Histochem Cytochem. 2005;53:1003–1009. doi: 10.1369/jhc.4A6568.2005. [DOI] [PubMed] [Google Scholar]
- Li Y, Dinsdale D, Glynn P. Protein domains, catalytic activity and subcellular distribution of neuropathy target esterase in mammalian cells. J Biol Chem. 2003;278:8820–8825. doi: 10.1074/jbc.M210743200. [DOI] [PubMed] [Google Scholar]
- Lotti M, Moretto A. Organophosphate-induced delayed polyneuropathy. Toxicol Rev. 2005;24:37–49. doi: 10.2165/00139709-200524010-00003. [DOI] [PubMed] [Google Scholar]
- Lucchesi JC. Dosage compensation in Drosophila and the “complex” world of transcriptional regulation. Bioessays. 1996;18:541–547. doi: 10.1002/bies.950180705. [DOI] [PubMed] [Google Scholar]
- Lush MJ, Li Y, Read DJ, Willis AC, Glynn P. Neuropathy target esterase and a homologous Drosophila neurodegeneration-associated mutant protein contain a novel domain conserved from bacteria to man. Biochem J. 1998;332:1–4. doi: 10.1042/bj3320001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meléndez A, Li W, Kalderon D. Activity, expression and function of a second Drosophila protein kinase A catalytic subunit gene. Genetics. 1995;141:1507–1520. doi: 10.1093/genetics/141.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel JJ, Scott JD. AKAP mediated signal transduction. Annu Rev Pharmacol Toxicol. 2002;42:235–257. doi: 10.1146/annurev.pharmtox.42.083101.135801. [DOI] [PubMed] [Google Scholar]
- Moretto A. Promoters and promotion of axonopathies. Toxicol Lett. 2000;112–113:17–21. doi: 10.1016/s0378-4274(99)00248-9. [DOI] [PubMed] [Google Scholar]
- Moser M, Stempfl T, Li Y, Glynn P, Büttner R, Kretzschmar D. Cloning and expression of the murine sws/NTE gene. Mech Dev. 2000;90:279–282. doi: 10.1016/s0925-4773(99)00239-7. [DOI] [PubMed] [Google Scholar]
- Moser M, Li Y, Vaupel K, Kretzschmar D, Kluge R, Glynn P, Buettner R. Placental failure and impaired vasculogenesis result in embryonic lethality for neuropathy target esterase-deficient mice. Mol Cell Biol. 2004;24:1667–1679. doi: 10.1128/MCB.24.4.1667-1679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlig-Versen M, da Cruz AB, Tschäpe J A, Moser M, Büttner R, Athenstaedt K, Glynn P, Kretzschmar D. Loss of Swiss cheese/neuropathy target esterase activity causes disruption of phosphatidylcholine homeostasis and neuronal and glial death in adult Drosophila. J Neurosci. 2005;25:2865–2873. doi: 10.1523/JNEUROSCI.5097-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgad S, Dudai Y, Cohen P. The protein phosphatases of Drosophila melanogaster and their inhibitors. Eur J Biochem. 1987;164:31–38. doi: 10.1111/j.1432-1033.1987.tb10988.x. [DOI] [PubMed] [Google Scholar]
- Poteet-Smith CE, Shabb JB, Francis SH, Corbin JD. Identification of critical determinants for autoinhibition in the pseudosubstrate region of type I alpha cAMP-dependent protein kinase. J Biol Chem. 1997;272:379–388. doi: 10.1074/jbc.272.1.379. [DOI] [PubMed] [Google Scholar]
- Rainier S, Bui M, Mark E, Thomas D, Tokarz D, Ming L, Delaney C, Richardson RJ, Albers JW, Matsunami N, Stevens J, Coon H, Leppert M, Fink JK. Neuropathy target esterase gene mutations cause motor neuron disease. Am J Hum Genet. 2008;82:780–785. doi: 10.1016/j.ajhg.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabb JB, Ng L, Corbin JD. One amino acid change produces a high affinity cGMP-binding site in cAMP-dependent protein kinase. J Biol Chem. 1990;265:16031–16034. [PubMed] [Google Scholar]
- Smith MI, Elove E, Frazier WH. The pharmacological action of certain phenol esters with special reference to the etiology of the so-called Ginger paralysis. Pub Health Rep. 1930;45:2509–2524. [Google Scholar]
- Taylor SS, Kim C, Vigil D, Haste NM, Yang J, Wu J, Anand GS. Dynamics of signaling by PKA. Biochim Biophys Acta. 2005;1754:25–37. doi: 10.1016/j.bbapap.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Torroja L, Chu H, Kotovsky I, White K. Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr Biol. 1999;9:489–492. doi: 10.1016/s0960-9822(99)80215-2. [DOI] [PubMed] [Google Scholar]
- Tschäpe JA, Hammerschmied C, Mühlig-Versen M, Athenstaedt K, Daum G, Kretzschmar D. The neurodegeneration mutant lochrig interferes with cholesterol homeostasis and Appl processing. EMBO J. 2002;21:6367–6376. doi: 10.1093/emboj/cdf636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann B, Chiorini JA, Ma Y, Kotin RM, Herberg FW. PrKX is a novel catalytic subunit of the cAMP-dependent protein kinase regulated by the regulatory subunit type I. J Biol Chem. 1999;274:5370–5378. doi: 10.1074/jbc.274.9.5370. [DOI] [PubMed] [Google Scholar]