

Abstract
 An efficient nine-step total synthesis of the annulated indole natural products (±)-cis-trikentrin A and (±)-herbindole A was accomplished via an intermolecular Diels–Alder cycloaddition using our recently developed indole aryne (indolyne) methodology as the key step. This strategy provides rapid access into the trikentrins and the related herbindoles and represents the first application of this methodology to natural products total synthesis. The required 6,7-indolyne precursor was readily constructed by means of the Bartoli indole synthesis with substituted nitrobenzenes and vinyl magnesium bromide.
An efficient nine-step total synthesis of the annulated indole natural products (±)-cis-trikentrin A and (±)-herbindole A was accomplished via an intermolecular Diels–Alder cycloaddition using our recently developed indole aryne (indolyne) methodology as the key step. This strategy provides rapid access into the trikentrins and the related herbindoles and represents the first application of this methodology to natural products total synthesis. The required 6,7-indolyne precursor was readily constructed by means of the Bartoli indole synthesis with substituted nitrobenzenes and vinyl magnesium bromide.
Although the indole nucleus is ubiquitous in nature, annulated indoles at any of the benzenoid positions are uncommon. The trikentrins1 and the structurally similar herbindoles1e,i,k,2d represent fascinating examples of such 6,7-annulated indole natural products (Figure 1). The trikentrins were isolated by Capon1a from the marine sponge Trikentrion flabelliforme and display antibacterial activity. The more recently discovered herbindoles by Scheuer1e from the Australian sponge Axinella sp. possess both cytotoxic and antifeedant properties. The interesting biological profiles of these molecules combined with their uncommon structural motifs make them attractive targets for total synthesis.
Figure 1.
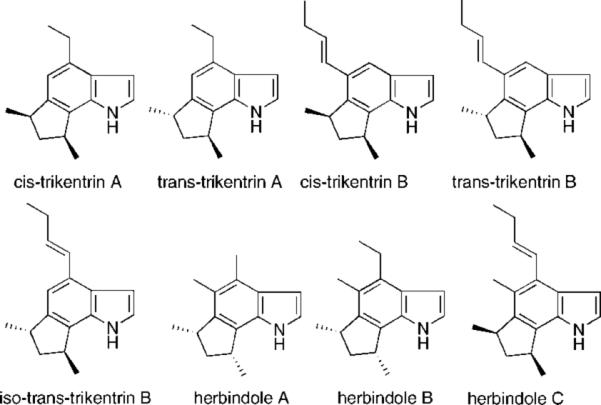
Trikentrin and herbindole natural products.
The significant challenges associated with the construction of these complex systems are reflected in the many distinct approaches that have been reported for their racemic1b-d,f-l (cis-trikentrin A, trans-trikentrin A, cis-trikentrin B, iso-trans-trikentrin B, herbindole A, herbindole B) and enantioselective2 total synthesis. The first total synthesis of (±)-cis-trikentrin A made use of radical cyclization1b,d,h to form the indane skeleton followed by indole ring annulation. Subsequent total synthesis efforts for the trikentrins and herbindoles involved intramolecular allene cycloaddition,1c heteroaromatic azadiene Diels–Alder reactions,1f intermolecular Heck coupling,1g intermolecular quinone monoimine Diels–Alder cyclization,1i,k electrocyclic divinylpyrroline ring closure,1j and ring contraction,1l whereas the enantioselective trikentrin and herbindole syntheses relied on pyrrole indolization2a-d or intramolecular allene cycloaddition strategies.2e
We recently discovered a new class of aryne derived from all three benzenoid positions of the indole nucleus and examined their cycloaddition behavior with furan (Scheme 1).3 It occurred to us that an intermolecular indolyne cycloaddition approach should provide a rapid entry into annulated indole natural products including the trikentrins, herbindoles, and teleocidins.4 We now present a completely different and efficient route for the total synthesis of (±)-cis-trikentrin A and (±)-herbindole A based on this new methodology.
Scheme 1.
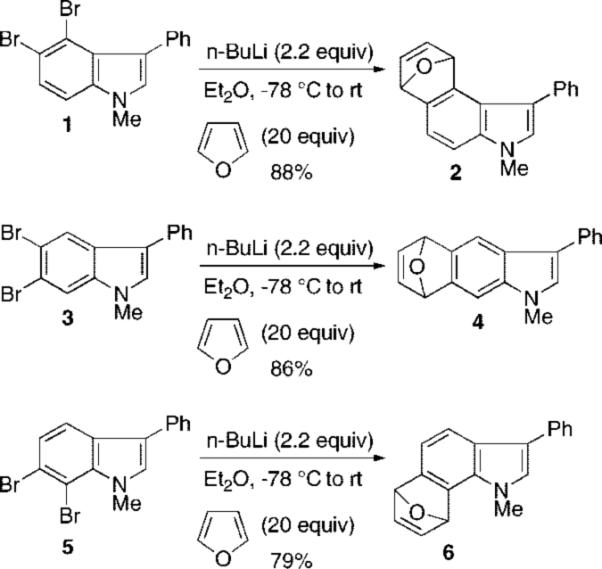
Indole Aryne Cycloadditions with Furan
We required the synthesis of 6,7-dibromoindole 11 as a precursor of the corresponding indolyne via halogen–metal exchange. We originally generated indole arynes from all three benzenoid positions by metal–halogen exchange-elimination of the corresponding o-dibromoindoles 1, 3, and 5 followed by trapping with furan or symmetrical 2,5-disubstituted furans. These in turn were prepared from the o-dibromohydrazines via Fischer indole chemistry. However, repeated efforts to synthesize o-dihaloindoles unsubstituted at the 2- and 3-position via Fischer chemistry gave disappointing results.
Our revised plan was influenced by Bartoli's observation that such unsubstituted indoles can be obtained in good yields from the reaction of nitrobenzenes and vinyl Grignard reagents.5 The synthesis of trikentrin A thus began with commercially available 4-ethylaniline 7 (Scheme 2). Nitration was accomplished in 96% yield in one pot by a literature procedure.6 Diazotization with t-BuONO followed by bromination catalyzed by CuBr2 was carried out in 82% yield to afford the o-dibromide 9.7 Application of the Bartoli indole synthesis (CH2CHMgBr, 3 equiv; THF, −40 °C) proceeded uneventfully and gave the desired indole 10 in 52% yield. In other cases reported by Bartoli, highly variable yields are found with only o-substituted nitrobenzenes. We were able to find only a few other examples of trisubstutited nitrobenzenes and no examples of 2,3-dihalo nitrobenzenes used in this procedure.1c,5 By comparison, the best indole synthesis yield reported by Bartoli is 67% with 2-methyl nitrobenzene.5d The NH group of the indole was then protected as its TBS group (KHMDS, TBSOTf, THF, −78 °C).
Scheme 2.
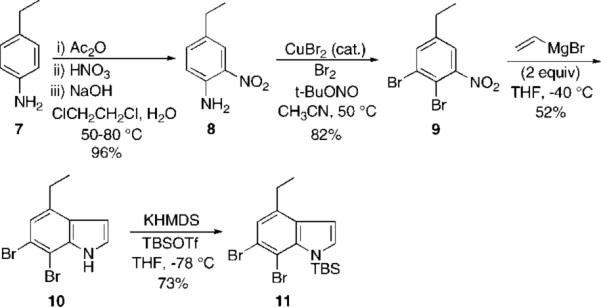
cis-Trikentrin A: Bartoli Indole Synthesis
With the desired indole in hand, metal–halogen exchange with n-BuLi (2.2 equiv, Et2O, −78 °C) in the presence of cyclopentadiene in ether or THF resulted in mainly deprotonation of cyclopentadiene and only a low yield of the desired cycloadduct.
Changing the solvent to toluene, however, nearly completely suppressed the acid–base reaction as observed by Coe with other benzyne cycloadditions8 and cleanly gave the desired cycloadduct 12 in 77% isolated yield (Scheme 3). Attempts to carry out the cycloaddition with the unprotected indole N–H or its anion with various simple counterions (Li+, Na+, or K+) met with failure. Osmylation of 12 (cat. OsO4/NMO, THF/H2O, 9:1) followed by oxidative cleavage of the diol 13 (NaIO4, THF/H2O (3:1)) afforded the dialdehyde 14 in 87% yield for the two steps. Several methods were attempted for converting the dialdehyde into the required cis-dimethyl groups, including Wolff–Kishner reduction, without success. Finally, 14 was converted into its corresponding dithioacetal 15 (excess EtSH, BF3·OEt2, −78 °C) with concomitant desilylation in 91% yield. Raney nickel reduction afforded in nine steps synthetic (±)-cis-trikentrin A, which was identical in all respects to the physical data reported for this racemic compound.1a
Scheme 3.
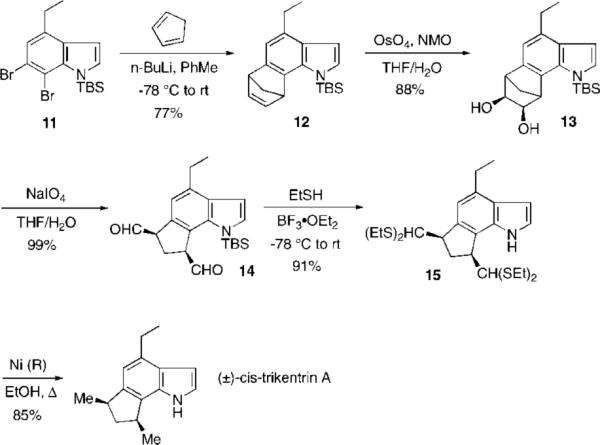
cis-Trikentrin A: 6,7-Indolyne Tactic
Encouraged by the success with the trikentrin synthesis, we turned our attention to the total synthesis of the cytotoxic herbindole A (Scheme 4). The approach entirely parallels that of of cis-trikentrin A. The intriguing issue presented by this synthesis was whether for electronic and steric reasons the required tetrasubstituted nitrobenzene 18 would be viable as a substrate in the Bartoli synthesis.
Scheme 4.
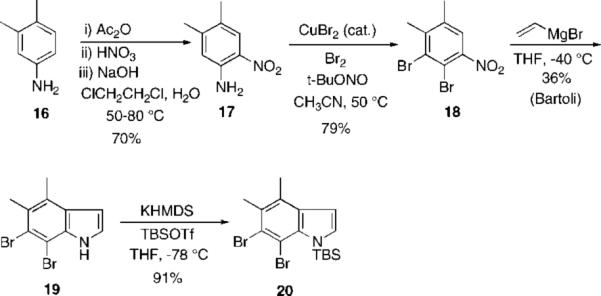
Herbindole A: Bartoli Indole Synthesis
Starting again with commercially available 3,4-dimethylaniline 16, a highly regioselective nitration procedure as described above gave the desired compound 17 in 70% yield, with the remaining material consisting mainly of the other easily separated nitroaniline isomer. Diazotization and bromination as before gave the o-dibromide 18 in 79% yield. Gratifyingly, the Bartoli protocol afforded the desired indole 19, albeit in a modest 36% yield. Although this example gave a lower yield than with trikentrin A (36% vs 52%), we ascribe this observation to the greater substitution of the nitrobenzene, and it is known that this process is highly sensitive to substitution patterns and substituent effects.5 The remaining steps have mostly comparable yields. Although attempts to improve the yield by modifying the reaction conditions proved ineffective, it is important to note that there is only one other report in the literature of polysubstituted nitrobenzenes participating in this reaction, with much lower yields.1g The presence of the bromine substitutents in our case appears to attenuate the apparently adverse electronic effects due to the remaining meta and para methyl substituents. N-Silylation as shown above gave the desired 6,7-indolyne precursor 20.
With the desired aryne precursor in hand, generation of the 6,7-indolyne followed by the Diels–Alder reaction gave the desired cycloadduct in an even higher 88% yield than observed with cis-trikentrin (Scheme 5). Compound 21 was carried through to the target as described above. Thus oxidative cleavage (67%, two steps), thioacetalization (81%), and Raney nickel reduction (96%) afforded racemic herbindole A in nine steps from the aniline 16. Herbindole A also exhibited the same physical and spectroscopic data (except for optical rotation) as that reported for the authentic samples.1e
Scheme 5.
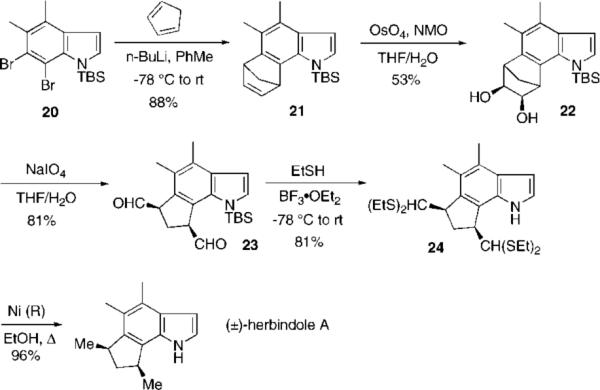
Herbindole A: 6,7-Indolyne Tactic
Finally, we observed in the case of herbindole B that increasing the electron density of the aromatic ring still further results in an even lower yield of the Bartoli indole product (Scheme 6).
Scheme 6.
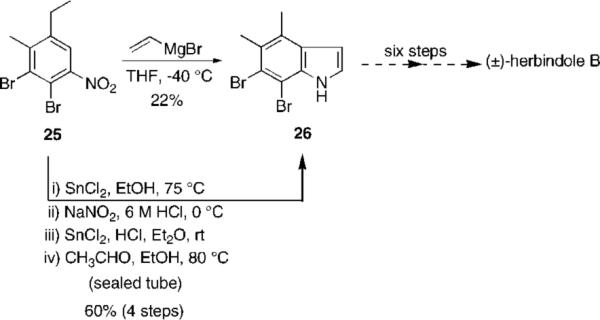
Herbindole B: Bartoli Indole Synthesis
Multiple electron-donating groups appear to exacerbate this effect. It has been observed by Blechert1g that the application of the Bartoli protocol to highly substituted alkyl nitrobenzenes gives very low yields of the desired indole system and thus appears to represent a limitation of the use of this methodology. However, by carrying 25 through an alternate Fischer indole regime, the yield of 26 is improved to 60%.
In summary, we have demonstrated the utility of the indole aryne cyloaddition reaction to readily access annulated indole natural products. This approach combined with the Bartoli indole synthesis provides for an especially efficient synthesis of the trikentrins and herbindoles. Further synthetic applications of our indolyne cycloadditon methodology to annulated indole natural products (e.g., teleocidins) are in progress and will be reported in due course.
Acknowledgment
We acknowledge support of this work by the National Institutes of Health, Grant R01 GM069711.
Footnotes
Supporting Information Available: 1H and 13C NMR data for all compounds reported and experimental details for their preparation. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Capon RJ, MacLeod JK, Scammells PJ. Tetrahedron. 1986;42:6545–6550. [Google Scholar]; b MacLeod JK, Monahan LC. Tetrahedron Lett. 1988;29:391–392. [Google Scholar]; c Yasukouchi T, Kanematsu K. Tetrahedron Lett. 1989;30:6559–6562. [Google Scholar]; d MacLeod JK, Monahan LC. Aust. J. Chem. 1990;43:329–337. [Google Scholar]; e Herb R, Carroll AR, Yoshida WY, Scheuer PJ, Paul VJ. Tetrahedron. 1990;46:3089. [Google Scholar]; f Boger DL, Zhang M. J. Am. Chem. Soc. 1991;113:4230–4234. [Google Scholar]; g Widenau P, Monse B, Blechert S. Tetrahedron. 1995;51:1167–1176. [Google Scholar]; h MacLeod JK, Ward A, Willis AC. Aust. J. Chem. 1998;51:177–187. [Google Scholar]; i Jackson SK, Banfield SC, Kerr MA. Org. Lett. 2005;7:1215–1218. doi: 10.1021/ol047498k. [DOI] [PubMed] [Google Scholar]; j Huntley RJ, Funk RL. Org. Lett. 2006;8:3403–3406. doi: 10.1021/ol061259a. [DOI] [PubMed] [Google Scholar]; k Jackson SK, Kerr MA. J. Org. Chem. 2007;72:1405–1411. doi: 10.1021/jo062350v. [DOI] [PubMed] [Google Scholar]; l Silva LF, Craveiro MV. Org. Lett. 2008;10:5417–5420. doi: 10.1021/ol8023105. [DOI] [PubMed] [Google Scholar]
- 2.a Muratake H, Natsume M. Tetrahedron Lett. 1989;30:5771–5772. [Google Scholar]; b Muratake H, Watanabe M, Goto K, Natsume M. Tetrahedron. 1990;46:4179–4192. [Google Scholar]; c Muratake H, Seino T, Natsume M. Tetrahedron Lett. 1993;34:4815–4818. [Google Scholar]; d Muratake H, Mikawa A, Seino T, Natsume M. Chem. Pharm. Bull. 1994;42:854–864. [Google Scholar]; e Lee M, Ikeda I, Kawabe T, Mori S, Kanematsu K. J. Org. Chem. 1996;61:3406–3416. [Google Scholar]
- 3.a Buszek KR, Luo D, Kondrashov M, Brown N, Vander-Velde D. Org. Lett. 2007;9:4135–4137. doi: 10.1021/ol701595n. [DOI] [PubMed] [Google Scholar]; b Brown N, Luo D, VanderVelde D, Yang S, Brassfield A, Buszek KR. Tetrahedron Lett. 2009;50:63–65. doi: 10.1016/j.tetlet.2008.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Nakatsuka S, Matsuda T, Goto T. Tetrahedron Lett. 1986;27:6245–6248. [Google Scholar]; b Nakatsuka S, Masuda T, Asano O, Terame T, Goto T. Tetrahedron Lett. 1986;27:4327–4330. [Google Scholar]; c Nakatsuka S, Matsuda T, Goto T. Tetrahedron Lett. 1987;38:3671–3674. [Google Scholar]
- 5.a Bartoli G, Palmieri G. Tetrahedron Lett. 1989;30:2129–2132. [Google Scholar]; b Bartoli G, Bosco M, Dalpozzo R, Palmieri G, Marcantoni E. J. Chem. Soc., Perkin Trans. 1. 1991:2757–2761. [Google Scholar]; c Bosco M, Dalpozzo R, Bartoli G, Palmieri G, Petrini M. J. Chem. Soc., Perkin Trans. 2. 1991:657–663. [Google Scholar]; d Dalpozzo R, Bartoli G. Curr. Org. Chem. 2005;9:163–178. [Google Scholar]
- 6.O'Neil BM, Ratto JE, Good KL, Tahmassebi DC, Helquist SA, Morales JC, Kool ET. J. Org. Chem. 2002;67:5869–5875. doi: 10.1021/jo025884e. [DOI] [PubMed] [Google Scholar]
- 7.Doyle MP, Van Lente MA, Mowat R, Fobare WF. J. Org. Chem. 1980;45:2570–2575. [Google Scholar]
- 8.Coe JW, Wirtz MC, Bashore CG, Candler J. Org. Lett. 2004;6:1589–1592. doi: 10.1021/ol049655l. [DOI] [PubMed] [Google Scholar]