

Abstract
BCL6 is a transcriptional repressor protein that is expressed in a developmentally regulated fashion during B-cell maturation. Specifically, BCL6 is required for formation of germinal centers in response to T-cell dependent antigen activation. Germinal center B-cells feature the ability to tolerate rapid proliferation and simultaneous genetic recombination. Genetic lesions that cause constitutive expression of BCL6 are commonly associated with diffuse large B-cell lymphomas (DLBCL). Recent studies show that BCL6 contributes to the germinal center phenotype by directly represses genes involved in sensing or responding to DNA damage including ATR, TP53 and CDKN1A. The CHEK1 protein is activated through phosphorylation by the ATR kinase domain in response to DNA damage. Activated CHEK1 can phosphorylate and modulate the activity a number of proteins including p53, providing a link between ATR sensing of DNA damage and p53 checkpoint activity. Herein we show that BCL6 can directly bind to a DNA consensus element in the CHEK1 promoter and repress its expression in normal and malignant B-cells. DLBCL cells can be killed by a specific BCL6 peptide inhibitor (BPI) that interferes with corepressor binding to the BCL6 BTB domain. BPI could reactivate CHEK1 in DLBCL cells, suggesting that its induction might contribute to BPI anti-lymphoma effects. Therefore, BCL6 can suppress multiple genes involved in a common pathway sensing, transducing and responding to genotoxic stress.
Keywords: Lymphoma, germinal center, transcriptional repression, affinity maturation
Introduction
Germinal centers (GCs) are transient and dynamic cellular compartments that form within secondary lymphoid organs after T-cell dependent antigenic challenge. The function of GCs is to generate de novo high affinity antibodies against specific antigens[1]. In order to produce such antibodies GC B-cells must undergo clonal expansion and simultaneously mutate and rearrange their immunoglobulin loci[1]. The latter process is mediated by the enzyme activation induced cytosine deaminase (AID), which introduces point mutations and double strand breaks during somatic hypermutation and class switch recombination respectively [2]. Although preferentially affecting the immunoglobulin genes, other transcriptionally active loci can be genetically altered by AID during GC affinity maturation[3]. When such mutations are introduced into oncogenes or tumor suppressors there is potential for oncogenic events that could result in lymphomagenesis[3]. Given the numbers of clones of cells generated by GCs, it seems likely that such potentially oncogenic hits occur frequently during immune reactions in normal individuals. It is therefore not surprising that many B-cell lymphomas arise from B-cells originating from or that have transited through the GC compartment.
Although perilous, the GC B-cell phenotype of physiological genomic instability may serve as an adaptive mechanism in response to evolutionary pressure to facilitate the survival of complex multicellular organisms that must fend off infections for many years until reproduction is complete. Presumably, rapid generation of high affinity antibodies could neutralize the potential lethal threat of successive and continuous exposure to microbial species and variants. However, eukaryotic organisms have also evolved numerous failsafe mechanisms to specifically protect the genome and prevent the genetic recombination that occurs during the GC reaction. DNA damage sensing proteins such as those triggered by the ATR protein and transduced by the CHEK1 protein play a critical role in maintaining genomic integrity[4, 5]. ATR and CHEK1 can trigger tumor suppressor checkpoints for example by phosphorylating the p53 tumor suppressor protein, which induces its activity as a transcription factor[6, 7]. P53 regulates numerous genes involved in regulation of cell survival and proliferation such as BAX and CDKN1A respectively[8, 9]. ATR pathway activation can be triggered by cellular conditions such as the proliferation and DNA damage typically occurring in GC B-cells[5]. Therefore, these cells must have evolved mechanisms to attenuate DNA damage and replicative checkpoints. It is likely that these mechanisms also play a central role in lymphomagenesis so that in the case of GCs, normal and malignant biological mechanisms are tightly linked.
In order to enter the GC reaction, mature B-cells must up-regulate the BCL6 transcriptional repressor[10]. BCL6 is required for formation of GCs, since BCL6 deficient mice fail to form these structures[11, 12]. As a consequence these animals are deficient in affinity maturation of immunoglobulins in response to T-cell dependent antigen stimulus. Emerging data suggest that BCL6 facilitates the GC phenotype by selectively attenuating certain DNA damage sensing pathways[13–15]. Specifically, BCL6 can reduce the ability of centroblasts and lymphoma cells to sense DNA damage, as demonstrated by a relative deficiency in phosphorylation of H2AX and delayed repair of double strand breaks in response to gamma-radiation[13]. This is due in part to BCL6 direct repression of the ATR gene, with consequent loss of the usual kinase dependent activation of ATR targets that normally occurs in response to DNA damage[13]. BCL6 can also directly repress the TP53 and CDKN1A genes, thus severely impairing the function of cellular DNA damage checkpoints at multiple levels [14, 15] (Fig. 1).
Figure 1. BCL6 regulation of a DNA damage pathway in germinal center cells.

BCL6 is expressed when B-cells enter the germinal center reaction. BCL6 can directly repress ATR, CHEK1 (shown herein), TP53 and CDKN1A, all of which form part of a sequential pathway that senses, transduces and triggers checkpoints in response to DNA damage. Repression of these genes facilitates clonal expansion and survival of B-cells in spite of their undergoing mutagenesis as a consequence of class switch recombination and somatic hypermutation. B-cells in turn can overcome this effect through several pathways. Increasing levels of genotoxic stress can degrade BCL6 through the actions of ATM, and CD40 signaling through NFkB can rapidly overcome BCL6 mediated transcriptional repression by disrupting its interaction with N-CoR corepressor, and can transcriptionally downregulate BCL6 through induction of the IRF4 transcription factor, which represses the BCL6 locus.
If sustained these effects of BCL6 could lead to ongoing proliferation and mutagenesis. This danger is underlined by the fact that constitutive expression of BCL6 in mice leads to formation of diffuse large B-cell lymphomas (DLBCLs), and genetic lesions that deregulate BCL6 expression are commonly associated with human DLBCLs[16–18]. Therefore, B-cells have evolved several mechanisms to overcome these effects of BCL6. For example, increasing levels of DNA damage were shown to trigger BCL6 proteolytic degradation through a pathway dependent on the ATM kinase protein[19]. During the GC reaction proliferating BCL6 positive B-cells called centroblasts migrate towards a region of the GC rich in T-cells, dendritic cells and macrophages[1]. Interaction with T-cells leads to triggering of the CD40 receptor present on the surface of B-cells. CD40 signaling has both immediate and delayed BCL6 inhibitory effects, both of which are mediated through NFkB[13, 20]. Thus, NfKB can rapidly disrupt the interaction between BCL6 and the N-CoR corepressor, which is required for BCL6 to repress ATR[13]. Accordingly CD40 signaling could induce expression of ATR in GC B-cells[13]. CD40 signaling through NFkB can also induce expression of the IRF4 transcription factor, which can in turn repress transcription of BCL6, leading to downregulation of BCL6 mRNA and protein levels[20]. Loss of IRF4 binding elements in the BCL6 promoter is associated with DLBCL and constitutive BCL6 expression[20]. Collectively, these data indicate a critical role for BCL6 in controlling DNA damage responses in germinal center B-cells (Fig. 1). Herein, we extend these findings by demonstrating that BCL6 can directly repress the CHEK1 gene, which is a critical mediator of the ATR-dependent DNA damage-signaling pathway.
Materials and Methods
Primary cells and cell lines
Ramos, cells were grown in RPMI 1640 media containing 2 mM L-glutamine and 10% fetal bovine serum (FBS, Gemini Bio-Products, Woodland, CA). LY1 and LY7 cells were grown in Iscove’s medium supplemented with 10% FBS. Germinal center B-cells were obtained from routine human tonsillectomy specimens from the Montefiore Children’s Hospital with approval of the Albert Einstein College of Medicine and Montefiore Hospital Institutional Review Boards and in accordance with the Helsinki protocols. After mincing, tonsilar mononuclear cells were isolated by HISTOPAQUE®-1077 (Sigma) density centrifugation. Centroblasts were separated by magnetic cell separation using the MidiMACS system (Miltenyi Biotec, Auburn, CA) following published protocols [21]. The purity of the isolated B-cell populations was determined by FACScan (Beckton Dickinson, Franklin Lake, NJ) analysis. Centroblasts (CBs) were CD77+ and CD38high. Antibodies used for FACS analysis were: anti-IgD-FITC, CD27-FITC, CD38-PE (BD Pharmingen, San Diego, CA) and anti-CD77 plus anti-MURM-FITC (Immunotech, Warrenale, PA).
BCL6 shRNA and BPI experiments
BCL6 knockdown was achieved using a lentiviral system as described in [13]. Briefly, B-cells were transduced with lentivirus containing a BCL6 shRNA hairpin or a scrambled control sequence in triplicate. 24 hours after transduction mRNA was extracted for QPCR of CHEK1 and BCL6 (to verify knockdown). Ly1 DLBCLs cells were also exposed to the BPI inhibitor peptide described in [22]or vehicle control for 8 hours, after which mRNA was extracted for measurement of CHEK1 mRNA abundance.
Real Time PCR
RNA was prepared from cells using TRIzol (Invitrogen, Carlsbad, CA). cDNA was prepared using Superscript III First Strand cDNA synthesis kit (Invitrogen) and detected by SyberGreen (Applied Biosystems, Foster city, CA) on an Opticon2 thermal cycler (MJ Research, Waltham, MA). We normalized gene expression to GAPDH and expressed values relative to control using the DDCT method. QPCR primers for CHEK1 were: CHEK1F- 5′-AGCGGTTGGTCAAAAGAATG-3′ and CHEK1-R: 5′-TGTCTGCATCCAATTTGGTAA-3′. GAPDH and BCL6 primers were as previously reported [13].
Chromatin Immunoprecipitation (ChIP)
Triplicate ChIP-on-chip was performed as previously described [23] in Ramos cells using the above-mentioned BCL6 and actin (as non-specific IgG control) antibodies. Enrichment of the known BCL6 target gene CCL3 was validated before and after ligation-mediated PCR amplification of genomic fragments, which were then labeled and co-hybridized with their respective input samples to a custom genomic array representing the CHEK1 genomic locus with overlapping 50-mer oligonucleotides (Nimblegen Systems, Madison, WI). The array design and complete results are available on the Gene Expression Omnibus (GEO) website accession number GSE7673. Specific BCL6 binding to genomic regions was detected by determining the fold enrichment of a five-oligonucleotide sliding window over input. BCL6 binding was confirmed by quantitative real-time PCR single locus ChIP (QChIP) as previously described [23]. CHEK1 binding was verified using the following primers: CHEK1-F: 5′-CCGCCGTCCTTAAATCTCTT-3′ and CHEK1-R: 5′-AAAAGGGTGACGTGGAGATG-3′.
Results and Discussion
In order to identify candidate BCL6 target genes, we previously performed a ChIP-on-chip experiment in Ramos B-cell lymphoma cells using an oligonucleotide microarray representing 24,000 promoters [23]. Among promoters identified as potential binding sites for BCL6 was that of CHEK1, suggesting that BCL6 might bind and repress this gene. In order to more precisely define the localization of BCL6 binding on the CHEK1 locus, we again performed ChIP-on-chip using BCL6 antibodies or actin antibodies as negative control. This time the BCL6 or actin products were co-hybridized with the corresponding input samples to a custom high-density tiling array covering the CHEK1 locus with overlapping 50-mer oligonucleotides. A cluster of five consecutive probes was specifically enriched by BCL6 antibodies, and corresponded to a region of the CHEK1 promoter upstream of the transcriptional start site between base pairs −422 and −367 (Fig. 2A). The DNA sequence contained within this region included a motif consistent with known BCL6 binding elements[24–26] (Fig. 2A). In order to validate binding of BCL6, single locus quantitative ChIP (QChIP) was performed using primers designed to amplify this region of the CHEK1 promoter (Fig. 2A). BCL6 antibodies enriched this sequence approximately seven-fold vs. a non-specific immunoglobulin control (Fig. 2B), confirming the presence of BCL6 at or near this binding element. In order to determine whether BCL6 was repressing CHEK1 mRNA transcription, we performed BCL6 loss of function studies in primary GC B-cells and in two B-cell lymphoma cell lines. BCL6 was depleted using a previously validated shRNA lentivirus[13] that downregulates its expression within 24 hours vs. a scrambled control (data not shown and [13]). In both primary B-cells and lymphoma cell lines, transduction with BCL6 shRNA but not scrambled control induced CHEK1 expression, indicating that CHEK1 is a bone fide BCL6 target gene (Figure 3).
Figure 2. BCL6 binds to a single site in the CHEK1 promoter.

Panel A: shows the result of ChIP-on-chip performed in Ramos cells using a custom tilting array covering the CHEK1 locus with overlapping oligonucleotides covering chromosome 11 base pairs 124,996,221 to 125,031,806. The black line corresponds to enrichment of probes using BCL6 antibodies and the gray lines to a non-specific control antibody. The Y axis represents the fold enrichment vs. input. The X axis represents the location of probes along the CHEK1 locus. The bar and line representation shows the exon/intron structure of the CHEK1 gene. The white bar and black arrows beneath the enrichment peak show the location of primers used to validate BCL6 binding in single locus ChIP (panel B). The nucleotide sequence corresponding to the peak enrichment region is shown below and covers base pairs −422 to −367. The bold and underlined sequence is similar to known BCL6 consensus elements. This experiment was repeated three times. Panel B: shows the result of single locus quantitative ChIP for BCL6 or non-specific IgG performed in Ly1 cells. The Y axis represents the fold enrichment for BCL6 vs. IgG control. This experiment was performed in triplicate.
Figure 3. BCL6 represses CHEK1.

BCL6 was downregulated in centroblasts, Ly1 and Ly7 cells by delivering an shRNA via lentiviral transduction. A scrambled shRNA (Scr.) as well as non-transduced cells were used as negative controls. Real-time PCR was performed after 24 hours to measure CHEK1 mRNA abundance. The Y-axis represents fold induction of CHEK1 mRNA by BCL6 shRNA vs. scrambled shRNA. These experiments were performed in triplicate.
CHEK1 is tyrosine kinase that can be activated by phosphorylated of serine 345 by ATR[27]. CHEK1 in turn can activate p53 by phosphorylating it on ser 20 [7]. CHEK1 is thus a link in the DNA damage pathway dependent on ATR and which involves activation of p53 and its downstream targets (Figure 1). In previous experiments where we evaluated the impact of BCL6 on the ATR pathway, we found that BCL6 depletion in primary GC B-cells induced CHEK1 protein level[13]. In contrast, BCL6 depletion did not affect CHEK1 protein levels in lymphoma cell lines. This could be due to the fact that activation of CHEK1 by ATR leads to its proteolyic degradation [28]. Accordingly, we found that depletion of BCL6 by inducing ATR, could greatly enhance phosphorylation of CHEK1 on Ser345[13], which could induce CHEK1 ubiquitylation and degradation[28]. Altogether, in both primary B-cells and lymphoma cells, BCL6 gain and loss of function showed that CHEK1 phosphorylation on ser 345 was greatly impaired in the presence of BCL6 in both primary B-cells and cell lines. Therefore, BCL6 can affect CHEK1 function both through direct repression and via suppression of ATR induced activation. In addition to phosphorylation of p53, CHEK1 plays a critical role in regulating cell cycle progression through S and G2/M phase at least in part by regulating the function of cdc25A and cdc25C proteins [29] and reviewed in [30]. By attenuating CHEK1 activity through ATR and direct repression, BCL6 could impair the ability of p53 to become fully activated, as well as facilitate progression through the S and G2/M phase of the cell cycle.
BCL6 is a member of the BTB/POZ – zinc finger class of transcriptional repressors. BCL6 recruits the N-CoR and SMRT corepressors through a unique lateral groove motif formed by homodimerization of its BTB domain[31]. A specific BCL6 peptide inhibitor (BPI) designed to bind to the BTB lateral groove can compete with these corepressors and reactivate BCL6 target genes[22]. As a consequence, administration of BPI to immunized mice blocks formation of germinal centers, similar to the BCL6 null phenotype. BPI can also kill BCL6 expressing DLBCL cells[22]. Not all BCL6 target genes are dependent on BTB domain recruitment of SMRT and N-CoR. For example, PRDM1, a BCL6 target gene involved in plasma cell differentiation is regulated through a different biochemical mechanism involving the MTA3/NuRD corepressor complex[32, 33]. However, genes from the DNA damage pathway including ATR, TP53 and CDKN1A are all induced by BPI and are thus dependent on N-CoR and SMRT [13, 34]. Likewise, CHEK1 mRNA transcription was also induced by BPI when administered to DLBCL cells (Figure 4). These data suggest that an entire axis of BCL6 regulated DNA damage pathway genes starting from ATR and including CHEK1, TP53 and CDKN1A are under the control of this biochemical mechanism, and collectively contribute to the germinal center B-cell phenotype imposed by BCL6 in normal and malignant B-cells.
Figure 4. BPI can reactivate expression of CHEK1.
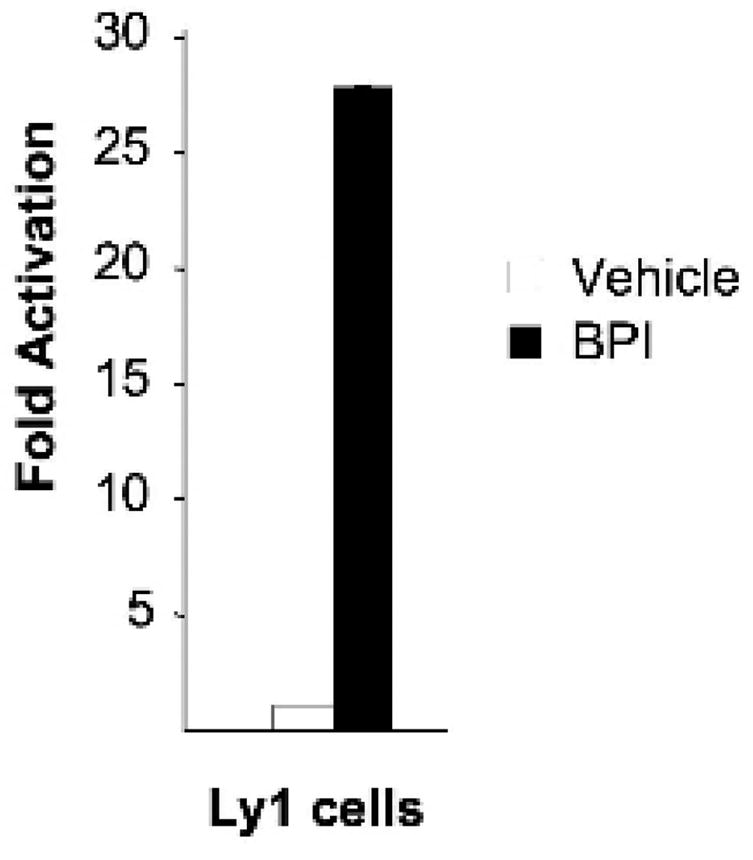
Ly1 cells were treated with BPI or vehicle for 8 hours followed by real-time PCR to measure CHEK1 mRNA abundance. The Y axis represents fold induction of CHEK1 by BPI vs. vehicle. This experiment was performed in quadruplicate.
From the therapeutic standpoint, re-activation of this BCL6 DNA damage targets contributes to effective killing of DLBCL cells, since depletion of ATR from DLBCL cells protected cells from dying after BCL6 loss of function[13]. The anti-lymphoma effects of DNA damaging agents such as radiation and chemotherapy are partly dependent on induction of DNA damage checkpoints. BCL6 protects DLBCL cells from cell death induced these agents at least in part by repressing ATR and p53 [13, 14]. Combinations of BPI with chemotherapy drugs can thus result in enhanced killing of lymphoma cells [34]. Reactivation of the ATR-CHEK1-TP53-CDKN1A DNA damage pathway likely plays an important role in this chemosensitization effect. In summary, the ability of BCL6 to impose the GC B-cell phenotype on activated B-cells is dependent on its transcriptional repression of a DNA damage pathway that includes CHEK1. This system has built in control mechanisms to avoid the danger of prolonged genomic instability during the GC reaction. When these control mechanisms fail, often due to constitutive expression of BCL6, lymphomagenesis ensues. However, BCL6 repression of this pathway can be overcome by therapeutic targeting of the biochemical mechanism through which BCL6 controls expression of these genes (i.e. BPI targeting of the BCL6 BTB domain). Therefore, the same mechanism used by B-cells to generate high affinity antibodies can serve as a therapeutic target in B-cell lymphomas.
Acknowledgments
SMR is supported by a Cancer Research Institute Fellowship. JMP is supported by a pre-doctoral fellowship from the National Cancer Center. AMM is supported by NCI R01 CA104348, the G&P foundation, Chemotherapy foundation, and The Leukemia & Lymphoma Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.MacLennan IC. Germinal centers. Annu Rev Immunol. 1994;12:117–39. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 2.de Yebenes VG, Ramiro AR. Activation-induced deaminase: light and dark sides. Trends Mol Med. 2006;12:432–9. doi: 10.1016/j.molmed.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Pasqualucci L, Neumeister P, Goossens T, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412:341–6. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 4.Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis. 2006;21:3–9. doi: 10.1093/mutage/gei063. [DOI] [PubMed] [Google Scholar]
- 5.Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007;6:953–66. doi: 10.1016/j.dnarep.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Tibbetts RS, Brumbaugh KM, Williams JM, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–7. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shieh SY, Ahn J, Tamai K, et al. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 8.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 9.Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–4. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 10.Allman D, Jain A, Dent A, et al. BCL-6 expression during B-cell activation. Blood. 1996;87:5257–68. [PubMed] [Google Scholar]
- 11.Ye BH, Cattoretti G, Shen Q, et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet. 1997;16:161–70. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- 12.Dent AL, Shaffer AL, Yu X, et al. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–92. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 13.Ranuncolo SM, Polo JM, Dierov J, et al. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat Immunol. 2007;8:705–14. doi: 10.1038/ni1478. [DOI] [PubMed] [Google Scholar]
- 14.Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–9. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 15.Phan RT, Saito M, Basso K, et al. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol. 2005;6:1054–60. doi: 10.1038/ni1245. [DOI] [PubMed] [Google Scholar]
- 16.Cattoretti G, Pasqualucci L, Ballon G, et al. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell. 2005;7:445–55. doi: 10.1016/j.ccr.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 17.Baron BW, Anastasi J, Montag A, et al. The human BCL6 transgene promotes the development of lymphomas in the mouse. Proc Natl Acad Sci U S A. 2004;101:14198–203. doi: 10.1073/pnas.0406138101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye BH, Lista F, Lo Coco F, et al. Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science. 1993;262:747–50. doi: 10.1126/science.8235596. [DOI] [PubMed] [Google Scholar]
- 19.Phan RT, Saito M, Kitagawa Y, et al. Genotoxic stress regulates expression of the proto-oncogene Bcl6 in germinal center B cells. Nat Immunol. 2007;8:1132–9. doi: 10.1038/ni1508. [DOI] [PubMed] [Google Scholar]
- 20.Saito M, Gao J, Basso K, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell. 2007;12:280–92. doi: 10.1016/j.ccr.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 21.Klein U, Tu Y, Stolovitzky GA, et al. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci U S A. 2003;100:2639–44. doi: 10.1073/pnas.0437996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polo JM, Dell’Oso T, Ranuncolo SM, et al. Specific peptide interference reveals BCL6 transcriptional and oncogenic mechanisms in B-cell lymphoma cells. Nat Med. 2004;10:1329–35. doi: 10.1038/nm1134. [DOI] [PubMed] [Google Scholar]
- 23.Polo JM, Juszczynski P, Monti S, et al. Transcriptional signature with differential expression of BCL6 target genes accurately identifies BCL6-dependent diffuse large B cell lymphomas. Proc Natl Acad Sci U S A. 2007;104:3207–12. doi: 10.1073/pnas.0611399104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baron BW, Stanger RR, Hume E, et al. BCL6 encodes a sequence-specific DNA-binding protein Genes Chromosomes. Cancer. 1995;13:221–4. doi: 10.1002/gcc.2870130314. [DOI] [PubMed] [Google Scholar]
- 25.Deweindt C, Albagli O, Bernardin F, et al. The LAZ3/BCL6 oncogene encodes a sequence-specific transcriptional inhibitor: a novel function for the BTB/POZ domain as an autonomous repressing domain. Cell Growth Differ. 1995;6:1495–503. [PubMed] [Google Scholar]
- 26.Chang CC, Ye BH, Chaganti RS, Dalla-Favera R. BCL-6, a POZ/zinc-finger protein, is a sequence-specific transcriptional repressor. Proc Natl Acad Sci U S A. 1996;93:6947–52. doi: 10.1073/pnas.93.14.6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Q, Guntuku S, Cui XS, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang YW, Otterness DM, Chiang GG, et al. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19:607–18. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 29.Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 30.Lam MH, Rosen JM. Chk1 versus Cdc25: chking one’s levels of cellular proliferation. Cell Cycle. 2004;3:1355–7. doi: 10.4161/cc.3.11.1225. [DOI] [PubMed] [Google Scholar]
- 31.Ahmad KF, Melnick A, Lax S, et al. Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol Cell. 2003;12:1551–64. doi: 10.1016/s1097-2765(03)00454-4. [DOI] [PubMed] [Google Scholar]
- 32.Fujita N, Jaye DL, Geigerman C, et al. MTA3 and Mi-2/NuRD Complex Regulate Cell Fate During B-Lymphocyte Differentiation. Cell. 2004;119:75–86. doi: 10.1016/j.cell.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 33.Parekh S, Polo JM, Shaknovich R, et al. BCL6 programs lymphoma cells for survival and differentiation through distinct biochemical mechanisms. Blood. 2007;110:2067–74. doi: 10.1182/blood-2007-01-069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cerchietti L, Polo J, DaSilva G, et al. BCL6, p53 and Molecular Targeted Therapy in B-Cell Lymphomas. Blood. 2005:106. [Google Scholar]