

Abstract
Objective
The initiation of atherosclerosis is in part dependent on the hemodynamic shear stress environment promoting a pro-inflammatory phenotype of the endothelium. Previous studies demonstrated increased expression of ER stress protein and unfolded protein response (UPR) regulator, GRP78, within all vascular cells in atherosclerotic lesions and its regulation in the endothelium by several atherosclerotic stressors; however regulation of GRP78 by shear stress directly has not been established.
Method and Results
Using an in vitro model to simulate human arterial shear stress waveforms, atheroprone or atheroprotective flow was applied to human endothelial cells. GRP78 was found to be significantly upregulated (3-fold) in a sustained manner under atheroprone, but not atheroprotective flow up to 24-hours. This response was dependent on both sustained activation of p38, as well integrin α2β1. Increased GRP78 correlated with the activation of the ER stress sensing element (ERSE1) promoter by atheroprone flow as a marker of the UPR. Shear stress regulated GRP78 through increased protein stability when compared to other flow regulated proteins, such as connexin-43 and VCAM-1. Increased endothelial expression of GRP78 was also observed in atheroprone versus atheroprotective regions of C57BL6 mice.
Conclusions
This study supports a role of the hemodynamic environment in preferentially inducing GRP78 and the UPR in atheroprone regions, prior to lesion development, and suggests a potential atheroprotective (i.e., pro-survival), compensatory effect in response to ER stress within atherosclerotic lesions.
Keywords: endothelial, GRP78, shear stress, atherosclerosis, unfolded protein response
Introduction
Atherosclerosis is a focal inflammatory disease that develops preferentially in areas of disturbed flow, where variations in shear stress has been shown to alter the phenotypes of endothelial cells toward either an atheroprone or atheroprotective state in vitro and in vivo1, 2. Therefore, hemodynamic-induced shear stress provides a major mechanical signal, which causes the overlying endothelium to become at risk for the promotion of atherosclerosis.
Of many proteins of interest, the chaperone protein, glucose regulated protein 78 (GRP78), a common marker for endoplasmic reticulum (ER) stress, is preferentially expressed in advanced atherosclerotic lesions 3 and on the fibrous cap surface in ApoE-KO mice 4. Further, cell-surface associated GRP78 has been speculated to serve a protective role in atheroprone environments by inhibiting tissue factor through direct binding to the endothelium overlying the plaque5.
Hyperhomocysteinemia is associated with increased risk of cardiovascular disease possibly by limiting the antioxidant activity and causing ER stress, leading to the activation of GRP78 4. ER stress is further linked to oxidative stress through peroxynitrite-induced GRP78 expression3. ER stress is present at every stage of atherosclerosis, even preceding free cholesterol accumulation 6. This suggests cardiovascular risk factors induce greater GRP78 expression and further, there exist localized conditions that cause this stress even prior to lesion development (e.g., hemodynamic shear stress)6.
GRP78 is involved in a number of processes, but has been well studied as the “master regulator” of the unfolded protein response (UPR). GRP78 binds the three ER stress sensors PERK, IRE1, and ATF6 and keeps them in an inactive form 7. Upon accumulation of unfolded proteins, GRP78 releases the stress sensing elements, allowing ATF6 to translocate to the nucleus and bind to the endoplasmic reticulum stress element (ERSE) and increase the transcription of genes to alleviate the ER stress. The early response to ER stress is the attenuation of protein synthesis by the PERK pathway via ATF4. ATF4 along with XBP1 (downstream of IRE1) have recently been found to regulate many important inflammatory genes, including IL-8, which is regulated by atherogenic levels of shear stress 2, 8. Currently, GRP78 is known to be induced by many hallmark atherosclerotic stressors, such as excess cholesterol, oxidized phospholipids, oxidative stress, peroxynitrite, and homocysteine 9-12. GRP78-/- mice are embryonic lethal and show increased apoptosis, and high levels of unfolded proteins may lead to disassociation of GRP78 from UPR mediators causing JNK and NFκB activation or caspase-mediated apoptosis13-15. Currently little is known about the role of the UPR in ECs or in the context of flow or atherosclerosis.
Here we tested the hypothesis that hemodynamic shear stress in regions of atherosclerosis, independent of other risk factors, regulates GRP78 expression in vivo and in vitro. It is hypothesized that GRP78 upregulation in the endothelium may provide a protective compensatory effect in response to ER stress within early or developing atherosclerotic lesions.
Methods
Animals
Tissue sections from wild type mice (C57BL6, n=4; Jackson Laboratory) between 8-10 weeks of age and 8 or 20 week old ApoE-/- mice (Jackson Laboratory, n=2) were obtained for immunoflouorescence staining for GRP78.
In Vitro Hemodynamic Flow Model and Analysis
Passage two human umbilical vein endothelial cells were used to investigate the in vitro role of shear stress patterns on the induction of the ER stress via a cone-and-plate flow device16. Upon completion of the flow experiments, the cells were immediately collected for western blotting or real-time reverse transcriptase polymerase chain reaction.
ERSE Luciferase Assay
Passage 7-9 bovine aortic endothelial cells were transfected with GRP78-ERSE1-Luc, GRP78-M-ERSE1-Luc, CHOP-ERSE1-Luc, and CHOP-M-ERSE1-luc (provided by Dr. Glembotski, San Diego State University)17.
For complete Methods please see www.ahajournals.org.
Results
Endothelium GRP78 is Differentially Expressed in Atheroprone and Atheroprotective Regions In Vivo
GRP78 has been found to be highly expressed in macrophages, smooth muscle and endothelial cells of atherosclerotic lesions 3, 6. We found a similar intense GRP78 staining in a 20-week plaque of an ApoE-/- mouse compared to the surrounding endothelium and tissue, especially on the surface of the cap (Figure 1G). This finding prompted an investigation of differences that might exist in atheroprotective relative to atheroprone regions prior to the initiation of atherosclerosis. To assess this, aortic tissue from C57BL/6 mice was stained for GRP78 in the aortic arch, where lesions develop preferentially on the inner arch, while the outside is relatively protected 18. Figure 1A shows the entire section of the aorta where more intense GRP78 staining was observed in the inner relative to the outer arch. Both the endothelium and the underlying smooth muscle cells along the inner arch express more GRP78 relative to outer arch (Figure 1B-1D) and the thoracic aorta, which is relatively free from lesion development (Figures 1E-1F). Moreover, staining for GRP78 was found at intercostal branch points along the aorta of an 8-week, ApoE-/- mouse (Figure 1H), common areas of disturbed flow, whereas, the surrounding endothelium thought to be in a more atheroprotective portion of the descending aorta, shows little GRP78 expression. Since greater levels of GRP78 expression are observed in atheroprone areas of the vasculature, we hypothesized that increased expression of GRP78 might be due in part to the local differences in the hemodynamic shear stress environment.
Figure 1. GRP78 is differentially expressed in atheroprone areas and within atherosclerotic lesions of mice aorta.
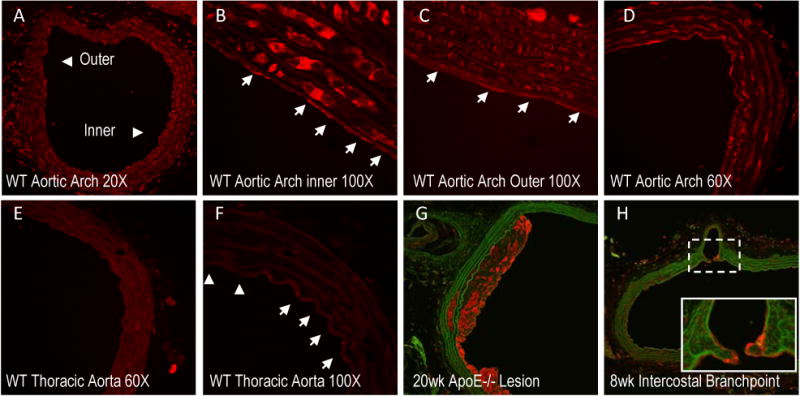
Histological sections of the aorta were stained for GRP78 (red) and matrix (green/autoflorescence) in C57BL/6 (A-F) or ApoE-/- (G-H) mice. Representative images of the aortic arch of C57BL/6 mice show the inner arch (atheroprone) and the outer arch (atheroprotective) (A-D). White arrows (100× images) indicate individual ECs. Staining was compared to protected regions of the thoracic aorta (E-F). GRP78 expressed in a 20-week lesion (G) and 8 week cross-section (H) along the descending aorta (intersecting at an intercostal branch) of an ApoE-/- mouse.
Endothelial GRP78 Regulated by Atheroprone Hemodynamics In Vitro
To test our hypothesis and corroborate the observations found in vivo, the role of hemodynamic shear stress on the regulation on GRP78 in ECs was investigated in vitro using atheroprone and atheroprotective flow patterns derived directly from human carotid circulation (Figure 2A) 19. To assess a more sustained phenotype, ECs were exposed to atheroprone or atheroprotective flow for 24-hours (Figure 2B). GRP78 was increased under atheroprone compared to atheroprotective flow and the time-matched static control. To assess the temporal regulation of GRP78 by arterial hemodynamics, ECs were exposed to both flow paradigms from 4 to 24-hours and subsequently assayed for GRP78 protein expression. Although GRP78 was elevated under both flow paradigms 4-hours after the onset of flow (Prone: p=0.03; Protective: p=0.17) the atheroprone flow resulted in a significant and sustained increase in GRP78 after 6-hours and up to 24-hours when compared to atheroprotective flow (Fig. 2C). In contrast, GRP78 expression under atheroprotective flow from 6 to 24-hours was no different than the time-matched static controls (p>0.3; Fig. 2C). The heightened expression levels of GRP78 following atheroprone flow at the onset and longer time points suggests shear stress is initiating and sustaining the activation of the UPR pathway, respectively.
Figure 2. Atheroprone shear stress differentially regulates GRP78 and activates the UPR pathway.
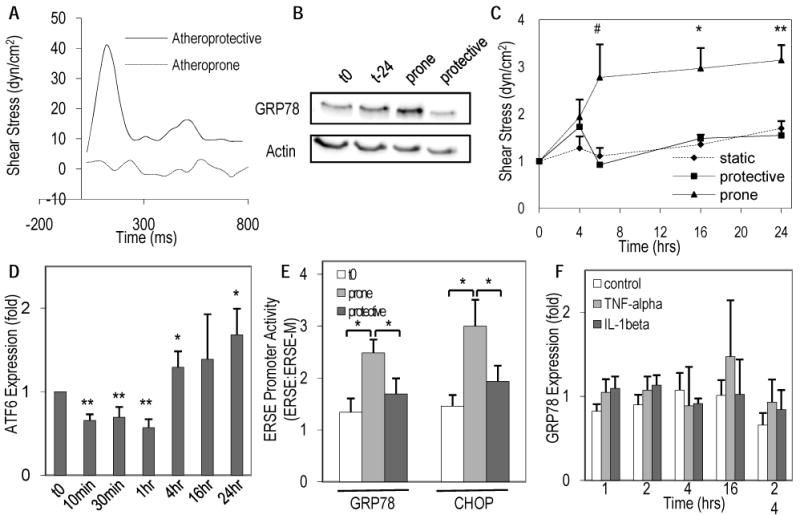
(A) Human hemodynamic shear stress profiles from an atheroprone and atheroprotective regions were applied to EC monolayers in a cyclical manner (waveform from one cardiac cycle shown). (B) Representative blots of GRP78 protein expression at flow onset (t0) and 24-hours static time-matched (Tm), atheroprone, or atheroprotective flow normalized to total actin. (C) Changes in protein expression of GRP78 as a function of exposure to atheroprone or protective flow from 4 to 24-hours or Tm control were analyzed using densitometry (# p=.058, * p<0.05, ** p≤0.002 when compared to protective flow or T0, n=3-6). (D) Changes in the p90-ATF6 following exposure to atheroprone flow from 10 minutes to 24-hours relative to time zero (* p<0.05, ** p<0.005; n=3-8). (E) Increase in ERSE1 promoter-luciferase activity derived from either the GRP78 or CHOP genes compared to initial (t0) static controls following 6-hours of atheroprone or atheroprotective flow (normalized to mutant ERSE1 under the same conditions, * p<0.05; n=3-6). (F) Relative changes in GRP78 protein expression was measured after exposure to cytokines IL-1β (5ng/ml) or TNF-α (10ng/ml) from 1 to 24-hours (n=3).
Atheroprone Flow Induces the UPR through ATF6
GRP78 is known to directly bind to p90 ATF6 (90kDa-form) regulating its activity and its transcription depends on the induction of ATF6 in a positive feedback manner20. p38 was found to phosphorylate ATF6 allowing it to translocate to the nucleus (as a 50kDa form) where it can upregulate genes controlled by the ER stress sensing element (ERSE), including chaperone proteins (GRP78, GRP94) and UPR proteins (ATF6, PERK, IRE1) 21, 22. Fig 2D shows the early time course of p90 ATF6 expression from 10-minutes to 1-hour under atheroprone flow. There is an immediate decrease in the expression of p90 ATF6 up to 1-hour after onset of flow, similar to the exposure to tunicamycin, a potent ER-stress inducer 21, suggesting activation of the UPR by flow. Longer exposure to atheroprone flow increased ATF6 expression at 4 and 24-hours.
To further assess the activation of the UPR under atheroprone flow the ERSE1 promoter derived from the GRP78 gene and CHOP gene or a mutated promoter from each gene (ERSE-M) driving a luciferase reporter were transfected into ECs prior to flow. The ERSE-M reporter does not allow ATF6 to bind17. Under atheroprone flow there was a significant increase in both the GRP78 and CHOP ERSE1 promoter activity normalized to the ERSE-M control compared to the initial conditions in static culture or atheroprotective shear stress (Fig 2E, p<0.05, one-tailed t-test). This induction of the ERSE promoter by atheroprone flow via ATF6 along with increased expression of ER stress proteins GRP78 and ATF6 indicates the activation of the UPR.
GRP78 is not Regulated by Pro-inflammatory Cytokines
Since higher levels of GRP78 were found in atherosclerosis 4, 6, we wanted to determine whether its upregulation could result from cytokine stimulation in the plaque (i.e., released by activated cells in the plaque) rather than solely being regulated by the hemodynamic environment. ECs were exposed to TNF-α or IL-1β from 1 to 24-hours and compared to untreated time-matched controls. Neither IL-1 nor TNF-α elicited an increase in GRP78 protein expression up to 24-hours of exposure (p>0.3; Fig. 2F); thus indicating a prominent role for hemodynamic stresses.
Shear stress does not affect cell-surface GRP78 function
GRP78 provides a dual functions either in the ER or on the cell surface where it has multiple binding partners, including tissue factor5. Bhattacharjee et al. found GRP78 to be antithrombotic by binding cell-surface tissue factor to reduce clotting times5. To assess whether the increase in GRP78 expression under atheroprone flow was contributing to an antithrombotic function, a single stage-clotting assay was performed on ECs pretreated with 24-hours of atheroprone flow. Samples were mixed ±anti-GRP78 (Santa Cruz, N-terminus), and a single-stage clotting assay was performed and compared to untreated control (for complete methods please see www.ahajournals.org). No significant difference was found between the two samples (Prone: 86.19±14.00s, Prone+anti-GRP78: 78.18±17.68s, n=3, p=0.5). Therefore, atheroprone-induced GRP78 expression appears to participate in sensing unfolded proteins in the ER leading to the release and activation of the UPR-mediators instead of cell-surface reactivity.
Shear Stress Regulates EC mRNA and Protein Half-life of GRP78
To determine if the increase in protein expression was the result of shear stress differentially regulating GRP78 gene expression, changes in mRNA were assessed following exposure to atheroprone or atheroprotective flow for 1 to 16-hours (Figure 3A). Both flow paradigms stimulated similar transient increases in GRP78 mRNA up to 4-hours of flow that returned to basal levels by 16-hours. This regulation was not consistent with the atheroprone flow-induced protein expression.
Figure 3. Shear stress patterns differentially regulate protein stability.
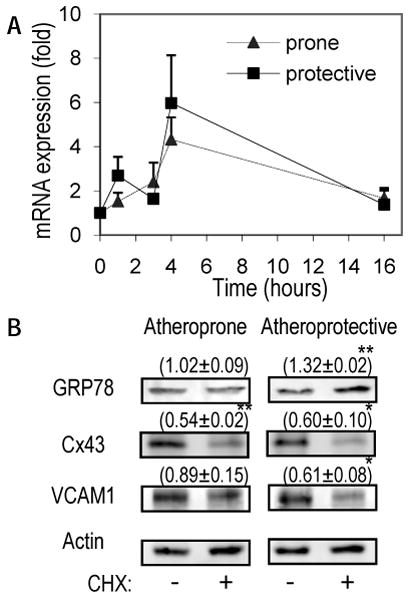
(A) GRP78 mRNA levels were measured by real time RT-PCR following exposure to atheroprone or atheroprotective flow from 1 to 16-hours (n=3-6). (B) ECs were preconditioned with 4-hours of atheroprone or atheroprotective flow followed by exposure to DMSO control or cycloheximide (CHX; 10ug/ml) for 2 additional hours under flow. Representative western blots for GRP78, Cx43, and VCAM1 after the combined 6-hours of flow and normalized to actin from three independent experiments; mean±se. (*p<0.05, **p<0.001)
ER stress can lead to increased mRNA and protein stability to UPR effectors, including GRP78 23. Since the differential expression of GRP78 was not found to be regulated solely by mRNA transcription, the stability of GRP78 was assessed by treating the cells with cyclohexamide (CHX) to block synthesis of new protein. ECs were pretreated with 4-hours of atheroprone or atheroprotective flow, after which cells were either exposed to CHX or vehicle (DMSO) control for 2-hours without interrupting flow. After incubation with CHX or DMSO, the cells were collected and analyzed using western blots to assess the stability of GRP78 protein expression in addition to known flow regulated proteins, such as VCAM-1 and connexin-43 (Cx43) as seen in Figure 3B2. Total Cx43 protein was found to degrade when treated with CHX under both flow conditions, however degradation of VCAM-1 was only observed under atheroprotective flow. Following atheroprone flow, GRP78 was found to have a higher protein half-life (i.e., stability) compared to flow-regulated proteins that degrade (Cx43, VCAM), as its expression did not change after 2-hours of CHX treatment. Interestingly, CHX prevented the decrease in GRP78 following 6-hours of atheroprotective flow (see Fig 2C), which is observed as increase in GRP78 expression (Fig 3B) relative to the vehicle control. This suggests that the restoration of GRP78 to basal levels under atheroprotective flow is dependent on the new synthesis of an unidentified protein for UPR control.
Both p38 and α2β1 were Necessary for GRP78 Regulation Under Atheroprone Flow
p38 is involved in major pathways involved with atherosclerosis, and a previous study had identified p38 as a proximal regulator of GRP78 in human HEp3 cells 24. We therefore, investigated p38 activation as a possible mechanism by which GRP78 was regulated by atheroprone flow. Figure 4A and 4B shows that flow immediately increased p38 activity significantly after 10-minutes under both flow paradigms, compared to the static condition, in a transient manner up to 60-minutes (p<0.05; Fig 4A), though the relative activity was similar between flow patterns after 1-hour (p=0.17). The activity of p38 remained similar between both flow types after the onset of flow for up to 24-hours (Fig 4B), although significantly increased compared to static time-zero (p<0.03).
Figure 4. Regulation of GRP78 under atheroprone flow is dependent on p38.

(A) Early and (B) late transient activation of p38 phosphorylation following exposure to atheroprone or atheroprotective flow. Data reported as fold change relative to initial p38 activity and normalized to total p38. (C) Pretreatment with specific p38 inhibitor, SB202190, blocked the induction of GRP78 following 24-hours of atheroprone flow. (D) GRP78 protein expression in ECs following exposure to ER stressors DTT, thapsigarin (Tg), or tunicamycin (Tm) for 6 or 24-hours in the presence or absence of SB202190. (n=5-6, ** p<0.001, *p<0.02).
Blocking p38 activity with 1μM SB202190, a potent p38 inhibitor, blocked the atheroprone-induced increase in GRP78 (p<0.05) after 24-hours (Figure 4C). Controls with the same p38 inhibitor were run in parallel for 24-hours and showed no difference from time-matched static controls without the inhibitor.
To determine whether p38 was involved in shear stress responsive pathways or part of UPR signaling, ECs were treated with three established ER stress inducers in combination with SB202190. The three drugs, 4mM DTT, 1.5μg/ml thapsigargin (Tg) and 10μg/ml tunicamycin (Tm) each increased ER stress as measured by the increase in GRP78 protein, but this was not blocked by the addition of SB202190. Therefore, the atheroprone flow increased in GRP78 in endothelial cells is dependent on p38 activity, whereas drug-induced ER stress is independent of this pathway.
Integrin ligation is an important component of EC responsiveness to shear stress and may be a means of adaptive mechanotransduction 25. Recent work has linked the integrin α2β1 to p38 activation 25, 26. Here we tested whether α2β1 ligation is upstream of p38 and GRP78 regulation in response to atheroprone flow. Pretreatment for 30 minutes and throughout the flow experiment with the α2β1 blocking antibody R2-8C8 (1:800; gift from Dr. Mark Ginsberg, UCSD) inhibited atheroprone flow induced increase of GRP78 at 24-hours compared to control, non-blocking α2β1 antibody 12F1 (3μg/ml; gift from Dr. Virgil Woods, UCSD) and the time-matched static control (p<0.05; Figure 5A and 5B). Blocking α2β1 was also found to significantly lower (p<0.02) p38 activity by more than 20% after 24-hours of atheroprone flow (Fig 5C). Again, to see whether α2β1 is part of the shear stress-sensing pathway or necessary for ER stress, R2-8C8 and 12F1 were used in conjunction with 3 ER stress inducers (DTT, Tg, Tm) for various amount of time (Fig 5D). Blocking α2β1 did not reduce ER stress at any of the tested points. Therefore, ligation of α2β1 and activation of p38 are important shear stress signaling mechanisms in the long-term, sustained upregulation of GRP78.
Figure 5. Upregulation of GRP78 and p38 by atheroprone flow is dependent on α2β1.
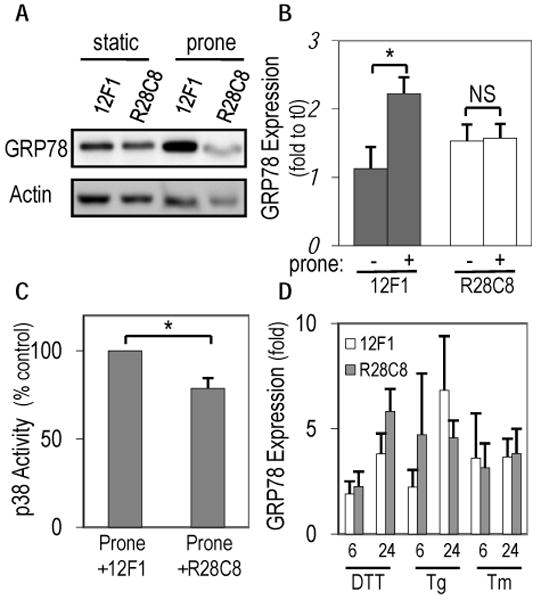
R2-8C8, an α2β1 blocking antibody, blocked atheroprone flow induced increase in GRP78 after 24-hours when compared to the non-blocking control, 12F1 as seen in representative western blots (A) or quantified using densitometry (B). (C) Reduction in p38 phosphorylation following 24-hours of atheroprone flow in the presence of blocking antibody R28C8, but not the control 12F1 (* p<0.02; NS, not significant n=6-9). (D) After treatment with R2-8C8 or 12F1, GRP78 expression was measured when challenged with ER stressors DTT, thapsigarin (Tg), or tunicamycin (Tm) for 6 or 24-hours.
Discussion
We present evidence for the differential control of hemodynamics in influencing EC expression of GRP78, an ER chaperone responsible for alleviating cell toxicity and a key regulator of the UPR. To our knowledge, this is the first instance of GRP78 protein expression to be mechanically responsive, and further is differentially regulated in atheroprone versus atheroprotective regions in vivo and regulated by shear stress in a waveform specific manner in vitro. The mechanisms by which atheroprone flow induces increases in GRP78 were dependent on p38 activity and α2β1. The differential regulation of GRP78 protein and the ERSE activation by the onset of atheroprone flow signify the induction of the UPR and its sustained activity. Inflammatory cytokines found in atheroprone environments had no effect on GRP78 expression in ECs and its regulation by known chemical agonists were independent of the p38 and α2β1 pathways. This result further accentuates the prominence of shear stress patterns in the atheroprone environment in regulating the basal endothelial phenotype.
GRP78 is shown here and elsewhere in early and late stages of atherosclerotic lesions in ApoE-/- mice 6. We now show that GRP78 is expressed at greater levels at intercostal branch points and the inner curvature of the aortic arch, arterial regions susceptible to the development atherosclerosis and correlate with disturbed flow patterns. Interestingly, the smooth muscle cells (SMC) in these areas also showed greater GRP78 staining. Although the immediate consequence of this observation is not known, it is interesting to speculate that hemodynamic regulation of ECs could mediate SMC behavior in atheroprone regions through cell-cell communication27. Atheroprone regions in vivo and hemodynamic relevant shear stresses in vitro have been shown to prime the endothelium and smooth muscle cells towards an activated, pro-inflammatory state2, 28. Atheroprone flow in vitro did not affect the clotting time, indicating that the induction of GRP78 by atheroprone flow likely combats ER stress as part of the UPR. Therefore, it is plausible that hemodynamic flow might be the earliest ER stressor in atheroprone environments.
Many survival pathways resulting from atheroprone stimuli converge on MAPKs, including the JNK and p38 cascades. p38 is activated by numerous atherosclerosis stressors such as oscillatory flow, reactive oxygen species, cholesterol, and TNF-α 29-32. Despite common convergence on MAPK signaling, cells are capable of controlling specific downstream effects by regulating subcellular localization, protein scaffolds, and extracellular matrix interactions 25, 33. Therefore, similar MAPK signaling activity diverge in downstream function as seen in the temporal regulation of the UPR, where inhibitors of p38 block GRP78 induction at 24-hours. This suggests differential regulators downstream of p38 and/or a protein serving as a CHX-sensitive negative regulator of GRP78, as we observed under atheroprotective flow.
Signals from biomechanical forces and ECM interactions converge and transduce via integrin ligation. α2β1 was found to regulate p38 in agreement with previous studies using purified matrices where p38 localized to focal adhesions, thus possibly providing specificity through subcellular localization 25. α2β1 can bind multiple ECM components which may be present in the cell derived matrix in cultured ECs or in vivo, which differs from the purified matrices used by Orr et al., possibly accounting for differences in activity levels 25. Integrins generally provide antiapoptotic/pro-survival signals from the ECM, where lack of integrin signaling, specifically the β1-tail, caused ECs to undergo apoptosis 34. These effects are often mediated through chaperone, heat-shock family proteins to which GRP78 belongs35. Collectively, this supports the protective role of α2β1-ECM in utilizing the UPR and GRP78 to counteract apoptosis.
The atheroprone flow-induced increase in GRP78 was linked in part to differential protein stability between the two flow paradigms. A previous study demonstrated that the adaptive response to long-term exposure to tunicamycin or thapsigargin increased protein and mRNA half-life of GRP78 and other UPR adaptive proteins, where apoptotic proteins like CHOP were less stable 23. In ECs, tunicamycin-induced ER stress was found to have no effect on mRNA stability36, and in our study GRP78 mRNA levels were transient and showed no difference between the two flow conditions. Therefore, we hypothesized the differences may be due to GRP78 protein stability. Studies with CHX treatment showed that GRP78 had greater stability under flow when compared to other flow-regulated proteins, Cx43 and VCAM1, also known to be increased by atheroprone flow2. CHX had no effect on GRP78 expression, implying that sustained expression at time points greater than 6-hours does not rely on transcription or the synthesis of new protein for its regulation. In contrast, CHX resulted in a ∼30% increase in GRP78 expression following atheroprotective flow. One possibility is that atheroprotective flow upregulates an unidentified CHX-sensitive protein that reduces levels of GRP78 through active degradation. The proposed model for hemodynamic regulation of GRP78 is summarized in Figure I (supplemental text). Mechanistically, these results suggest that atheroprone flow promotes protein stability (or prevents protein degradation) for important survival pathways including GRP78 to alleviate ER stress and apoptotic signaling.
GRP78 is considered the “master regulator” of the UPR due to its control of the ER stress sensing elements, ATF6, PERK, and IRE1. ATF6 is an important transcription factor that binds to the ERSE1 promoter to increase transcription of important UPR proteins including GRP78 and ATF6 itself. The initial response of ATF6 to the onset of atheroprone flow was similar to thapsigargin-induced ER stress—a significant decrease in expression 21. The authors concluded this to be an anti-apoptotic/survival mechanism that reduces the expression of CHOP while the cell attempts to alleviate stress. Prolonged exposure of thapsigargin induced an increase in ATF6 expression akin to results reported here under atheroprone flow 21. Further, this increase in ATF6 protein corresponds with the activation of the ERSE1 promoter via ATF6 signifying the onset of the UPR under atheroprone flow.
Atheroprone flow induces ER stress that may further aggravate the progression of atherosclerosis in these environments, while atheroprotective forces resolve the need for the UPR after 4-hours of flow. GRP78 provides numerous protective effects to minimize stress though UPR signaling, as well as, anti-apoptotic and anti-thrombotic signaling. Therefore, there is a link between hemodynamics, inflammation, apoptosis and the UPR. There are reports of the ability of atheroprotective flow to reduce inflammatory effects of cytokines37; however, this study suggests there may also be compensatory atheroprotective effects when challenged with atheroprone flow.
Acknowledgments
This work was supported by The Whitaker Foundation Biomedical Research grant RG-02-0853 and NIH Biomedical Research Partnership Grant R01-HL080956 to B.R.B. We thank Dr. Gail Macik, Gary Manuel and the University of Virginia coagulation laboratory for their assistance in using the START4 coagulation analyzer.
Footnotes
Disclosures: None
References
- 1.Passerini AG, Polacek DC, Shi C, Francesco NM, Manduchi E, Grant GR, Pritchard WF, Powell S, Chang GY, Stoeckert CJ, Jr, Davies PF. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci U S A. 2004;101:2482–2487. doi: 10.1073/pnas.0305938101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, Garcia-Cardena G, Gimbrone MA., Jr Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc Natl Acad Sci U S A. 2004;101:14871–14876. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu C, Bhattacharjee G, Boisvert W, Dilley R, Edgington T. In vivo interrogation of the molecular display of atherosclerotic lesion surfaces. Am J Pathol. 2003;163:1859–1871. doi: 10.1016/S0002-9440(10)63545-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou J, Werstuck GH, Lhotak S, de Koning AB, Sood SK, Hossain GS, Moller J, Ritskes-Hoitinga M, Falk E, Dayal S, Lentz SR, Austin RC. Association of multiple cellular stress pathways with accelerated atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient mice. Circulation. 2004;110:207–213. doi: 10.1161/01.CIR.0000134487.51510.97. [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharjee G, Ahamed J, Pedersen B, El-Sheikh A, Mackman N, Ruf W, Liu C, Edgington TS. Regulation of tissue factor--mediated initiation of the coagulation cascade by cell surface grp78. Arterioscler Thromb Vasc Biol. 2005;25:1737–1743. doi: 10.1161/01.ATV.0000173419.31242.56. [DOI] [PubMed] [Google Scholar]
- 6.Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 7.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 8.Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Baruch-Oren T, Berliner JA, Kirchgessner TG, Lusis AJ. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 9.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D, Tabas I. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 10.Kim R, Emi M, Tanabe K, Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11:5–13. doi: 10.1007/s10495-005-3088-0. [DOI] [PubMed] [Google Scholar]
- 11.Dickhout JG, Hossain GS, Pozza LM, Zhou J, Lhotak S, Austin RC. Peroxynitrite causes endoplasmic reticulum stress and apoptosis in human vascular endothelium: implications in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:2623–2629. doi: 10.1161/01.ATV.0000189159.96900.d9. [DOI] [PubMed] [Google Scholar]
- 12.Outinen PA, Sood SK, Pfeifer SI, Pamidi S, Podor TJ, Li J, Weitz JI, Austin RC. Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood. 1999;94:959–967. [PubMed] [Google Scholar]
- 13.Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–5697. doi: 10.1128/MCB.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Q, Kim YS, Lin Y, Lewis J, Neckers L, Liu ZG. Tumour necrosis factor receptor 1 mediates endoplasmic reticulum stress-induced activation of the MAP kinase JNK. EMBO Rep. 2006;7:622–627. doi: 10.1038/sj.embor.7400687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mauro C, Crescenzi E, De Mattia R, Pacifico F, Mellone S, Salzano S, de Luca C, D'Adamio L, Palumbo G, Formisano S, Vito P, Leonardi A. Central role of the scaffold protein tumor necrosis factor receptor-associated factor 2 in regulating endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2006;281:2631–2638. doi: 10.1074/jbc.M502181200. [DOI] [PubMed] [Google Scholar]
- 16.Blackman BR, Garcia-Cardena G, Gimbrone MA., Jr A new in vitro model to evaluate differential responses of endothelial cells to simulated arterial shear stress waveforms. J Biomech Eng. 2002;124:397–407. doi: 10.1115/1.1486468. [DOI] [PubMed] [Google Scholar]
- 17.Thuerauf DJ, Hoover H, Meller J, Hernandez J, Su L, Andrews C, Dillmann WH, McDonough PM, Glembotski CC. Sarco/endoplasmic reticulum calcium ATPase-2 expression is regulated by ATF6 during the endoplasmic reticulum stress response: intracellular signaling of calcium stress in a cardiac myocyte model system. J Biol Chem. 2001;276:48309–48317. doi: 10.1074/jbc.M107146200. [DOI] [PubMed] [Google Scholar]
- 18.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 19.Gelfand BD, Epstein FH, Blackman BR. Spatial and spectral heterogeneity of time-varying shear stress profiles in the carotid bifurcation by phase-contrast MRI. J Magn Reson Imaging. 2006;24:1386–1392. doi: 10.1002/jmri.20765. [DOI] [PubMed] [Google Scholar]
- 20.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 21.Hong M, Li M, Mao C, Lee AS. Endoplasmic reticulum stress triggers an acute proteasome-dependent degradation of ATF6. J Cell Biochem. 2004;92:723–732. doi: 10.1002/jcb.20118. [DOI] [PubMed] [Google Scholar]
- 22.Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol. 2004;16:653–662. doi: 10.1016/j.ceb.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66:1702–1711. doi: 10.1158/0008-5472.CAN-05-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol. 2005;169:191–202. doi: 10.1083/jcb.200410073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivaska J, Reunanen H, Westermarck J, Koivisto L, Kahari VM, Heino J. Integrin alpha2beta1 mediates isoform-specific activation of p38 and upregulation of collagen gene transcription by a mechanism involving the alpha2 cytoplasmic tail. J Cell Biol. 1999;147:401–416. doi: 10.1083/jcb.147.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hastings NE, Simmers MB, McDonald OG, Wamhoff BR, Blackman BR. Atherosclerosis-prone hemodynamics differentially regulates endothelial and smooth muscle cell phenotypes and promotes pro-inflammatory priming. Am J Physiol Cell Physiol. 2007;293:C1824–1833. doi: 10.1152/ajpcell.00385.2007. [DOI] [PubMed] [Google Scholar]
- 28.Walpola PL, Gotlieb AI, Cybulsky MI, Langille BL. Expression of ICAM-1 and VCAM-1 and monocyte adherence in arteries exposed to altered shear stress. Arterioscler Thromb Vasc Biol. 1995;15:2–10. doi: 10.1161/01.atv.15.1.2. [DOI] [PubMed] [Google Scholar]
- 29.Ono H, Ichiki T, Ohtsubo H, Fukuyama K, Imayama I, Iino N, Masuda S, Hashiguchi Y, Takeshita A, Sunagawa K. CAMP-response element-binding protein mediates tumor necrosis factor-alpha-induced vascular cell adhesion molecule-1 expression in endothelial cells. Hypertens Res. 2006;29:39–47. doi: 10.1291/hypres.29.39. [DOI] [PubMed] [Google Scholar]
- 30.Chen XL, Grey JY, Thomas S, Qiu FH, Medford RM, Wasserman MA, Kunsch C. Sphingosine kinase-1 mediates TNF-alpha-induced MCP-1 gene expression in endothelial cells: upregulation by oscillatory flow. Am J Physiol Heart Circ Physiol. 2004;287:H1452–1458. doi: 10.1152/ajpheart.01101.2003. [DOI] [PubMed] [Google Scholar]
- 31.Dobreva I, Zschornig O, Waeber G, James RW, Widmann C. Cholesterol is the major component of native lipoproteins activating the p38 mitogen-activated protein kinases. Biol Chem. 2005;386:909–918. doi: 10.1515/BC.2005.106. [DOI] [PubMed] [Google Scholar]
- 32.Huot J, Houle F, Marceau F, Landry J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ Res. 1997;80:383–392. doi: 10.1161/01.res.80.3.383. [DOI] [PubMed] [Google Scholar]
- 33.Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 34.Meredith JE, Jr, Fazeli B, Schwartz MA. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polanowska-Grabowska R, Simon CG, Jr, Falchetto R, Shabanowitz J, Hunt DF, Gear AR. Platelet adhesion to collagen under flow causes dissociation of a phosphoprotein complex of heat-shock proteins and protein phosphatase 1. Blood. 1997;90:1516–1526. [PubMed] [Google Scholar]
- 36.Berger BJ, Muller TS, Buschmann IR, Peters K, Kirsch M, Christ B, Prols F. High levels of the molecular chaperone Mdg1/ERdj4 reflect the activation state of endothelial cells. Exp Cell Res. 2003;290:82–92. doi: 10.1016/s0014-4827(03)00316-1. [DOI] [PubMed] [Google Scholar]
- 37.Garin G, Abe J, Mohan A, Lu W, Yan C, Newby AC, Rhaman A, Berk BC. Flow antagonizes TNF-alpha signaling in endothelial cells by inhibiting caspase-dependent PKCzeta processing. Circ Res. 2007;101:97–105. doi: 10.1161/CIRCRESAHA.107.148270. [DOI] [PubMed] [Google Scholar]