

Abstract
Background
A majority of mutations within the amyloid β (Aβ) region of the amyloid precursor protein (APP) gene cause inherited forms of intracerebral haemorrhage. Most of these mutations may also cause cognitive impairment, but the Arctic APP mutation is the only known intra-Aβ mutation to date causing the more typical clinical picture of Alzheimer's disease (AD).
Objective
To describe features of one Swedish and one American family with the previously reported Arctic APP mutation.
Subjects
Affected and non-affected carriers of the Arctic APP mutation from the Swedish and American families were investigated clinically. In addition, one brain from each family was investigated neuropathologically.
Results
The clinical picture, with age at disease onset in the sixth to seventh decade of life and dysfunction in multiple cognitive areas, is indicative of AD and similar to the phenotype for other AD APP mutations. Several affected mutation carriers displayed general brain atrophy and reduced blood flow of the parietal lobe, as demonstrated by magnetic resonance imaging and single photon emission computed tomography. One Swedish and one American case with the Arctic APP mutation have come to autopsy, neither of which showed any signs of haemorrhage but revealed severe congophilic angiopathy, region-specific neurofibrillary tangle pathology as well as abundant amyloid plaques. Intriguingly, a majority of plaques from both of these cases had a characteristic ring-like character.
Conclusions
Overall, our findings corroborate that the Arctic APP mutation causes a clinical and neuropathological picture compatible with AD.
Keywords: Familial Alzheimer's disease, APP gene mutations, Arctic mutation, cerebral amyloid angiopathy, dementia, genealogy
Introduction
Alzheimer's disease (AD), the most common neurodegenerative disorder, is neuropathologically characterized by extracellular deposition of amyloid-β peptide (Aβ) into plaques and intraneuronal accumulation of abnormal tau protein as neurofibrillary tangles.
Autosomal dominant forms of AD may explain approximately 5% of all disease cases. AD causing mutations have been identified in three genes; the amyloid precursor protein (APP) gene on chromosome 21 1-3, the presenilin (PS) 1 gene on chromosome 14 and the PS 2 gene on chromosome 1 4. To date, close to 20 APP mutations, about 150 PS1 mutations and 11 PS2 mutations have been described (for an overview, see http://www.molgen.ua.ac.be/ADMutations/). A majority of the known APP mutations are located in the vicinity of the cleavage sites for β- and γ-secretases and are believed to cause increased levels of Aβ by affecting its enzymatic cleavage from APP. Also PS1 and PS2 mutations result in elevated Aβ, as has been demonstrated both in vivo and in vitro 5, 6. The convergence of AD mutations towards APP metabolism has been explained by the identification of presenilin as an essential component of the complex that mediates γ-secretase cleavage 7.
Several APP mutations have been identified within the Aβ sequence. Massive amyloid accumulation in brain vessel walls, cerebral haemorrhages and parenchymal amyloid plaques can be demonstrated in carriers of the Flemish mutation (APP A692G) 8. Typically, patients with the Flemish mutation clinically present in their 40's either with symptoms related to cerebrovascular events or with cognitive dysfunction 9-11. Some mutation carriers even develop a progressive dementia compatible with AD both clinically and neuropathologically 8, 11.
Carriers of the Dutch mutation (APP E693Q) also develop cerebral amyloid angiopathy, but only rarely amyloid plaque pathology, in their 40's and 50's. Clinically, the Dutch mutation is mainly characterized by focal symptoms related to recurrent strokes 12. Most mutation carriers also develop a clinical picture of dementia, which sometimes precedes but more often follows the initial cerebrovascular event.
A third intra-Aβ APP mutation has been described in an American family. The Iowa mutation (APP D694N) affects carriers in their 50's or 60's, causing a neuropathological picture of discrete brain atrophy, severe cerebral amyloid angiopathy with numerous small infarcts and haemorrhages of the brain parenchyma as well as extensive deposition of amyloid plaques and neurofibrillary tangles 13. The clinical picture is one of a progressive dementia with speech impairment without any apparent focal symptoms of cerebrovascular events 13.
Finally, yet another APP mutation located within the Aβ sequence was identified in a Swedish family with onset of AD in the 50's 14. The Arctic mutation (APP E693G) had originally been identified in an American family 15, but the significance of the mutation was unclear at the time. Interestingly, the Arctic mutation has been found to cause diminished instead of increased Aβ40 and Aβ42 levels in conditioned media from transfected cells and in plasma from mutation carriers 16. As recently shown, this paradoxical feature of the Arctic mutation can be explained by an increased formation of Aβ protofibrils. Such intermediate Aβ species are promoted by the Arctic APP mutation and were found to not be appropriately recognized by the Aβ antibodies under the non-denaturing conditions in commonly used ELISAs 17. Based on the Arctic mutation and other findings, it has been suggested that the neurotoxic effects in the AD brain are exerted mainly by oligomeric and protofibrillar forms of Aβ 18, whereas the mature Aβ fibrils in plaques may be less noxious remnants of the disease process 19.
The identification of a Swedish and an American family with the Arctic mutation in the APP gene have been previously published 14, 15 and we here report a more detailed account on the clinical and neuropathological picture of affected members in these same families.
Material and methods
Subjects / clinical information
The Swedish family with the Arctic mutation originates from a small village in northern Sweden and the pedigree extends over five generations (fig. 1a). Information on affected individuals was gained through hospital records, interviews of family members (many of whom had remained in the same geographical area), collection of information from parish registers and historical archives.
Fig. 1.
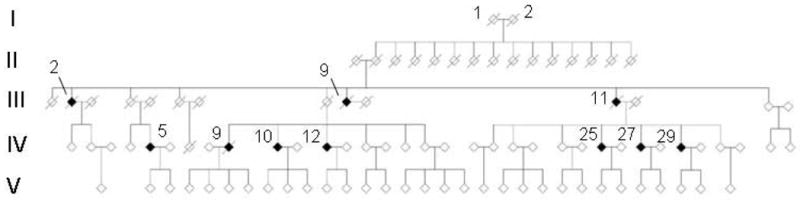

a) Pedigree of the Swedish family with the Arctic APP mutation. b) Pedigree of the American family with the Arctic APP mutation. Generations are marked with roman and certain individuals with greek numbers, which refer to those given in the text.
Affected (n= 5) and unaffected (n= 1) mutation carriers as well as non-mutation carriers (n= 5) were investigated with physical examination, routine blood tests, neuropsychological assessment, magnetic resonance imaging (MRI), electroencephalogram (EEG) and single photon emission computed tomography (SPECT).
In the American family, there had been six persons affected with dementia over four generations. The family is descended from Swedish immigrants and is thus likely to be related to the Swedish family with the same mutation. One family member (fig. 1b, subject III:2) had autopsy-proven AD, but did not have the Arctic mutation and is therefore not included in the analyses. This individual was most likely suffering from a sporadic form of AD.
For brain imaging (MRI and SPECT) and electrophysiological investigation (EEG), only individuals of the Swedish family have been evaluated.
The respective institutional review boards approved of this study. Moreover, informed consent for the genetic analyses was obtained from all participants and for the neuropathological work from close relatives of the two subjects autopsied.
Genetic analyses
Genetic analyses were carried out on subjects from whom informed consent had been obtained. On these, sequencing of the exon 17 of the APP gene was performed according to methods previously described 14.
Neuropsychological assessment
On subjects from the Swedish family, a comprehensive set of neuropsychological tests was performed by an experienced neuropsychologist (O.A.). The tests assessed global cognitive function (FSIQ) 20, verbal abstraction (Similarities) 20, verbal fluency (FAS) 21, visuospatial construction (Block Design) 20, copying of geometrical designs 21, verbal (RAVLT) 21 and visuospatial episodic memory 21, as well as executive function (Digit Symbol) 20 and cognitive speed (TMTA) 21.
For the American family, only one individual underwent neuropsychological evaluation. Apart from MMSE, the tests performed were: Mattis Dementia Rating Score, Trails A, Trails B, Clinical Dementia Rating (CDR), Hamilton Depression Scale, NYU paragraph recall and Lawton Brody Activities of Daily Living.
Magnetic Resonance Imaging
MRI of the brain was performed using a Vision 1.5 Tesla system (Siemens AG, Munich, Germany). Five mm thick transaxial slices were acquired and processed to generate T2-weighted and proton density weighted images as well as fluid attenuated inversion recovery sequence (FLAIR) images. Degree of atrophy and white matter changes were evaluated visually from hard copy images.
Single photon emission computed tomography
SPECT was performed using a three-headed Picker gamma camera (Marconi Medical Systems, Coventry, UK). The regional cerebral blood flow (rCBF) was measured using the tracer compound HMPAO marked with 99Tc. The assessment followed a standardized protocol, in which the patient was scanned for 20 min. The reduction in rCBF was evaluated visually from hard copy images.
Electroencephalogram
Quantitative EEG was performed in the morning with the subject awake and with eyes closed. Data were recorded using the Nervus system (Tangagreining HF, Reykjavik, Iceland), according to the 10/20 system. Frequency analysis was performed using Fast Fourier Transformation. A global rating of the pathological changes was undertaken.
Neuropathological Analysis
To date, one case from the Swedish family and one case from the American family have been autopsied. The Swedish case (fig. 1a, subject IV:10) was 62 years old and died after six years of disease duration. The American case (fig. 1b, subject III:1) was 72 years old and died after 16 years of disease duration. In order to harmonize the staining conditions, brain tissues from both cases were stained simultaneously.
For both cases, sections from various cortical, subcortical and brainstem regions were prepared from paraffin-embedded blocks and fixed in buffered 4% formaldehyde. The sections were stained with hematoxylin-eosin (HE), Nissl, Bielschowsky silver, Gallyas silver impregnation and Congo Red / thioflavine S (ThioS). In addition, immunohistochemical stainings was performed according to a regular protocol for immunohistochemistry 22 with a panel of monoclonal and polyclonal antibodies against Aβ as well as against non-phosphorylated and phosphorylated forms of tau (AT8, Innogenetics, Ghent, Belgium).
Sections from the Swedish case were immunostained by the Aβ mAb 6E10 (Signet, Dedham, MA, USA) and with polyclonal antibodies specific for Aβx-40 and Aβ x-42 23. In addition, sections from this case were stained by the phospho-tau mAb AT8 (Innogenetics, Ghent, Belgium). Tissue from the American case was stained with the Aβ mAb 5D10 (Athena Neuroscience, San Francisco, CA, USA), with Aβ x-40 and Aβ x-42 as well as with the tau mAb tau-2 (Sigma Immunochemicals, St. Louis, MO, USA).
Semiquantitative assessment of silver-positive neurofibrillary tangles, neuritic plaques and thioS-positive blood vessels was done following the CERAD protocol 24.
Results
The Swedish family descended from ancestors I:1 and I:2 (fig. 1), who were born in 1853 and 1862, respectively. The mode of inheritance was compatible with an autosomal dominant disorder with absolute penetrance, as the rate of affected offspring was close to 50%.
Clinical features
In the Swedish family, age at disease onset ranged from 52 years to 62 years with a mean age of 56.9 (+/- 1.1) years. No gender difference in age at onset was observed. Disease duration of the deceased cases with mutation (n=6) was 5.7 (+/- 0.3) years, and for mutation-bearing affected individuals still alive (n=6) 8.8 (+/- 1.3) years. An insidious loss of memory for recently acquired information was the presenting symptom in nine of the ten affected cases, whereas headache and fatigue were the first symptoms for one individual (table 1). Furthermore, spatial and temporal disorientation, dysphasia and dyspraxia occurred early in the course of the disease. Psychiatric symptoms such as anxiety, paranoia and hallucinations were observed in some cases. In several patients, myoclonus and rigidity were late symptoms (table 1).
Table 1.
Clinical features of affected individuals in the Swedish Arctic APP mutation family
| Family ID | First symptom | Other symptoms | Psychiatric symptoms | Motor symptoms | Disease duration |
|---|---|---|---|---|---|
| III:2 | Memory1 | Dyspraxia | Hallucinations | Rigidity | 11 years2 |
| III:9 | Memory1 | Disorientation, dyspraxia | Hallucinations, paranoia, anxiety | Myoclonia | 6 years2 |
| III:11 | Memory1 | Disorientation, dyspraxia | Suspiciousness, anxiety, paranoia, hallucination | Rigidity | 6 years2 |
| IV:5 | Memory1 | Dyspraxia, dysphasia, dysphagia, | Reserved | Rigidity, myoclonia | 9 years2 |
| IV:9 | Memory1 | Disorientation, dysphasia, dyspraxia | Anxiety, restlessness | Rigidity | 5 years2 |
| IV:10 | Memory1 | Disorientation, Dyspraxia, dysphasia, dysphagia | Lack of initiative, aggressiveness | Rigidity, myoclonia | 6 years2 |
| IV:12 | Memory1 | Tiredness, disorientation | - | - | 3 years |
| IV:25 | Headache, fatigue | Memory1, dysphasia, disorientation | Suspiciousness, anxiety, delusion, hallucination | - | 7 years |
| IV:27 | Memory1 | Depression, disorientation | - | - | 7 years |
| IV:29 | Memory1 | Disorientation, dysphasia | Anxiety | - | 3 years |
Memory impairment for recently acquired information
Deceased
In the American family, four affected mutation-carriers (subjects I:1, II:3, II:4 and III:1) had a mean age at onset of 58.8 years and a mean age at death of 71.8 years. The index case (fig. 1b, subject III:1) presented with disorientation and insomnia at the age of 56. During the disease course, there were no psychiatric symptoms, rigidity or myoclonus but the patient suffered from a grand mal seizure at the age of 65 before dying at the age of 72. This individual's offspring (fig. 1b, subject IV:1) developed disorientation and disrupted sleep at the age of 59 and subsequently developed dementia.
None of the affected cases, neither in the Swedish nor in the American family, had a history of cerebrovascular events and focal neurological signs were not observed in any of the examined subjects.
Neuropsychological assessments
Six of the affected cases from the Swedish family, one presymptomatic mutation carrier and five non-mutation carriers, were examined. All six affected subjects demonstrated a clear reduction in multiple cognitive domains, e.g. in global cognitive function, verbal abilities, visuospatial performance, episodic memory and attention/cognitive speed, whereas simple motor performance was unremarkable (table 2). The mean degree of cognitive decline vs. the only examined healthy non-mutation carrier in the same family amounted to 1-2 SDs or more.
Table 2.
Neuropsychological test result (M±SD) for carriers of the Arctic APP mutation in clinical and preclinical stage of dementia as well as healthy non-carriers.
| Test | Demented carriers (n=6) | Non symptomatic carrier (n=1) | Non carriers (n=5) |
|---|---|---|---|
| FSIQ, iq score | 73.7±6.8 | 93 | 92.4±19.1 |
| Vocabulary, no correct | 13.4±8.9 | 25 | 19.4±4.8 |
| Information, raw score | 12.8±2.2 | 20 | 17.6±7.1 |
| Similarities, raw score | 11.5±5.8 | 17 | 17.6±3.9 |
| Boston Naming, no correct | 51.3±7.0 | 52 | 53.8±4.9 |
| FAS word fluency, no correct | 30.3±17.2 | 31 | 41.8±14.8 |
| Figure Classification, raw score | 10.3±9.3 | 20 | 17.1±6.7 |
| Block Design, raw score | 11.5±8.4 | 22 | 29.4±16.1 |
| Rey-Osterrieth Copy, raw score | 24.2±13.7 | 35 | 35.3±1.1 |
| Digit Span Forward, mean score | 5.3±0.6 | 6.9 | 6.1±0.6 |
| Corsi Span, mean score | 4.0±1.7 | 5.6 | 6.1±0.9 |
| Rey AVL, total score | 17.0±4.2 | 29 | 44.4±8.7 |
| Rey-Osterrieth Memory, raw score | 7.1±9.1 | 14.5 | 21.0±7.3 |
| Word Recognition, d-prime | 1.7±0.9 | 2.8 | 3.3±0.3 |
| Face Recognition, no correct | 3.7±2.3 | 9 | 10.2±2.5 |
| Digit Symbol, no correct | 19.0±10.7 | 37 | 42.8±10.8 |
| Trail-Making A, sec's | 111±140 | 36 | 35±16 |
| Trail-Making B, sec's | 191±65 | 82 | 96±23 |
| Simple Reaction Time, msec | 259±34 | 236 | 250±26 |
| Finger-Tapping dx, no taps | 71±5 | 56 | 55±2 |
| Finger-Tapping sin, no taps | 69±2 | 50 | 48±0 |
| Finger-Tapping alternating, no of taps | 64±6 | 45 | 59±6 |
Neuroimaging
In the Swedish family, MRI and SPECT were performed on six of the affected mutation carriers, and on five of the healthy non-mutation carriers. No cerebral infarcts were seen, but slight-moderate white matter changes could be demonstrated for three of the affected cases (table 3). One of the affected individuals demonstrated a pronounced general brain atrophy, a marked reduction of temporo-parietal blood flow, but no changes in the white matter. Another subject (fig. 1a, subject IV:10) had a moderate parietal lobe atrophy but no atrophies in other brain regions. For this case, extensive white matter changes were seen.
Table 3.
Magnetic Resonance Imaging, rCBF and qEEG findings in members of the Arctic mutation family. Symptomatic subjects are marked with an asterisk.
| Age/Sex | Carrier status | MRI | Atrophy | SPECT | Temporal | Parietal | qEEG | MMSE |
|---|---|---|---|---|---|---|---|---|
| WMC | Frontal | |||||||
| 1) 51/M | +/- | + | 0 | 0 | 0 | 0 | 20/30 2) | |
| 52/F | +/- | 0 | 0 | 0 | 0 | + | 0 | 28/30 |
| 1) 56/F | +/- | ++ | ++ | 0 | + | + | 0 | 29/30 2) |
| 1) 58/M | +/- | 0 | +++ | 0 | +++ | +++ | ++ | 10/30 2) |
| 1) 60/M | +/- | ++ | + | 0 | 0 | ++ | +++ | 1/30 |
| 66/M | +/- | + | 0 | 0 | 0 | + | (+) | 27/30 |
| 48/M | -/- | 0 | 0 | 0 | 0 | 0 | 0 | 30/30 |
| 52/M | -/- | 0 | 0 | 0 | 0 | + | + | 29/30 |
| 53/F | -/- | 0 | 0 | 0 | + | 0 | ++ | 27/30 |
| 60/M | -/- | 0 | 0 | + | + | + | 0 | 26/30 |
| 62/F | -/- | 0 | 0 | 0 | + | + | 0 | 28/30 |
Symptomatic subjects
Treatment with an acetylcholinesterase inhibitor.
WMC: 0 = no changes; + = slight changes; ++ = moderate changes; +++ = pronounced changes
Atrophy: 0 = no atrophy + = slight atrophy; ++ = moderate atrophy; +++ = pronounced atrophy
rCBF: 0 = no reduction; + = slight reduction; ++ = moderate reduction; +++ = pronounced reduction
qEEG: 0 = no changes; + = slight changes; ++ = moderate changes; +++ = pronounced changes (mean frequency, increased slow wave activity)
MMSE and age recorded at the time for the different investigations.
Neuropathology
The Swedish patient that came to autopsy (fig. 1a, subject IV:10) presented with memory impairment at the age of 62. Subsequent symptoms included spatial disorientation and apathy. He responded temporarily to treatment with acetylcholinesterase inhibitors (tacrine and rivastigmine) and died at the age of 68.
The brain weighed 1385 g. A gross examination revealed a focal moderate atrophy of the parietal superior lobulus with no signs of infarcts or haemorrhages. Microscopically, there was a general widening of the perivascular spaces and severe congophilic angiopathy of parenchymal, leptomeningeal and hippocampal vessels (fig. 2a). Moreover, ring-formed plaques without amyloid core pathology were abundant in neocortex and stained strongly for the C-terminal Aβx-40 and Aβx -42 mAbs (fig. 2b), but only weakly against the N-terminal antibodies 6E10 (not shown). In addition, the ring-formed plaques were positive for silver impregnation techniques but negative for Congo Red (fig. 3b). Silver positive tangles could be demonstrated in the hippocampus and also to a lesser extent in neocortex. Tangles positive for the phospho-tau specific AT8 mAb were present in neocortical pyramidal neurons in low to moderate numbers. Neuronal loss was mainly seen in the limbic areas and to a lesser extent in the neocortex.
Fig. 2.
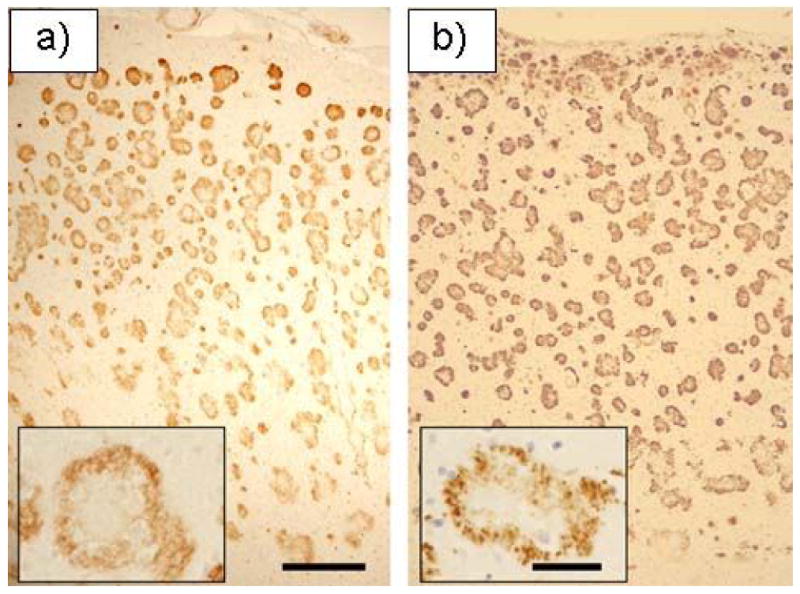
Immunohistochemistry by the Aβ x-42 mAb on tissue from frontal neocortex of the Swedish (a) and the American (b) Arctic APP mutation case. Note that the amyloid pathology is spread throughout the cortical thickness. Also, for both cases, the amyloid plaques have a characteristic non-cored ring form (a and b inserts). Size bars= 250 μm and 50 μm (inserts).
Fig. 3.
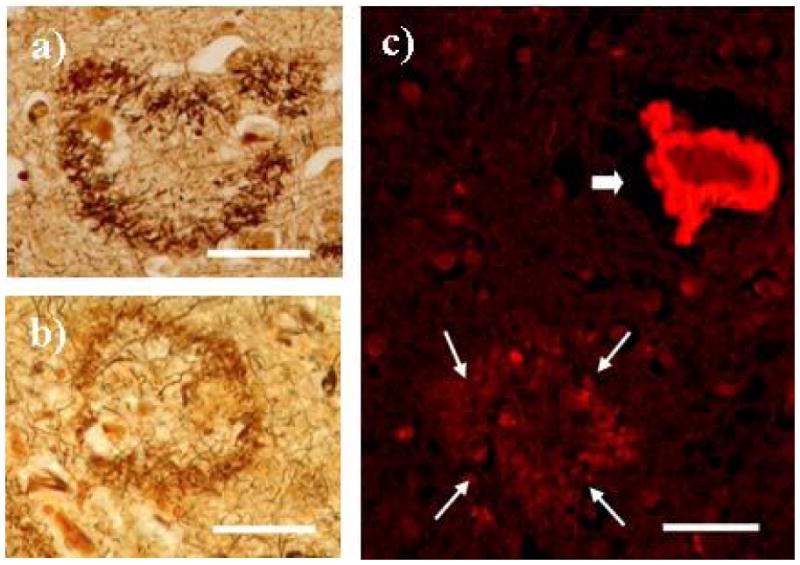
Bielschowsky silver impregnation staining indicate neuritic features of the ring-formed plaques in the Swedish (a) and American (b) Arctic APP mutation case. c) Only arteries (thick arrow) were stained with Congo Red, demonstrating congophilic angiopathy. The ring-formed plaques (thin arrows) were negative for Congo Red. Size Bars = 50μm.
The American autopsy case (fig. 1b, subject III:1) presented with disorientation and insomnia at the age of 57. For the latter case, no psychiatric symptoms, rigidity or myoclonus were present during the disease course. The patient died at the age of 72.
The brain of the American case weighed 822 g. Macroscopically, there was marked atrophy of the frontal, temporal and parietal lobes. Numerous lacunae in the range of 0.1 to 0.3 cm were seen throughout the white matter and ventral to the left putamen. On the microscopic level, a moderate diffuse neuronal loss was present throughout the cerebral cortex, with a more severe cell loss accompanied by reactive gliosis in certain regions, such as amygdala, parahippocampal gyrus and hippocampus. A patchy spongiosis of the external granule cell layer was evident. Further, both Aβ-immunopositive (10D5) and thioS-positive amyloid angiopathy was observed, most notably in the hippocampus and parahippocampal gyrus (not shown). As for the Swedish case, there were ring-formed amyloid plaques mainly in the neocortex that were immunopositive for the Aβx -40 and Aβx -42 mAbs (fig. 2a). These plaques consisted of a crown of delicate neurites surrounding pale brown fibrillary material corresponding to neuritic plaques without amyloid cores (fig. 3a). Finally, frequent neurofibrillary tangles, neuritic plaques and dystrophic neurites were mainly seen in the various neocortical layers (not shown). The neuropathological description has been somewhat restricted due to the clinical nature of this account but will be described more in detail in a separate manuscript (Bogdanovic, in manuscript).
Discussion
We here describe a Swedish and an American family with an inherited form of dementia caused by the Arctic mutation in the APP gene. Affected individuals typically present with memory impairment between 52 and 65 years of age, after which their cognitive status is slowly deteriorating with additional symptoms such as disorientation, dysphasia and dyspraxia. Except for late stage myoclonus and rigidity, no remarkable motor symptoms can be seen and since also no signs or symptoms indicating cerebrovascular events are evident, the clinical features can be considered as indistinguishable from AD. Moreover, the AD phenotype in carriers of other APP or PS mutations is generally characterized by a typical pattern of multiple cognitive dysfunction during the early disease stages. The neuropsychological test results for affected carriers of the Arctic mutation illustrate that the same pattern of cognitive impairments can be seen also for this APP mutation. In addition to profound disturbances in episodic memory and deficits in attention and cognitive speed, we could also demonstrate marked impairments of verbal and visuospatial functions among the diseased cases. Thus, the clinical picture in affected carriers of the Arctic mutation appears to be in good correspondence with the cognitive phenotype of AD, familial as well as sporadic.
The structural and functional imaging of members of the Swedish family with the Arctic mutation showed that white matter changes were present in a majority of mutation carriers but not in non-carriers of similar ages. The most evident imaging pattern in cases with the Arctic mutation revealed atrophy and reduction of blood flow in parietal lobes without any corresponding changes in temporal and frontal lobes (fig. 2a).
In addition to a severe amyloid angiopathy in subarachnoidal and parenchymal vessels, both the Swedish (fig. 1a, subject IV:10) and the American (fig. 1b, subject III:1) autopsy case was neuropathologically characterised by the presence of severe congophilic angiopathy, amyloid plaques and neurofibrillary pathology. For both cases, the plaques displayed a specific ring-like form lacking a core and being strongly positive for C-terminal Aβ antibodies. In addition, these plaques could be stained by silver impregnation techniques, which indicate that they have assumed a neuritic-like pathology. This unusual plaque feature highlights the possibility that the amyloid core pathology is not obligate for the neurodegeneration in AD and that other forms of Aβ may be more significant in terms of neurotoxicity. Conflicting results exist as to how well soluble Aβ serves as a clinicopathological marker 23, 25 but Aβ intermediates, such as oligomers or protofibrils, may exert the neurotoxic effects seen in the AD brain. Interestingly, in mice Aβ oligomers have been demonstrated to disrupt synaptic plasticity 19 and cause neurotoxicity 18 at concentrations comparable to those found in human brains.
The presence of plaques without an amyloid core and negative for Congo Red is not unique to brains with the Arctic APP mutation. A similar plaque variant is a striking neuropathologic feature in brains from individuals with a genomic deletion of exon 9 of the presenilin 1 gene 26. This so called cotton wool plaque is characterized by its large size (up to 120 μm in diameter) and its lack of a thioS-positive core 27. Finally, also the intra-Aβ Dutch APP mutation has been described to cause a similar non-cored Congo Red-negative type of amyloid plaques 12.
Since a thorough neuropsychological evaluation had been performed on the Swedish subject that eventually came to autopsy (fig. 1a, subject IV:10), we had the opportunity to relate the neuropathological picture with the cognitive aberrations caused by the Arctic mutation. Notably, the neuropsychological test results of this individual were compatible with the more advanced pathology in the right vs. left hemisphere. Moreover, the negative influence on speed of performance (z<-3) in this subject is compatible with knowledge-based rather than personality-based difficulties, which is in accordance with the finding of a more aggravated pathology in posterior as compared to anterior brain regions. By and large, the observations on this subject are in agreement with the clinical and neuropathological criteria for a moderate stage of AD, albeit with some asymmetry in cognition. This overall evaluation was further strengthened by the patient's clear response to treatment with an acetyl cholinesterase inhibitor.
The Arctic APP mutation leads to a severe amyloidosis of the brain, as do other mutations within the Aβ region. However, in carriers of the Arctic mutation, no brain haemorrhage has so far been observed, neither clinically nor by adopting modern imaging techniques. This is in stark contrast to both the Dutch and the Flemish APP mutations, for which a conspicuous amyloid angiopathy cause cerebral haemorrhage, probably due to a local weakening of the vessel wall. It is not understood why e.g. the Arctic and the Dutch mutations, both affecting APP amino acid 693, lead to disparate phenotypes. However, it could be speculated that an exchange of the electrically charged glutamic acid to the polar and hydrophilic glutamine renders the Dutch Aβ peptide more prone to adhere to and disrupt the intima of the vessel wall as compared to wild type Aβ or to Arctic Aβ (in which the glutamic acid instead is replaced by the nonpolar and hydrophobic glycine).
In conclusion, our clinical and neuropathological findings suggest that the Arctic APP mutation cause a picture of Alzheimer's disease with brain deposition of a particular ring-formed type of amyloid plaque. These findings indicate marked phenotypical differences between this and other various intra-Aβ APP mutations.
References
- 1.Chartier-Harlin MC, Crawfort F, Houlden H, et al. Early-onset Alzheimer's disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 2.Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein gene associated with hereditary Alzheimer's disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- 3.Mullan M, Crawford F, Axelman K, et al. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–7. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 4.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 5.Borchelt DR, Thinakaran G, Eckman CB, et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–13. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 6.Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nature Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 7.Xia W, Ray WJ, Ostaszewski BL, et al. Presenilin complexes with the C-terminal fragments of amyloid precursor protein at the sites of amyloid beta-protein generation. Proc Natl Acad Sci U S A. 2000;97:9299–304. doi: 10.1073/pnas.97.16.9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cras P, van Harskamp F, Hendriks L, et al. Presenile Alzheimer dementia characterized by amyloid angiopathy and large amyloid core type senile plaques in the APP 692Ala-->Gly mutation. Acta Neuropathol (Berl) 1998;96:253–60. doi: 10.1007/s004010050892. [DOI] [PubMed] [Google Scholar]
- 9.Levy E, Carman M, Fernandez-Madrid I, et al. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 10.Hendriks L, van Duijn CM, Cras P, et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the β-amyloid precursor protein gene. Nat Genet. 1992;1:218–221. doi: 10.1038/ng0692-218. [DOI] [PubMed] [Google Scholar]
- 11.Roks G, Van Harskamp F, De Koning I, et al. Presentation of amyloidosis in carriers of the codon 692 mutation in the amyloid precursor protein gene (APP692) Brain. 2000;123(Pt 10):2130–40. doi: 10.1093/brain/123.10.2130. [DOI] [PubMed] [Google Scholar]
- 12.Bornebroek M, Haan J, Maat-Schieman ML, Van Duinen SG, Roos RA. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): I--A review of clinical, radiologic and genetic aspects. Brain Pathol. 1996;6:111–4. doi: 10.1111/j.1750-3639.1996.tb00793.x. [DOI] [PubMed] [Google Scholar]
- 13.Grabowski T, Cho H, Vonsattel J, Rebeck G, Greenberg S. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- 14.Nilsberth C, Westlind-Danielsson A, Eckman CB, et al. The “Arctic” (E693G) mutation in the Aβ region of APP causes Alzheimer's disease by increasing Aβ protofibril formation. Nat Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 15.Kamino K, Orr HT, Payami H, et al. Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. Am J Hum Genet. 1992;51:998–1014. [PMC free article] [PubMed] [Google Scholar]
- 16.Stenh C, Nilsberth C, Hammarback J, Engvall B, Naslund J, Lannfelt L. The Arctic mutation interferes with processing of the amyloid precursor protein. Neuroreport. 2002;13:1857–60. doi: 10.1097/00001756-200210280-00005. [DOI] [PubMed] [Google Scholar]
- 17.Stenh C, Englund H, Lord A, et al. Amyloid-beta oligomers are inefficiently measured by enzyme-linked immunosorbent assay. Ann Neurol. 2005;58:147–50. doi: 10.1002/ana.20524. [DOI] [PubMed] [Google Scholar]
- 18.Dahlgren K, Manelli A, Stine WJ, Baker L, Krafft G, LaDu M. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 19.Walsh D, Klyubin I, Fadeeva J, et al. Naturally secreted oligomers of amyloid-beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 20.Wechsler D. Wechsler Adult Intelligence Scale – Revised: Manual. New York: Psychological Corporation; 1981. [Google Scholar]
- 21.Lezak M. Neuropsychological Assessment. 3rd. New York: Oxford University Press; 1995. [Google Scholar]
- 22.Nochlin D, Mackenzie A, Bryant E, Norwood T, Sumi S. A simple method of rapid freezing adequately preserves brain tissue for immunocytochemistry, and light and electron microscopic examination. Acta Neuropathol. 1993;86:645–650. doi: 10.1007/BF00294305. [DOI] [PubMed] [Google Scholar]
- 23.Näslund J, Haroutunian V, Mohs R, et al. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 24.Mirra SS, Heyman A, McKeel D. The Consortium to establish a registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 25.Ingelsson M, Fukumoto H, Newell K, et al. Early Abeta accumulation and progressive synaptic loss, gliosis and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 26.Verkkoniemi A, Somer M, Rinne JO, et al. Variant Alzheimer's disease with spastic paraparesis: clinical characterization. Neurology. 2000;54:1103–9. doi: 10.1212/wnl.54.5.1103. [DOI] [PubMed] [Google Scholar]
- 27.Tabira T, Chui de H, Nakayama H, Kuroda S, Shibuya M. Alzheimer's disease with spastic paresis and cotton wool type plaques. J Neurosci Res. 2002;70:367–72. doi: 10.1002/jnr.10392. [DOI] [PubMed] [Google Scholar]