

Abstract
The striking clinical benefit of PTH in osteoporosis began a new era of skeletal anabolic agents. Several studies have been performed, new studies are emerging out and yet controversies remain on PTH anabolic action in bone. This review focuses on the molecular aspects of PTH and PTHrP signaling in light of old players and recent advances in understanding the control of osteoblast proliferation, differentiation and function.
Keywords: PTH, PTHrP, PTH1R, osteoblast, signaling
I. INTRODUCTION
The skeleton serves a structural function-providing mobility, support, and protection for the body; a reservoir function- being a storehouse for essential minerals calcium and phosphorus; an endocrine function-regulating extracellular mineral concentrations by responding to hormonal signals; immune function and cellular regeneration function housing multi-potent stem cells. Maintaining a strong and healthy skeleton is a complex process that integrates multiple endocrine, cellular and immune signals. Therefore, bone is an active organ whose structure is constantly changing. Old bone breaks down and new bone is formed on a continuous basis through the process of remodeling. Remodeling involves a coordinated action of a number of cells which work in concert in what is referred to as the Basic Muticellular Unit (BMU, Fig. 1) (see review) [1].
Figure 1.

Bone remodeling. A scheme illustrating how the osteoclasts derive from hematopoietic stem cells through coordinated interaction of diverse factors; and osteoblasts from bone marrow stromal cells. The initial organic matrix (osteoid) is later calcified to form mature bone within the cavity created by osteoclasts with inclusion of osteoblasts as osteocytes within lacunae and lining cells. BMSC; bone marrow stromal cell; RANK, receptor activator of nuclear factor-κB ligand; RANKL, RANK ligand; M-CSF, macrophage-colony-stimulating factor; cFMS, cell-surface receptor in hematopoietic precursors.
In the remodeling process bone is destroyed or resorbed by osteoclasts and then laid down by osteoblasts. The lifespan of a single BMU is about 6–9 months during which several generations of osteoclasts (average life of about 2 weeks) and osteoblasts (average life of about 3 months) are formed. BMUs operate at an average speed of 25 μm/day; one single BMU replaces about 0.025 mm3 of bone during its life span and about 10% of the skeleton is replaced each year. A new BMU may be formed every 2–5 years at the same site, thus the whole skeleton is replaced several times during the lifespan of an individual without changing much the size or the shape of bone. In contrast, during fetal life and childhood, bone is also continuously replaced; however, this process of bone modeling increases the mass and the size of the skeleton.
Osteoblasts and osteoclasts respond to multiple autocrine, paracrine and endocrine factors and to mechanical stimulation affecting the whole skeleton (systemic) or affecting only a small region of the skeleton (local). Osteoblasts precursors and mature osteoblasts release local factors which regulate osteoclast differentiation and activity within the BMU and thus bone resorption and bone formation are tightly coupled [1]. Since remodeling occurs in adult life, abnormalities in remodeling are the primary cause of adult bone diseases. Therefore, it is critically important to understand the remodeling process by examining the roles/actions of factors controlling the activities of osteoblasts and osteoclasts.
In adults, bone mass is determined by the net result of bone remodeling. The entire process is tightly regulated by the hormonal milieu and the local bone environment. Decreased bone mass, osteopenia or osteoporosis, may result from accelerated resorption or defective formation of bone matrix, both of which are regulated by multiple positive and negative humoral local signals. Osteopenia may result from alterations in hormone action, such as loss of normal estrogen production in post-menopausal women, excessive production of parathyroid hormone (PTH) as in primary or secondary hyperparathyroidism, or glucocorticoid excess as a consequence of chronic steroid use in immunosuppressive therapy. Recent research has emphasized establishing a more complete understanding of the hormonal regulation of bone and developing anabolic agents with therapeutic potential for the treatment of low bone mass. The goal of this paper is to focus on the putative mechanism(s) of action of PTH and PTH related peptide (PTHrP) on bone formation.
II. PTH, PTHrP AND THEIR RECEPTOR
PTH and PTHrP are distinct polypeptides that show limited overall sequence homology (16%) [2]. Because significant homology is clustered within their N-termini, 9 amino acid residues out of their 1–13 sequences are identical. PTH and PTHrP can bind to and activate a common G-protein coupled receptor, the PTH/PTHrP receptor or PTH1R [3] which is expressed in PTH and PTHrP target cells, such as osteoblasts in bone and renal tubular cells in kidney. The extreme differences in their primary sequences also suggest that PTH and PTHrP sub serve distinct biological functions although PTHrP can mimic many of the functions of PTH. While PTH is synthesized by and secreted from the parathyroid glands, PTHrP is synthesized and expressed by various tissues such as skin, blood vessels, smooth muscles, tooth buds, growth plate chondrocytes, bone, kidney and neuronal and glial tissues. The role of PTHrP in endochondral bone formation has been studied extensively. Targeted disruption of PTHrP or PTH1R gene in mice resulted in perinatal death with gross skeletal abnormalities [4–7] whereas heterozygous PTHrP+/− demonstrated a reduction in trabecular bone volume [8] (Table 1).
Table 1.
PTH and PTHrP
| PTH | PTHrP | |
|---|---|---|
| Structure |
|
|
| Function | Ca and Phosphate homeostasis |
|
| Secretion | Endocrine | Paracrine/Autocrine |
| Target Tissues | Osteoblast Renal tubular cells | Chondrocytes, osteoblasts placental cells, skin, hair follicles, brain, teeth |
| Receptor | A common PTH/PTHrP Receptor (or PTH1R) | |
The PTH/PTHrP receptor is a G protein-coupled receptor with 7 transmembrane spanning domains. The PTH/PTHrP receptor is encoded by a multi-exonic gene (Fig 2) with potential for alternate splicing and alternate promoter usage that was characterized in human, rat and mouse [9]. Understanding the physiological roles, molecular and cellular actions of PTH and PTHrP began when PTH1R was first cloned in 1990s [3, 10]. The gene encoding the PTH1R is located on chromosome 3 in humans and the gene involved in its synthesis has a total of 14 exons. The amino terminal region of PTH(1–34) and PTHrP(1–34) interacts with the J- domain, the functional portion of the receptor that contains the seven transmembrane spanning helices and the connecting loops [11]. The carboxy terminal portion of PTH and PTHrP binds to the extracellular N-domain of the receptor promoting association of the biologically active amino terminal of the ligand to J-domain [12] (Fig. 2).
Figure 2.

Primary sequence of the rat PTH/PTHrP receptor [9,11]. Different colors represent sequences encoded by different exons and predicted extracellular, trans-membrane spanning and cytoplasmic domains are shown. Exon nomenclature is as follow: S for exon encoding signal peptide, E1, E2 and E3 for exons encoding the extracellular extension, M1, M2, M3, M4, M5a, M5b, M6/7a and M7b for exons encoding the trans-membrane domains and portions of their connecting loops and T for the exon encoding the cytoplasmic tail. The J-domain refers to the trans-membrane domains and the connecting loops.
Following receptor-agonist interaction inactivation of PTH1R occurs via phosphorylation which eventually becomes desensitized, internalized and recycled [13, 14]. Using phosphorylation- deficient PTH1R mice recent studies demonstrate inability of PTH to normally internalize the receptor resulting in sustained increase in cAMP concentration in these animals [15, 16]. The impact of deficient internalization on bone development, maturation and turnover is not yet known.
Genetic manipulation of PTH1R using homologous recombination and transgenic technology revealed important physiological role for the PTH1R in bone development and bone cell differentiation [17–19]. PTH1R knock out mice have decreased trabecular bone and increased thickness of cortical bone during fetal development [19]. Blomstrand chondrodysplasia, an autosomal recessive disorder caused by inactivating PTH1R mutations, also showed intra-uterine death and severe skeletal abnormalities [18]. Histomorphometric analysis of bone from a patient with Jansen’s metaphyseal chondrodysplasia, which is caused by an activating mutation of PTH1R [17], shows loss of cortical bone without any loss of trabecular bone [20]. Transgenic mice expressing constitutively active PTH1R in osteoblasts under the control of Col-1A promoter increased the trabecular bone dramatically and decreased cortical bone thus illustrating both the anabolic and catabolic actions mediated by PTH1R [21]. PTH1R is expressed in mature and pre-oseoblasts during rat fetal development [22], in osteoblastic osteosarcoma cell [23], and normal osteoblastic cells [24].
Additional receptors for PTH and PTHrP have been described. PTH2R [25–27], cloned by sequence homology to PTH1R, was first characterized as a receptor for PTH only since it does not bind PTHrP, however, the hypothalamic tuberoinfundibular peptide (TIP39) was later characterized as a high affinity ligand for PTH2R. PTH2R therefore may not be involved in bone metabolism. PTH3R, which is identified in zebrafish [28] and sea bream enterocytes [29] preferentially binds PTHrP. PTH3R, which is not seen in mammals, may represent an important phylogenetic step in the development of the PTH/PTHrP system during evolution of species.
III. ANABOLIC AND CATABOLIC ACTIONS OF PTH AND PTHrP ON BONE
PTH binds to cells of the osteoblast lineage [30] and produces both anabolic and catabolic effects. The fact that PTH has dual effects depending on its administration method raises important questions about its mechanisms of action on bone formation and resorption. It was hypothesized that PTH and PTHrP’s anabolic and catabolic effects on osteoblasts take place by activating different signaling cascades from PTH1R (reviewed in [31]). Traditionally PTH was known to be catabolic to the human skeleton as severe osteoporosis and osteitis fibrosa cystica may complicate long standing hyperparathyroidism. In 1932 Selye reported the PTH’s ability to stimulate osteogenesis [32]. Subsequently the anabolic effects of PTH have been examined in greater detail [33–46]. In randomized clinical trial PTH was shown to be useful to prevent fracture in osteoporotic subjects [37, 43, 46]. It was initially noted that PTH could increase bone mass in rats [32, 33]. Now it is well recognized that intermittent administration of PTH and PTHrP has net anabolic effects on bone [34, 35, 39, 42, 44, 45]. Once a day subcutaneous injection of PTH increased bone mass in patients with osteoporosis [36] and in ovariectomized monkeys [38]. The capability of PTH to augment bone formation is dependent upon the hormone being administered in a way that yields a transient peak blood level [40, 41]. The increased bone formation is largely due to a rise in osteoblast number as a result of increased proliferation and differentiation of osteoblasts in vitro and in vivo [45–53], decrease in osteoblast apoptosis [54, 55], and activation of bone lining cells[35, 56]. A mechanism involving cell-cell contact in PTH induced osteoblast proliferation has also been suggested [57].
PTH stimulates proliferation of rat and human osteosarcoma-derived osteoblast like cells [49]. The involvement of PTHrP in intramembranous bone formation has also been suggested in an experimental rabbit model [58] and in cells of osteoblast lineage [59]. Interestingly, the response to PTH and PTHrP is largely dependent on the cell lines, the species, and differentiation stage of osteoblasts [45, 60, 61]. Earlier studies have shown that PTHrP stimulates proliferation of chondrocytes, primary spongiosa of rat trabecular bone in vivo and inhibits osteoblast proliferation in vitro [62, 63]. Interruption of PTHrP signaling in vivo in mice caused a reduction in the number of proliferative chondrocytes as a well as premature differentiation of these cells [4, 5]. In fact PTH and PTHrP play important roles at multiple stages during bone development through their effects on cell survival [55]. While PTHrP prevents apoptosis of immature pre-confluent mesenchymal cells, it induces apoptosis of more mature post- confluent cells [64]. These data suggested that PTHrP is required for normal turnover of bone cells, and activation of apoptosis in mature osteoblasts could represent a potential mechanism whereby cells no longer forming matrix are cleared to provide access for new cells. Besides PTH’s clinical use for the treatment of osteoporosis, its potential for the application in fracture healing, tissue engineering and implant integration are being explored [65–68].
Continuous administration of PTH or PTHrP induces bone resorption by activating osteoclasts indirectly through their actions on osteoblastic cells [69]. When osteoclasts were physically separated from osteoblasts they did not respond to PTH [70]; however, PTH induced resorption only after adding osteoblasts or osteoblast like cells to the cultured osteoclasts [71]. Although osteoblastic cells mediate osteoclastic responsiveness to PTH [71], some studies indicated that osteoclasts from several species possess functional PTH receptors. The presence of PTH1R in human osteoclast was initially suggested by Langub [72] and more recently by Dempster et al [73]. PTH stimulated osteoclast activity in the absence of osteoblasts, proposing direct and indirect actions of PTH on osteoclasts. In addition to osteoclastic bone resorption PTH is also a potent activator of osteoclastic mobility [71].
The various actions of PTH on cells of the osteoblast lineage affect the complex process of bone remodeling; the molecular mechanism(s) responsible for the coordinated coupling of osteoblastogenesis and osteoclastogenesis in bone turnover has been partially elucidated. Several effects of PTH on osteoclast formation are mediated by its actions on the production of the receptor activator of nuclear factor-κB ligand (RANKL) [74] and its soluble decoy receptor for RANKL, osteoprotegerin (OPG) [75, 76]. PTH stimulates RANKL and inhibits OPG mRNA expression [77] and dependent on the stage of differentiation of the osteoblastic cells [78, 79]. Finally, PTH induced osteoblastic expression of monocyte chemoattractant protein-1 (MCP-1) in recruitment and differentiation of osteoclast precursors suggested a role for MCP-1 and enhanced bone remodeling that accompanies the anabolic effects of PTH [80]. The anabolic effectiveness of PTH and how it relates to osteoblast physiology has been recently reviewed [81, 82].
IV. PTH AND PTHrP REGULATED SIGNALING PATHWAYS
Activation of the PTH1R by PTH or PTHrP causes an acute increase in several intracellular signaling molecules which include the 2 classic G protein signaling cascades, adenylate cyclase (AC) and phospholipase C (PLC) [10], and several recently recognized signaling proteins through protein-protein interactions [83–85]. Stimulation of the AC signaling cascade leads to activation of protein-kinase A (PKA) and phosphorylation of transcription factors, such as CREB, which regulates transcription of PTH target genes. Stimulation of the PLC signaling cascade leads to accumulation of inositol trisphosphate (IP3) and diacylglycerol (DAG); IP3 increases intracellular calcium concentration and DAG activates protein kinase C (PKC); and consequently genomic effects through transcription factors regulated by the calcium and PKC (Fig 3). Shortly after the molecular cloning of the PTH/PTHrP receptor it was noticed that relatively high agonist concentrations (micromolar) and/or high receptor density are required for efficient activation of PLC; this is in contrast to PTH1R activation of AC which occurs at physiologic (sub-nanomolar) agonist concentrations in the same cell host [86]. The higher agonist requirement for activation of PLC is intriguing since the physiologic PTH concentrations in the serum are sub-nanomolar; this raised the hypothesis that the PLC pathway may be physiologically relevant when the local agonist concentrations are elevated; this may occur at site of high local PTHrP secretion, such as in the developing growth plate. Interestingly, it has been shown that the Na+/H+ exchanger regulatory factor (NHERF) binds to a PDZ domain within the carboxy-terminal tail of PTH1R and shifts its signaling efficiency from AC to PLC through efficient coupling of the PTH1R-NHERF complex to Gq/11, which stimulates PLC, and Gi, which inhibits AC (Fig. 3) [87]. NHERF may play an important role in PTH inhibition of phosphate transport in the polarized renal tubular cells, which express high levels of NHERF [88]. A role for NHERF was also suggested for PTH1R desensitization [89]. Recently, the phosphorylation-deficient PTH1R, constructed by mutating the phosphorylation site on its carboxy-terminal tail, was shown to couple more efficiently to PLC than the wild type receptor [16, 90]; this finding suggests that activation of PLC may be a physiological process that normally occurs but that desensitizes rapidly through the process of receptor phosphorylation.
Figure 3.
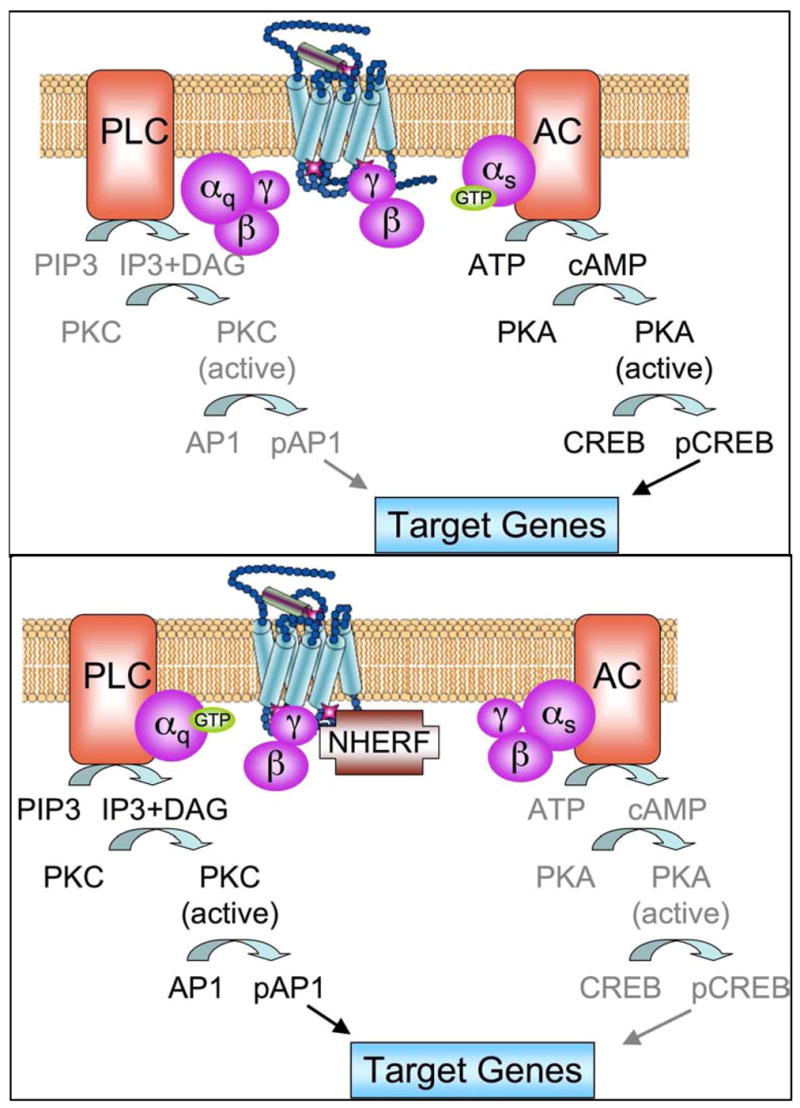
A schema for the signaling pathway regulated by NHERF. Upper panel shows the dominant cAMP signaling pathways in most cells. Lower panel illustrates how NHERF shift signaling from adenylate cyclase (AC) dominance to phospholipace C (PLC) dominance.
PTH mediated activation of PKC may also occur via non-phospholipase C mediated pathway [91] and PTH can increase intracellular calcium via stimulation of extracellular calcium influx through PKC-dependent and/or cAMP-dependent actions on calcium channels [92, 93].
While PTH(1–34) activates both AC and PLC several investigators have shown that shorter PTH analogs with intact N-terminus have signaling specificity that favor stimulation of cAMP accumulation [94–97]. Substitution of few residues within PTH(1–34) or its shorter fragment with non-natural amino acids have improved affinity and potency of the shorter fragments with more selectivity for signaling through Gs-adenylate cyclase [98]. Deletion of the first 2 residues of PTH resulted in a competitive antagonist [99]. Other investigators reported stimulation of PKC by N-terminally-truncated PTH analogs in vitro [100]. In recent years using different analogs of PTH the mechanisms of ligand binding to the PTH1R has been a subject of intense study. These studies provide new clues for understanding the selectivity, structure, stability, bioreactivity and overall topology of the biomolecular complex [101–105]. Using several pharmacological and biophysical approaches recent studies have also shown that PTH and PTHrP have characteristic individual effects on the conformation of the PTH1R [105]; this observation suggests that PTH and PTHrP actions mediated by the same receptor may be distinct.
The second protein target for cAMP, the cAMP-guanine nucleotide exchange factor (cAMP-GEF)/Epac, which is a Rap1 specific GEF (Rap-1/B-Raf) [106, 107], was also found to be functional in osteoblastic cells; EPAC may be involved in PTH and PTHrP signaling in osteoblasts and may mediate some of its anabolic actions [108, 109].
PTH and PTHrP added to cell lines expressing recombinant PTH1R transiently stimulated ERK1/2 phosphorylation [110]. However, in normal osteoblasts and in several osteoblastic osteosarcoma cell lines PTH inhibited ERK1/2 phosphorylation [111, 112]. It was shown that PTH1R activation may have opposing effects on the ERK-MAPK pathway in osteoblast depending on whether they are proliferating or already differentiated [45, 61, 112]. In differentiated osteosblastic cells PTH (or PTHrP) inhibited ERK1/2 phosphorylation and induced growth arrest of differentiated osteoblastic cells [61]. On the other hand, when added to proliferating osteoblasts PTH (or PTHrP) did not inhibit ERK1/2 phosphorylation and, in fact, they promoted cell growth [45]. Interestingly, several G-protein coupled receptors (GPCRs) became associated with the p42/p44 MAPK (ERK) module when activated by their cognate ligands through an internalization process that involves receptor phosphorylation and recruitment of β-arrestins [113]. PTH mediated activation of β-arrestin leads to desensitization of the cAMP response and at the same time activation of the ERK1/2 signaling cascade [110, 114]. Studies with β-arrestin null mice showed decreased bone mass following PTH treatment [115]; this suggested that β-arrestins limit osteoclastogenesis during intermittent PTH administration. PTH was also shown to stimulate p38-MAPK in early differentiating osteoblastic cells and increase mineralization at later stages has also been suggested [116]. Finally, PTH and PTHrP induced MAPK phosphatase-1, an enzyme responsible for dephosphorylation of MAPKs [117–119]. These studies indicate an important role for MAPKs in the effects of PTH and PTHrP on osteoblasts.
The Wnt signaling pathway has recently been demonstrated to play an important role in bone cell function and several studies revealed a possible role for Wnt signaling in PTH actions in bone. Canonical Wnt signaling is transduced by a receptor complex comprised of a member of the family of seven transmembrane domain receptors known as Frizzleds, and the co-receptor lipoprotein receptor related proteins 5 or 6 (LRP5/6) [120–123]. The resulting signal leads to the activation of the cytoplasmic protein disheveled (Dsh), disrupting the protein complex of axin, adenomatous polyposis coli (APC), and glycogen synthase kinase-3b (GSK-3b). As a result, phosphorylated β-catenin translocates to the nucleus, forms a complex with T-cell factor/Lymphoid enhancer factor (TCF/LEF), which then regulates the transcription of Wnt target genes [124, 125]. In rat distal metaphyseal bone in vivo and osteoblastic UMR 106 cells PTH and PTHrP treatment results in the differential regulation of the receptor complex, up-regulate the mRNA expression of LRP6 and FZD-1 and decreases LRP5 and Dkk-1 [126]. PTH and PTHrP also increase the levels of β-catenin and induces the further downstream signaling response [126]. These results suggest a possible role for Wnt signaling in PTH actions in bone. PTH enhances TCF-dependent transactivation and inactivation of GSK-3β in human osteoblastic cell line SaOS2 [127]. This may inhibit bone resorption by a negative impact on RANKL expression on osteoblasts. Studies in mice lacking LRP5 have shown that PTH increases femoral cortical bone mass suggesting that this effect does not require Wnt signaling; the latter may facilitate the anabolic response in cancellous bone [128–130]. Overall, these studies support a role for LRP5 mediated Wnt signaling in the anabolic effect of PTH in bone. A very recent study in rat reveals a novel signaling pathway of PTH by demonstrating that a ternary complex containing PTH, PTH1R, and LRP6 promotes rapid phosphorylation of LRP6, stabilization of β-catenin in osteoblasts with a concurrent increase in bone formation in a distinct manner from that of the canonical Wnt signaling [131].
V. MEDIATORS OF BONE TISSUE REGENERATION: PTH AND PTHrP TARGETS
Mouse and human genetics have provided a greater understanding of how stem cells differentiate into chondrocytes, osteoblasts and osteoclasts and the complexities of the skeletal biology [132]. Full understanding of bone cell physiology and bone formation depends upon characterization of the molecular events occurring during the differentiation and proliferation of bone cells. Studies demonstrate that during the process of differentiation progenitor cells acquire specific phenotype under the control of regulatory factors that regulate induction and transcription of an array of temporally expressed genes. It appears that PTH1R actions may involve a number of growth factors, transcription factors, and various other regulatory genes (Table 2).
Table 2.
Known mediators of PTH regulation
| Growth Factors | Transcription Factors | Signaling Pathways | Regulatory Molecules | Cell-Cycle Proteins |
|---|---|---|---|---|
| TGF-β IGF-I and II BMPs bFGF |
CREB AP-1 Runx2 |
cAMP PKA PKC Rap-1 Wnt MKP-1 MAPK |
MMP-13 Collagen-I ALKP OCN, OPN OPG RANKL MCSF SOX4, IL6 Bcl2 Connexin Nurr1 Nurr77 |
CDK1 (cdc2) CDK2 p27 p21 p16 Cyclin D1 |
1. Growth factor regulation
Local cytokines and growth factors are recognized as important modulators of the action of hormones on bone cells. Cytokines and growth factors are also important mediators of cell-to-cell and matrix-to-cell communications. They can change the number of receptors for calciotropic hormones on bone cells, while these hormones modulate the production rate of these factors by bone cells [133]. Research performed during the past several years implicates important roles for a variety of growth factors and cytokines that affect osteoblasts or their precursors during bone development, remodeling, or repair. Of these, three families of growth factors, the transforming growth factor beta (TGF-β), insulin-like growth factors (IGFs), and bone morphogenetic proteins (BMPs), are important local regulators of osteogenesis. TGF-β inhibits osteoblasts proliferation and stimulates the production of bone extracellular matrix (ECM) proteins, including type I collagen, fibronectin, and osteocalcin [134]. It has been also implicated as a coupling factor that coordinates the processes of bone resorption and subsequent bone formation [135]. Modifications of the production of IGF-I, or of one of its binding proteins, could be responsible for low bone formation during ageing has been suggested [133]. Most systemic and local cytokines are active in bone; however, the existence of a bone-specific cytokine(s) has been questioned. Studies suggest that interleukin-6 (IL-6) production by osteoblasts is responsible for increased bone resorption after ovariectomy in mice. The importance of the events that link these growth factors to nuclear proteins in response to glucocorticoids, sex steroids, prostaglandin E2 (PGE2), PTH, or PTHrP are now being appreciated.
Several laboratories have provided convincing evidences that PTH-induced stimulation of bone formation involves local IGF-I, IGF-II and TGF-β and growth factors, which may act as mediators of the physiological effects of PTH in bone [136]. PTH stimulates the release of IGF-I from fetal rat and mouse calvariae [50], and IGF-I and IGF-II from mouse calvariae [137]. While short time PTH exposure of cultured osteoblastic cells did not alter mRNA level of IGF-I [138], increased transcription or peptide levels were noted in rat bone cultures[139] and osteoblasts after long exposure in vivo [140]. Increased IGF-I by PTH treatment promoted differentiation and survival of osteoblasts [141, 142]. Since IGF-I and IGF-II stimulated osteoblastic cell proliferation, the effect of PTH on the release of these and other growth factors may mediate coupling of bone formation to bone resorption [137]. The anabolic actions of PTH on bone were suppressed in IGF-I knock out mice [143, 144]. Recent studies indicated that IGFI receptor null mutation in mature osteobasts lead to decreased bone formation, in part because IGF-IR expression in mature osteoblasts was required for PTH stimulation of osteoprogenitor cell proliferation and differentiation [145]. IGF-I deficiency substantially reduced the stimulatory effects of PTH on the expression of RANKL, possibly resulting in decreased osteoclast formation [145].
In rat osteosacoma cells TGF-β altered PTH receptors and responsiveness in osteoblasts [146]. PTH stimulation of osteoclastic bone resorption resulted in the liberation of significant amounts of TGF-β from the matrix [147]. It was reported that PTH regulated TGF-βI and TGF- βII synthesis in human fetal osteoblasts [148] and amplified the anabolic effects of TGF-β by accelerating the transcriptional activity of Smad3 which exerts anti-apoptotic effects in mouse osteoblastic cells [149]. The actions of TGF- β, such as proper folding and secretion, are modulated by the latent TGF-β binding proteins (LTBPs). PTH stimulation of LTBP-1 mRNA expression in rat and mouse preosteoblastic cells has been suggested to be important for PTH action via TGF-β in bone remodeling [150].
Studies demonstrated that PTHrP can direct osteoblastic, rather then adipogenic, commitment of mesenchymal cells, and showed that PTHrP action involves enhanced gene expression of the BMP IA receptor, which facilitates BMP2 action in enhancing osteoblastogenesis in pluripotent mesenchymal cells [151]. Other growth factors that could act to mediate an anabolic effect of PTH include FGF-2. In cultured osteoblastic cells PTH increased FGF-2 mRNA and FGF-2 receptor mRNA [152]. In FGF-2 null mice the effect of PTH on cancellous bone volume was largely diminished [153]. Using co-cultured bone marrow cells from wild type and FGF-2 null mice it was demonstrated that PTH stimulation of osteoclast formation and bone resorption requires endogenous FGF-2 synthesis by osteoblasts [154].
2. Regulation of transcription factors
Tissue specific transcription factors are nuclear proteins that regulate the transcription of multiple genes that are expressed in a particular tissue type as part of highly regulated program through a larger regulatory protein complex. Experiments inducing loss of function of AP-1 proteins in mice and cells have provided important insights into their role in skeletogenesis [155]. More studies implied that the AP-1 transcription factors play an important role in bone development [155–157] and in mediating certain osteogenic stimuli of PTH [158]. The protooncogene c-fos, a member of the AP-1 transcription factor, is regulated by PTH both in vitro and in vivo. Intermittent PTH injections caused anabolic effects in wild type mice and catabolic effects in the osteosclerotic c-fos- null mice [159]. In response to PTH the cAMP response element binding protein (CREB) is phosphorylated at serine 133 [160] which then modulates the transcription of PTH target genes [161]. Thus PTH and PTHrP mediate transactivation of CREB [162], stimulation of c-fos promoter activity via phospho-CREB [160], and increased expression of c-jun and c-fos in osteoblasts [163, 164], which mediate transcription of genes containing AP-1 promoter elements. PTH and PTHrP stimulate the expression of other AP-1 family members which include c-jun, fosB, JunB, fra1, and fra2 [61, 163, 165–167].
Within the bone cell lineage, Runx2 (Cbfa1/AML3) plays an important role as a central regulator of bone and for maintaining the osteophenotype [168]. Cbfa1 or RunX2 is the first osteoblast specific transcription factor to be identified. It is expressed in mesenchymal cells that develop into chondrocytes or osteoblasts [169, 170]. Homozygous RunX2 null mice show normal skeletal patterning, but the skeleton is cartilaginous as osteoblast differentiation does not occur in these animals [171, 172]. Runx2 mediates the temporal activation and/or repression of cell growth and phenotypic genes as osteoblasts progress through stages of differentiation [173, 174]. Runx2 target genes include regulators of cell growth control (such as BMP/TGF-β, IGF I and II), components of the bone extracellular matrix, angiogenesis, and signaling proteins (Wnt and Src) required for development of the osteoblast phenotype [158, 173, 175–177]. A more recent study suggests an important function for ERK-MAPK pathway in bone formation that involves Runx2 phosphorylation and transcriptional activity [178]. The role of MAPKs in osteoblast differentiation was evaluated in transgenic mice expressing dominant negative (dn) or constitutively active (ca) MAPK kinase (MEK) under the control of the osteocalcin promoter (TgMEK-dn and TgMEK-ca, respectively) [178]. These mice have no apparent phenotype; however, when crossed with the heterozygous Runx2 knockout mice (Runx2 +/−) the TgMEK-ca transgene rescued the hypomorphic clavicles and the undemineralized calvaria of the Runx2 haploinsufficiency, whereas TgMek-dn transgene worsened the Runx2 haploinsufficiency phenotype.
Being recognized as a key transcription factor that determines osteoblast phenotype and differentiation [179–181], the effects of PTH on Runx2 have been investigated by several investigators. Studies with osteoblastic cell lines and cultured rat metatarsal bones indicated a rapid and transient increase in Runx2 mRNA, protein levels and activity following PTH treatment [181]. Transgenic osteoporotic female mice overexpressing Runx2 in osteoblasts, under the control of the osteocalcin promoter, had a blunted response to the anabolic actions of PTH [182]. This observation indicates that high Runx2 expression in osteoblasts in vivo abolishes the anabolic effects of PTH, and suggests that PTH anabolic actions are not additive to those of Runx2 or that PTH actions may converge on Runx2. Runx2 involvement in the anabolic actions of PTH was further supported by the finding that the level of expression of Runx2 is critical for PTH actions on bone in vivo [182]. Runx2 transactivation by a PKA dependent mechanism was shown to be required for PTH stimulation of the collagenase 3 promoter in vitro [183]. PTH may influence the phosphorylaion of a PKA consensus site in the activation domain 3 of Runx2 since PTH decreases the activity of Runx2 bearing mutations within the P/S/T domain fused to the DNA binding domain of the yeast Gal4 transcription factor [183]. Although it is not yet known if this phosphorylation event takes place in intact cells, this same site is phosphorylated by purified PKA in vitro [182]. These data suggest that PTH stimulates the collagenase 3 promoter by a PKA-dependent pathway that phosphorylates Runx2 and up-regulates c-Fos and c-Jun via phosphorylation of CREB.
TWIST, a basic helix-loop-helix transcription factor, also plays a role in osteoprogenitor cells. It is expressed in preosteoblasts and is upregulated by the fibroblast growth factor (FGF2) [184]. SaOS2 cells overexpressing TWIST maintain a more osteoprogenitor phenotype. In contrast, antisense oligonucleotides of TWIST increase alkaline phosphatase and type I collagen expression in these cells [185]. Recent studies show that Id1 and TWIST may regulate differentiation of mesenchymal cells into osteoblasts by controlling BMP signaling [186]. The regulation of TWIST by PTH1R is not yet known.
Msx2, a homologue of the Drosophila muscle segment Msh gene, is expressed in preosteoblasts [187] and in early developing bone [168]. Msx2 deficiency leads to a marked delay in ossification of the skull bones [188]. Since Runx2 is down-regulated in Msx2 mutant mice, Msx2 has been speculated as an upstream regulator of Runx2. In this regard it has been shown that PTH inhibits osteogenic vascular calcification in diabetic LDL receptor - deficient mice via induction of osteopontin expression and suppression of Msx2 expression [189].
The expression and function of the Distal-less (Dlx) gene family, are being studied by several investigators. Overexpression of Dlx5 stimulates osteoblast differentiation [190]. Dlx5−/−/Dlx6− − mice have severe craniofacial and limb defects, which may be due to defects in osteoblast maturation [190]. Gene expression studies performed in cells from different stages of the osteoblast lineage showed that Dlx2, Dlx5 and Dlx6 are expressed most strongly in less mature osteoblasts, whereas Dlx3 is highly expressed in differentiated osteoblasts. PTH1R regulation of Dlx genes is not yet reported.
SOX-4 is a member of a gene family (SOX and SRY) comprising transcription factors that bind to DNA through their high mobility group (HMG)-type binding domain and are involved in skeletal development [191]. Sox-4 mRNA was detected in osteoblast-like cells of both human and rodent origin. In OHS cells, physiological concentrations (10−10-10−9M) of hPTH(1–84) and hPTH(1–34), but not hPTH(3–84), stimulated Sox-4 mRNA expression in a time-dependent manner [192]. SOX4 mRNA is increased in bone biopsies from patients with primary hyperparathyroidism. [193].
3. PTH and PTHrP regulated genes
MMP-13, a matrix metalloproteinase, is expressed as a late-differentiation gene in osteoblasts, and is primarily responsible for the degradation of extracellular bone matrix components (type I, II, and III fibrillar collagens). MMP-13 expression, which promotes bone turnover, is regulated by PTH [165]. PTH (and/or PTHrP) was shown to decrease the expression of alkaline phosphatase [23], type I collagen [194], osteopontin [195], osteonectin [196], and osteoprotegerin [77]. In contrast PTH (or PTHrP) increases the expression of tissue inhibitors of metalloproteinases [197], tissue plasminogen activator [198], IL-6 [112, 199], IL-18 [200], the regulator of G-protein signaling (RGS)[201], macrophage-colony stimulating factor (M-CSF) [202] as well as the orphan receptors, Nurr1 and Nur77 [203, 204].
PTH and PTHrP regulates the expression of Connexins, the gap junction proteins required for cell-cell communications. Connexin43 and connexin45 are present in osteoblastic cells and their expression varies between osteoblastic cell lines [205]. PTH increases connexin43 expression [206, 207]; this effect is developmental stage specific in UMR 106-01 cells [206].
Bcl2 has been hypothesized to be a key molecule that mediates the antiapoptotic effects of PTH in osteoblasts by turning mature osteoblasts into long-living osteoblasts, resulting in enhanced bone formation [54, 180, 208]. Bcl2 null mice have a high bone mass associated with suppressed osteoclasts; these finding suggested that Bcl2 is critical for osteoclast maturation. Evidence also suggested that Bcl2 is not required for the PTH anabolic action [209].
Recent studies suggest that the Ephrins receptor tyrosine kinases and their cell-surface-anchored ephrin ligands, which are versatile regulators of tissue morphogenesis and cell-cell communication, play bidirectional signaling role between osteoblasts and osteoclasts [210]. The forward signaling through ephrinB2, expressed in osteoclasts, interacting with its receptor, EphB4, expressed in osteoblasts, enhances osteoblast activity; and the reverse EphB4-ephrinB2 singaling suppresses osteoclasts [210]. It was shown that EphrinB2 is produced by osteoblasts (primary cultures of mouse calvarial osteoblasts and 2 osteoblastic cell lines, UMR106 and Kusa 4b10), that ephrinB2 production is regulated in a dose-dependent manner by both PTH and PTHrP in vitro and in vivo, and that blockade of ephrinB2/EphB4 interaction inhibits the mineralization of Kusa 4b10 cells [211]. Since osteoblasts express EphB4 [210], the receptor of ephrinB2, this observation raises the interesting hypothesis that PTH and PTHrP enhance signaling through this system within osteoblasts by increasing the expression of ephrinB2 [211].
Osteocytes, a heterogeneous cell population of terminally differentiated osteoblasts, express PTH1R [212]. Sclerostin, a product of the Sost gene and a negative regulator of the canonical Wnt signaling [213] is expressed in osteocytes [214] and is involved in PTH control of osteoblastogenesis [215]. The expression of constitutively active PTH1R in osteocytes increases bone mass, decreases the expression of sclerostin, and increases Wnt signaling [216].
4. PTH, PTHrP and osteoblast cell cycle
Progression through the cell cycle is strictly regulated by the periodic and phase specific abundance and activity of a defined set of proteins. Two classes of proteins make up the protein-kinase complexes involved in the biochemical control of the cell cycle. The cell division kinases (CDKs), also referred to as cyclin-dependent kinases, are the catalytic subunits of these complexes, whereas the cyclins function as the regulatory subunits. Cyclins undergo dramatic fluctuations in abundance during progression of the cell cycle, and thus regulate activation of the holoenzyme [217]. The G1 phase preceding the DNA synthesis (S) phase and the mechanisms, which drive the cells across the restriction point, are crucial for the cell’s fate towards division, differentiation, senescence, or apoptosis. Cyclin D1 is a positive regulator of cell cycle progression through the G1 phase of the cycle [218, 219]. Following its association with CDK4 and CDK6 the activated complex phosphorylates key substrates (Rb family) to facilitate transcription of target genes necessary for DNA replication [219, 220]. Cell cycle progression is also controlled by CDK inhibitors (CDKIs) Cip/Kip (p21, p27, p57) and INK4 (p15, p16, p18, p19) which negatively regulate G1 phase progression by forming complexes with CDKs and preventing S phase entry [221, 222]. CDKIs are therefore critical mediators of anti proliferative signals that arrest the cell cycle.
The role of the intracellular signaling pathways activated by PTH and PTHrP, which regulate the cell cycle in osteoblasts, are yet to be defined. PTH has been shown to inhibit osteoblast proliferation by increasing p27 levels and inhibiting CDK2 activity [223]. Gene array analysis showed differential expressions of cyclins, Bcl-2 family members, as well as p53 during differentiation of a mouse calvarial-derived cell line to acquire an osteoblast like phenotype [224]. PTHrP, on the other hand, has been shown to target cyclin D1, CDK1 p21, p27 and p16 protein in differentiated osteoblasts and induced G1 phase growth arrest [61]. In differentiated osteoblasts, PTHrP reduces CDK1 activity and down-regulates cyclin D1 level possibly via decreased phosphorylation and stabilization of JunB, a member of AP-1 transcription factor [61]. In this study JunB silencing partially rescued PTHrP down-regulation of cyclin D1 [61]; this observation revealed the link between JunB, cyclin D1 and PTHR1 actions in bone. Cyclin D1 has been suggested as a potential target for AP-1 in fibroblasts and in other cell systems such as chondrocytes [225–228].
Osteoblast proliferation is an integral and early component of bone accrual. PTH stimulation of CDK1 expression is associated with TE-85 cell proliferation, an osteoblast precursor cell line [229]. The proliferative effect of PTHrP in MC3T3 calvarial osteoblasts is associated with increased or sustained levels of cyclin D1 expression; thus implies that PTHrP action on osteoblast cell cycle is dependent on the maturation stage of osteoblast [45]. It was shown that PTHrP requires the presence of growth factor/s for optimal activity, suggesting that PTHrP acts on proliferating cells that are at a favorable condition to undergo cell division. This study also suggested that the serum concentration is critical for the response to PTHrP stimulation.
As discussed above, Runx2 plays an important role in osteoblast maturation. The increased level of Runx2 in growth-arrested/serum-deprived cells has been shown to attenuate growth and support the non-dividing state by causing a delay in G1 in immature osteoblasts [230, 231]. It was hypothesized that the presence of growth factors, which is also a requisite under normal physiological conditions, may suppress Runx2 levels during proliferative expansion of pre-osteoblasts and is permissive for the PTHrP effect. Furthermore, new information has emerged on the role of cyclin D1 in Runx2 expression, phosphorylation and function [232]. Cyclin D1-CDK4, besides controlling G1 phase of the cell cycle, regulate Runx2 protein level by targeting it to ubiquitin mediated proteasomal degradation [232]. Thus, both ERK1/2 phosphorylation and cyclin D1 activity are likely to play a role in PTH mediated regulation of RunX2 in the process of bone formation [45]. Using a tissue engineered bone growth model in vivo, it was shown that PTH induces cyclin D1 expression in immature ectopic ossicles; this illustrates the pro-proliferative action of PTH and no new bone was noted at this stage. In contrast, PTH down-regulates cyclin D1 in the more differentiated ossicles with a significant increase in bone formation [45]. A recent study also showed increased proliferation of osteoblast precursors in the earlier phase of PTH treatment and an increased bone formation at later stages [53]. Increased osteoblast proliferation by PTH treatment was also reported in rats [31, 233] and mice [234]. PTH daily administration in human increases osteoblast number and osteoblast apoptosis [46]. In contrast to these studies, Wang et al reported that PTH does not influence cell proliferation in neonatal calvarial cells and suggested that the anabolic effect of intermittent PTH in vivo may result from an enhancement of the progenitor population to an osteogenic fate, resulting in a higher proportion of cells that achieve full osteoblast differentiation [235, 236]. The data of Dobnig and Turner proposed that the PTH anabolic effect does not involve stimulation of osteoblast progenitor proliferation in rats although the osteoblast number is dramatically increased [35]; these investigators suggested activation of bone lining cells.
These studies establish the concept that the cell cycle regulatory proteins are important regulators of PTH and PTHrP anabolic action in bone. Clearly, additional studies are required to determine quantitatively how much osteoblast cell proliferation/differentiation contributes to this action. The state of the field is exploratory and studies using more controlled designs, with high likelihood of mechanistic insight are needed to clarify the dependence of proliferation on PTH1R anabolic action.
VI. SUMMARY AND CONCLUSIONS
In recent years new advances in research with significant developments have occurred in bone biology. PTH1R is being recognized as an important regulator of bone remodeling, and has been demonstrated to mediate paradoxical (catabolic and anabolic) functions in vivo and in vitro. The anabolic effects of exogenous PTH depend upon its method of administration; whereas activation of the PTH1R by endogenous PTH or PTHrP plays an important role in the physiology of bone. The biological response of osteoblasts to PTH can be modulated by diverse mechanisms, including reduction of PTH1R on the cell surface, loss of receptor signaling efficiency, and changes in post-receptor nuclear responses.
In addition to the classical PTH mediated signaling through AC/PKA and PLC/PKC pathways, several novel PTH1R targets are being recognized which are essential for bone homeostasis. Activation of osteoblasts by PTH involves growth factors, transcription factors, MAPKs, several regulatory molecules, and cell cycle regulatory proteins. Although a large number of in-vitro, in-vivo and human studies have been performed, it is clear that the mechanisms involved in PTH regulation of osteoblast function is poorly understood and only partly characterized. Furthermore, elucidating the signaling mechanisms has been complicated by discrepancies between in vitro and in vivo model systems. A further definition of the cellular and molecular actions of PTH and PTHrP on osteoblasts continues to evolve. Several critical steps in the actions of PTH beyond receptor activation are being recognized and more are yet to be discovered; which may become novel therapeutic targets for metabolic bone diseases such as osteoporosis.
Acknowledgments
The authors were partially supported by the grants from The National Institute of Health, DE016865 (to NSD) and DK062286 (to ABA), during writing of this manuscript
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takahashi N, Udagawa N, Takami M, Suda T. Principles of Bone Biology. Academic Press; San Diego: 2002. pp. 109–126. [Google Scholar]
- 2.Schluter KD. News Physiol Sci. 1999;14:243–249. doi: 10.1152/physiologyonline.1999.14.6.243. [DOI] [PubMed] [Google Scholar]
- 3.Juppner H, Abou-Samra AB, Freeman M, Kong XF, Schipani E, Richards J, Kolakowski LF, Jr, Hock J, Potts JT, Jr, Kronenberg HM, et al. Science. 1991;254(5034):1024–1026. doi: 10.1126/science.1658941. [DOI] [PubMed] [Google Scholar]
- 4.Amizuka N, Warshawsky H, Henderson JE, Goltzman D, Karaplis AC. J Cell Biol. 1994;126(6):1611–1623. doi: 10.1083/jcb.126.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VL, Kronenberg HM, Mulligan RC. Genes Dev. 1994;8(3):277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 6.Karaplis A. Principles of Bone Biology. Academic Press; San Diego: 1996. pp. 1189–1201. [Google Scholar]
- 7.Karaplis AC, Kronenberg HM. Vitam Horm. 1996;52:177–193. doi: 10.1016/s0083-6729(08)60411-2. [DOI] [PubMed] [Google Scholar]
- 8.Amizuka N, Karaplis AC, Henderson JE, Warshawsky H, Lipman ML, Matsuki Y, Ejiri S, Tanaka M, Izumi N, Ozawa H, Goltzman D. Dev Biol. 1996;175(1):166–176. doi: 10.1006/dbio.1996.0104. [DOI] [PubMed] [Google Scholar]
- 9.Kong XF, Schipani E, Lanske B, Joun H, Karperien M, Defize LH, Juppner H, Potts JT, Jr, Segre GV, Kronenberg HM, et al. Biochem Biophys Res Commun. 1994;200(3):1290–1299. doi: 10.1006/bbrc.1994.1591. [DOI] [PubMed] [Google Scholar]
- 10.Abou-Samra AB, Juppner H, Force T, Freeman MW, Kong XF, Schipani E, Urena P, Richards J, Bonventre JV, Potts JT, Jr, et al. Proc Natl Acad Sci U S A. 1992;89(7):2732–2736. doi: 10.1073/pnas.89.7.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Juppner HW, Gardella TJ, Brown EM. Endocrinology. Philadelphia: Elsvier; 2000. pp. 969–998. [Google Scholar]
- 12.Hoare SR, Gardella TJ, Usdin TB. J Biol Chem. 2001;276(11):7741–7753. doi: 10.1074/jbc.M009395200. [DOI] [PubMed] [Google Scholar]
- 13.Chauvin S, Bencsik M, Bambino T, Nissenson RA. Mol Endocrinol. 2002;16(12):2720–2732. doi: 10.1210/me.2002-0049. [DOI] [PubMed] [Google Scholar]
- 14.Tawfeek HA, Qian F, Abou-Samra AB. Mol Endocrinol. 2002;16(1):1–13. doi: 10.1210/mend.16.1.0760. [DOI] [PubMed] [Google Scholar]
- 15.Bounoutas GS, Tawfeek H, Frohlich LF, Chung UI, Abou-Samra AB. Endocrinology. 2006;147(10):4674–4679. doi: 10.1210/en.2006-0301. [DOI] [PubMed] [Google Scholar]
- 16.Tawfeek HA, Abou-Samra AB. Endocrinology. 2008;149(8):4016–4023. doi: 10.1210/en.2007-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schipani E, Kruse K, Juppner H. Science. 1995;268(5207):98–100. doi: 10.1126/science.7701349. [DOI] [PubMed] [Google Scholar]
- 18.Jobert AS, Zhang P, Couvineau A, Bonaventure J, Roume J, Le Merrer M, Silve C. J Clin Invest. 1998;102(1):34–40. doi: 10.1172/JCI2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lanske B, Amling M, Neff L, Guiducci J, Baron R, Kronenberg HM. J Clin Invest. 1999;104(4):399–407. doi: 10.1172/JCI6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parfitt AM, Schipani E, Rao DS, Kupin W, Han ZH, Juppner H. J Clin Endocrinol Metab. 1996;81(10):3584–3588. doi: 10.1210/jcem.81.10.8855805. [DOI] [PubMed] [Google Scholar]
- 21.Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, Kronenberg HM, Baron R, Schipani E. J Clin Invest. 2001;107(3):277–286. doi: 10.1172/JCI11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee K, Deeds JD, Segre GV. Endocrinology. 1995;136(2):453–463. doi: 10.1210/endo.136.2.7835276. [DOI] [PubMed] [Google Scholar]
- 23.Majeska RJ, Rodan GA. Calcif Tissue Int. 1982;34(1):59–66. doi: 10.1007/BF02411210. [DOI] [PubMed] [Google Scholar]
- 24.Partridge NC, Kemp BE, Veroni MC, Martin TJ. Endocrinology. 1981;108(1):220–225. doi: 10.1210/endo-108-1-220. [DOI] [PubMed] [Google Scholar]
- 25.Usdin TB, Gruber C, Bonner TI. J Biol Chem. 1995;270(26):15455–15458. doi: 10.1074/jbc.270.26.15455. [DOI] [PubMed] [Google Scholar]
- 26.Usdin TB, Bonner TI, Harta G, Mezey E. Endocrinology. 1996;137(10):4285–4297. doi: 10.1210/endo.137.10.8828488. [DOI] [PubMed] [Google Scholar]
- 27.Juppner H. Bone. 1999;25(1):87–90. doi: 10.1016/s8756-3282(99)00110-6. [DOI] [PubMed] [Google Scholar]
- 28.Rubin DA, Juppner H. J Biol Chem. 1999;274(40):28185–28190. doi: 10.1074/jbc.274.40.28185. [DOI] [PubMed] [Google Scholar]
- 29.Rotllant J, Guerreiro PM, Redruello B, Fernandes H, Apolonia L, Anjos L, Canario AV, Power DM. Cell Tissue Res. 2006;323(2):333–341. doi: 10.1007/s00441-005-0070-7. [DOI] [PubMed] [Google Scholar]
- 30.Rouleau MF, Mitchell J, Goltzman D. Endocrinology. 1988;123(1):187–191. doi: 10.1210/endo-123-1-187. [DOI] [PubMed] [Google Scholar]
- 31.Hock JM, Fitzpatrick LA, Bilezikian JP. Principles of Bone Biology. San Diego: Academic Press; 2002. pp. 463–482. [Google Scholar]
- 32.Selye H. Endocrinology. 1932;16:547. [Google Scholar]
- 33.Beauer W, Aub JC. Albright F J Exp Med. 1929;49:145–162. doi: 10.1084/jem.49.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dempster DW, Cosman F, Parisien M, Shen V, Lindsay R. Endocr Rev. 1993;14(6):690–709. doi: 10.1210/edrv-14-6-690. [DOI] [PubMed] [Google Scholar]
- 35.Dobnig H, Turner RT. Endocrinology. 1995;136(8):3632–3638. doi: 10.1210/endo.136.8.7628403. [DOI] [PubMed] [Google Scholar]
- 36.Sone T, Fukunaga M, Ono S, Nishiyama T. Miner Electrolyte Metab. 1995;21(1–3):232–235. [PubMed] [Google Scholar]
- 37.Cosman F, Nieves J, Woelfert L, Shen V, Lindsay R. J Bone Miner Res. 1998;13(6):1051–1055. doi: 10.1359/jbmr.1998.13.6.1051. [DOI] [PubMed] [Google Scholar]
- 38.Brommage R, Hotchkiss CE, Lees CJ, Stancill MW, Hock JM, Jerome CP. J Clin Endocrinol Metab. 1999;84(10):3757–3763. doi: 10.1210/jcem.84.10.6039. [DOI] [PubMed] [Google Scholar]
- 39.Schiller PC, D’Ippolito G, Roos BA, Howard GA. J Bone Miner Res. 1999;14(9):1504–1512. doi: 10.1359/jbmr.1999.14.9.1504. [DOI] [PubMed] [Google Scholar]
- 40.Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH. N Engl J Med. 2001;344(19):1434–1441. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 41.Frolik CA, Black EC, Cain RL, Satterwhite JH, Brown-Augsburger PL, Sato M, Hock JM. Bone. 2003;33(3):372–379. doi: 10.1016/s8756-3282(03)00202-3. [DOI] [PubMed] [Google Scholar]
- 42.Horwitz MJ, Tedesco MB, Gundberg C, Garcia-Ocana A, Stewart AF. J Clin Endocrinol Metab. 2003;88(2):569–575. doi: 10.1210/jc.2002-021122. [DOI] [PubMed] [Google Scholar]
- 43.Cosman F, Lindsay R. Curr Osteoporos Rep. 2004;2(1):5–11. doi: 10.1007/s11914-004-0008-0. [DOI] [PubMed] [Google Scholar]
- 44.Pettway GJ, Schneider A, Koh AJ, Widjaja E, Morris MD, Meganck JA, Goldstein SA, McCauley LK. Bone. 2005;36(6):959–970. doi: 10.1016/j.bone.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 45.Datta NS, Pettway GJ, Chen C, Koh AJ, McCauley LK. J Bone Miner Res. 2007;22(7):951–964. doi: 10.1359/jbmr.070328. [DOI] [PubMed] [Google Scholar]
- 46.Lindsay R, Zhou H, Cosman F, Nieves J, Dempster DW, Hodsman AB. J Bone Miner Res. 2007;22(4):495–502. doi: 10.1359/jbmr.070104. [DOI] [PubMed] [Google Scholar]
- 47.Herrmann-Erlee MP, Heersche JN, Hekkelman JW, Gaillard PJ, Tregear GW, Parsons JA, Potts JT., Jr Endocr Res Commun. 1976;3(1):21–35. doi: 10.3109/07435807609057738. [DOI] [PubMed] [Google Scholar]
- 48.DeBartolo TF, Pegg LE, Shasserre C, Hahn TJ. Calcif Tissue Int. 1982;34(5):495–500. doi: 10.1007/BF02411291. [DOI] [PubMed] [Google Scholar]
- 49.MacDonald BR, Gallagher JA, Russell RG. Endocrinology. 1986;118(6):2445–2449. doi: 10.1210/endo-118-6-2445. [DOI] [PubMed] [Google Scholar]
- 50.Canalis E, Centrella M, Burch W, McCarthy TL. J Clin Invest. 1989;83(1):60–65. doi: 10.1172/JCI113885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishida S, Yamaguchi A, Tanizawa T, Endo N, Mashiba T, Uchiyama Y, Suda T, Yoshiki S, Takahashi HE. Bone. 1994;15(6):717–723. doi: 10.1016/8756-3282(94)90322-0. [DOI] [PubMed] [Google Scholar]
- 52.Partridge NC, Li X, Qin L. Ann N Y Acad Sci. 2006;1068:187–193. doi: 10.1196/annals.1346.024. [DOI] [PubMed] [Google Scholar]
- 53.Pettway GJ, Meganck JA, Koh AJ, Keller ET, Goldstein SA, McCauley LK. Bone. 2008;42(4):806–818. doi: 10.1016/j.bone.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. J Clin Invest. 1999;104(4):439–446. doi: 10.1172/JCI6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manolagas SC. Endocr Rev. 2000;21(2):115–137. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 56.Leaffer D, Sweeney M, Kellerman LA, Avnur Z, Krstenansky JL, Vickery BH, Caulfield JP. Endocrinology. 1995;136(8):3624–3631. doi: 10.1210/endo.136.8.7628402. [DOI] [PubMed] [Google Scholar]
- 57.van der Plas A, Nijweide PJ. Bone. 1988;9(2):107–111. doi: 10.1016/8756-3282(88)90111-1. [DOI] [PubMed] [Google Scholar]
- 58.Kartsogiannis V, Moseley J, McKelvie B, Chou ST, Hards DK, Ng KW, Martin TJ, Zhou H. Bone. 1997;21(5):385–392. doi: 10.1016/s8756-3282(97)00180-4. [DOI] [PubMed] [Google Scholar]
- 59.Suda N, Gillespie MT, Traianedes K, Zhou H, Ho PW, Hards DK, Allan EH, Martin TJ, Moseley JM. J Cell Physiol. 1996;166(1):94–104. doi: 10.1002/(SICI)1097-4652(199601)166:1<94::AID-JCP11>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 60.Nijweide PJ, Burger EH, Feyen JH. Physiol Rev. 1986;66(4):855–886. doi: 10.1152/physrev.1986.66.4.855. [DOI] [PubMed] [Google Scholar]
- 61.Datta NS, Chen C, Berry JE, McCauley LK. J Bone Miner Res. 2005;20(6):1051–1064. doi: 10.1359/JBMR.050106. [DOI] [PubMed] [Google Scholar]
- 62.Onyia JE, Miller B, Hulman J, Liang J, Galvin R, Frolik C, Chandrasekhar S, Harvey AK, Bidwell J, Herring J, Hock JM. Bone. 1997;20(2):93–100. doi: 10.1016/s8756-3282(96)00350-x. [DOI] [PubMed] [Google Scholar]
- 63.Beier F, LuValle P. Mol Endocrinol. 2002;16(9):2163–2173. doi: 10.1210/me.2001-0103. [DOI] [PubMed] [Google Scholar]
- 64.Chen HL, Demiralp B, Schneider A, Koh AJ, Silve C, Wang CY, McCauley LK. J Biol Chem. 2002;277(22):19374–19381. doi: 10.1074/jbc.M108913200. [DOI] [PubMed] [Google Scholar]
- 65.Liu X, Pettway GJ, McCauley LK, Ma PX. Biomaterials. 2007;28(28):4124–4131. doi: 10.1016/j.biomaterials.2007.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barnes GL, Kakar S, Vora S, Morgan EF, Gerstenfeld LC, Einhorn TA. J Bone Joint Surg Am. 2008;90(Suppl 1):120–127. doi: 10.2106/JBJS.G.01443. [DOI] [PubMed] [Google Scholar]
- 67.Jeon JH, Puleo DA. Biomaterials. 2008;29(26):3591–3598. doi: 10.1016/j.biomaterials.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Q, Cuartas E, Mehta N, Gilligan J, Ke HZ, Saltzman WM, Kotas M, Ma M, Rajan S, Chalouni C, Carlson J, Vignery A. Tissue Eng Part A. 2008;14(2):237–246. doi: 10.1089/tea.2007.0261. [DOI] [PubMed] [Google Scholar]
- 69.Teitelbaum SL. Science. 2000;289(5484):1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 70.Chambers TJ, Fuller K, McSheehy PM, Pringle JA. J Pathol. 1985;145(4):297–305. doi: 10.1002/path.1711450403. [DOI] [PubMed] [Google Scholar]
- 71.McSheehy PM, Chambers TJ. Endocrinology. 1986;118(2):824–828. doi: 10.1210/endo-118-2-824. [DOI] [PubMed] [Google Scholar]
- 72.Langub MC, Monier-Faugere MC, Qi Q, Geng Z, Koszewski NJ, Malluche HH. J Bone Miner Res. 2001;16(3):448–456. doi: 10.1359/jbmr.2001.16.3.448. [DOI] [PubMed] [Google Scholar]
- 73.Dempster DW, Hughes-Begos CE, Plavetic-Chee K, Brandao-Burch A, Cosman F, Nieves J, Neubort S, Lu SS, Iida-Klein A, Arnett T, Lindsay R. J Cell Biochem. 2005;95(1):139–148. doi: 10.1002/jcb.20388. [DOI] [PubMed] [Google Scholar]
- 74.Shiotani A, Takami M, Itoh K, Shibasaki Y, Sasaki T. Anat Rec. 2002;268(2):137–146. doi: 10.1002/ar.10121. [DOI] [PubMed] [Google Scholar]
- 75.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Cell. 1998;93(2):165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 76.Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, Kanno T, Murakami A, Tsuda E, Morinaga T, Higashio K. Endocrinology. 1998;139(3):1329–1337. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 77.Lee SK, Lorenzo JA. Endocrinology. 1999;140(8):3552–3561. doi: 10.1210/endo.140.8.6887. [DOI] [PubMed] [Google Scholar]
- 78.Gori F, Hofbauer LC, Dunstan CR, Spelsberg TC, Khosla S, Riggs BL. Endocrinology. 2000;141(12):4768–4776. doi: 10.1210/endo.141.12.7840. [DOI] [PubMed] [Google Scholar]
- 79.Atkins GJ, Kostakis P, Pan B, Farrugia A, Gronthos S, Evdokiou A, Harrison K, Findlay DM, Zannettino AC. J Bone Miner Res. 2003;18(6):1088–1098. doi: 10.1359/jbmr.2003.18.6.1088. [DOI] [PubMed] [Google Scholar]
- 80.Li X, Qin L, Bergenstock M, Bevelock LM, Novack DV, Partridge NC. J Biol Chem. 2007;282(45):33098–33106. doi: 10.1074/jbc.M611781200. [DOI] [PubMed] [Google Scholar]
- 81.Compston JE. Bone. 2007;40(6):1447–1452. doi: 10.1016/j.bone.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 82.Jilka RL. Bone. 2007;40(6):1434–1446. doi: 10.1016/j.bone.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mahon MJ, Shimada M. FEBS Lett. 2005;579(3):803–807. doi: 10.1016/j.febslet.2004.12.056. [DOI] [PubMed] [Google Scholar]
- 84.Shimada M, Mahon MJ, Greer PA, Segre GV. Endocrinology. 2005;146(5):2336–2344. doi: 10.1210/en.2004-1637. [DOI] [PubMed] [Google Scholar]
- 85.Mahon MJ, Bonacci TM, Divieti P, Smrcka AV. Mol Endocrinol. 2006;20(1):136–146. doi: 10.1210/me.2005-0169. [DOI] [PubMed] [Google Scholar]
- 86.Bringhurst FR, Juppner H, Guo J, Urena P, Potts JT, Jr, Kronenberg HM, Abou-Samra AB, Segre GV. Endocrinology. 1993;132(5):2090–2098. doi: 10.1210/endo.132.5.8386606. [DOI] [PubMed] [Google Scholar]
- 87.Mahon MJ, Donowitz M, Yun CC, Segre GV. Nature. 2002;417(6891):858–861. doi: 10.1038/nature00816. [DOI] [PubMed] [Google Scholar]
- 88.Mahon MJ, Cole JA, Lederer ED, Segre GV. Mol Endocrinol. 2003;17(11):2355–2364. doi: 10.1210/me.2003-0043. [DOI] [PubMed] [Google Scholar]
- 89.Sneddon WB, Syme CA, Bisello A, Magyar CE, Rochdi MD, Parent JL, Weinman EJ, Abou-Samra AB, Friedman PA. J Biol Chem. 2003;278(44):43787–43796. doi: 10.1074/jbc.M306019200. [DOI] [PubMed] [Google Scholar]
- 90.Miedlich SU, Abou-Samra AB. Am J Physiol Endocrinol Metab. 2008;295(3):E665–671. doi: 10.1152/ajpendo.00036.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takasu H, Guo J, Bringhurst FR. J Bone Miner Res. 1999;14(1):11–20. doi: 10.1359/jbmr.1999.14.1.11. [DOI] [PubMed] [Google Scholar]
- 92.Yamaguchi DT, Hahn TJ, Iida-Klein A, Kleeman CR, Muallem S. J Biol Chem. 1987;262(16):7711–7718. [PubMed] [Google Scholar]
- 93.Yamaguchi DT, Kleeman CR, Muallem S. J Biol Chem. 1987;262(31):14967–14973. [PubMed] [Google Scholar]
- 94.Carter PH, Shimizu M, Luck MD, Gardella TJ. J Biol Chem. 1999;274(45):31955–31960. doi: 10.1074/jbc.274.45.31955. [DOI] [PubMed] [Google Scholar]
- 95.Luck MD, Carter PH, Gardella TJ. Mol Endocrinol. 1999;13(5):670–680. doi: 10.1210/mend.13.5.0277. [DOI] [PubMed] [Google Scholar]
- 96.Takasu H, Gardella TJ, Luck MD, Potts JT, Jr, Bringhurst FR. Biochemistry. 1999;38(41):13453–13460. doi: 10.1021/bi990437n. [DOI] [PubMed] [Google Scholar]
- 97.Shimizu M, Carter PH, Khatri A, Potts JT, Jr, Gardella TJ. Endocrinology. 2001;142(7):3068–3074. doi: 10.1210/endo.142.7.8253. [DOI] [PubMed] [Google Scholar]
- 98.Shimizu M, Potts JT, Jr, Gardella TJ. J Biol Chem. 2000;275(29):21836–21843. doi: 10.1074/jbc.M909861199. [DOI] [PubMed] [Google Scholar]
- 99.Mahaffey JE, Rosenblatt M, Shepard GL, Potts JT., Jr J Biol Chem. 1979;254(14):6496–6498. [PubMed] [Google Scholar]
- 100.Whitfield JF, Isaacs RJ, Chakravarthy B, Maclean S, Morley P, Willick G, Divieti P, Bringhurst FR. J Bone Miner Res. 2001;16(3):441–447. doi: 10.1359/jbmr.2001.16.3.441. [DOI] [PubMed] [Google Scholar]
- 101.Dean T, Khatri A, Potetinova Z, Willick GE, Gardella TJ. J Biol Chem. 2006;281(43):32485–32495. doi: 10.1074/jbc.M606179200. [DOI] [PubMed] [Google Scholar]
- 102.Dean T, Linglart A, Mahon MJ, Bastepe M, Juppner H, Potts JT, Jr, Gardella TJ. Mol Endocrinol. 2006;20(4):931–943. doi: 10.1210/me.2005-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Potetinova Z, Barbier JR, Suen T, Dean T, Gardella TJ, Willick GE. Biochemistry. 2006;45(37):11113–11121. doi: 10.1021/bi060500q. [DOI] [PubMed] [Google Scholar]
- 104.Potts JT, Gardella TJ. Ann N Y Acad Sci. 2007;1117:196–208. doi: 10.1196/annals.1402.088. [DOI] [PubMed] [Google Scholar]
- 105.Dean T, Vilardaga JP, Potts JT, Jr, Gardella TJ. Mol Endocrinol. 2008;22(1):156–166. doi: 10.1210/me.2007-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Nature. 1998;396(6710):474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 107.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. Science. 1998;282(5397):2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 108.Fujita T, Meguro T, Fukuyama R, Nakamuta H, Koida M. J Biol Chem. 2002;277(25):22191–22200. doi: 10.1074/jbc.M110364200. [DOI] [PubMed] [Google Scholar]
- 109.Datta NS, D’Silva NJ, Abou-Samra AB. J Bone Miner Res. 2008;23:S139. [Google Scholar]
- 110.Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, Lefkowitz RJ. J Biol Chem. 2006;281(16):10856–10864. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- 111.Verheijen MH, Defize LH. Endocrinology. 1995;136(8):3331–3337. doi: 10.1210/endo.136.8.7628368. [DOI] [PubMed] [Google Scholar]
- 112.Chen C, Koh AJ, Datta NS, Zhang J, Keller ET, Xiao G, Franceschi RT, D’Silva NJ, McCauley LK. J Biol Chem. 2004;279(28):29121–29129. doi: 10.1074/jbc.M313000200. [DOI] [PubMed] [Google Scholar]
- 113.Pouyssegur J. Science. 2000;290(5496):1515–1518. doi: 10.1126/science.290.5496.1515. [DOI] [PubMed] [Google Scholar]
- 114.Gensure RC, Gardella TJ, Juppner H. Biochem Biophys Res Commun. 2005;328(3):666–678. doi: 10.1016/j.bbrc.2004.11.069. [DOI] [PubMed] [Google Scholar]
- 115.Ferrari SL, Pierroz DD, Glatt V, Goddard DS, Bianchi EN, Lin FT, Manen D, Bouxsein ML. Endocrinology. 2005;146(4):1854–1862. doi: 10.1210/en.2004-1282. [DOI] [PubMed] [Google Scholar]
- 116.Rey A, Manen D, Rizzoli R, Ferrari SL, Caverzasio J. Bone. 2007;41(1):59–67. doi: 10.1016/j.bone.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 117.Qin L, Li X, Ko JK, Partridge NC. J Biol Chem. 2005;280(4):3104–3111. doi: 10.1074/jbc.M409846200. [DOI] [PubMed] [Google Scholar]
- 118.Aghaloo TL, Pirih FQ, Shi A, Bezouglaia O, Tetradis S. J Periodontol. 2006;77(1):21–30. doi: 10.1902/jop.2006.77.1.21. [DOI] [PubMed] [Google Scholar]
- 119.Datta NS, Kolailat R, Pettway GJ, Berry JE, McCauley LK. J Bone Miner Res. 2007;22:S137. doi: 10.1359/jbmr.070328. [DOI] [PubMed] [Google Scholar]
- 120.Hsieh JC, Rattner A, Smallwood PM, Nathans J. Proc Natl Acad Sci U S A. 1999;96(7):3546–3551. doi: 10.1073/pnas.96.7.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. Nature. 2000;407(6803):535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- 122.Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. Nature. 2001;411(6835):321–325. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- 123.Mao J, Wang J, Liu B, Pan W, Farr GH, 3rd, Flynn C, Yuan H, Takada S, Kimelman D, Li L, Wu D. Mol Cell. 2001;7(4):801–809. doi: 10.1016/s1097-2765(01)00224-6. [DOI] [PubMed] [Google Scholar]
- 124.Wodarz A, Nusse R. Annu Rev Cell Dev Biol. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- 125.Uusitalo M, Heikkila M, Vainio S. Exp Cell Res. 1999;253(2):336–348. doi: 10.1006/excr.1999.4710. [DOI] [PubMed] [Google Scholar]
- 126.Kulkarni NH, Halladay DL, Miles RR, Gilbert LM, Frolik CA, Galvin RJ, Martin TJ, Gillespie MT, Onyia JE. J Cell Biochem. 2005;95(6):1178–1190. doi: 10.1002/jcb.20506. [DOI] [PubMed] [Google Scholar]
- 127.Suzuki A, Ozono K, Kubota T, Kondou H, Tachikawa K, Michigami T. J Cell Biochem. 2008;104(1):304–317. doi: 10.1002/jcb.21626. [DOI] [PubMed] [Google Scholar]
- 128.Kharode YP, Sharp MC, Milligan CL, Pirrello JM, Selim SF, Bodine P. J Bone Miner Res. 2006;21:S114. [Google Scholar]
- 129.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, Li J, Maye P, Rowe DW, Duncan RL, Warman ML, Turner CH. J Biol Chem. 2006;281(33):23698–23711. doi: 10.1074/jbc.M601000200. [DOI] [PubMed] [Google Scholar]
- 130.Iwaniec UT, Wronski TJ, Liu J, Rivera MF, Arzaga RR, Hansen G, Brommage R. J Bone Miner Res. 2007;22(3):394–402. doi: 10.1359/jbmr.061118. [DOI] [PubMed] [Google Scholar]
- 131.Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, Cao X. Genes & Development. 2008;22:2968–2979. doi: 10.1101/gad.1702708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Karsenty G. Nature. 2003;423(6937):316–318. doi: 10.1038/nature01654. [DOI] [PubMed] [Google Scholar]
- 133.de Vernejoul MC, Cohen-Solal M, Orcel P. Curr Opin Rheumatol. 1993;5(3):332–338. doi: 10.1097/00002281-199305030-00012. [DOI] [PubMed] [Google Scholar]
- 134.Roberts AB. Miner Electrolyte Metab. 1998;24(2–3):111–119. doi: 10.1159/000057358. [DOI] [PubMed] [Google Scholar]
- 135.Centrella M, Horowitz MC, Wozney JM, McCarthy TL. Endocr Rev. 1994;15(1):27–39. doi: 10.1210/edrv-15-1-27. [DOI] [PubMed] [Google Scholar]
- 136.Linkhart TA, Mohan S, Baylink DJ. Bone. 1996;19(1 Suppl):1S–12S. doi: 10.1016/s8756-3282(96)00138-x. [DOI] [PubMed] [Google Scholar]
- 137.Linkhart TA, Mohan S. Endocrinology. 1989;125(3):1484–1491. doi: 10.1210/endo-125-3-1484. [DOI] [PubMed] [Google Scholar]
- 138.Locklin RM, Khosla S, Turner RT, Riggs BL. J Cell Biochem. 2003;89(1):180–190. doi: 10.1002/jcb.10490. [DOI] [PubMed] [Google Scholar]
- 139.McCarthy TL, Centrella M, Canalis E. Endocrinology. 1989;124(3):1247–1253. doi: 10.1210/endo-124-3-1247. [DOI] [PubMed] [Google Scholar]
- 140.Watson P, Lazowski D, Han V, Fraher L, Steer B, Hodsman A. Bone. 1995;16(3):357–365. doi: 10.1016/8756-3282(94)00051-4. [DOI] [PubMed] [Google Scholar]
- 141.Conover CA. Growth Horm IGF Res. 2000;10(Suppl B):S107–110. doi: 10.1016/s1096-6374(00)80020-9. [DOI] [PubMed] [Google Scholar]
- 142.Grey A, Chen Q, Xu X, Callon K, Cornish J. Endocrinology. 2003;144(11):4886–4893. doi: 10.1210/en.2003-0350. [DOI] [PubMed] [Google Scholar]
- 143.Miyakoshi N, Kasukawa Y, Linkhart TA, Baylink DJ, Mohan S. Endocrinology. 2001;142(10):4349–4356. doi: 10.1210/endo.142.10.8436. [DOI] [PubMed] [Google Scholar]
- 144.Bikle DD, Sakata T, Leary C, Elalieh H, Ginzinger D, Rosen CJ, Beamer W, Majumdar S, Halloran BP. J Bone Miner Res. 2002;17(9):1570–1578. doi: 10.1359/jbmr.2002.17.9.1570. [DOI] [PubMed] [Google Scholar]
- 145.Wang Y, Nishida S, Boudignon BM, Burghardt A, Elalieh HZ, Hamilton MM, Majumdar S, Halloran BP, Clemens TL, Bikle DD. J Bone Miner Res. 2007;22(9):1329–1337. doi: 10.1359/jbmr.070517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Seitz PK, Zhu BT, Cooper CW. J Bone Miner Res. 1992;7(5):541–546. doi: 10.1002/jbmr.5650070510. [DOI] [PubMed] [Google Scholar]
- 147.Raisz LG. Ciba Found Symp. 1988;136:226–238. doi: 10.1002/9780470513637.ch14. [DOI] [PubMed] [Google Scholar]
- 148.Wu Y, Kumar R. J Bone Miner Res. 2000;15(5):879–884. doi: 10.1359/jbmr.2000.15.5.879. [DOI] [PubMed] [Google Scholar]
- 149.Sowa H, Kaji H, Iu MF, Tsukamoto T, Sugimoto T, Chihara K. J Biol Chem. 2003;278(52):52240–52252. doi: 10.1074/jbc.M302566200. [DOI] [PubMed] [Google Scholar]
- 150.Kwok S, Qin L, Partridge NC, Selvamurugan N. J Cell Biochem. 2005;95(5):1002–1011. doi: 10.1002/jcb.20453. [DOI] [PubMed] [Google Scholar]
- 151.Chan GK, Miao D, Deckelbaum R, Bolivar I, Karaplis A, Goltzman D. Endocrinology. 2003;144(12):5511–5520. doi: 10.1210/en.2003-0273. [DOI] [PubMed] [Google Scholar]
- 152.Hurley MM, Tetradis S, Huang YF, Hock J, Kream BE, Raisz LG, Sabbieti MG. J Bone Miner Res. 1999;14(5):776–783. doi: 10.1359/jbmr.1999.14.5.776. [DOI] [PubMed] [Google Scholar]
- 153.Hurley MM, Okada Y, Xiao L, Tanaka Y, Ito M, Okimoto N, Nakamura T, Rosen CJ, Doetschman T, Coffin JD. Biochem Biophys Res Commun. 2006;341(4):989–994. doi: 10.1016/j.bbrc.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 154.Okada Y, Montero A, Zhang X, Sobue T, Lorenzo J, Doetschman T, Coffin JD, Hurley MM. J Biol Chem. 2003;278(23):21258–21266. doi: 10.1074/jbc.M302113200. [DOI] [PubMed] [Google Scholar]
- 155.Wagner EF. Ann Rheum Dis. 2002;61(Suppl 2):ii40–42. doi: 10.1136/ard.61.suppl_2.ii40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Angel P, Karin M. Biochim Biophys Acta. 1991;1072(2–3):129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 157.Grigoriadis AE, Wang ZQ, Wagner EF. Trends Genet. 1995;11(11):436–441. doi: 10.1016/s0168-9525(00)89142-8. [DOI] [PubMed] [Google Scholar]
- 158.Selvamurugan N, Chou WY, Pearman AT, Pulumati MR, Partridge NC. J Biol Chem. 1998;273(17):10647–10657. doi: 10.1074/jbc.273.17.10647. [DOI] [PubMed] [Google Scholar]
- 159.Demiralp B, Chen HL, Koh AJ, Keller ET, McCauley LK. Endocrinology. 2002;143(10):4038–4047. doi: 10.1210/en.2002-220221. [DOI] [PubMed] [Google Scholar]
- 160.Pearman AT, Chou WY, Bergman KD, Pulumati MR, Partridge NC. J Biol Chem. 1996;271(41):25715–25721. doi: 10.1074/jbc.271.41.25715. [DOI] [PubMed] [Google Scholar]
- 161.Gonzalez GA, Montminy MR. Cell. 1989;59(4):675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 162.Tyson DR, Swarthout JT, Jefcoat SC, Partridge NC. Endocrinology. 2002;143(2):674–682. doi: 10.1210/endo.143.2.8626. [DOI] [PubMed] [Google Scholar]
- 163.Clohisy JC, Scott DK, Brakenhoff KD, Quinn CO, Partridge NC. Mol Endocrinol. 1992;6(11):1834–1842. doi: 10.1210/mend.6.11.1480173. [DOI] [PubMed] [Google Scholar]
- 164.McCauley LK, Koh AJ, Beecher CA, Rosol TJ. Endocrinology. 1997;138(12):5427–5433. doi: 10.1210/endo.138.12.5587. [DOI] [PubMed] [Google Scholar]
- 165.Scott DK, Brakenhoff KD, Clohisy JC, Quinn CO, Partridge NC. Mol Endocrinol. 1992;6(12):2153–2159. doi: 10.1210/mend.6.12.1337147. [DOI] [PubMed] [Google Scholar]
- 166.Koe RC, Clohisy JC, Tyson DR, Pulumati MR, Cook TF, Partridge NC. Calcif Tissue Int. 1997;61(1):52–58. doi: 10.1007/s002239900294. [DOI] [PubMed] [Google Scholar]
- 167.McCauley LK, Koh-Paige AJ, Chen H, Chen C, Ontiveros C, Irwin R, McCabe LR. Endocrinology. 2001;142(5):1975–1981. doi: 10.1210/endo.142.5.8157. [DOI] [PubMed] [Google Scholar]
- 168.Lian JB, Stein GS, Stein JL, van Wijnen AJ. J Cell Biochem Suppl. 1998;30–31:62–72. [PubMed] [Google Scholar]
- 169.Franceschi RT. Crit Rev Oral Biol Med. 1999;10(1):40–57. doi: 10.1177/10454411990100010201. [DOI] [PubMed] [Google Scholar]
- 170.Ducy P, Schinke T, Karsenty G. Science. 2000;289(5484):1501–1504. doi: 10.1126/science.289.5484.1501. [DOI] [PubMed] [Google Scholar]
- 171.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Cell. 1997;89(5):755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 172.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cell. 1997;89(5):765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 173.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Cell. 1997;89(5):747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 174.Ducy P, Starbuck M, Priemel M, Shen J, Pinero G, Geoffroy V, Amling M, Karsenty G. Genes Dev. 1999;13(8):1025–1036. doi: 10.1101/gad.13.8.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ito Y, Miyazono K. Curr Opin Genet Dev. 2003;13(1):43–47. doi: 10.1016/s0959-437x(03)00007-8. [DOI] [PubMed] [Google Scholar]
- 176.Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, Hassan MQ, Gaur T, Lengner CJ, Young DW. Rev Endocr Metab Disord. 2006;7(1–2):1–16. doi: 10.1007/s11154-006-9001-5. [DOI] [PubMed] [Google Scholar]
- 177.Reinhold MI, Naski MC. J Biol Chem. 2007;282(6):3653–3663. doi: 10.1074/jbc.M608995200. [DOI] [PubMed] [Google Scholar]
- 178.Ge C, Xiao G, Jiang D, Franceschi RT. J Cell Biol. 2007;176(5):709–718. doi: 10.1083/jcb.200610046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Xiao G, Jiang D, Thomas P, Benson MD, Guan K, Karsenty G, Franceschi RT. J Biol Chem. 2000;275(6):4453–4459. doi: 10.1074/jbc.275.6.4453. [DOI] [PubMed] [Google Scholar]
- 180.Bellido T, Ali AA, Plotkin LI, Fu Q, Gubrij I, Roberson PK, Weinstein RS, O’Brien CA, Manolagas SC, Jilka RL. J Biol Chem. 2003;278(50):50259–50272. doi: 10.1074/jbc.M307444200. [DOI] [PubMed] [Google Scholar]
- 181.Krishnan V, Moore TL, Ma YL, Helvering LM, Frolik CA, Valasek KM, Ducy P, Geiser AG. Mol Endocrinol. 2003;17(3):423–435. doi: 10.1210/me.2002-0225. [DOI] [PubMed] [Google Scholar]
- 182.Merciris D, Marty C, Collet C, de Vernejoul MC, Geoffroy V. Am J Pathol. 2007;170(5):1676–1685. doi: 10.2353/ajpath.2007.061069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Selvamurugan N, Pulumati MR, Tyson DR, Partridge NC. J Biol Chem. 2000;275(7):5037–5042. doi: 10.1074/jbc.275.7.5037. [DOI] [PubMed] [Google Scholar]
- 184.Rice DP, Aberg T, Chan Y, Tang Z, Kettunen PJ, Pakarinen L, Maxson RE, Thesleff I. Development. 2000;127(9):1845–1855. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- 185.Lee MS, Lowe GN, Strong DD, Wergedal JE, Glackin CA. J Cell Biochem. 1999;75(4):566–577. doi: 10.1002/(sici)1097-4644(19991215)75:4<566::aid-jcb3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 186.Hayashi M, Nimura K, Kashiwagi K, Harada T, Takaoka K, Kato H, Tamai K, Kaneda Y. J Cell Sci. 2007;120(Pt 8):1350–1357. doi: 10.1242/jcs.000067. [DOI] [PubMed] [Google Scholar]
- 187.Ryoo HM, Hoffmann HM, Beumer T, Frenkel B, Towler DA, Stein GS, Stein JL, van Wijnen AJ, Lian JB. Mol Endocrinol. 1997;11(11):1681–1694. doi: 10.1210/mend.11.11.0011. [DOI] [PubMed] [Google Scholar]
- 188.Satokata I, Ma L, Ohshima H, Bei M, Woo I, Nishizawa K, Maeda T, Takano Y, Uchiyama M, Heaney S, Peters H, Tang Z, Maxson R, Maas R. Nat Genet. 2000;24(4):391–395. doi: 10.1038/74231. [DOI] [PubMed] [Google Scholar]
- 189.Shao JS, Cheng SL, Charlton-Kachigian N, Loewy AP, Towler DA. J Biol Chem. 2003;278(50):50195–50202. doi: 10.1074/jbc.M308825200. [DOI] [PubMed] [Google Scholar]
- 190.Li H, Marijanovic I, Kronenberg MS, Erceg I, Stover ML, Velonis D, Mina M, Heinrich JG, Harris SE, Upholt WB, Kalajzic I, Lichtler AC. Dev Biol. 2008;316(2):458–470. doi: 10.1016/j.ydbio.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Nissen-Meyer LS, Jemtland R, Gautvik VT, Pedersen ME, Paro R, Fortunati D, Pierroz DD, Stadelmann VA, Reppe S, Reinholt FP, Del Fattore A, Rucci N, Teti A, Ferrari S, Gautvik KM. J Cell Sci. 2007;120(Pt 16):2785–2795. doi: 10.1242/jcs.003855. [DOI] [PubMed] [Google Scholar]
- 192.Reppe S, Rian E, Jemtland R, Olstad OK, Gautvik VT, Gautvik KM. J Bone Miner Res. 2000;15(12):2402–2412. doi: 10.1359/jbmr.2000.15.12.2402. [DOI] [PubMed] [Google Scholar]
- 193.Reppe S, Stilgren L, Olstad OK, Brixen K, Nissen-Meyer LS, Gautvik KM, Abrahamsen B. Bone. 2006;39(1):189–198. doi: 10.1016/j.bone.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 194.Partridge NC, Dickson CA, Kopp K, Teitelbaum SL, Crouch EC, Kahn AJ. Mol Endocrinol. 1989;3(2):232–239. doi: 10.1210/mend-3-2-232. [DOI] [PubMed] [Google Scholar]
- 195.Noda M. Endocrinology. 1989;124(2):612–617. doi: 10.1210/endo-124-2-612. [DOI] [PubMed] [Google Scholar]
- 196.Termine JD, Kleinman HK, Whitson SW, Conn KM, McGarvey ML, Martin GR. Cell. 1981;26(1 Pt 1):99–105. doi: 10.1016/0092-8674(81)90037-4. [DOI] [PubMed] [Google Scholar]
- 197.Cook TF, Burke JS, Bergman KD, Quinn CO, Jeffrey JJ, Partridge NC. Arch Biochem Biophys. 1994;311(2):313–320. doi: 10.1006/abbi.1994.1243. [DOI] [PubMed] [Google Scholar]
- 198.Catherwood BD, Titus L, Evans CO, Rubin J, Boden SD, Nanes MS. Endocrinology. 1994;134(3):1429–1436. doi: 10.1210/endo.134.3.8119183. [DOI] [PubMed] [Google Scholar]
- 199.Greenfield EM, Gornik SA, Horowitz MC, Donahue HJ, Shaw SM. J Bone Miner Res. 1993;8(10):1163–1171. doi: 10.1002/jbmr.5650081003. [DOI] [PubMed] [Google Scholar]
- 200.Raggatt LJ, Qin L, Tamasi J, Jefcoat SC, Jr, Shimizu E, Selvamurugan N, Liew FY, Bevelock L, Feyen JH, Partridge NC. J Biol Chem. 2008;283(11):6790–6798. doi: 10.1074/jbc.M709909200. [DOI] [PubMed] [Google Scholar]
- 201.Miles RR, Sluka JP, Santerre RF, Hale LV, Bloem L, Boguslawski G, Thirunavukkarasu K, Hock JM, Onyia JE. Endocrinology. 2000;141(1):28–36. doi: 10.1210/endo.141.1.7229. [DOI] [PubMed] [Google Scholar]
- 202.Weir EC, Lowik CW, Paliwal I, Insogna KL. J Bone Miner Res. 1996;11(10):1474–1481. doi: 10.1002/jbmr.5650111014. [DOI] [PubMed] [Google Scholar]
- 203.Tetradis S, Bezouglaia O, Tsingotjidou A. Endocrinology. 2001;142(2):663–670. doi: 10.1210/endo.142.2.7926. [DOI] [PubMed] [Google Scholar]
- 204.Tetradis S, Bezouglaia O, Tsingotjidou A, Vila A. Biochem Biophys Res Commun. 2001;281(4):913–916. doi: 10.1006/bbrc.2001.4459. [DOI] [PubMed] [Google Scholar]
- 205.Steinberg TH, Civitelli R, Geist ST, Robertson AJ, Hick E, Veenstra RD, Wang HZ, Warlow PM, Westphale EM, Laing JG, et al. EMBO J. 1994;13(4):744–750. doi: 10.1002/j.1460-2075.1994.tb06316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Schiller PC, Roos BA, Howard GA. J Bone Miner Res. 1997;12(12):2005–2013. doi: 10.1359/jbmr.1997.12.12.2005. [DOI] [PubMed] [Google Scholar]
- 207.Civitelli R, Ziambaras K, Warlow PM, Lecanda F, Nelson T, Harley J, Atal N, Beyer EC, Steinberg TH. J Cell Biochem. 1998;68(1):8–21. doi: 10.1002/(sici)1097-4644(19980101)68:1<8::aid-jcb2>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 208.Gubrij I, Ali AA, Chambers TM, Berryhil SB, Liu X, Roberson P, O’Brien CA, Weinstein RS, Manolagas SC, Jilka RL. J Bone Miner Res. 2003;18:S136. [Google Scholar]
- 209.Yamashita J, Datta NS, Chun YH, Yang DY, Carey AA, Kreider JM, Goldstein SA, McCauley LK. J Bone Miner Res. 2008;23(5):621–632. doi: 10.1359/JBMR.071211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, Suda T, Matsuo K. Cell Metab. 2006;4(2):111–121. doi: 10.1016/j.cmet.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 211.Allan EH, Hausler KD, Wei T, Gooi JH, Quinn JM, Crimeen-Irwin B, Pompolo S, Sims NA, Gillespie MT, Onyia JE, Martin TJ. J Bone Miner Res. 2008;23(8):1170–1181. doi: 10.1359/jbmr.080324. [DOI] [PubMed] [Google Scholar]
- 212.Fermor B, Skerry TM. J Bone Miner Res. 1995;10(12):1935–1943. doi: 10.1002/jbmr.5650101213. [DOI] [PubMed] [Google Scholar]
- 213.Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. J Biol Chem. 2005;280(20):19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 214.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA. EMBO J. 2003;22(23):6267–6276. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, Jilka RL. Endocrinology. 2005;146(11):4577–4583. doi: 10.1210/en.2005-0239. [DOI] [PubMed] [Google Scholar]
- 216.O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH, Jilka RL, Weinstein RS, Manolagas SC, Bellido T. PLoS ONE. 2008;3(8):e2942. doi: 10.1371/journal.pone.0002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Pines J. Semin Cell Biol. 1994;5(6):399–408. doi: 10.1006/scel.1994.1047. [DOI] [PubMed] [Google Scholar]
- 218.Xiong Y, Connolly T, Futcher B, Beach D. Cell. 1991;65(4):691–699. doi: 10.1016/0092-8674(91)90100-d. [DOI] [PubMed] [Google Scholar]
- 219.Matsushime H, Ewen ME, Strom DK, Kato JY, Hanks SK, Roussel MF, Sherr CJ. Cell. 1992;71(2):323–334. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- 220.Xiong Y, Zhang H, Beach D. Cell. 1992;71(3):505–514. doi: 10.1016/0092-8674(92)90518-h. [DOI] [PubMed] [Google Scholar]
- 221.Sherr CJ. Cell. 1994;79(4):551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 222.Hirama T, Koeffler HP. Blood. 1995;86(3):841–854. [PubMed] [Google Scholar]
- 223.Onishi T, Hruska K. Endocrinology. 1997;138(5):1995–2004. doi: 10.1210/endo.138.5.5146. [DOI] [PubMed] [Google Scholar]
- 224.Beck GR, Jr, Zerler B, Moran E. Cell Growth Differ. 2001;12(2):61–83. [PubMed] [Google Scholar]
- 225.Brown JR, Nigh E, Lee RJ, Ye H, Thompson MA, Saudou F, Pestell RG, Greenberg ME. Mol Cell Biol. 1998;18(9):5609–5619. doi: 10.1128/mcb.18.9.5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Pestell RG, Albanese C, Reutens AT, Segall JE, Lee RJ, Arnold A. Endocr Rev. 1999;20(4):501–534. doi: 10.1210/edrv.20.4.0373. [DOI] [PubMed] [Google Scholar]
- 227.Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. EMBO J. 2000;19(9):2056–2068. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Beier F, Ali Z, Mok D, Taylor AC, Leask T, Albanese C, Pestell RG, LuValle P. Mol Biol Cell. 2001;12(12):3852–3863. doi: 10.1091/mbc.12.12.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Onishi T, Zhang W, Cao X, Hruska K. J Bone Miner Res. 1997;12(10):1596–1605. doi: 10.1359/jbmr.1997.12.10.1596. [DOI] [PubMed] [Google Scholar]
- 230.Pratap J, Galindo M, Zaidi SK, Vradii D, Bhat BM, Robinson JA, Choi JY, Komori T, Stein JL, Lian JB, Stein GS, van Wijnen AJ. Cancer Res. 2003;63(17):5357–5362. [PubMed] [Google Scholar]
- 231.Galindo M, Pratap J, Young DW, Hovhannisyan H, Im HJ, Choi JY, Lian JB, Stein JL, Stein GS, van Wijnen AJ. J Biol Chem. 2005;280(21):20274–20285. doi: 10.1074/jbc.M413665200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Shen R, Wang X, Drissi H, Liu F, O’Keefe RJ, Chen D. J Biol Chem. 2006;281(24):16347–16353. doi: 10.1074/jbc.M603439200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hock JM, Krishnan V, Onyia JE, Bidwell JP, Milas J, Stanislaus D. J Bone Miner Res. 2001;16(6):975–984. doi: 10.1359/jbmr.2001.16.6.975. [DOI] [PubMed] [Google Scholar]
- 234.Freitas PHL, Amizuka N, Li M, Takagi R, Maeda T. J Bone Miner Res. 2006;21:S321. [Google Scholar]
- 235.Wang YH, Liu Y, Buhl K, Rowe DW. J Bone Miner Res. 2005;20(1):5–14. doi: 10.1359/JBMR.041016. [DOI] [PubMed] [Google Scholar]
- 236.Wang YH, Liu Y, Rowe DW. Am J Physiol Endocrinol Metab. 2007;292(2):E594–603. doi: 10.1152/ajpendo.00216.2006. [DOI] [PubMed] [Google Scholar]