

Abstract
Photoredox catalysis and organocatalysis represent two powerful fields of molecule activation that have found widespread application in the areas of inorganic and organic chemistry, respectively. We merged these two catalysis fields to solve problems in asymmetric chemical synthesis. Specifically, the enantioselective intermolecular α-alkylation of aldehydes has been accomplished using an interwoven activation pathway that combines both the photoredox catalyst Ru(bpy)3Cl2 (where bpy is 2,2′-bipyridine) and an imidazolidinone organocatalyst. This broadly applicable, yet previously elusive, alkylation reaction is now highly enantioselective and operationally trivial.
Nature’s ability to convert solar energy to chemical energy in photosynthesis has inspired the development of a host of photoredox systems in efforts to mimic this process. Arguably the most studied one-electron photoredox catalyst has been Ru(bpy)3 2+ (where bpy is 2,2′-bipyridine): an inorganic complex that has facilitated important advances in the areas of energy storage, hydrogen and oxygen evolution from water, and methane production from carbon dioxide (1, 2). Given its proven ability to mediate electron transfer, it is surprising that Ru(bpy)3 2+ has not found a substantial application in organic synthesis, wherein a large number of fundamental reactions rely on the generation and exploitation of radicals or single-electron intermediates (3).
Over the past decade, the field of organocatalysis has grown at a dramatic pace, providing more than 130 chemical reactions that rapidly facilitate enantioselective C–C, C–O, C–N, and C–halogen bond formation (4, 5). Whereas a broad range of reaction types have recently succumbed to this mode of catalysis (including aldol, Friedel-Crafts, and cycloadditions), it is important to consider that nearly all organocatalytic bond constructions are restricted to two-electron pathways, wherein the highest occupied molecular orbital of an electron-rich substrate reacts with the lowest unoccupied molecular orbital of an electron-deficient partner. Recently, however, our laboratory introduced the concept of organo–singly occupied molecular orbital (SOMO) catalysis, a one-electron mode of activation that has enabled the development of several useful transformations (6–10).
Given the widespread success of both electron transfer catalysis and organocatalysis, we recently questioned whether it might be possible to merge these two powerful areas, with the goal of solving long-standing, yet elusive problems in chemical synthesis. More specifically, as a blue-print for reaction invention, we hoped to exploit the lessons of photoredox enzymatic catalysis (11), wherein a series of consecutive low-barrier, open-shell steps are energetically preferred to high-barrier, two-electron pathways. On this basis, we hypothesized that the enantioselective catalytic α-alkylation of aldehydes (12–15), a widely sought yet elusive transformation, might be brought to fruition via the marriage of inorganic electron transfer and organic catalysis (Fig. 1).
Fig. 1.

Merging amine catalysis and organometallic photoredox catalysis to enable asymmetric organic transformations. Me, methyl; R, generic organic substituent; FG, electron-withdrawing functional group.
We proposed that two interwoven catalytic cycles might be engineered to simultaneously generate an electron-rich enamine from the condensation of an aldehyde and an amine catalyst and an electron-deficient alkyl radical via reduction of an alkyl bromide with a Ru photoredox catalyst (Fig. 2). Given that electron-deficient radicals are known to rapidly combine with π-rich olefins to forge even the most elusive C–C bonds (16, 17), we hoped that this dual-catalysis mechanism would successfully converge to enable the direct coupling of aldehydes with α-bromo ketones or esters. As a critical design element, we presumed that the use of a suitable chiral amine catalyst would induce high enantioselectivity. Moreover, we recognized that the interaction of a SOMOphilic enamine with an electron-deficient radical is the converse mechanism to our previously described SOMO activation studies. As such, a complementary array of catalytic bond constructions should be possible.
Fig. 2.

Merging photoredox catalysis with organocatalysis. Proposed mechanism. t-Bu, tert-butyl.
A detailed description of our dual-catalysis aldehyde alkylation is presented in Fig. 2. It has long been established that Ru(bpy)3 2+ (1) will readily accept a photon from a variety of light sources to populate the *Ru(bpy)3 2+ (2) metal-to-ligand charge transfer (MLCT) excited state (1, 2). Although *Ru(bpy)3 2+ (2) can function as a reductant or an oxidant, we postulated that this high-energy intermediate would efficiently remove a single electron from a sacrificial quantity of enamine, to initiate our first catalytic cycle and provide the electron-rich Ru(bpy)3 + (3). Given that Ru(bpy)3 + (3) has been shown to be a potent reductant [−1.33 V versus saturated calomel electrode (SCE) in CH3CN] (18), we anticipated that single-electron transfer (SET) to the α-bromocarbonyl substrate 4 would rapidly furnish the electron-deficient alkyl radical 5 while returning Ru(bpy)3 2+ (1) to the catalytic cycle (E1/2 for phenacyl bromide = −0.49 V versus SCE in CH3CN, where E1/2 is the half reduction potential) (19–22). As a central design consideration, we recognized that the redox potentials of Ru(bpy)3 2+ can be readily fine-tuned by ligand modification (1).
Concurrent with this photoredox pathway, the organocatalytic cycle would begin with condensation of the imidazolidinone catalyst 6 and the aldehyde substrate 7 to form enamine 8. At this stage, we expected the two catalytic cycles to intersect via the addition of the SOMOphilic enamine 8 to the electron-deficient alkyl radical 5, thereby achieving the key alkylation step. This coupling event would concomitantly produce an electron-rich α-amino radical 9, a single-electron species that has a low barrier to oxidation (−0.92 to −1.12 V versus SCE in CH3CN) (23). Once again, convergence of our catalytic cycles should ensure SET from α-amino radical 9 to the *Ru(bpy)3 2+ (2) excited state to produce the iminium ion 10 and regenerate the active reductant, Ru(bpy)3 + (3)—a step that would close the photoredox cycle (24). Hydrolysis of the resulting iminium 10 would reconstitute the amine catalyst 6 while delivering the requisite enantioenriched α-alkyl aldehyde product.
From the outset, we understood that the utility of this alkylation reaction would rely on the identification of an amine catalyst that could generically enforce high levels of enantiocontrol in the coupling of the pivotal π-rich enamine with a diverse array of electron-deficient radicals. On the basis of density functional theory (DFT) calculations (25, 26), we proposed that the imidazolidinone catalyst 6 should selectively form an enamine 8 (DFT-8), that projects the 2π electron system away from the bulky tert-butyl group, whereas the electron-rich olefin will selectively populate an (E)-configuration to minimize nonbonding interactions with the imidazolidinone ring (Fig. 2). In terms of enantiofacial discrimination, the calculated DFT-8 structure also reveals that the methyl group on the catalyst system will effectively shield the Re face of the enamine, leaving the Si face exposed for enantioselective radical addition. We have found that the trans methyl, tert-butyl 2,5-disubstituted imidazolidinone 6 is an excellent enamine catalyst for transformations performed at room temperature. Specifically, catalyst 6 provides excellent levels of kinetic enantiocontrol yet does not readily participate in enamine formation with the 2,2′-disubstituted aldehyde-alkylation adduct, a step that would erode product enantiopurity via epimerization.
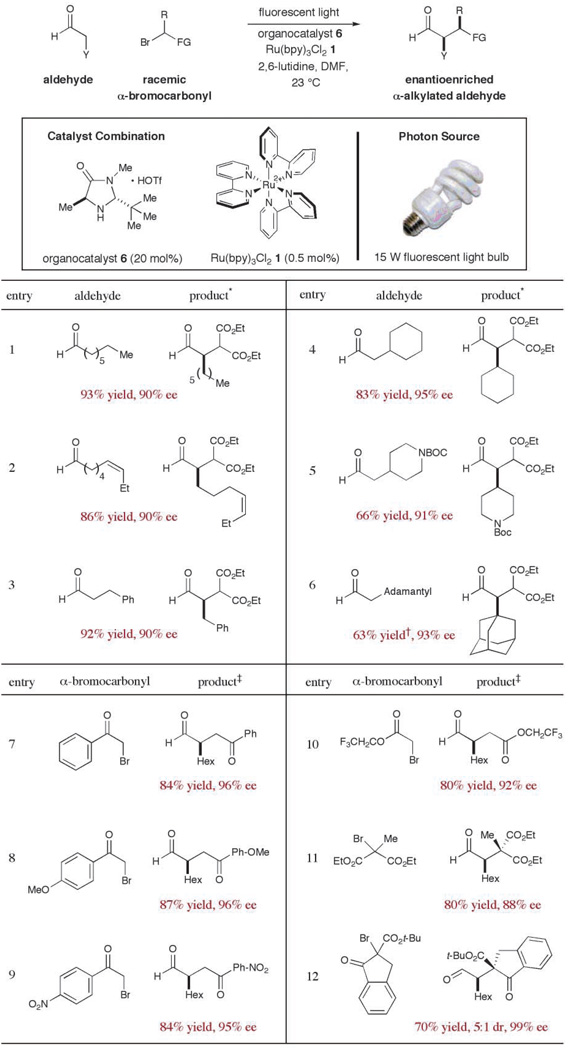
This new asymmetric alkylation protocol was first examined using octanal and bromo diethylmalonate as the coupling partners, along with a catalyst combination of Ru(bpy)3Cl2 (1) and imidazolidinone 6, and a 15-W fluorescent light source (Table 1) (27). To our great delight, preliminary studies revealed the successful execution of our dual-cycle design ideals to provide (R)-2-malonyloctanal with excellent levels of enantiocontrol and reaction efficiency [entry 1, 93% yield, 90% enantiomeric excess (ee)]. Experiments that probe the scope of the aldehyde component in this new alkylation reaction are summarized in Table 1 (entries 1 to 6). Chemical functionalities that are often prone to either oxidation or reduction (e.g., olefins, esters, carbamates, and arenes) were found to be inert to these mild redox conditions (entries 2 to 5, 66 to 92% yield, 90 to 95% ee). Moreover, the steric demand of the α-formyl substituent has little impact on the efficiency and enantioinduction of the alkylation process (entries 1 and 4, substituent is n-hexyl versus cyclohexyl, 83 to 93% yield, 90 to 95% ee), a point that is underscored by the successful use of adamantyl acetaldehyde (entry 6, 63% yield, 93% ee).
Table 1.
Survey of the bromide and aldehyde scope in the direct α-alkylation of aldehydes. Y, any organic substituent (alkyl, aryl, alkenyl, alkynyl, etc.); DMF, N,N′-dimethylformamide; Tf, triflate; Me, methyl; Et, ethyl; Hex, hexyl; Ph, phenyl; t-Bu, tert-butyl; Boc, tert-butyl carbamoyl.
 |
|---|
Reactions performed with diethyl bromomalonate.
40 mole percent of organocatalyst 6 was employed.
Reactions performed with octanal.
A broad array of electron-deficient α-bromo carbonyls can effectively serve as alkylating agents in this tandem catalysis manifold (Table 1, entries 7 to 12). For example, bromoacetophenone systems of diverse electronic orientation (p-OMe, p-NO2, p-H) provide almost identical selectivity and efficiency profiles (entries 7 to 9; 84 to 87% yield, 95 to 96%ee).Whereas α-bromo esters are readily tolerated (BrCH2CO2Et, 53% yield, 94% ee), we have found that superior yields are obtained with markedly electron-deficient carbonyls such as the trifluoroethyl ester (80% yield, 92% ee). As a testament to the versatility and power of one-electron mediated pathways, we have found that tertiary bromo-substituted alkylating agents can be readily employed to forge all-carbon quaternary centers, (entries 11 and 12, ≥70% yield, 88 to 99% ee). Moreover, racemic α-bromo radical precursors can be employed to generate quaternary stereocenters with appreciable levels of diastereocontrol (entry 12, 5:1 diastereomeric ratio), illustrating the capacity of the pivotal enamine intermediate to differentiate the enantiotopic faces of a trisubstituted carbon-centered radical. The sense of asymmetric induction observed in all cases (Table 1) is consistent with selective addition of the electron-deficient radical to the Si face of the enamine 8, in complete accord with the calculated structure DFT-8.
With respect to operational convenience, it is important to consider that this alkylation protocol does not require any heating or cooling, all of the components employed in this study (substrates, catalysts, and solvents) are commercially available and inexpensive, and a simple household 15-W fluorescent light bulb can be employed as a suitable light source. A 2-g alkylation was readily accomplished using the outlined procedure (entry 7).
We have conducted a series of control experiments and luminescence quenching studies to test the validity of our proposed dual-cycle pathway and gain further insight into the photonic requirements for metal-mediated redox catalysis. The control experiments were performed using octanal with α-bromoacetophenone or diethyl bromomalonate in the presence of various catalyst combinations and a 15-W fluorescent light source (unless otherwise stated). Several observations are of note: Rigorous exclusion of light failed to produce even trace quantities of the coupling adduct. Moreover, removal of Ru(bpy)3 2+ from our standard protocol resulted in <10% alkylation product over an extended timeframe (24 hours). High levels of reaction efficiency (>80%) can be obtained in the absence of Ru(bpy)3 2+ if a high-energy UV irradiation source (300 to 350 nm) is employed in a photobox environment. In this specific case, we assume that a monocyclic catalysis mechanism is operable wherein the α-bromocarbonyl is converted to the requisite electron-deficient radical via photolytic bond homolysis (as opposed to catalytic SET reduction). Execution of our standard reaction with a light source specifically tuned to the Ru(bpy)3 2+ MLCT absorption band (465 ± 20 nm full width at half maximum, 500 mW) resulted in a dramatic acceleration in overall rate (90 min) as compared with the use of a typical household 15-W fluorescent bulb (6 hours), which operates with a wide spectral window (~400 to 700 nm). The use of the same 465-nm photon source in the absence of Ru(bpy)3 2+ resulted in only trace product formation (<5%) (28). These experiments provide strong evidence of the participation of the *Ru(bpy)3 2+ (2) excited state in the catalytic cycle.
With respect to our luminescence quenching studies, it has long been established that certain electron-deficient C–Br bonds can quench the emission intensity of *Ru(bpy)3 2+ by SET (29). However, we did not observe a decrease in *Ru(bpy)3 2+ luminescence in the presence of α-bromoacetophenone or diethyl bromomalonate, a result that negates the possibility that *Ru(bpy)3 2+ (2) is participating as a reductant in our tandem catalysis sequence. In contrast, enamine 8 (pregenerated in stoichiometric quantities) does decrease the *Ru(bpy)3 2+ emission intensity with a small but significant Stern-Volmer constant of 10M−1 (see fig. S1) (30). These observations collectively support our mechanistic proposal that the *Ru(bpy)3 2+ (2) excited state behaves as an oxidant in our photoredox cycle.
We have also gained circumstantial evidence that enamine 8 is the organocatalytic intermediate that participates in the key bond-forming step. More specifically, exposure of 2-phenylcyclopropyl acetaldehyde to our standard reaction protocol resulted in clean conversion (83% yield) to the corresponding alkylation product (see supporting online material). Failure of this radical clock substrate to undergo cyclopropyl ring opening clearly indicates that a 3π electron SOMO activated intermediate is not operative in the organocatalytic cycle.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/1161976/DC1
Materials and Methods
Figs. S1 to S3
References and Notes
- 1.Kalyanasundaram K. Coord. Chem. Rev. 1982;46:159. [Google Scholar]
- 2.Juris A, et al. Coord. Chem. Rev. 1988;84:85. [Google Scholar]
- 3.Renaud P, Sibi MP, editors. Radicals in Organic Synthesis. Weinheim, Germany: Wiley-VCH; 2001. [Google Scholar]
- 4.Berkessel A, Gröger H, editors. Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis. Weinheim, Germany: Wiley-VCH; 2005. [Google Scholar]
- 5.Dalko PI, editor. Enantioselective Organocatalysis: Reactions and Experimental Procedures. Weinheim, Germany: Wiley-VCH; 2007. [Google Scholar]
- 6.Beeson TD, Mastracchio A, Hong JB, Ashton K, MacMillan DWC. Science. 2007;316:582. published online 28 March 2007 (10.1126/science. 1142696). [PubMed] [Google Scholar]
- 7.Jang H, Hong JB, MacMillan DWC. J. Am. Chem. Soc. 2007;129:7004. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]
- 8.Kim H, MacMillan DWC. J. Am. Chem. Soc. 2008;130:398. doi: 10.1021/ja077212h. [DOI] [PubMed] [Google Scholar]
- 9.A SOMO activation mechanism has also been reported for the α-oxidation of aldehydes (10).
- 10.Sibi M, Hasegawa M. J. Am. Chem. Soc. 2007;129:4124. doi: 10.1021/ja069245n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gray HB, Winkler JR. Annu. Rev. Biochem. 1996;65:537. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 12.An intramolecular α-formyl alkylation has been reported (13).
- 13.Vignola N, List B. J. Am. Chem. Soc. 2004;126:450. doi: 10.1021/ja0392566. [DOI] [PubMed] [Google Scholar]
- 14.For a catalytic enantioselective alkylation of racemic α-bromoesters, see (15).
- 15.Dai X, Strotman NA, Fu GC. J. Am. Chem. Soc. 2008;130:3302. doi: 10.1021/ja8009428. [DOI] [PubMed] [Google Scholar]
- 16.Renaud P, Schubert S. Synlett. 1990;624 1990. [Google Scholar]
- 17.Russell GA, Wang K. J. Org. Chem. 1991;56:3475. [Google Scholar]
- 18.Bock CR, et al. J. Am. Chem. Soc. 1979;101:4815. [Google Scholar]
- 19.Value was corrected from the Ag/Ag+ClO4 − electrode (20).
- 20.Tanner DD, Singh HK. J. Org. Chem. 1986;51:5182. [Google Scholar]
- 21.Ru(bpy)3 + has previously been shown to reduce phenacyl bromide (22).
- 22.Fukuzumi S, Mochizuki S, Tanaka T. J. Phys. Chem. 1990;94:722. [Google Scholar]
- 23.Wayner DDM, Dannenberg JJ, Griller D. Chem. Phys. Lett. 1986;131:189. [Google Scholar]
- 24.The possibility of direct one-electron reduction of the α-bromocarbonyl by the α-amino radical as a propagation step cannot be excluded.
- 25.DFT calculations were performed with the use of B3LYP/6-311+G(2d,2p)//B3LYP/6-31G(d).
- 26.A conformer that positions the enamine olefin toward the tert-butyl group was also found to be energetically relevant in these calculations. Because of the pseudo C2-symmetric nature of catalyst 6, this enamine conformer also exists with the Si face open and the Re face blocked in a manner similar to DFT-8.
- 27.Materials and methods are available as supporting material on Science Online.
- 28.No rate enhancement was observed in the absence of Ru(bpy)3 2+ with additive bpy or Bu4NCl.
- 29.Oishi S, Furuta N. Chem. Lett. 1978;7:45. [Google Scholar]
- 30.None of the remaining reaction components (aldehyde, amine catalyst, 2,6-lutidine, or 2,6-lutidinium bromide) quenched *Ru(bpy)3 2+.
- 31.We thank S. Bernhard for his assistance in performing quenching experiments, as well as many insightful discussions. Additionally, we thank T. J. Rainey for performing DFT calculations. Financial support was provided by the NIH General Medical Sciences (grant R01 GM078201-01-01) and gifts from Merck, Amgen, and Bristol-Myers Squibb. D.A.N. is grateful for a NIH National Service Research Award fellowship (F32GM076816).