

Abstract
Reported herein are thermochemical studies of hydrogen atom transfer (HAT) reactions involving transition metal H-atom donors MIILH and oxyl radicals. [FeII(H2bip)3]2+, [FeII(H2bim)3]2+, [CoII(H2bim)3]2+ and RuII(acac)2(py-imH) [H2bip = 2,2’-bi-1,4,5,6-tetrahydropyrimidine, H2bim = 2,2’-bi-imidazoline, acac = 2,4-pentandionato, py-imH = 2-(2’-pyridyl)-imidazole)] each react with TEMPO (2,2,6,6-tetramethyl-1-piperidinoxyl) or tBu3PhO• (2,4,6-tri-tert-butylphenoxyl) to give the deprotonated, oxidized metal complex MIIIL, and TEMPOH or tBu3PhOH. Solution equilibrium measurements for the reaction of [CoII(H2bim)3]2+ with TEMPO show a large, negative ground-state entropy for hydrogen atom transfer, −41 ± 2 cal mol−1 K−1. This is even more negative than the ΔSoHAT = −30 ± 2 cal mol−1 K−1 for the two iron complexes and the ΔSoHAT for RuII(acac)2(py-imH) + TEMPO, 4.9 ± 1.1 cal mol−1 K−1, as reported earlier. Calorimetric measurements quantitatively confirm the enthalpy of reaction for [FeII(H2bip)3]2+ + TEMPO, thus also confirming ΔSoHAT. Calorimetry on TEMPOH + tBu3PhO• gives ΔHoHAT = −11.2 ± 0.5 kcal mol−1 which matches the enthalpy predicted from the difference in literature solution BDEs. A brief evaluation of the literature thermochemistry of TEMPOH and tBu3PhOH supports the common assumption that ΔSoHAT ≈ 0 for HAT reactions of organic and small gas-phase molecules. However, this assumption does not hold for transition metal based HAT reactions. The trend in magnitude of |ΔSoHAT| for reactions with TEMPO, RuII(acac)2(py-imH) << [FeII(H2bip)3]2+ = [FeII(H2bim)3]2+ < [CoII(H2bim)3]2+, is surprisingly well predicted by the trends for electron transfer half-reaction entropies, ΔSoET, in aprotic solvents. This is because both ΔSoET and ΔSoHAT have substantial contributions from vibrational entropy, which varies significantly with the metal center involved. The close connection between ΔSoHAT and ΔSoET provides an important link between these two fields and provides a starting point from which to predict which HAT systems will have important ground-state entropy effects.
Introduction
The transfer of a hydrogen atom, reaction 1, is one of the most fundamental chemical transformations. It is a cornerstone of organic free-radical chemistry, from combustion to the in
| (1) |
vitro and in vivo action of antioxidants.1 In recent years, it has become clear that this reaction is also involved in a variety of metal-mediated oxidations, including coordination complexes, metalloenzyme active sites, and metal-oxide surfaces.2-5 For example, both plants and animals employ lipoxygenases to catalyze the selective hydroperoxidation of 1,4 diene units in fatty acids by using hydrogen transfer to an iron(III) hydroxide species.6 Reaction 1 has also been implicated in catalysis by other metalloenzymes such as cytochrome P450,7 methane monooxygenases8 and class I ribonucleotide reductases.3a,9 Cobaloximes, cobalt-porphyrins, and chromium cyclopentadienyl compounds effect chain transfer in living radical polymerizations using reaction 1.10 Developing a fundamental understanding of hydrogen transfer is thus broadly important.
Reaction 1, which we will call hydrogen atom transfer (HAT),11 is part of a broad class of processes involving proton and electron transfer, often called proton-coupled electron transfer (PCET).12-14 Theoretical treatments of PCET, like their antecedent theories of electron transfer (ET)15 and proton transfer (PT),16 use free energies (ΔG) as measures of reaction driving force.5 In contrast, analyses of HAT reactions have typically used the enthalpic driving force, as in the Bell-Evans-Polanyi equation (BEP) that relates the activation barrier to the ΔH.1a,17 ΔHo for an HAT reaction is the difference in bond dissociation enthalpies (BDEs) between the reactant A–H and the product B–H. In our view, the BEP correlation is a primary historical reason why chemists have focused on BDEs, rather than bond dissociation free energies (BDFEs).
The focus on enthalpies to understand HAT reactions is surprising since reactivity typically is correlated with free energies, as in linear free energy relationships (LFERs).18 These treatments are equivalent when the entropies of reaction are close to zero (when ΔSo = 0, ΔGo = ΔHo), as has been assumed in most treatments of hydrogen atom transfer (with a few exceptions19). The assumption that ΔSo ≈ 0 is also part of the foundation for the increasingly common determination of BDEs from solution pKa and E1/2 values, as popularized by Bordwell and co-workers.20-25
The assumption that ΔSo is ≈ 0 appears to hold for HAT reactions of small molecules in the gas phase26 and of larger organics in solution,27 but HAT reactions of two iron complexes have recently been shown to have very large |ΔSoHAT|.28 For instance, H-atom transfer from [FeII(H2bip)3]2+ to TEMPO (H2bip = 2,2’-bi-1,4,5,6-tetrahydropyrimidine) has ΔSo4 = −30 ± 2 cal mol−1 K−1.28 In this case, TΔSo4 = −8.9 kcal mol−1 at 298 K, a change in Keq of 4 × 106. This large |ΔSoHAT| originates primarily from a change in vibrational entropy upon redox change at the iron center,28 which suggests that HAT reactions of other metal systems may also have large values of |ΔSo|.
Herein, we report calorimetric and equilibrium measurements of ground state enthalpies and entropies for a series of HAT reactions.29 These reactions involve iron, cobalt, and ruthenium complexes with unsaturated nitrogen ligands, of the general type shown in eq 2. These thermodynamic measurements are used to elucidate trends in the magnitude of ground-state entropies for hydrogen atom transfer reactions, ΔSoHAT.
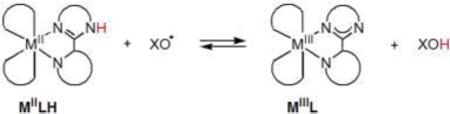 |
(2) |
Results
I. Equilibrium studies
A. CoII(H2bim) + TEMPO
[CoII(H2bim)3]2+ [CoII(H2bim); H2bim = 2,2’-bi-2-imidazoline; 10 mM] reacts with the stable nitroxyl radical TEMPO (3−15 equiv) in CD3CN to give an equilibrium mixture with [CoIII(Hbim)(H2bim)2]2+ [CoIII(Hbim)] and TEMPOH (eq 3; N-N = H2bim). Equilibrium is reached within approximately 48 hours at 298 K under these conditions. In the reverse direction,
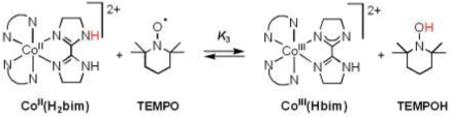 |
(3) |
CoIII(Hbim) plus excess TEMPOH gives complete formation of CoII(H2bim) and TEMPO over the course of 4 hours. This reaction has been very briefly described in a previous report.30 The equilibrium constant K3 has been determined by integrating 1H NMR spectra of reaction mixtures. All four species have easily observable 1H NMR spectra over the temperature range studied, even though CoII(H2bim) and TEMPO are both paramagnetic. The average for 3 experiments gives K3 = (5.9 ± 0.8) × 10−3 at 298 K, ΔGo3 = 3.0 ± 0.4 kcal mol−1 (Table 1). This value for ΔGo3 is within the error of the previous estimate (+0.3 ± 3 kcal mol−1) derived from the relevant pKa and Eo values.30 The large error in the previous estimate is due to the poor electrochemical response of CoIII(H2bim).31
Table 1.
Equilibrium constants and ground state thermodynamics for hydrogen atom transfer reactions in MeCN.
| AH + B | Methoda | Keq (298 K) | ΔGoHAT (kcal mol−1) | ΔHoHAT (kcal mol−1) | ΔSoHAT (cal mol−1 K−1) |
|---|---|---|---|---|---|
| FeII(H2bip) + TEMPO | VHb | 1.7 ± 0.3 | −0.3 ± 0.2 | −9.4 ± 0.6 | −30 ± 2 |
| Cal | -- | -- | −8.9 ± 0.6 | -- | |
| BDFE | ∼2 | −0.5 ± 1 | -- | -- | |
| FeII(H2bim) + TEMPO | VHb | (2.0 ± 0.3) × 10−4 | 5.0 ± 0.2 | −4.1 ± 0.3 | −30 ± 2 |
| BDFE | ∼0.9 × 10−4 | 5.5 ± 1.0 | -- | -- | |
| RuII(py-imH) + TEMPO | VHc | (1.8 ± 0.2) × 103 | −4.4 ± 0.1 | −3.0 ± 0.3 | 4.9 ± 1.1 |
| BDFE | (2.0 ± 1.5) × 103 | −4.5 ± 0.4 | -- | -- | |
| CoII(H2bim) + TEMPO | VH | (5.9 ± 0.8) × 10−3 | 3.0 ± 0.4 | −9.3 ± 0.4 | −41 ± 2 |
| BDFE | ∼0.6 | 0.3 ± 3d | -- | -- | |
| RuII(py-imH) + tBu3PhO• | BDE | -- | -- | −15 ± 1 | -- |
| RuII(hfac)2(py-imH) + tBu3PhO• | VH | 0.062 ± 0.013 | 1.6 ± 0.1 | -- | -- |
| CoII(H2bim) + tBu3PhO• | BDE | -- | -- | -24 ± 4 | -- |
| TEMPOH + tBu3PhO• | Cal | -- | -- | −11.2 ± 0.5 | -- |
| BD(F)E | -- | −10 ± 1 | −11.5 ± 1.4 | −2 ± 3 |
Method: VH (van't Hoff) = Temperature dependence of Keq from van't Hoff plots; Cal = calorimetric; BDFE/BDE: ΔGoHAT = BDFE[AH] - BDFE[BH] and/or ΔHoHAT = BDE[AH] - BDE[BH], using values from Table 2.
Data from reference 28.
Data from reference 32.
Estimated from BDFEs derived from pKa and Eo values, see Table 2.
K3 was measured from 274 − 313 K and found to vary by an order of magnitude over this 40 °C range (Figure 1A). Van't Hoff analysis yields ΔHo3 = −9.3 ± 0.4 kcal mol−1 and ΔSo3 = −41 ± 2 cal mol−1 K−1 (Table 1). For each sample, after measurements were complete at the high and low temperatures, the NMR tubes were allowed to re-equilibrate at room temperature (294 K). In each case, the ratio of species re-adjusted to values consistent with the predicted K3 at 294 K (Figure 1A), indicating that this is a true equilibrium. Over a week at these concentrations, there is no observable decrease in the cobalt mass balance relative to the NMR integration standard, though there is slight decomposition (ca. 5%) of the excess TEMPOH.
Figure 1.
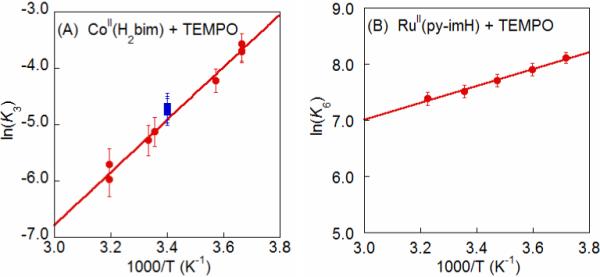
Van't Hoff plots (A) for CoII(H2bim) + TEMPO (eq 3) (●), with (■) indicating reactions that were initially run at high and low temperatures and then re-equilibrated back to 294 K; and (B) for the much less temperature dependent RuII(py-imH) + TEMPO ⇄ RuIII(py-im) + TEMPOH (eq 6). Figure B adapted, with permission, from reference 32b.
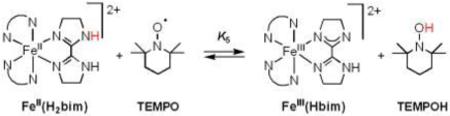
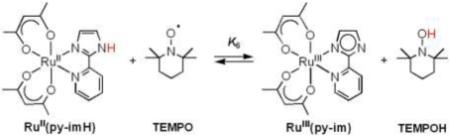
B. [FeII(H2bip)3]2+, [FeII(H2bim)3]2+, and RuII(acac)2(py-imH) + TEMPO
Equilibrium constants have been reported for the reactions of TEMPO with [FeII(H2bip)3]2+ [FeII(H2bip)], [FeII(H2bim)3]2+ [FeII(H2bim)], and RuII(acac)2(py-imH) [RuII(py-imH))] (eqs 4 - 6; H2bip = 2,2’-bi-1,4,5,6-tetrahydropyrimidine; acac = 2,4-pentanedionato, py-imH = 2-(2’-pyridyl)imidazole).28,32 In the iron systems, K4 and K5 in MeCN were determined both by static methods (as above) and from the ratio of the opposing second-order rate constants; the ruthenium K6 was measured by UV-vis spectroscopic titration. The temperature dependence of these equilibrium constants yield the ΔHo and ΔSo values given in Table 1. K6 is much less temperature dependent than either the iron or cobalt systems discussed above, varying by barely a factor of 1.5 over the 41°C temperature range examined 269 − 310 K (Figure 1B). For the ruthenium hexafluoro-acac derivative [RuII(hfac)2(py-imH)], HAT to the stable and isolable33 free radical tBu3PhO• (eq 7): K7 = 0.062 ± 0.013 at 298 K (hfac = 1,1,1,5,5,5-hexafluoro-2,4-pentanedionato, CF3C(O)CHC(O)CF3).32
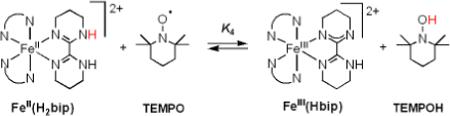 |
(4) |
 |
(5) |
 |
(6) |
| (7) |
II. Calorimetry
Solution calorimetry experiments have been done to independently confirm the ground-state enthalpy values determined from the van't Hoff analyses above. A Setaram C-80 Calvet calorimeter was outfitted with a pair of Hastelloy dual chamber reversal cells and run under isothermal conditions. In a typical experiment, separate solutions of FeII(H2bip) and TEMPO were thermally equilibrated under nitrogen in separate chambers of one cell. Excess TEMPO (8−34 equiv) were used to ensure that the reaction went to completion. The second cell contained an identical volume of MeCN and acted as a reference. Inversion of the calorimeter mixed the solutions and initiated the reaction. The heat flux signal changes rapidly after the reaction is initiated and then gradually returns to its equilibrium value (Figure 2). Integration of this signal over the course of several hours gives the total heat released, which can be converted to ΔHo using the reagent concentrations. Both reagents started as solutions in order to avoid the contributions from the heat of solution for the solid reagent. Instead, the heats of dilution were measured, which are typically much smaller contributions to the overall heat flux.34
Figure 2.
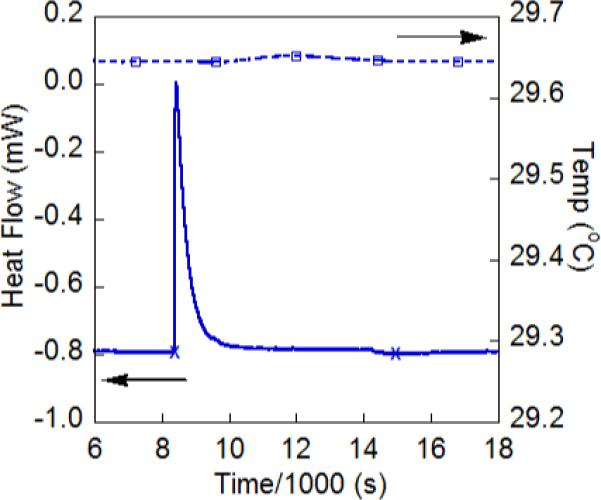
Heat flow curve ( ) and sample temperature (
) and sample temperature ( ) for the reaction between 3.2 mM FeII(H2bip) + 0.10 M TEMPO in MeCN. The “x” marks indicate the integration limits used to extract the enthalpy of reaction.
) for the reaction between 3.2 mM FeII(H2bip) + 0.10 M TEMPO in MeCN. The “x” marks indicate the integration limits used to extract the enthalpy of reaction.
The average of three measurements of the heat of reaction of FeII(H2bip) and TEMPO (eq 4) gave ΔHo4 = −8.9 ± 0.6 kcal mol−1. The observed heat of reaction was found to be independent of [TEMPO], indicating that heat of dilution for TEMPO, ΔHodil[TEMPO], is small. ΔHodil[FeII(H2bip)] was measured independently and also found to be negligible. The value of ΔHo4 from calorimetry is in excellent agreement with that determined previously from van't Hoff analysis of K4, −9.4 ± 0.6 kcal mol−1. Thus, calorimetry provides a direct and independent confirmation of the heat of reaction and also, because ΔGo4 is well known, the entropy of reaction, ΔSo = −30 ± 2 kcal mol−1.
Attempts to measure other heats of metal HAT reactions unfortunately all proved problematic. For the reaction of FeIII(Hbim) and TEMPOH, the exoergic direction for reaction 5, 1H NMR and UV-Vis spectra of product mixtures after calorimetric measurements showed decomposition of the iron product. This decomposition was found to be strongly exothermic, and overwhelmed the small endothermic signal expected. The cobalt/TEMPO reaction (eq 3) is too slow to be reliably measured directly by the Calvet calorimeter apparatus, so the reaction of CoII(H2bim) with tBu3PhO• was investigated instead. This reaction cleanly forms the HAT products CoIII(Hbim) and tBu3PhOH over the few minutes required for kinetic measurements. On the multiple-hour timescale of the calorimetry experiment, however; the UV-Vis spectra showed further reaction of the excess tBu3PhO•. This further reactivity results in calorimetric molar reaction enthalpy values that vary linearly with the amount of excess tBu3PhO• present, and this heat signal masks the enthalpy of the simple HAT reaction. Conditions with stoichiometric reagents or with excess CoII(H2bim) were also unsuccessful. Calorimetric measurements of the reactions of RuII(py-imH) with TEMPO (eq 6) or tBu3PhO• gave irreproducible heat flux signals, with large shifts in the baseline heat flux before and after mixing. Similar baseline shifts were also observed in the heat of dilution of RuII(py-imH) experiments. These experiments unfortunately have to be run at the edge of the sensitivity of the calorimeter due to the low solubility of RuII(py-imH) in MeCN.
As part of these studies, the heat of H-atom transfer from TEMPOH to tBu3PhO• to form TEMPO + tBu3PhOH was measured (eq 8). These products are quantitatively formed and the
| (8) |
product mixture is stable overnight at 30 °C, by 1H NMR and UV-Vis spectra. The directly measured heats of dilution in MeCN (ΔHodil) for both TEMPOH and tBu3PhO• are small, on the order of 2% of the total heat for the HAT reaction. The average of three experiments gave ΔHo8[calorimetry] = −11.2 ± 0.5 kcal mol−1.
Discussion
I. Overview of Hydrogen Atom Transfer Thermochemistry
Hydrogen atom transfer (HAT11,29) reactions in solution (eq 9) can be written as the sum of two pseudo-half-reactions, eqs 10 and 11. This is analogous to describing an electron transfer (ET) process as two ET half-reactions, except that in HAT these are complete reactions since they are not relative to a reference electrode. For HAT, each half-reaction is the definition of the bond dissociation enthalpy (BDE) or bond dissociation free energy (BDFE). Strictly speaking, BDEs and BDFEs are gas-phase quantities but it is convenient to consider analogs in solution, BDEs and BDFEs in solvent ‘s’. The driving force for eq 9 is therefore the difference between the BDFEs (or BDEs if working with enthalpy) of the two half reactions, eq 12.
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
The BDFEs and BDEs are related by the entropy of reaction ΔSos (eq 13), which is the difference between absolute entropies of the component species in solution (eq 14). So[AH]s is the sum of the gas-phase entropy of AH (So[AH]g) and the entropy of solvation of AHg (ΔSosolv[AH]s), eq 15. Analogous definitions apply to So[H•]s and So[A•]s. So[AH]g has contributions from the entropy of translations (depending on mass), rotations (depending on moment of inertia), vibrations (depending on frequencies), and electron spins.37 ΔSosolv[AH]s will vary given the polarity and hydrogen-bonding ability of the solvent used.38
Organic HAT reactions, as noted above, have for many decades been analyzed using BDEs and the Bell-Evans-Polanyi (BEP) correlation of activation energies with ΔHoHAT.1a,17,39 The differences between the BEP enthalpy treatment and more typical linear free-energy relationships (LFERs)18,19 have not previously been of serious concern because HAT reactions have typically been assumed to have ΔSos close to zero.26 This assumption derives from the parallel assumption that (for most species) AH and A• have similar absolute entropies (eq 16) because they have similar mass, size, and charge. As discussed elsewhere, equation 16 appears
| (16) |
to hold for small gas phase molecules and for solution phase organic compounds26,27 but does not hold for reactions of FeII(H2bip), FeII(H2bim),28 and (as reported here) CoII(H2bim). The magnitude of ΔSoHAT has not been extensively explored for transition metal complexes.40 The four different transition metal hydrogen atom donors studied here allow us to study the metal-based trends associated with ground-state entropies, and to determine if large ΔSoHAT is a general phenomenon for transition metal complexes.
II. Bond Dissociation Enthalpies and Free Energies
TEMPOH/TEMPO and tBu3PhOH/tBu3PhO• are convenient and common hydrogen atom transfer reagents, and they are reference points for much of the thermochemistry described here. It is therefore important to assess the ‘best’ current values for their O-H BDFEs and BDEs, as is done in the following sections and summarized in Table 2. In addition, the excellent agreement among the different approaches to these values supports the validity of each method.
Table 2.
Bond dissociation free energies (BDFEs) and enthalpies (BDEs) in MeCN.
| AH | BDEMeCN (kcal mol−1) | BDFEMeCN (kcal mol−1) | Reference |
|---|---|---|---|
| tBu3PhOH | 83 ± 1 | 77 ± 1 | see text, 34 |
| TEMPOH | 71.5 ± 0.5 | 66.5 ± 0.5 | see text, 23, 35 |
| FeII(H2bip) | 62.0 ± 1.7a | 66.0 ± 1.7b | 28, 30 |
| FeII(H2bim) | 67 ± 2a | 72 ± 2b | 36 |
| RuII(py-imH) | 68 ± 1a | 62 ± 1b | 32 |
| RuII(hfac)2(py-imH) | -- | 79.6 ± 1b | 32 |
| CoII(H2bim) | 62 ± 1a,c | 69.5 ± 0.9c | see text |
IIa. Solution BDEs of tBu3PhOH from Calorimetry
The literature on gas phase and solution phase BDEs of phenols is extensive.41 Data are available in a variety of solvents, and it is important in comparisons and thermochemical cycles to use values in the same solvent.42 In 1969, calorimetry determined the heat of transfer of two H atoms from diphenylhydrazine to 2 tBu3PhO• in both benzene and CCl4.34 When coupled with the known solution heats of formation for PhNHNHPh and PhNNPh (the latter having been re-evaluated since 1969),43 this gives ΔHof[tBu3PhO•]s - ΔHof[tBu3PhOH]s, (28.09 ± 0.08 and 28.01 ± 0.12 kcal mol−1 for C6H6 and CCl4 respectively) which can be converted to a BDEs using eq 17.
| (17) |
The last term in eq 17, ΔHof[H•]s, is the sum of the gas-phase heat of formation of H• (ΔHof[H•]g = 52.103 ± 0.001 kcal mol−1)44 and its enthalpy of solvation (ΔHosolv[H•]s). The solvation of H2 is considered to be a good model for solvation of H•45 so ΔHosolv[H•]s can be approximated by ΔHosolv[H2]s. ΔHosolv[H2]s in benzene and CCl4 are not known, but should be similar to those in toluene and 1,2-dichloroethane (1.38 and 1.81 kcal mol−1, respectively).46,47 Using these values in eq 17 gives solution BDEs for tBu3PhOH of 81.6 ± 0.4 kcal mol−1 in benzene and 82.0 ± 0.5 kcal mol−1 in CCl4.
The BDE of tBu3PhOH in MeCN differs from the values in benzene and CCl4 by the differences in solvation (eq 18). For H•, the difference in solvation between benzene and MeCN
| (18) |
is very small, 0.18 kcal mol−1.46 Ingold and coworkers have proposed that the difference in solvation between a phenol and its radical is due primarily to differences in hydrogen bonding to the solvent41b which they estimate using Abraham's empirical hydrogen-bond strength model.48,49 For AH = tBu3PhOH, the Ingold/Abraham H-bonding model estimates {ΔHosolv[A•]MeCN - ΔHosolv[AH]MeCN} - {ΔHosolv[A•]C6H6 - ΔHosolv[AH]C6H6} ≈ 1.3 kcal mol−1, 50 and therefore BDE[tBu3PhOH]MeCN = 83 ± 1 kcal mol−1.
IIb. BDE and BDFE of tBu3PhOH in MeCN from Gas Phase Data
The BDEMeCN and BDFEMeCN for tBu3PhOH can also be determined from gas phase values and estimates for the appropriate solvation terms (eqs 18, 19). The gas phase BDE of tBu3PhOH has been critically reviewed41c and found to be 8.8 ± 0.95 kcal mol−1 weaker than BDE[PhOH]g, which is 88.7 ± 0.5 kcal mol−1.41a Therefore, the BDE[tBu3PhOH]g = 79.9 ± 1.1 kcal mol−1.
| (19) |
| (20) |
ΔHosolv[tBu3PhO•]MeCN - ΔHosolv[tBu3PhOH]MeCN has been estimated by two methods. The Ingold/Abraham H-bond model gives 1.43 kcal mol−1. 49 Alternatively, ΔHosolv[tBu3PhO•]MeCN - ΔHosolv[tBu3PhOH]MeCN can be evaluated computationally. Bakalbassis, et al. found that chemically accurate values of BDEs for several phenols could be obtained with the (RO)B3LYP level of theory using a non-standard basis set (see Experimental for details).51 In our laboratory, this method yields BDE[tBu3PhOH]g,DFT = 79.3, in good agreement with the experimental value above. Application of a polarizable continuum solvation model (PCM) yields the slightly negative value for ΔHosolv[tBu3PhO•]MeCN - ΔHosolv[tBu3PhOH]MeCN of −0.4 kcal mol−1. This is a consequence of the larger dipole moment of tBu3PhO•. Together, eq 19, the BDE[tBu3PhOH]g of 79.9 kcal mol−1 and the average of the two solvation models above (0.5 ± 1.3 kcal mol−1) give BDE[tBu3PhOH]MeCN = 82 ± 2 kcal mol−1, consistent with the independently-determined calorimetric value above.
The bond dissociation free energy for tBu3PhOH can also be determined using eq 20, starting with BDFE[tBu3PhOH]g = 71.8 ± 1 kcal mol−1.52 As above, PCM calculations were used to compute ΔGosolv[tBu3PhO•]MeCN - ΔGosolv[tBu3PhOH]MeCN = −0.4 kcal mol−1 (see Experimental for details). Ingold's model gives similarly small values. When used in eq 20, with ΔGosolv[H•]MeCN ≅ ΔGosolv[H2]MeCN = 5.12 kcal mol−1,46 the BDFE[tBu3PhOH]MeCN is 77 ± 1 kcal mol−1.
IIc. BDFE of tBu3PhOH in MeCN from pKa and Eo Measurements
An alternative measure of BDFEMeCN is obtained from a thermochemical cycle using a pKa and an Eo (eq 21). Bordwell
| (21) |
and co-workers popularized the method in DMSO,22 and Tilset worked out the cycle for MeCN.20 The two reported values for Eo[tBu3PhO•] in MeCN vs. Cp2Fe+/0, −707 mV53 and −689 mV,54 average to −0.70 V. The pKa in DMSO for tBu3PhOH (17.8)55 can be converted into a value of 27.5 in MeCN using the linear relationship found by Kutt and coworkers.56 Putting these values into eq 21 gives BDFE[tBu3PhOH]MeCN = 76 ± 1.3 kcal mol−1. The measured equilibrium constant for reaction of tBu3PhO• with RuII(hfac)2(py-imH), equation 7 above, provides another measure of BDFE[tBu3PhOH]MeCN, since the BDFE of the ruthenium complex is known from its pKa and an Eo values.32 Application of eq 12 to this data gives a BDFE[tBu3PhOH]MeCN of 78 ± 1 kcal mol−1. The close agreement among these three BDFE values (77 ± 1, 76 ± 1, and 78 ± 1 kcal mol−1) supports the validity of all three approaches and indicates a consensus BDFE[tBu3PhOH]MeCN of 77 ± 1 kcal mol−1 (Table 2).
IId. BDFE and BDE of TEMPOH in MeCN
A calorimetric study of the reaction of TEMPO with diphenylhydrazine in benzene,33 following the analysis above for tBu3PhOH, gives57 BDE[TEMPOH]C6H6 = 70.0 ± 0.8 kcal mol−1 and BDE[TEMPOH]MeCN = 71.5 ± 0.9 kcal mol−1. This BDE can be converted to a BDFE of 66.4 ± 0.5 kcal mol−1 via eqs 13 and 14 assuming that So[TEMPO]MeCN ≈ So[TEMPOH]MeCN (an example of eq 16) and ΔGosolv[H•]MeCN = 5.12 kcal mol−1. This BDFE[TEMPOH]MeCN can also be calculated directly using the known pKa and Eo values (eq 21), which gives 66.9 ± 0.5 kcal mol−1 (see Supporting Information). BDFE[TEMPOH]MeCN is also determined by the equilibrium constants for reactions of FeII(H2bip),28 FeII(H2bim),28 or RuII(py-imH)32 with TEMPO (the BDFEs for each metal complex independently determined from pKa and Eo values). The average value from these equilibration experiments, 66.5 ± 0.5 kcal mol−1, is essentially the same as the values from the TEMPOH pKa and Eo and from calorimetry. This agreement supports the consensus BDFE value (Table 2) and the assumption that So[TEMPO]MeCN ≈ So[TEMPOH]MeCN.
IIe. Solution BDFE and BDE of Metal Complexes in MeCN
For each of the four metal complexes examined here, the BDFEMeCN was determined using eq 21 and the relevant pKa and Eo values (which have been previously reported32,30). As noted above, these values are consistent with the measured equilibrium constants and the BDFEs of TEMPOH and tBu3PhOH. The BDEMeCN (enthalpies) shown in Table 2 were calculated from the BDFEMeCNs and the experimentally determined ΔSoMeCN for reaction with TEMPO, attributing all of this ΔSo to the metal complex ‘half reaction’ (eq 11).
III. Comparison between Calorimetry and van't Hoff methodologies
The calorimetric experiments confirm the enthalpy of reaction of FeII(H2bip) + TEMPO (eq 4) measured by solution equilibrium and kinetic data. The ΔHo4[calorimetry] = −8.9 ± 0.6 kcal mol−1, is within error of ΔHo4[van't Hoff] = −9.4 ± 0.6 kcal mol−1.28 This quantitative agreement confirms the large negative ΔSo = −30 ± 3 cal mol−1 K−1.28 Similarly, the calorimetric heat of H-atom transfer from TEMPOH to tBu3PhO• (eq 8), ΔHo8[calorimetry] = −11.2 ± 0.5 kcal mol−1, agrees with the value calculated from the literature solution BDEs in MeCN (Table 2), −11.5 ± 1.4 kcal mol−1. Calorimetric studies of RuII(py-imH), FeIII(Hbim), and CoII(H2bim) plus TEMPO were unsuccessful because the reactions did not meet the experimental requirements of high solubilities and good long-term stability of the reaction mixtures. For these systems, the van't Hoff methodology is easier and more reliable for determining the ground state thermodynamics, as long as equilibrium constants are measurable.
IV. Origins of the ΔSo for H-atom transfer
The four reactions of transition metal H-atom donors with the same atom acceptor, TEMPO, have widely varying values of ΔSo (Table 1). The reaction of RuII(py-imH) shows only a small positive entropy, ΔSo6 = 4.9 cal mol−1 K−1. The reactions with FeII(H2bip), FeII2bim), CoII(H2bim) show much larger negative values: ΔSoHAT = −30, −30, and −41 cal mol−1 K−1 respectively. These are very substantial values of |ΔSoHAT|, in contradiction with the common assumption that the entopic contribution to HAT is not significant. Thinking of these reactions as the sum of two quasi-half reactions (eqs 9-11), ΔSoHAT ≠ 0 requires that So[AH]s ≠ So[A•]s (in contradiction to eq 16) for either the metal or organic redox couple.
The independent measurements of the BDEMeCN and the BDFEMeCN in Section II give (after subtracting TSo[H•]MeCN), {So[TEMPO]MeCN - So[TEMPOH]MeCN} = 1 ± 4 cal mol−1 K−1 and {So[tBu3PhO•]MeCN - So[tBu3PhOH]MeCN} = 5 ± 7 cal mol−1 K−1. In addition, HAT from TEMPOH to tBu3PhO• (eq 8), has ΔHo8 = −11.2 ± 0.5 kcal mol−1 and ΔGo = −10.5 ± 1.3 kcal mol−1 so ΔSo8 = −2 ± 3 cal mol−1 K−1. The difference between ΔSo8 and {So[TEMPO]MeCN - So[TEMPOH]MeCN} further limits the entropy for {So[tBu3PhO•]MeCN - So[tBu3PhOH]MeCN} to 3 ± 5 cal mol−1 K−1. These data demonstrate that So[AH]s – So[A•]s ≈ 0 for both TEMPOH and tBu3PhOH. Therefore, the unusual entropy contributions in our HAT reactions come from the metal redox couple.
The metal redox couples for reactions 3 − 6 show the following trend for |ΔSo|: RuII(py-imH) << FeII(H2bip) ≈ FeII(H2bim) < CoII(H2bim). The Fe and Co reactions have negative entropies, indicating that the MIII(HL) complex is more ordered than MII(H2L). For the iron systems, previous experimental and computational studies28 showed that the large |ΔSo| originates primarily from changes in the vibrational entropy (ΔSovib) upon oxidation of the iron. The calculations showed that the primary contributors are ca. 30 low-frequency (ν ≤ kT = 207 cm−1 at 298 K) torsions and bends that change frequency between the FeII and FeIII compounds.28 Alternative origins of the large |ΔSo| such as ion pairing or solvent effects were ruled out.
For CoII(H2bim) + TEMPO, the ΔSoHAT (−41 ± 2 cal mol−1 K−1) is even more negative than for the two iron reactions. In solution, CoII(H2bim) is entirely high-spin, while CoIII(Hbim) is entirely low-spin.31 In the idealized octahedral case, this is a change in multiplicity from a 12-fold degenerate 4T1g CoII electronic state to a non-degenerate 1A1g CoIII state, an electronic entropy of Rln(12) or ΔSoelec = −4.9 cal mol−1 K−1. This maximum value of ΔSoelec (spin-orbit coupling and the D3 symmetry of CoII(H2bim) will lower the degeneracy) is still a minor contribution to the observed ΔSoHAT. The Co and Fe complexes are very similar: in structure (in both systems the M-N bond lengths are ∼0.1 Å shorter in the MIII derivative);31,36 in acidity (similar pKa values in MeCN); and in hydrogen bonding (the ΔGo for formation of a hydrogen-bonded adduct between TEMPOH and either CoIII(Hbim) or FeIII(Hbim) differs by less than 0.1 kcal mol−1).28,58 This similarity suggests a common vibrational origin for the entropy in both systems. The entropy would be expected to be larger for Co, since the high-spin to low-spin conversion should cause even larger frequency changes.67,60a An increase of ca. 10 cal mol−1 K−1 for the addition of a spin-change is not unreasonable based on the electron transfer entropies discussed below. The ruthenium complexes, with a 4d transition metal, are all low-spin and have stronger bonds and therefore fewer low-frequency vibrational modes. With fewer modes ≤ kT, the vibrational entropy will be much reduced, as observed: RuII(py-imH) + TEMPOH has ΔSoHAT = 4.9 ± 1.1 cal mol−1 K−1. Thus the trend for |ΔSoHAT|, RuII(py-imH) << FeII(H2bip) ≈ FeII(H2bim) < CoII(H2bim), is consistent with a dominant role for vibrational entropy in these reactions.
V. Trends in Electron Transfer Entropies
The entropies of electron transfer half reactions (ΔSoET, eq 22) have been determined for a wide range of complexes and found to depend on the nature of the metal center, the associated redox change, the coordinating ligands, and the surrounding solvent and counterions.59-65 We note that, by convention, the ET half-reaction in eq 22 is written as a reduction, opposite to the way the HAT reactions are written here (eqs 3-8, 11, 12).
| (22) |
Vibrational entropy has been shown to be important in both electron transfer66,67 and spin-equilibrium processes,68,69 when there are changes in metal-ligand bonding upon redox or spin change. Richardson and Sharpe estimated that in the gas phase the vibrational contribution to the total electron transfer entropy ranges from as low as 9% in RuO4 (where there are few vibrations and many are at high frequency) up to 42% in [Fe(CN)6]4-.67a Vibrational entropy is a substantial part of the measured ΔSoET = 29 ± 3 cal mol−1 K−1 for the FeII(H2bim)/FeIII(H2bim) redox couple,28 and of the ΔSoHAT observed for the HAT reactions of FeII(H2bim) and FeII(H2bip). These entropies are close in magnitude because they both arise from the FeIII complexes being more rigid and having fewer low-frequency vibrational modes than FeII.28 In general, when vibrational entropy is a significant contributor, there should be a strong parallel between the entropies for ET and HAT reactions in a given system.
Differences in electron transfer entropies between redox two couples, when measured in the same solvent for reagents of the same charge, are primarily due to vibrational and electronic entropies.60a For example, {ΔSoET[Co(tacn)3]3+/2+solv - ΔSoET [Ru(tacn)3]3+/2+solv} is 16 ± 3 cal mol−1 K−1 for solv = DMSO, acetone, water and four other solvents (tacn = 1,4,7-triazacyclononane).60a,70,71 This difference is independent of solvent because the electronic and vibrational entropies do not depend on the solvent. {ΔSovib[Co(tacn)3]3+/2+ - ΔSovib[Ru(tacn)3]3+/2+} has been estimated as 12.7 cal mol−1 K−1, and the remainder of the difference can be accounted for by ΔSoelec.60a
The results described in this report indicate that, when comparing one transition metal system to another, the same trends are observed in the half-reaction entropies for both ΔSoET and |ΔSoHAT|. The CoIII/CoII couples have the largest ΔSoET, with the exception of a few lanthanide and actinide complexes,59d,64,72 because they involve a spin-state change (low-spin CoIII to high-spin CoII) in addition to an oxidation state change. ΔSoET for CoIII/CoII couples ranges from 30 − 50 cal mol−1 K−1 in organic solvents.59f,60,61,67,72,73 For similar complexes, ΔSoET values for Co derivatives are typically 10 − 20 cal mol−1 K−1 larger than those for the Fe analogs. In our measurements of HAT entropies, the Co reaction has the largest |ΔSoHAT| (ca. −41 cal mol−1 K−1), 11 cal mol−1 K−1 more negative than the iron analogs in the same reaction with TEMPO. FeIII/II couples have ΔSoET of typically 15 − 30 cal mol−1 K−1 in organic solvents (except for those with an accompanying spin change).59,60,67 RuIII/RuII couples have ΔSoET in a similar range as FeIII/FeII couples (∼10−30 cal mol−1 K−1 59,60,66a,67,72). Comparing complexes in the same solvent, low-spin FeIII/II couples are quite similar to the RuIII/II analogs, while iron couples that exhibit spin-equilibrium or are high-spin only have larger ΔSoET than the related RuIII/II couples (probably due to larger vibrational entropies).74
Thus the trend for ΔSoET is Co > high-spin Fe > low-spin Fe ≈ Ru.59b This is the same as the trend in –ΔSoHAT described above for HAT (recall that the half reactions are by convention written in opposite directions for ET (eq 22) and HAT (eq 10), so the signs are opposite). This trend is a result of the major contribution of ΔSovib, for both HAT and ET reactions of transition metal complexes. The close connection between ΔSoET and ΔSoHAT provides valuable insight in cases where only one or the other has been measured. In particular, it suggests that ground-state entropy effects will be important for HAT reactions of high-spin first-row transition metals.
VI. Relevance to Biological Systems and HAT Analyses
The observation of significant ground-state entropies for HAT reactions appears to be general for first-row transition metal coordination complexes. This leads to a significant temperature dependence of ΔG. For instance, in a reaction with ΔSo = −30 cal mol−1 K−1 such as observed for the Fe systems above, ΔG shifts by more than 1 kcal mol−1 between 5 and 45°C and Keq shifts by almost an order of magnitude. This effect does not appear to have been incorporated into most applications of modern PCET theories to either small molecule or enzymatic systems.19 In particular, variation in ΔG indicates changes in the shape of the free energy surface which should affect processes involving hydrogen tunneling. Does the variation in ΔG play a role, for instance, in the unusual temperature dependence of the kinetic isotope effect for HAT from a fatty acid to the non-heme iron center in lipoxygenase enzymes? Since large values of |ΔSoET| have been observed in biological systems,75,76 the close connection between ΔSoET and ΔSoHAT described here suggests that there are significant entropic contributions in HAT and PCET reactions of metalloproteins.
Conclusions
Ground state entropy changes for hydrogen atom transfer reactions, ΔSoHAT, vary substantially depending on the reaction. Values reported vary from −41 ± 2 cal mol−1 K−1 for HAT from a cobalt(II) 2,2’-bi-2-imidazoline complex to the nitroxyl radical TEMPO (eq 3), to – 2 ± 3 cal mol−1 K−1 for HAT from TEMPOH to the stable aryloxyl radical tBu3PhO• (eq 8). These values have been determined by Van't Hoff analysis of equilibrium data, by calorimetric measurements, and using thermochemical cycles. These data and those from previous reports show that the magnitude of |ΔSoHAT| for reactions with TEMPO have the following trend: CoII(H2bim) > FeII(H2bip) = FeII(H2bim) > RuII(py-imH) ≳ tBu3PhOH ≈ 0. The analysis presented here supports the long-standing assumptions that ΔSoHAT ≈ 0 and that So[AH] ≈ So[A•] for HAT reactions of organic and small gas phase molecules, but not for transition metal complexes. Analyses of transition metal HAT reactions need to take into account the frequently large reaction entropies and should not be based just on bond dissociation enthalpies. The trend in ΔSoHAT for the metal complexes is the same as that observed for electron transfer half-reaction entropies in aprotic solvents ΔSoET, and the magnitudes of these values are often similar as well. This striking analogy is a result of both the HAT and ET values being significantly influenced by vibrational entropy contributions. The more extensive database of electron transfer entropies therefore provides guidelines for initial predictions of hydrogen transfer entropies.
Experimental Section
General Considerations
All manipulations were carried out under anaerobic conditions in MeCN using standard high-vacuum line and nitrogen-filled glovebox techniques unless otherwise noted. NMR spectra were acquired on Bruker Avance-500, DRX-499, Avance-300, or Avance-301 spectrometers. Static UV-Visible spectra were obtained using either a Hewlett-Packard 5483 spectrophotometer equipped with an eight-cell holder thermostatted with a Thermo-Neslab RTE-740 waterbath, or using a Shimadzu UV-2401 PC dual beam instrument. Spectra are reported as λmax, nm [ε, M−1 cm−1] and were blanked relative to pure MeCN. Air-sensitive samples were prepared in the glovebox and their spectra taken using either quartz cuvettes attached to Teflon-stoppered valves (Kontes) or injectable screw-capped cuvettes with silicone/PFTE septa (Spectrocell). Septa were replaced after each experiment. Rapid kinetic measurements were taken using an OLIS USA stopped-flow instrument equipped with the OLIS-rapid scanning monochromator and UV-Vis detector, and thermostatted by a Neslab RTE-111 waterbath. All errors are reported as ±2σ̵ based on fits weighted with the errors propagated from experimental measurements.
Materials
Low water content CH3CN (< 10 ppm H2O; Allied Signal / Burdick and Jackson brand) was taken from a steel keg sparged with Ar and dispensed through the glovebox. CD3CN (Cambridge Isotopes Laboratories) was dried by stirring overnight with CaH2, vacuum transferring and stirring briefly (< 1 hr) over P2O5, vacuum transferring back over CaH2 for ca. 30 min and then storing in the glove box free of drying agent. Other solvents were dried using a ”Grubbs-type” Seca Solvent System installed by GlassContour.77 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO; Acros Organic and Aldrich) was sublimed at room temperature under static vacuum before use. [FeII(H2bip)3][ClO4]2, (FeII(H2bip));31 [FeIII(H2bip)2(Hbip)][ClO4]2 (FeIII(Hbip));31 [CoII(H2bim)3][ClO4]2, (CoII(H2bim));31 [CoIII(Hbim)(H2bim)2][ClO4]2 (CoIII(Hbim));31 [RuII(acac)2(py-imH)], (RuII(py-imH));32 [RuIII(acac)2(py-im)], (RuIII(pyim));32 2,2,6,6-tetramethyl-1-piperidinylhydroxide (TEMPOH);28,78 and 2,4,6-tri-tert-butyl phenoxyl (tBu3PhO•)33 were prepared and characterized following literature procedures. All other reagents were purchased from Aldrich and used as received. Caution: The perchlorate salts used herein are potentially explosive and should be handled with care in small quantities only. They should not be heated when dry or subjected to friction or shock, such as scratching with a non-Teflon-coated spatula.
Solution Keq measurements
Equilibrium experiments for FeII(H2bip) and FeII(H2bim) + TEMPO have been previously reported.28 K3 for CoII(H2bim) + TEMPO ⇄ CoIII(Hbim) + TEMPOH were directly measured by 1H NMR spectroscopy. A set of J-Young capped NMR tubes were charged with varying amounts of TEMPO (5 − 10 mg, 0.03 − 0.06 mmol), leaving one tube empty to act as a ‘time zero’ spectrum. To each of these tubes was added a 0.4 mL aliquot of a stock solution of CoII(H2bim) (11 mM in CD3CN containing 3−10 μL CH2Cl2 as an integration standard). After mixing, the tubes were quickly removed from the glovebox and placed in a thermal bath. Changes in the NMR spectrum were monitored over the course of 2−7 days depending on the temperature. Data acquisition was stopped when the integrations for each species remained constant for a minimum of 12 hours. The tubes were then placed at room temperature (21 °C) and allowed to re-equilibrate for 2−7 days, again monitored by 1H NMR spectroscopy. Reliable integrations were obtained using Mestre-C by manually phasing and baselining the spectrum from +60 to −3 ppm, as well as adjusting the phase and bias for each integral. Integrations were found to be reproducible to ± 3−5% based on the standard deviation of three spectra taken in rapid succession on a tube at equilibrium. K4 values were obtained from the ratio of the following integration regions: CoII(H2bim) 24.7 − 21.2 ppm, CoIII(Hbim) 4.85 − 2.68 ppm, TEMPO 21.2 − 10.8 ppm, TEMPOH 1.73 − 1.34 ppm, from at least two spectra.
The equilibrium constant for [RuII(hfac)2(py-imH)] + tBu3PhO• ⇄ [RuIII(hfac)2(py-im)] + tBu3PhOH (eq 7) was measured by UV–vis titration using the method used in reference 32 to determine K7, using a solution of RuII(hfac)2(py-imH) (0.027 mM, 2.5 mL) titrating with tBu3PhO• (6.7 mM) until 10 equiv (10 μL = 1 equiv). The UV–vis data were analyzed using the absorbance at 481 nm to determine an equilibrium constant K7 = 0.062 ± 0.013 (ΔGo7 = 1.6 ± 0.1 kcal mol−1) from the average of two runs. Since ΔGo7 = BDFE[RuII(hfac)2(py-imH)]MeCN – BDFE(tBu3PhOH)MeCN and BDFE[RuII(hfac)2(py-imH)] = 79.6 ± 1.0 (from Eo and pKa values32), this yields BDFE(tBu3PhOH)MeCN = 78 ± 1.
Calorimetry
Experiments were done on a Setaram C-80 Calvet Calorimeter outfitted with a pair of Hastelloy C276 Reversal Mixing cells (utilizing the larger 2.5 mL reaction cup and graphited Teflon seals) under isothermal conditions. In the calorimeter, each cell is surrounded by an electrical heater coil. The difference in current required to maintain a constant temperature between the two cells is related to the heat evolved from the reaction. The methodology used is a modification of literature procedures.34,35 The calorimeter was set at 30.0 °C and had an actual sample temperature of 29.6 ± 0.1 °C. The calorimeter had been previously calibrated with a Joule-effect vessel, and the calibration was checked intermittently using the standardized aqueous heat of solution for KCl.
In a glovebox, the inner cup of one cell charged with 5−15 mg of limiting reagent in 2.0 mL of MeCN and covered with a Hastelloy cap. The outer chamber was charged with 1.0 mL of the reagent in slight excess (1.1 − 2.0 equiv) and the remainder of the cell assembled. See Table 3 for typical concentrations. The reference cell was assembled under identical conditions using a total of 3.0 mL of MeCN. The sealed cells were removed from the glovebox and thermally equilibrated in the calorimeter until both the flux and the temperature had reached steady-state (ca. 1−2 hours). This was deemed the experimental baseline, and the reaction was initiated by rotation of the calorimeter body, which inverted the cells and allowed the two solutions to mix. The heat flux was then recorded until the baseline was once-again achieved. The resulting fluxogram was integrated using SetSoft-2000 to give the heat evolved (Table 3). After the reaction was complete, the sample cell was removed from the calorimeter and returned to the glovebox. The final reaction mixture was diluted in a Kontes-valve cuvette (0.1 mL sample in ca. 2.0 mL MeCN) and an optical spectrum was obtained, to determine that the reaction had gone to completion and remained uncontaminated by air. Reactions were repeated a minimum of three times to achieve the desired level of reproducibility. Control reactions for the heats of dilution were measured for each reagent under similar concentration regimes to the actual reaction conditions. In most cases these were found to be small effects.
Table 3.
Standard conditions and integration times for calorimetry reactions.
| Reaction | Inner Chamber | Outer Chamber | Integration time |
|---|---|---|---|
| FeII(H2bip) + TEMPO a | 2.8 mM FeII(H2bip) |
23 − 109 mM TEMPO a |
∼ 10000 s |
| RuII(py-imH) + tBu3PhO• | 5 − 10 mM RuII(py-imH) |
11 mM tBu3PhO• |
∼ 2000 s |
| RuII(py-imH) + TEMPO | 10 mM RuII(py-imH) |
11 mM TEMPO |
∼ 4000 s |
| CoII(H2bim) + tBu3PhO• | 6.2 mM CoII(H2bim) |
4.4 mM tBu3PhO• |
∼ 8000 s |
| TEMPOH + tBu3PhO• | 11 mM TEMPOH |
14.7 mM tBu3PhO• |
∼ 4000 s |
At 29.6 °C, FeII(H2bip) + TEMPO has Keq = 1.7 ± 0.3. In order to ensure that the reaction went to completion, 8 − 34 equiv of TEMPO were used.
Calculations
All calculations were performed using Gaussian03.79 tBu3PhOH and tBu3PhO• were optimized in C1 symmetry and geometries were confirmed to be local minima by vibrational analysis. Following literature precedent,51 the B3LYP functional was used with the 6−31+G basis set with additional (p) polarization functions on hydrogen atoms only. Radical species were computed using the restricted-open shell (RO) formalism. Bakalbassis, et al. found this (RO)B3LYP/6−31+G(,p) method (the non-standard basis set nomenclature indicates that polarization functions are included for hydrogen atoms only and not heavy atoms) produced chemically accurate values of BDE for several phenolic compounds.51 Solution enthalpies and free energies were obtained from geometry optimizations and frequency analyses including a polarizable continuum model (PCM)80 of acetonitrile, as implemented in Gaussian03.81 Free energies of solution, ΔGsolv, were also obtained from PCM single point calculations on the gas phase optimized geometries, with the inclusion of the SCFVAC keyword; for these calculations atomic radii from the United Atom Topological Model (UAHF) were used. The values of ΔGosolv[tBu3PhO•]MeCN - ΔGosolv[tBu3PhOH]MeCN computed via these two methods are essentially identical (within ca. 0.01 kcal mol−1).
Supplementary Material
Acknowledgements
We gratefully acknowledge financial support from the U.S. National Institutes of Health (grant GM50422), the National Research Council of Canada (NSERC) (for a fellowship to EAM), the UW Department of Chemistry, and the Office of Science, Office of Basic Energy Sciences under contract DE-AC06 RLO 1830. We thank Drs. T. S. Autrey, D. M. Camaioni, and A. J. Karkamkar for assistance with the calorimetry experiments, C. Isborn and Dr. X. Li for assistance with PCM calculations, and J. J. Warren for his thoughtful comments.
Footnotes
Supporting Information Available. Detailed calorimetric traces, integrations, reaction spectra, the complete reference 79, and calculated data are provided. This material is available free of charge via the internet at http://pubs.acs.org.
Footnotes
- 1.a Kochi JK, editor. Free Radicals. Wiley; New York: 1973. [Google Scholar]; Ingold KU. p. 67ff. especially. Chapter 2.; Russell GA. pp. 275–331. Chapter 7.; O'Neal H, Benson SW. pp. 275–359. Chapter 17.; b Fischer H, editor. Radical Reaction Rates in Liquids. Springer-Verlag; Berlin: (Landolt-Börnstein New Series). [Google Scholar]; a 1984. subvol. a-e.; b 1994. subvol. A-E.; c Hendry DG, Mill T, Piszkiewicz L, Howard JA, Eigenmann HK. J. Phys. Chem. Ref. Data. 1974;3:937–978. [Google Scholar]; d Fossey J, Lefort D, Sorba J. Free Radicals in Organic Chemistry. Wiley; New York: 1995. [Google Scholar]; e Leffler JE. An Introduction to Free Radicals. Wiley; New York: 1993. Chapters 7−8. [Google Scholar]; f Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford University Press; New York: 1999. [Google Scholar]; g Sies H, editor. Oxidative Stress: Oxidants and Antioxidants. Academic; New York, NY: 1991. [DOI] [PubMed] [Google Scholar]; h Foote CS, Valentine JS, Liebman J, Greenberg A, editors. Active Oxygen in Chemistry. Blackie, Chapman and Hall; Glasgow: 1995. [Google Scholar]
- 2.Hynes JT, Klinman JP, Limback H-H, Schowen RL, editors. Hydrogen-Transfer Reactions. Wiley-VCH; Weinheim: 2007. b See also footnotes 6−14.
- 3.a Stubbe J, van der Donk WA. Chem. Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]; b Pesavento RP, van der Donk WA. Adv. Protein Chem. 2001;58:317–385. doi: 10.1016/s0065-3233(01)58008-0. [DOI] [PubMed] [Google Scholar]; c Marsh ENG. BioEssays. 1995;17:431–441. doi: 10.1002/bies.950170511. [DOI] [PubMed] [Google Scholar]; d Pierre JL, Thomas F. Compt. Rend. Chim. 2005;8:65–74. [Google Scholar]; e Fontecave M, Pierre JL. Comptes Rendus Acad. Sci. Ser. II C. 2001;4:531–538. [Google Scholar]; f Decker A, Chow MS, Kemsley JN, Lehnert N, Solomon EI. J. Am. Chem. Soc. 2006;128:4719–4733. doi: 10.1021/ja057378n. [DOI] [PubMed] [Google Scholar]
- 4.a Labinger JA. J. Mol. Catal. A. 2004;220:27–35. [Google Scholar]; b Labinger JA. Catal. Lett. 1988;1:371–376. [Google Scholar]; c Limberg C. Angew. Chem. Int. Ed. 2003;42:5932–5954. doi: 10.1002/anie.200300578. [DOI] [PubMed] [Google Scholar]
- 5.a Hammes-Schiffer S. Acc. Chem. Res. 2001;34:273–281. doi: 10.1021/ar9901117. [DOI] [PubMed] [Google Scholar]; b Cukier RI. J. Phys. Chem. B. 2002;106:1746–1757. [Google Scholar]; c Kuznetsov AM, Ulstrup J. Can. J. Chem. 1999;77:1085–1096. [Google Scholar]; d Krishtalik LI. Biochim. Biophys. Acta. 2000;1458:6–27. doi: 10.1016/s0005-2728(00)00057-8. [DOI] [PubMed] [Google Scholar]; e Hatcher E, Soudackov A, Hammes-Schiffer S. Chem. Phys. 2005;319:93–100. doi: 10.1063/1.1814635. [DOI] [PubMed] [Google Scholar]; f Cukier RI. ACS Symp. Series. 2004;883:145–158. [Google Scholar]
- 6.a Liang Z-X, Klinman JP. Curr. Opin. Struct. Bio. 2004;14:648–655. doi: 10.1016/j.sbi.2004.10.008. [DOI] [PubMed] [Google Scholar]; b Hatcher E, Soudackov AV, Hammes-Schiffer S. J. Am. Chem. Soc. 2004;126:5763–5775. doi: 10.1021/ja039606o. [DOI] [PubMed] [Google Scholar]; c Lehnert N, Solomon EI. J. Biol. Inorg. Chem. 2003;8:294–305. doi: 10.1007/s00775-002-0415-6. [DOI] [PubMed] [Google Scholar]; d Goldsmith CR, Stack TDP. Inorg. Chem. 2006;45:6048–6055. doi: 10.1021/ic060621e. [DOI] [PubMed] [Google Scholar]; e Costas M, Mehn MP, Jensen MP, Que L. Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 7.a Kaizer J, Klinker EJ, Oh NY, Rohde J-U, Song WJ, Stubna A, Kim J, Munck E, Nam W, Que L., Jr. J. Am. Chem. Soc. 2004;126:472–473. doi: 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]; b de Visser SP, Kumar D, Cohen S, Shacham R, Shaik S. J. Am. Chem. Soc. 2004;126:8362–8363. doi: 10.1021/ja048528h. [DOI] [PubMed] [Google Scholar]; c Kumar D, de Visser SP, Shaik S. J. Am. Chem. Soc. 2004;1260:5072–5073. doi: 10.1021/ja0318737. [DOI] [PubMed] [Google Scholar]; d Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 8.a Ragsdale SW. Chem. Rev. 2006;106:3317–3337. doi: 10.1021/cr0503153. [DOI] [PubMed] [Google Scholar]; b Baik M-H, Newcomb M, Friesner RA, Lippard SJ. Chem. Rev. 2003;103:2385–2419. doi: 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]; c Brazeau BJ, Austin RN, Tarr C, Groves JT, Lipscomb JD. J. Am. Chem. Soc. 2001;123:11831–11837. doi: 10.1021/ja016376+. [DOI] [PubMed] [Google Scholar]
- 9.a Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem. Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]; b Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Phil. Trans. R. Soc. B. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gridnev AA, Ittel SD. Chem. Rev. 2001;101:3611–3659. doi: 10.1021/cr9901236. [DOI] [PubMed] [Google Scholar]
- 11.The terminology in the PCET area is in flux. A number of papers, including some of ours, define HAT very narrowly.12,13b-d A recent major review defines HAT as a reaction in which “both the transferring electron and proton come from the same bond” (ref 12a, p. 5024). This mechanistic distinction is, however, often problematic to apply in practice. Therefore in cases, when the intimate details are not at issue, we prefer a broad definition of HAT that encompasses all processes involving concerted movement of a proton and an electron (e− + H+ ≡ H•) in a single kinetic step, when both the proton and the electron originate from the same reactant and travel to the same product.13a The metal-containing reactions 4−7 here are HAT in the broad definition but excluded under the narrower ones, because the transferred H+ forms an N-H σ-bond while the e− formally adds to a different orbital, a metal π-symmetry t2g-type orbital. The organic reaction of tBu3ArO• + TEMPOH (eq 8), under the definition quoted above12a, could be considered HAT in the forward direction but not in the reverse direction (assuming that the phenolic H lies in the plane of the aromatic ring).
- 12.a Huynh MHV, Meyer TJ. Chem. Rev. 2007;107:5004–5064. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cukier RI, Nocera DG. Annu. Rev. Phys. Chem. 1998;49:337–369. doi: 10.1146/annurev.physchem.49.1.337. [DOI] [PubMed] [Google Scholar]; c Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem. Rev. 2003;103:2167–2202. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]; d Meyer TJ, Huynh MHV. Inorg. Chem. 2003;42:8140–8160. doi: 10.1021/ic020731v. [DOI] [PubMed] [Google Scholar]; e Lebeau EL, Binstead RA, Meyer TJ. J. Am. Chem. Soc. 2001;123:10535–10544. doi: 10.1021/ja000517a. [DOI] [PubMed] [Google Scholar]; f Chen X, Bu Y. J. Am. Chem. Soc. 2007;129:9713–9720. doi: 10.1021/ja071194m. [DOI] [PubMed] [Google Scholar]; g Cowlet RE, Bontchev RP, Sorrell J, Sarrachino O, Feng Y, Wang H, Smith JM. J. Am. Chem. Soc. 2007;129:2424–2425. doi: 10.1021/ja066899n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a Mayer JM. Annu. Rev. Phys. Chem. 2004;55:363–390. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]; b Mayer JM, Hrovat DA, Thomas JL, Borden WT. J. Am. Chem. Soc. 2002;124:11142–11147. doi: 10.1021/ja012732c. [DOI] [PubMed] [Google Scholar]; c Litwinienko G, Ingold KU. Acc. Chem. Res. 2007;40:222–230. doi: 10.1021/ar0682029. [DOI] [PubMed] [Google Scholar]; d Tishchenko O, Truhlar DG, Ceulemans A, Nguyen MT. J. Am. Chem. Soc. 2008;130:7000–7010. doi: 10.1021/ja7102907. [DOI] [PubMed] [Google Scholar]
- 14.a Reference 12a. Manner VW, DiPasquale AG, Mayer JM. J. Am. Chem. Soc. 2008;130:7210–7211. doi: 10.1021/ja801672w.Warren JJ, Mayer JM. J. Am. Chem. Soc. 2008;130:2774–2776. doi: 10.1021/ja711057t.Mayer JM, Rhile IJ. Biochim. Biophys. Acta. 2004;1655:51–58. doi: 10.1016/j.bbabio.2003.07.002.Mayer JM, Rhile IJ, Larsen FB, Mader EA, Markle TF, DiPasquale AG. Photosynth. Res. 2006;87(1):3–20. 21–24. doi: 10.1007/s11120-005-8164-3.Mayer JM, Mader EA, Roth JP, Bryant JR, Matsuo T, Dehestani A, Bales BC, Watson EJ, Osako T, Valliant-Saunders K, Lam W-H, Hrovat DA, Borden WT, Davidson ER. J. Mol. Catal. A. 2006;251:24–33.
- 15.a Marcus RA, Sutin N. Biochim. Biophys. Acta, Rev. Bioenerg. 1985;811:265–322. [Google Scholar]; b Marcus RA, Sutin N. Inorg. Chem. 1975;14:213–216. [Google Scholar]
- 16.Kiefer PM, Hynes JT. J. Phys. Chem. A. 2004;108:11809–11818. and refs. therein. [Google Scholar]
- 17. cf. [Google Scholar]; a Shaik SS, Schlegel HB, Wolfe S. Theoretical Aspects of Physical Organic Chemistry: The SN2 Reaction. John Wiley & Sons, Inc.; New York: 1992. [Google Scholar]; b Knox JH. Advan. Chem. Ser. Vol. 76. American Chemical Society; 1968. Rate constants in the gas-phase oxidation of alkanes and alkyl radicals. pp. 1–22. [Google Scholar]; c Cohen N, Benson SW. J. Phys. Chem. 1987;91:171–175. [Google Scholar]; d Senkan SM, Quam D. J. Phys. Chem. 1992;96:10837–10842. [Google Scholar]; e Tsang W. In: Energetics of Organic Free Radicals. Simões JAM, Greenberg A, Liebman JF, editors. Blackie; New York: 1996. pp. 22–58. Chapter 2. [Google Scholar]
- 18.a Lowry TH, Richardson KE. Mechanism and Theory in Organic Chemistry. Harper & Row Publishers; San Francisco: 1976. [Google Scholar]; b Smith MB, March J. March's Advanced Organic Chemistry. 5th ed. Wiley-Interscience; New York: 2001. [Google Scholar]
- 19.The temperature dependence of ΔGPCET is discussed in:Pu J, Gao J, Truhlar DG. Chem. Rev. 2006;106:3140–3169. doi: 10.1021/cr050308e.Gao J, Ma S, Major DT, Nam K, Pu J, Truhlar DG. Chem. Rev. 2006;106:3188–3209. doi: 10.1021/cr050293k.Costentin C, Robert M, Savéant J-M. J. Am. Chem. Soc. 2007;129:9953–9963. doi: 10.1021/ja071150d.Costentin C, Robert M, Savéant J-M. J. Am. Chem. Soc. 2006;128:4552–4553. doi: 10.1021/ja060527x.Markle TF, Rhile IJ, DiPasquale AG, Mayer JM. PNAS. 2008;105:8185–8190. doi: 10.1073/pnas.0708967105.Rhile IJ, Markle TF, Nagao H, DiPasquale AG, Lam OP, Lockwood MA, Rotter K, Mayer JM. J. Am. Chem. Soc. 2006;128:6075–6088. doi: 10.1021/ja054167+.Costentin C. Chem. Rev. 2008;108:2145–2179. doi: 10.1021/cr068065t.
- 20.Tilset M. The thermodynamics of organometallic systems involving electron transfer paths. In: Balzani V, editor. Electron Transfer in Chemistry. Vol. 2. Wiley-VCH; Weinheim: 2001. pp. 677–713. [Google Scholar]
- 21.Wiberg KB, Foster G. J. Am. Chem. Soc. 1961;83:423–429. (thermochemical cycle is on p. 425).Eberson L. Acta Chem. Scand. 1963;17:2004–2018.Juan B, Scharz J, Breslow R. J. Am. Chem. Soc. 1980;102:5741–5748. and references therein.
- 22.Leading references:Bordwell FG, Cheng J-P, Ji G-Z, Satish AV, Zhang X. J. Am. Chem. Soc. 1991;113:9790–9795.Bordwell FG, Cheng J-P, Harrelson JA., Jr. J. Am. Chem. Soc. 1988;110:1229–1231.Bordwell FG, Satish AV, Zhang S, Zhang X-A. Pure & Appl. Chem. 1995;67(5):735–740.Cheng JP, Liu B, Zhao YY, Wen Z, Sun YK. J. Am. Chem. Soc. 2000;122:9987–9992.
- 23.Bordwell FG, Liu W-Z. J. Am. Chem. Soc. 1996;118:10819–10823. [Google Scholar]
- 24.A subset of the references using BDE's are:Parker VD, Handoo KL, Roness F, Tilset M. J. Am. Chem. Soc. 1991;113:7493–7498.Tilset M, Parker VD. J. Am. Chem. Soc. 1990;112:2843–2843.Tilset M, Parker VD. J. Am. Chem. Soc. 1989;111:6711–6717.Borovik AS. Acc. Chem. Res. 2005;38:54–61. doi: 10.1021/ar030160q.Zhang J, Grills DC, Huang KW, Fujita E, Bullock RM. J. Am. Chem. Soc. 2005;127:15684–15685. doi: 10.1021/ja0555724.Carrell TG, Bourles E, Lin M, Dismukes GC. Inorg. Chem. 2003;42:2849–2858. doi: 10.1021/ic025977e.Astruc D. Acc. Chem. Res. 2000;33:287–298. doi: 10.1021/ar9901319.Wang D, Angelici RJ. J. Am. Chem. Soc. 1996;118:935–942.Eisenberg DC, Norton JR. Isreali J. Chem. 1991;31:55–66.Simões JAM, Beauchamp JL. Chem. Rev. 1990;90:629–688.
- 25.For a few studies using bond dissociation free energies (BDFEs), see:Fu X, Wayland BB. J. Am. Chem. Soc. 2005;127:16460–16467. doi: 10.1021/ja054548n. and references therein. Miedaner A, Raebinger JW, Curtis CJ, Miller SM, DuBois DL. Organometallics. 2004;23:2670–2679.Ellis WW, Miedaner A, Curtis CJ, Gibson DH, DuBois DL. J. Am. Chem. Soc. 2002;124:1926–1932. doi: 10.1021/ja0116831.
- 26.Blanksby SJ, Ellison GB. Acc. Chem. Res. 2003;36:255–263. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- 27.a Lucarini M, Pedulli GF, Cipollone M. J. Org. Chem. 1994;59:5063–5070. [Google Scholar]; b Lucarini M, Pedrielli P, Pedulli GF, Valgimigli L, Gigmes D, Tordo P. J. Am. Chem. Soc. 1999;121:11546–11553. [Google Scholar]
- 28.Mader EA, Davidson ER, Mayer JM. J. Am. Chem. Soc. 2007;129:5153–5166. doi: 10.1021/ja0686918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The reactions examined here are net hydrogen atom transfers. Other work in progress in our laboratories indicates that they follow a concerted HAT mechanism as well, but this is independent of the thermochemical results here. See references 28, 31.
- 30.Roth JP, Yoder JC, Won TJ, Mayer JM. Science. 2001;294:2524–2526. doi: 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]
- 31.Yoder JC, Roth JP, Gussenhoven EM, Larsen AS, Mayer JM. J. Am. Chem. Soc. 2003;125:2629–2640. doi: 10.1021/ja0273905. [DOI] [PubMed] [Google Scholar]
- 32.a Wu A, Masland J, Swartz RD, Kaminsky W, Mayer JM. Inorg. Chem. 2007;46:11190–11201. doi: 10.1021/ic7015726. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wu A, Mayer JM. J. Am. Chem. Soc. 2008;130:14745–14754. doi: 10.1021/ja805067h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manner VW, Markle TF, Freudenthal J, Roth JP, Mayer JM. Chem. Commun. 2008;2:256–258. doi: 10.1039/b712872j. [DOI] [PubMed] [Google Scholar]
- 34.Mahoney LR, Ferris FC, Ingold KU. J. Am. Chem. Soc. 1969;91:3883–3889. [Google Scholar]
- 35.Mahoney LR, Mendenhall GD, Ingold KU. J. Am. Chem. Soc. 1973;95:8610–8614. [Google Scholar]
- 36.Roth JP, Lovell S, Mayer JM. J. Am. Chem. Soc. 2000;122:5486–5498. [Google Scholar]
- 37.Benson SW. Thermochemical Kinetics. Wiley; New York: 1968. [Google Scholar]
- 38.Lynden-Bell RM, Rasaiah JC. J Chem. Phys. 1997;107:1981–1991. [Google Scholar]
- 39.McMillen DF, Golden DM. Annu. Rev. Phys. Chem. 1982;33:493–532. [Google Scholar]
- 40.Wayner DDM, Parker VD. Acc. Chem. Res. 1993;26:287–294. and references therein. Tang L, Papish ET, Abramo GP, Norton JR, Baik MH, Friesner RA, Rappe A. J. Am. Chem. Soc. 2003;125:10093–10102. doi: 10.1021/ja034927l. 2006, 128, 11314.
- 41.a dos Santos RMB, Cabral BJC, Simoes JAM. Pure Appl. Chem. 2007;79:1369–1382. [Google Scholar]; b Mulder P, Korth H-G, Pratt DA, DiLabio GA, Valgimigli L, Pedulli GF, Ingold KU. J. Phys. Chem. A. 2005;109:2647–2655. doi: 10.1021/jp047148f. [DOI] [PubMed] [Google Scholar]; c dos Santos RMB, Simoes JAM. J. Phys. Chem. Ref. Data. 1998;27:707–739. [Google Scholar]
- 42.One reason that the BDE[PhOH]soln is still being debated in the literature revolves around the magnitude of solvation energies in different solvents and how best to interconvert between them. These effects are potentially magnified when dealing with transition metal complexes because of the increased polarity changes between oxidations states.
- 43.Pratt DA, Blake JA, Mulder P, Walton JC, Korth H-G, Ingold KU. J. Am. Chem. Soc. 2004;126:10667–10675. doi: 10.1021/ja047566y. [DOI] [PubMed] [Google Scholar]
- 44.NIST Standard Reference Database 69 . In: NIST Chemistry WebBook. Linstrom PJ, Mallard W, editors. National Institute of Standards and Technology; Gaithersburg MD: Jun, 2005. 20899. [Google Scholar]
- 45.Parker VD. J. Am. Chem. Soc. 1992;114:7458–7462.J. Am. Chem. Soc. 1993;115:1201. and correction.Roduner E. Radiat. Phys. Chem. 2005;72:201–206.Roduner E, Bartels DM. Ber. Bunsen-Ges. Phys. Chem. 1992;96:1037–1042.. It should be noted that the conversion between standard states in references (b) and (c) do not correctly account for the unit mol fraction standard state in solution or the conversion between 1 atm and 1 M standard states in the gas phase.
- 46.Using ΔH°solv[H•]s ≅ ΔH°solv[H2]s, ΔH°solv[H•]s = 1.56, 1.38 and 1.81 kcal mol−1 for MeCN, toluene, and ClCH2CH2Cl, respectively. ΔG°solv[H•]MeCN ≅ ΔG°solv[H2]MeCN = 5.12 kcal mol−1: Brunner E. J. Chem. Eng. Data. 1985;30:269–273.
- 47.Young LC, editor. Hydrogen and Deuterium. 5/6. Pergamon Press; New York: 1981. [Google Scholar]
- 48.Abraham MH, Grellier PL, Prior DV, Duce PP, Morris JJ, Taylor PJ. J.Chem. Soc., Perkin Trans. 2. 1989:699–711.Abraham MH, Grellier PL, Prior DV, Morris JJ, Taylor PJ. J. Chem. Soc., Perkin Trans. 2. 1990:521–529.Abraham MH, Grellier PL, Prior DV, Taft RW, Morris JJ, Taylor PJ, Laurence C, Berthelot M, Doherty RM, Kamlet MJ, Abboud J-LM, Sraidi K, Guihéneuf G. J. Am. Chem. Soc. 1988;110:8534–8536.Abraham MH, Platts JA. J. Org. Chem. 2001;66:3484–3491. doi: 10.1021/jo001765s.e This model is parameterized into the acidity (αH2) and the basicity (βH2) of the hydrogen bond donor and acceptor.
- 49.A review of several solvation models for PhOH41a suggests that the Ingold/Abraham H-bonding model is an overestimate of (ΔH°solv[A•]s - ΔH°solv[AH]s).
- 50.From ref. 41b: ΔH°solv[A•]MeCN - ΔH°solv[AH]MeCN - (ΔH°solv[A•]C6H6 - ΔH°solv[AH]C6H6 ) ≈ -χHbond×ΔH°Hbond,MeCN + χHbond×ΔH°Hbond,C6H6. χHbond is the fraction of hydrogen-bonded species in solution and logKHbond = 7.354×αH2,AH×βH2,solvent – 1.094. From Litwinienko G, Ingold KU. J. Org. Chem. 2003;68:3433–3438. doi: 10.1021/jo026917t. and ref. 48b: αH2(tBu3PhOH) = 0.24, βH2(MeCN) = 0.44, βH2(benzene) = 0.14.
- 51.Bakalbassis EG, Lithoxoidou AT, Vafiadis AP. J. Phys. Chem. A. 2003;107:8594–8606. [Google Scholar]
- 52.a Calculated from BDFE[tBu3PhOH]g = BDE[tBu3PhOH]g - T(S°[H•]g + S°[tBu3PhO•]g - S°[tBu3PhOH]g). S°[H•]g = 27.419 cal mol−1 K−144. S°[tBu3PhO•]g - S°[tBu3PhOH]g is assumed to be negligible based on the small values of the related entropies of formation {S°[benzyl radical]g - S°[toluene]g} and {S°[PhO•]g - S°[PhOH]g} (−0.47 and −0.8 cal mol−1 K−1). Entropies of formation from references 52(b)-(e). Curran H, Wu C, Marinov N, Pitz WJ, Westbrook CK, Burcat A. J. Phys.Chem. Ref. Data. 2000;29:463–517.Ruscic B, Boggs JE, Burcat A, Csaszar AG, Demaison J, Janoschek R, Martin JML, Morton ML, Rossi MJ, Stanton JF, Szalay PG, Westmoreland PR, Zabel F, Berces T. J. Phys. Chem. Ref. Data. 2005;34:573–656.Burcat A, Ruscic B. 2005. TAE Report No. 960; Technical Report, See also ftp://ftp.technion.ac.il/pub/supported/aetdd/thermodynamics.
- 53.Niyazymbetov ME, Evans DH. J. Chem. Soc. Perkin Trans. 2. 1993;7:1333–1338. [Google Scholar]
- 54.Grampp G, Landgraf S, Muresanu C. Electrochim. Acta. 2004;49:537–544. This value was reported vs. SCE and converted to vs. Cp2Fe+/0 by adding +0.4 V Connelly NG, Geiger WE. Chem. Rev. 1996;96:877–910. doi: 10.1021/cr940053x.
- 55.Bordwell FG, Cheng J-P. J. Am. Chem. Soc. 1991;133:1736–1743. [Google Scholar]
- 56.Kutt A, Leito I, Kaljurand I, Soovali L, Vlasov VM, Yagupolskii LM, Koppel IA. J. Org. Chem. 2006;71:2829–2838. doi: 10.1021/jo060031y. [DOI] [PubMed] [Google Scholar]
- 57.The reported calorimetric value35 for ΔH°f[TEMPO - TEMPOH]C6H6 of 16.52 ± 0.5 kcal mol−1 was corrected for the revised ΔH°f of azobenzene.43 The values of ΔH°f[H•]C6H6 and ΔH°f[H•]MeCN are given above, and S°[TEMPO]MeCN is taken as equal to S°[TEMPOH]MeCN for the reasons given above.
- 58.Mader EA, Mayer JM. In preparation.
- 59.a Turner JW, Schultz FA. J. Phys. Chem. B. 2002;106:2009–2017. [Google Scholar]; b Turner JW, Schultz FA. Inorg. Chem. 2001;40:5296–5298. doi: 10.1021/ic0013678. [DOI] [PubMed] [Google Scholar]; c Sharpe P, Kebarle P. J. Am. Chem. Soc. 1993;115:782–789. [Google Scholar]; d Kratochvil B, Knoeck J. J. Phys. Chem. 1966;70:944–946. [Google Scholar]; e Hupp JT, Weaver MJ. Inorg. Chem. 1983;22:2557–2564. [Google Scholar]; f Sahami S, Weaver MJ. J. Electroanal. Chem. Interfacial Electrochem. 1981;122:155–170. [Google Scholar]; g Hupp JT, Weaver MJ. Inorg. Chem. 1984;23:3639–3644. [Google Scholar]; h Yee EL, Weaver MJ. Inorg. Chem. 1980;19:1077–1079. [Google Scholar]
- 60.a Turner JW, Schultz FA. Inorg. Chem. 1999;38:358–364. [Google Scholar]; b Sahami S, Weaver MJ. J. Electroanal. Chem. Interfacial Electrochem. 1981;122:171–181. [Google Scholar]
- 61.a Lay PA, McAlpine NS, Hupp JT, Weaver MJ, Sargeson AM. Inorg. Chem. 1990;29:4322–4328. [Google Scholar]; b Koval CA, Gustafson RM, Reidsema CM. Inorg. Chem. 1987;26:950–952. [Google Scholar]; c Moattar F, Walton JR, Bennett LE. Inorg. Chem. 1983;22:550–553. [Google Scholar]
- 62.a Hupp JT, Weaver MJ. Inorg. Chem. 1984;23:256–258. [Google Scholar]; b Ogino H, Ogino K. Inorg. Chem. 1983;22:2208–2211. [Google Scholar]
- 63.Schmitz JEJ, Van der Linden JGM. Inorg. Chem. 1984;23:3298–3303. [Google Scholar]
- 64.Tabib J, Hupp JT, Weaver MJ. Inorg. Chem. 1986;25:1916–1918. [Google Scholar]
- 65.a Schmitz JEJ, Van der Linden JGM. Inorg. Chem. 1984;23:117–119. [Google Scholar]; b Crawford PW, Schultz FA. Inorg. Chem. 1994;33:4344–4350. [Google Scholar]; c Gao Y-D, Lipkowitz KB, Schultz FA. J. Am. Chem. Soc. 1995;117:11932–11938. [Google Scholar]; d Sharpe P, Kebarle P. J. Am. Chem. Soc. 1993;115:782–789. [Google Scholar]; e Ogino H, Nagata T, Ogino K. Inorg. Chem. 1989;28:3656–3659. [Google Scholar]; f Youngblood MP, Margerum DW. Inorg. Chem. 1980;19:3068–3072. [Google Scholar]; g Kadish KM, Das K, Schaeper D, Merrill CL, Welch BR, Wilson LJ. Inorg. Chem. 1980;19:2816–2821. [Google Scholar]; h George P, Hanania GIH, Irvine DH. Recl. Trav. Chim. Pays-Bas Belg. 1956;75:759–762. [Google Scholar]; i Blonk HL, Roelofsen AM, Frelink T, Anders MJ, Schmitz JEJ, Van der Linden JGM, Steggerda JJ. J. Phys. Chem. 1992;96:6004–6012. [Google Scholar]; j Weaver MJ, Nettles SM. Inorg. Chem. 1980;19:1641–1646. [Google Scholar]; k George P, Hanania GIH, Irvine DH. J. Chem. Soc. 1959:2548–2554. [Google Scholar]; l Zhu T, Su CH, Schaeper D, Lemke BK, Wilson LJ, Kadish KM. Inorg. Chem. 1984;23:4345–4349. [Google Scholar]; m Noviandri I, Brown KN, Fleming DS, Gulyas PT, Lay PA, Masters AF, Phillips L. J. Phys. Chem. B. 1999;103:6713–6722. [Google Scholar]; n Moulton R, Weidman TW, Vollhardt KPC, Bard AJ. Inorg. Chem. 1986;25:1846–1851. [Google Scholar]; o Ryan MF, Eyler JR, Richardson DE. J. Am. Chem. Soc. 1992;114:8611–8619. [Google Scholar]; p Fabbrizzi L, Mariani M, Seghi B, Zanchi F. Inorg. Chem. 1989;28:3362–3366. [Google Scholar]; q Fabbrizzi L, Perotti A, Profumo A, Soldi T. Inorg. Chem. 1986;25:4256–4259. [Google Scholar]; r Blackbourn RL, Hupp JT. Inorg. Chem. 1989;28:3786–3790. [Google Scholar]; s Curtis JC, Blackbourn RL, Ennix KS, Hu S, Roberts JA, Hupp JT. Inorg. Chem. 1989;28:3791–3795. [Google Scholar]
- 66.a Turner JW, Schultz FA. Coord. Chem. Rev. 2001;219−221:81–97. [Google Scholar]; b Goodwin HA. Top. Curr. Chem. 2004;233:59–90. [Google Scholar]
- 67.a Richardson DE, Sharpe P. Inorg. Chem. 1993;32:1809–1812. [Google Scholar]; b Richardson DE, Sharpe P. Inorg. Chem. 1991;30:1412–1414. [Google Scholar]
- 68.a Sorai M, Seki S. J. Phys. Chem. Solids. 1974;35:555–570. [Google Scholar]; b van Koningsbruggen PJ, Maeda Y, Oshio H. Top. Curr. Chem. 2004;233:259–324. [Google Scholar]; c Gutlich P, Goodwin HA. Spin Crossover in Transition Metal Compounds I. Vol. 233. Springer-Verlag Berlin; Berlin: 2004. Spin crossover - An overall perspective. pp. 1–47. [Google Scholar]; d Konig E. Struct. Bond. 1991;76:51–152. [Google Scholar]
- 69.Note that spin-crossover entropies are substantially larger than expected solely from the electronic multiplicities. For example, ΔS°elec is −5.4 cal mol−1 K−1 for high-spin 5T2g FeII ⇄ low-spin 1A1g FeII, much less than the measured ΔS°SCO = −21 cal mol−1 K−1 for FeII(H2bip).31
- 70.Similar differences in ΔS°ET are seen for hexaaquo (22 cal mol−1 K−1),62,72 trisethylenediamine (20 ± 4 cal mol−1 K−1),60a and tris-2−2′-bipyridine (19 ± 3 cal mol−1 K−1)59 complexes of Co and Ru.
- 71.On the other hand, the value of the ET entropy for a single complex is highly solvent dependent. ΔS°ET for [Ru(tacn)3]3+/2+, for example, is 26.2, 31.1, and 8.6 cal mol−1 K−1 in DMSO, acetone, and water, respectively (tacn = 1,4,7-triazacyclononane).60a, For aqueous [M(OH2)6]3+/2+ ions and related species, the strong hydrogen bonding can lead to large solvent contributions to ΔS°ET.67
- 72.Yee EL, Cave RJ, Guyer KL, Tyma PD, Weaver MJ. J. Am. Chem. Soc. 1979;101:1131–1137. [Google Scholar]
- 73.ΔS°ET for CoIII/CoII redox couples in protic media vary more widely (−6 to +60 cal mol−1 K−1)62 because other effects can contribute.67
- 74.For example, [Fe(phen)3]3+/2+ (phen = phenanthroline) is low-spin for both FeIII and FeII and has ΔS°ET = 25.4 ± 2 cal mol−1 K−1 in MeCN, similar to [Ru(bpy)3]3+/2+ (27 cal mol−1 K−1).59d,g [Fe(tacn)2]3+/2+ has ΔS°ET = 36.3 cal mol−1 K−1 in MeCN; 29.2 cal mol−1 K−1 when the spin-equilibrium contribution at FeII is removed.60a [Ru(tacn)2]3+/2+ = 27.9 cal mol−1 K−1 in MeCN.60a
- 75.a Farhangrazi ZS, Fosett ME, Powers LS, Ellis WR., Jr. Biochemistry. 1995;34:2866–2871. doi: 10.1021/bi00009a017. [DOI] [PubMed] [Google Scholar]; b Sailasuta N, Anson FC, Gray HB. J. Am. Chem. Soc. 1979;101:455–458. [Google Scholar]; c Ellis WR, Jr., Wang H, Blair DF, Gray HB, Chan SI. Biochemistry. 1986;25:161–167. doi: 10.1021/bi00349a023. [DOI] [PubMed] [Google Scholar]
- 76.a Taniguchi VT, Sailasuta-Scott N, Anson FC, Gray HB. Pure Appl. Chem. 1980;52:2275–2281. [Google Scholar]; b Battistuzzi G, Borsari M, Sola M. Eur. J. Inorg. Chem. 2001;2001:2989–3004. [Google Scholar]
- 77.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. http://www.glasscontour.com/index.html.
- 78.Ozinskas AJ, Bobst AM. Helv. Chim. Acta. 1980;63:1407–1411. [Google Scholar]
- 79.Frisch MJ, et al. Gaussian 03, Revision D.02. 2004. . See Supporting Information for full citation.
- 80.a Cancès MT, Mennucci B, Tomasi J. J. Chem. Phys. 1997;107:3032–3041. [Google Scholar]; b Cossi M, Barone V, Mennucci B, Tomasi J. Chem. Phys. Lett. 1998;286:253–260. [Google Scholar]; c Mennucci B, Tomasi J. J. Chem. Phys. 1997;106:5151–5158. [Google Scholar]
- 81.Cossi M, Scalmani G, Rega N, Barone V. J. Chem. Phys. 2002;117:43–54. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.