

Abstract
Increasing evidence implicates activation of NF-κB in a variety of glomerular diseases, but the mechanisms involved are unknown. Here, upregulation of NF-κB in the podocytes of transgenic mice resulted in glomerulosclerosis and proteinuria. Absence of the podocyte protein nephrin resulted in NF-κB activation, suggesting that nephrin negatively regulates the NF-κB pathway. Signal transduction assays supported a functional relationship between nephrin and NF-κB and suggested the involvement of atypical protein kinase C (aPKCζ/λ/ι) as an intermediary. We propose that disruption of the slit diaphragm leads to activation of NF-κB; subsequent upregulation of NF-κB-driven genes results in glomerular damage mediated by NF-κB-dependent pathways. In summary, nephrin may normally limit NF-κB activity in the podocyte, suggesting a mechanism by which it might discourage the evolution of glomerular disease.
NF-κB is a transcription factor activated by cell surface receptor signaling to meet stress and inflammatory responses, regulating key cellular processes such as inflammation, innate and adaptive immunity, and cell growth and survival.1 Five mammalian NF-κB proteins share a Rel homology domain with composition of the active dimer dictated by cell type and nature of inducing stimulus.2 Inactive NF-κB is sequestered in the cytoplasm bound to IκB3; phosphorylation of IκB releases active NF-κB, which translocates to the nucleus to induce an extensive range of target genes.4 RelA dimers are the most abundant and potent gene transactivators within the family.5
Induction of NF-κB signaling and specificity of transcriptional response are dependent on a complex interplay of pathways. Adaptor proteins p626 and MyD887 and intracellular messengers such as atypical protein kinase C (aPKCζ/ι)8 connect NF-κB with cell surface receptors. aPKCζ/ι activates NF-κB by either release from IκB9 or direct nuclear phosphorylation,10 whereas activation is severely impaired by aPKCζ deficiency.11 Expression of aPKCζ/ι inhibitory proteins such as Par4 can also abrogate NF-κB activation.12,13
Glomerular disease manifests as urinary protein leak resulting from malfunction of the glomerular filtration barrier. Podocytes separated by slit diaphragms are crucial in maintaining barrier integrity.14 Injury disrupts their actin cytoskeleton, causing foot process effacement; detachment from the glomerular basement membrane (GBM), with significant molecular reorganization of the slit diaphragms; and urinary protein loss.15
Nephrin, an Ig superfamily member, is a key slit diaphragm component and presumed adhesion molecule that contributes directly to a physical filtration barrier at the slit diaphragm.16,17 Nephrin mutations result in disruption of the actin cytoskeleton and severe glomerular disease. Downregulation of nephrin expression unrelated to gene mutations also occurs in podocyte injury,18,19 and mutations that are within the cytosolic tail and do not affect protein expression cause equivalent glomerular disease,20,21 supporting a role in cellular signaling. This is verified by evidence that nephrin serves as a signaling scaffold to recruit other podocyte proteins,22–25 is phosphorylated by Src-family kinases,26,27 and associates with adapters such as Nck to regulate the actin cytoskeleton.28,29
To our knowledge, the mechanism of NF-κB regulation in glomerular cells is unknown. Evidence for the involvement of NF-κB activation in glomerular and renal tubular cell injury is provided by correlative studies of human kidney disease30,31 and experimental disease models.31,32 Complement activates NF-κB and NF-κB–dependent gene transcription in podocytes in vitro,33 and upregulation of IL-1, IL-4, and TNF-α is detected in injured podocytes, supporting a direct role for NF-κB activation in human glomerular disease.30,31 Whether nephrin modulates NF-κB has not been investigated, but regulation of NF-KB activation by other Ig superfamily molecules involved in cell adhesion, namely N-Cam and L1Ig6, is well described.33,34
We investigated a causal link between NF-κB activation and renal glomerular damage by examining whether genetic inactivation of an upstream inhibitor of NF-κB activation could result in glomerular injury independent of immune cell activation. Considering its ubiquitous role in maintaining glomerular filtration, we then explored whether the receptor protein nephrin participates in the regulation of this process.
Results
Activation of NF-κB Results in Glomerulosclerosis and Proteinuria In Vivo
We first tested the hypothesis that NF-κB activation is a key response leading to glomerular damage. Because no renal phenotype has been reported in conjunction with mouse models resulting in NF-κB deficiency, we examined the significance of NF-κB activation in the kidney in vivo, focusing on models with constitutive activation of NF-κB resulting from removal of natural pathway inhibition. This conventional approach enables study of NF-κB overexpression while avoiding off-target effects.35
One such model involves genetic inactivation of the prostate apoptosis response 4 gene (Par4), which specifically binds and inhibits aPKCζ/λ/ι and is a critical negative regulator of the RelA/NF-κB pathway.12,13 Disruption of Par4 through homologous recombination in outbred (CD1) wild-type (WT) mice resulted in de-repression of aPKCζ/λ/ι and activation of NF-κB.36 Although Par4 is ubiquitously expressed, no clear systemic phenotype was identified. We therefore examined Par4−/− mice for renal abnormalities.
Par4−/− mice were studied at 2 wk, 3 mo, and 6 mo (n = 40). Although mice initially seemed normal, by 3 mo, the majority had developed significant proteinuria (Figure 1A). Analysis of kidneys by light microscopy at 2 wk showed mild mesangial expansion within glomeruli and ultrastructural evidence of foot process fusion indicative of a primary podocyte defect, supported by evidence of some podocyte loss on electron microscopy (Figures 1, Ba and C, c and d). At 3 mo, increasingly severe glomerulosclerosis was apparent with hyalinosis, tubular atrophy, pseudocysts, and luminal deposition of proteinaceous material (Figure 1Bc). At the ultrastructural level (Figure 1C), light microscopic changes were mirrored by progressive disruption of podocyte architecture and foot process fusion, with glomerular hypertrophy, abnormal duplication of the GBM, and hyaline deposits clearly apparent (Figure 1C, e and f). By 6 mo of age, glomerular disease was severe, and approximately 30% of animals had developed a coincident renal cystic tubular phenotype not dependent on the degree of proteinuria or gender (Figure 1B, g and h). Because crosses were performed on an outbred background, a degree of genetic heterogeneity was expected. Ultrastucturally, complete disruption of podocyte architecture was evident, with extensive foot process fusion, massive cell body swelling, sclerosis, collapse and folding of the mesangial matrix, and denudation of the diffusely thickened and corrugated GBM.
Figure 1.

Glomerular disease is present in Par4−/− mice. (A) A total of 4 μl of urine from 3-mo-old WT (WT1 and WT2) and Par4−/− (KO1 through 6) mice was suspended in Laemmli buffer and subjected to SDS-PAGE. Coomassie staining detected proteinuria in Par4−/− mice but not their WT counterparts. A band corresponding to albumin is shown (68 kD; black arrow). The variability in proteinuria resolved with increasing age of mice. (B) Hematoxylin and eosin staining of Par4−/− and WT glomeruli at 2 wk (a and b), 3 mo (c and d), and 6 mo (e and f). At 2 wk, only mild mesangial hypercellularity and glomerular enlargement is present in Par4−/− glomeruli, with preservation of the tubulointerstitial compartment (a). At 3 mo, marked mesangial expansion, glomerulosclerosis, and widening of the subcapsular space is evident, with some capsular adhesions (c). Significant disease progression occurs by 6 mo, with widespread scarring and atrophy of glomeruli, tubular atrophy, and pseudocysts containing proteinaceous material (arrow; E). In 25% of mice, coexisting renal tubular cysts were lined by simple epithelium (*), which also progressed in severity with age. Glomeruli and renal tubules were normal at all ages in WT controls (B, D, and F). (C) Par4−/− glomeruli were also abnormal at the ultrastructural level (c through h). In 2-wk-old mice (c and d), there is discontinuous podocyte foot process effacement and mild cell body swelling, but the GBM is normal. By 3 mo, there is marked effacement and swelling with hyaline deposits and vacuoles, glomerulosclerosis, mesangial expansion, and patchy blebbing of the GBM (e and f). By 6 mo, podocyte swelling, sclerosis, and foot process fusion is extensive, with diffuse thickening, corrugation, and denudation of the GBM. The mesangial matrix is folded and collapsed (g and h). By contrast, normal podocyte and glomerular architecture is seen in WT mice and littermates (a and b). Podocyte foot processes are well demarcated, and the mesangium is normal with preservation of the urinary space and no tubular cysts. Magnifications: ×40 in B; ×300 in C (a, c, e, and g); ×6000 in C (b, d, f, and h).
Glomerular disease was detected in >90% of mice and the concurrent cystic tubular phenotype in approximately 25%. Both phenotypes became progressively milder with subsequent crosses to WT (CD1) mice, as the increasing genetic heterogeneity allowed the epigenetic influence of different modifying loci to emerge. Littermates and WT mice with the same genetic background, however, showed no functional or morphologic renal phenotype (Figures 1, B, b, d, and f, and C, a and b). These data suggested direct activation of the aPKC/NF-κB axis results in glomerular and renal tubular disease in vivo.
Activation of RelA/NF-κB and aPKCζ Occurs in the Podocytes of Par4−/− Mice
To confirm whether aPKC/NF-κB pathway activation was present in Par4−/− podocytes to contribute to glomerulosclerosis, we examined the direct NF-κB target of this pathway, RelA, and phospho-aPKCζ expression in glomeruli of Par4−/− and WT mice at 2 wk, 3 mo, and 6 mo of age. Because activated RelA localizes to the nucleus, we investigated the subcellular localization in glomeruli for evidence for NF-κB activation in situ. Confocal immunofluorescence microscopy of cortical kidney sections showed cytoplasmic RelA expression in WT podocytes indicative of quiescence (Figure 2A, a, c, and e). Conversely, a shift to nuclear expression signified activation in Par4−/− podocytes (Figure 2A, b, d, and f). This activation was not detected in other Par4−/− glomerular cells or in WT glomeruli.
Figure 2.

RelA and aPKCζ/λ/ι are activated in Par4−/− podocytes. (A) Double-immunofluorescence staining of WT and Par4−/− glomeruli showing expression of RelA/NF-κB (a and b) and acetylated tubulin (c and d) in murine podocytes (white arrows), with image merge (e and f). RelA expression is cytoplasmic in WT podocytes (a and e), indicative of quiescence, and nuclear in Par4−/− podocytes (b and c), indicative of activation even at 2 wk of age. (B) Double-immunofluorescence staining of WT and Par4−/− glomeruli shows phospho-aPKCζ/λ/ι(Thr410/403) (a and b) and acetylated tubulin (c and d) in murine podocytes (white arrows), with image merge (e and f). Phospho-aPKC expression is predominately nuclear in WT podocytes with some cytoplasmic staining (a and e), whereas in Par4−/− glomeruli, the equilibrium shifts to cytoplasmic predominance (b and c) even at 2 wk of age. Magnification, ×63 (oil immersion lens).
Similarly, we confirmed the activation and localization of aPKCζ using a phospho-specific antibody. A shift in the equilibrium of phospho-aPKCζ(Thr410/403) expression from predominately nuclear in WT podocytes (Figure 2B, a, c, and e) to mainly cytoplasmic in Par4−/− (Figure 2B, b, d, and f) supported aPKC activation in Par4−/− glomeruli.
Downregulation of Wilms' tumor 1 (WT1) and nephrin was detected in Par4−/− but not WT glomeruli (Figure 3, A, B, G, and H), confirming podocyte dysregulation and loss. PAX2 expression was normal in glomeruli and tubules of Par4−/− mice (Figure 3, C through F), going against WT1-mediated podocyte disease.37–39 Accordingly, through its connection with slit diaphragm signaling,26–29 we examined whether nephrin might regulate NF-κB activation.
Figure 3.
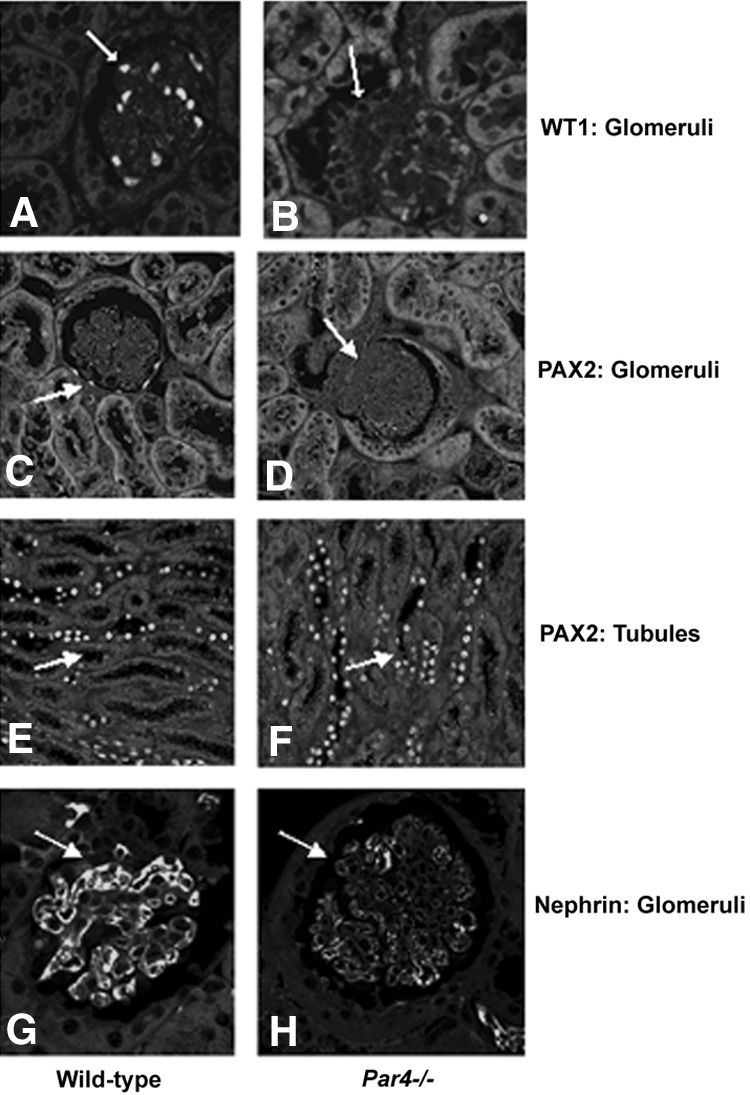
Immunohistochemical analysis of WT and Par4−/− glomeruli to show expression of podocyte markers WT1, PAX2, and nephrin. (A and B) Normal nuclear WT1 expression is seen in WT podocytes (A), whereas in Par4−/− glomeruli WT1 expression is virtually absent (B). (C and D) Nuclear PAX2 expression is absent in WT and Par4−/− glomeruli. (E and F) Renal tubular PAX2 expression was equivalent and is included as a control. (G and H) Nephrin expression is normal in WT podocytes (G) but significantly downregulated in Par4−/− glomeruli (H). Immunohistochemistry was performed on paired samples under identical experimental conditions. Data are shown for 3-mo-old mice, with abnormalities detected from 2 wk of age.
Loss of Nephrin Results in RelA/NF-κB Activation in Human Podocytes
To study the mechanism of NF-κB activation in podocytes in more detail, we turned to an in vitro model. Cell lysates from unstimulated WT human immortalized podocytes40 were first immunoblotted to identify components of the NF-κB signaling pathway. All five isoforms RelA, RelB, c-Rel, p105/p50, and p100/p52; IκBα and IκBβ; and the IKKα and IKKβ components of the IKK enzyme complex were detected, verifying the presence of their orthologs in murine podocytes.41 NF-κB isoform expression was then examined in nonstimulated mutant human podocytes (MT) with a constitutive 121delCT frameshift mutation lacking functional nephrin.42 Interestingly, we detected specific upregulation of the RelA/NF-κB isoform in MT cells compared with WT (Figure 4A). Reprobing of membranes with antibodies to other proteins associated with NF-κB including PTEN, PDK1, pJNK, p38, Par4, and BCL2 showed equivalent expression between WT and MT, verifying equal protein loading (data shown for BCL2). This indicated that NF-κB activation in MT podocytes was independent of Par4. Upregulation of phospho-IκB (Ser-32/36) but not total IκB in MT endorsed RelA/NF-B activation through release from the IκB complex (Figure 4B).
Figure 4.
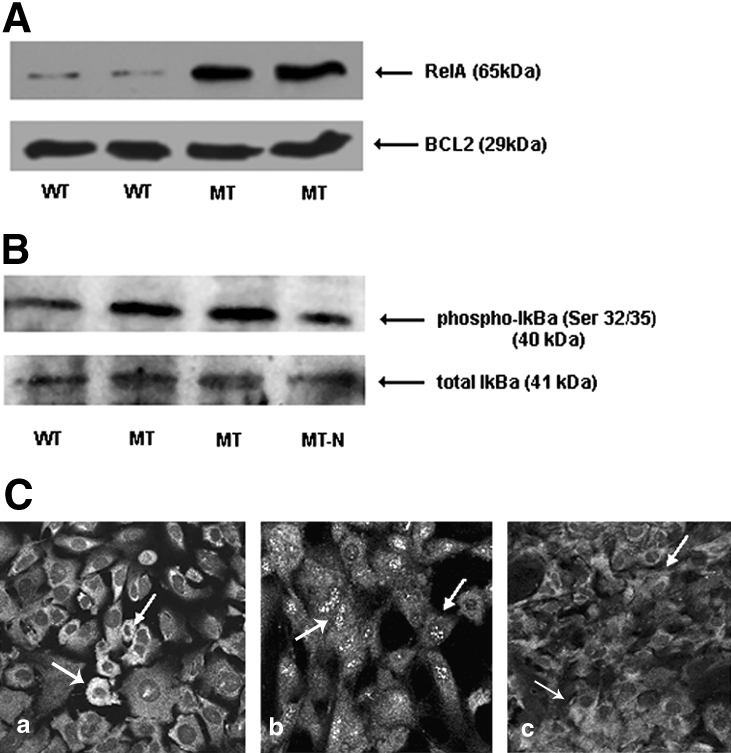
RelA/NF-κB is activated in nephrin-deficient human podocytes. (A) Western blot of WT, MT, and MT+N whole-cell protein extracts of human podocytes. All lanes were equally loaded with 20 μg of protein extract verified by both Ponceau S staining (data not shown) and reprobing of the membrane. Upregulation of RelA is present in MT cells, with rescue in MT+N. No difference was seen for p38, pJNK, BCL2, PTEN, or PDK1; data shown for BCL2. (B) Upregulation of phospho-IκB (Ser-32/36) was detected in nephrin-deficient MT cells, endorsing NF-κB activation through release from IκBα, with rescue occurring on re-introduction of nephrin expression (MT-N). Total IκBα expression is equivalent between WT and MT, as expected. (C) Immunofluorescence staining of differentiated podocytes. Expression of RelA is cytoplasmic in WT, indicative of quiescence (a, white arrows), whereas in mutant podocytes (MT) RelA translocates to the nucleus consistent with activation (b). Expression is rescued to the cytoplasm on re-introduction of nephrin by stable transfection (c; MT+N). Magnification, ×40.
We used immunofluorescence confocal microscopy to confirm RelA/NF-κB activation through its subcellular localization in WT and MT podocytes (Figure 4C). In WT cells, RelA/NF-κB was sequestered in the cytoplasm, indicative of quiescence. Conversely, expression was nuclear in MT, indicating activation. This was specific to nephrin, because re-introduction of WT nephrin in MT podocytes (MT+N) rescued RelA/NF-κB expression to the cytoplasm and to WT levels on immunoblot.
Absence of Nephrin Relocates Phospho-aPKCζ Expression to the Cytoplasm
Given that our mouse studies had implicated aPKCζ in the mechanism of RelA/NF-κB activation in Par4−/− podocytes, we also compared expression levels and subcellular localization of aPKCζ in WT and MT podocytes. Immunoblot of podocyte cell extracts showed upregulation of phospho-aPKC(Thr410/403) in MT podocytes, whereas total aPKC expression was equivalent (Figure 5A). Immunofluorescence confocal microscopy also detected predominately nuclear phospho-aPKCζ expression in WT, with a shift to the cytoplasm in MT podocytes (Figure 5B). Re-expression of WT nephrin in MT cells (MT+N) rescued phospho-aPKCζ to the cytoplasm, supporting a direct link between nephrin and aPKCζ expression.
Figure 5.
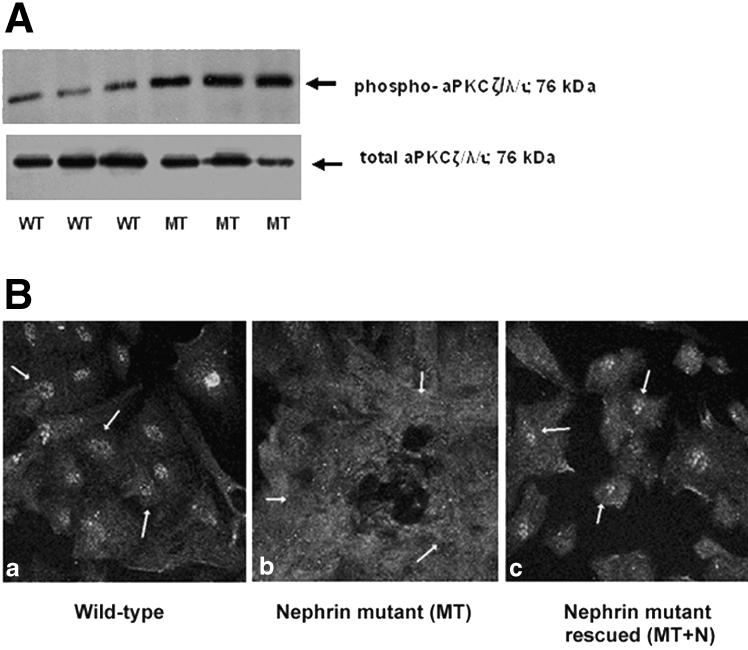
aPKCζ/λ/ι is activated in nephrin-deficient human podocytes. (A) Upregulation of phospho-aPKCζ/λ/ι(Thr410/403) is seen in nephrin-deficient podocytes on Western blot. Equal protein loading was verified by Ponceau S staining and reprobing with an antibody against total aPKCζ/λ/ι, which shows equivalent expression between WT and MT podocytes. (B) Immunofluorescence staining of cultured human podocytes shows nuclear sequestration of phospho-aPKCζ/λ/ι(Thr410/403) (white arrows, a) in WT podocytes and cytoplasmic expression in nephrin-deficient podocytes (MT; b). Re-introduction of nephrin in podocytes (MT+N) rescues the phenotype to predominantly nuclear expression (c). Magnification, ×40.
NF-κB Repression in Response to Nephrin Is Specific and Independent of Podocin
To substantiate a direct relationship between nephrin and NF-κB signaling, we designed an in vitro functional assay to test NF-κB promoter activity using a cis-acting NF-κB enhancer element linked to a luciferase reporter (pNF-κB). Co-transfection of B-gal, a cytomegalovirus (CMV)-driven constitutive reporter, was used to standardize transfections. Assays were performed in cultured MDCK cells and in human podocytes.
WT nephrin (pCMV-NPHS1; amino acids [AA] 1 through 1241) and two truncated nephrin mutants encompassing AA 1 through 1201 and AA 1 through 1160 were used for experiments. Equivalent expression and membrane localization of all WT and mutant nephrin proteins were verified by immunocytochemistry and immunoblot (data not shown). Truncation mutants shorter than R1160X did not demonstrate equivalent expression to WT nephrin. The R1160X mutant mimicked the most C-terminal mutation detected in human nephrotic syndrome to date,20,21 which removes a key functional region43,44 that includes three Nck SH2-binding motifs28,29 that may act cooperatively to stimulate actin reorganization.45,46
Transfection of WT nephrin consistently repressed pNF-κB reporter activity to between half and two thirds of basal (P < 0.001), with the sequentially truncated mutants resulting in a graded reduction in pNF-κB repression, indicating specificity to nephrin (Figure 6A), and raising the possibility of multiple binding sites for cytoplasmic adaptor proteins linking nephrin to NF-κB signaling.
Figure 6.

pNF-κB reporter activity is repressed by nephrin. (A) pNF-κB reporter activity was significantly repressed (P < 0.001) by co-transfection with full-length WT nephrin NPHN AA 1 through 1241. Sequential truncation to produce mutant nephrin proteins NPHN AA 1 through 1201 and NPHN AA 1 through 1160 resulted in a graded reduction in the repression of pNF-KB reporter activity. (B) Full-length podocin (POD) did not alter pNF-κB reporter activity or its repression by WT NPHN, indicating that this observation was specific to and independent of podocin. (C) pNF-κB reporter was also examined in human podocytes (MT). Significant (P < 0.001) repression of reporter activity was again seen with both transient and stable transfections of WT NPHN. This confirmed observations made in MDCK cells.
AP1 was activated by nephrin, in keeping with previous studies,43,46 whereas nephrin had no effect on pNFAT (data not shown). Podocin, a key nephrin interactor47 that is known to enhance nephrin-mediated AP1 activation in vitro44,45 had no effect on nephrin-mediated repression of pNF-κB, indicating this was independent of podocin (Figure 6B). Findings in MDCK cells were mirrored by transient and stable transfection of WT nephrin into human podocytes, which also resulted in significant repression of pNF-κB activity (P < 0.001; Figure 6C).
Nephrin-Mediated Repression of pNF-κB Is Abrogated by Dominant Negative Pathway Intermediaries
A number of molecules adapt between cell surface receptors and NF-κB. We therefore used three different dominant negative pathway intermediaries linked to NF-κB to validate findings.
A key step in RelA/NF-κB activation is the phosphorylation of the IκB complex.4 Transfection of dominant negative IκBα (pCMV-IκM; Clontech, Mountain View, CA) squelched pNF-κB reporter activity and blocked nephrin-mediated repression of pNF-κB (Figure 7A), corroborating NF-κB activation through release from cytoplasmic IκB.
Figure 7.

Pathway mapping of nephrin-mediated repression of NF-κB. (A) Repression of pNF-κB by NPHN is abrogated by co-transfection with dominant negative IκBα (IκM) and rescued by co-transfection with WT IκB in MDCK cells. (B) Repression of pNF-κB by NPHN is abrogated by co-transfection with dominant negative aPKCζ (PKC-DN) in MDCK cells. (C) Repression of pNF-κB by WT NPHN is abrogated by increasing concentrations of dominant negative 14-3-3 (DN1 = 4 μg/μl; DN2 = 6 μg/μl) in MDCK cells.
In a separate experiment, we examined the effect of co-transfection of dominant negative aPKCζ. This abrogated nephrin-induced repression of pNF-κB, substantiating a role for aPKCζ as a pathway intermediary (Figure 7B). An interaction between aPKCζ and cytosolic nephrin was confirmed by GST immunoprecipitation (Figure 8A) and co-localization in WT podocytes (data not shown).
Figure 8.
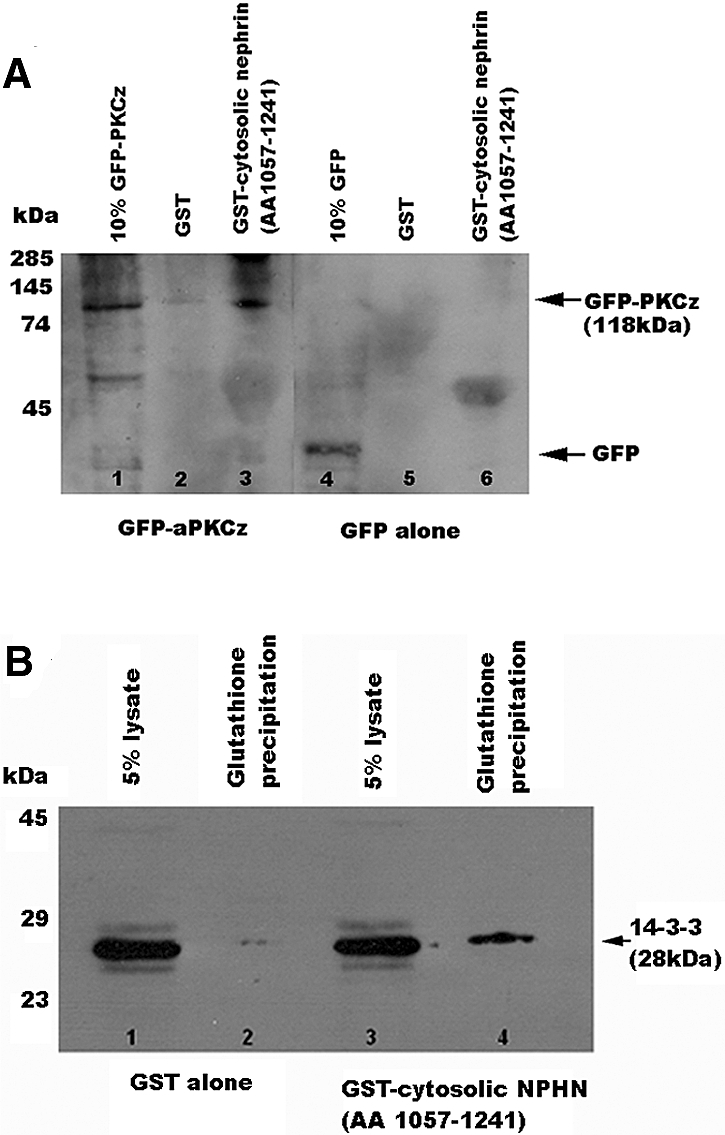
aPKCζ and 14-3-3θ bind cytosolic nephrin (AA 1057 through 1241). (A) Successful glutathione precipitation of native aPKCζ from MDCK cells by cytosolic nephrin (AA 1057 through 1241). Blots were probed with aPKCζ (rabbit polyclonal). Lanes 1 and 2, GST alone, no precipitation of 14-3-3θ (lane 2); lanes 3 and 4, precipitation of endogenous 14-3-3θ by the GST-cytosolic nephrin (AA 1057 through 1241) fusion protein in lane 4. (B) Successful glutathione precipitation of native 14-3-3θ from MDCK cells by cytosolic nephrin (AA 1057 through 1241). Blots were probed with 14-3-3θ (rabbit polyclonal; Santa Cruz Biotechnology). Lanes 1 and 2, GST alone, no precipitation of 14-3-3θ (lane 2); lanes 3 and 4, precipitation of endogenous 14-3-3θ by the GST-cytosolic NPHN (AA 1057 through 1241) fusion protein (lane 4).
Surprising, dominant negative 14-3-3, an intracellular adapter that modulates RelA/NF-κB signaling through an independent mechanism,48 also blocked nephrin-induced repression of pNF-κB (Figure 7C). The significance is unclear, although yeast two-hybrid screening revealed that 14-3-3 interacts with nephrin (data not shown), confirmed by GST immunoprecipitation (Figure 8B) and co-localization (data not shown).
Discussion
NF-κB deviates from the classical receptor signal paradigm through an ability to respond to cellular stress and microbial pathogens, both implicated in the pathogenesis of glomerular disease. Its stimuli and target genes are extraordinarily diverse, differing greatly depending on cell and stimulus context. Consequently, tight regulation of NF-κB activity is a prerequisite for ensuring that coordinated processing and integration of stimuli occur.
Although the significance of NF-κB activation in glomerular disease is incompletely understood, a series of correlations between the activation of NF-κB and onset of both immune- and non–immune-mediated glomerular injury implicate dysregulation of the NF-κB pathway in its pathogenesis.30–33 In response to this, we asked whether a direct causal link between NF-κB activation and glomerular disease exists in vivo and whether the key podocyte protein nephrin might regulate this pathway.
We postulated that NF-κB activation may result in podocyte injury. To test this, we examined the kidneys of a mouse model with genetic inactivation of Par4, which negatively regulates the RelA/NF-κB pathway through inhibition of aPKC.12,13 Par4−/− mice exhibited hyperactivation of NF-κB and, in keeping with our hypothesis, developed a primary podocyte defect within the first 2 wk of life. Rapid progression to severe glomerular disease combined with aPKC/RelA/NF-κB activation in podocytes indicated pathway control may be essential for integrity of the glomerular filtration barrier.
RelA/NF-κB activation in Par4−/− podocytes and glomerular proteinuria in Par4−/− mice connected aberrant slit diaphragm signaling with induction of NF-κB–driven genes and podocyte injury. Constitutive activation of RelA/NF-κB in nephrin mutant podocytes, rescued by WT nephrin, endorsed a direct link between nephrin deficiency and RelA/NF-κB activation in vitro. Signal transduction assays demonstrated nephrin-mediated repression of pNF-κB, corroborating cell culture findings. Nephrin downregulation in Par4−/− podocytes might be distal to NF-κB–induced transcription; however, identification of aPKCζ and 14-3-3 as nephrin-binding partners illustrated the likely complexity of pathway interactions. Both are directly associated with NF-κB activation as well as regulation of the actin cytoskeleton in podocytes.49–51 The corollary of results in the Par4−/− model and in vitro supports a hypothesis that nephrin may also exert a negative tone on NF-κB activity in vivo.
Downregulation of developmental markers WT1 and nephrin in Par4−/− glomeruli was consistent with podocyte injury and loss. Although Par4 co-represses transcription with WT152, absence of other WT1-linked developmental abnormalities and normal expression of WT1 targets PAX2 and BCL2 in Par4−/− podocytes precluded WT1 as a direct cause. Notably, the cystic tubular phenotype supported a direct Par4 effect, because this is a crucial factor in the regulation of renal tubular cell death and survival, although renal carcinoma was not detected.53 Par4 deficiency also resulted in mild T cell proliferation,54 although after the onset of kidney disease. The lack of an overt immunologic phenotype or inflammatory cell infiltrate in Par4−/− glomeruli also indicated systemic immunologic derangement was an unlikely primary cause.
The shift in aPKCζ expression may be associated with its ability to act as a molecular switch,55 playing a beneficial role in the nucleus through combinatorial transcriptional co-repression or co-activation10 but moving to the cytoplasm to phosphorylate IκB56 and release RelA/NF-κB as occurs in renal tubular cell injury.57 aPKCζ and NF-κB directly regulate cytoskeletal stability,58–60 which may augment podocyte damage. Upregulation of RelA/NF-κB in nephrin-deficient podocytes indicated abnormal NF-κB turnover, usually under tight dynamic control.61 Total IκB expression was equivalent, indicating NF-κB degradation was intact; however, loss of pathway modulation by nephrin would lead to upregulation, supporting its key role in the regulation of the process.
Although our data indicated that RelA/NF-κB activation causes podocyte damage, the identity of inducing signals remains unknown. The association between NF-κB and stress and innate immunity provides a link between podocyte injury and NF-κB activation stimuli potentially originating from cytokines or microbial pathogens.62,63 LPS-induced proteinuria is also associated with NF-κB activation in podocytes.64,65 Because receptors linked to innate immunity, including Toll-like receptor 4, are constitutively expressed on podocytes,66,67,68 it is tempting to speculate that these might also participate in NF-κB activation in podocytes outside the LPS model. RelA/NF-κB activation may represent an end point for a number of convergent pathways in glomerular damage, with regulation by nephrin constituting an essential cellular survival mechanism. Further support for this concept comes from clinical observations that glucocorticoids and angiotensin inhibitors commonly used to limit proteinuria downregulate NF-κB and upregulate nephrin.69,70
In summary, we propose that nephrin maintains a negative tone on NF-κB activation signals, triggering its nuclear translocation in podocytes. As illustrated in Figure 9, either primary or secondary nephrin deficiency could remove this brake, allowing inappropriate induction of NF-κB transcriptional targets that are able to cause cellular injury directly or indirectly. Our data verify NF-κB activation as an important mediator of aberrant slit diaphragm signaling and provide a novel insight into the regulation of this ancient pathway in nonimmune cells.
Figure 9.

RelA/NF-κB activation may lead to podocyte injury. (A) Opposition of aberrant slit diaphragm signaling by nephrin, no NF-κB activation. (B) Absence of nephrin allows NF-κB activation and podocyte injury.
Concise Methods
Plasmids and Generation of cDNA Constructs
Full-length pcDNA3-NPHS1 (gift from Prof. K. Trygvasson, Karolinska Institute, Stockholm, Sweden) was used to generate constructs. Bsu36I and Xba1 digestion excised cDNA corresponding to the cytosolic tail encoding AA 1142 through 1241. cDNA encoding AA 1142 through 1160 and AA 1142 through 1201 was amplified by Pfu PCR using gene-specific primers and subcloned in frame to encode mutant nephrin proteins truncated at R1160 and W1201. Full-length NPHS2 cDNA was amplified from QUICK-Clone kidney cDNA (Clontech, Mountain View, CA) by Pfu PCR and subcloned as an EcoRI/EcoRI fragment into pcDNA-HA (in-house). Dominant negative 14-3-3 is described by Thorson et al.71 Dominant negative pcDNA-HA-PKCζ is described by Diaz-Meco et al.72 pcDNA-GST-NPHS1 was generated from pcDNA-FLAG-NPHS1 by EcoRI excision of cDNA corresponding to AA 1057 through 1241, blunt ending, Xho1 digestion, and ligation into pcDNA-GST (gift from Dr. S. Parkinson, Cancer Research UK, London, UK). GFP-PKCζ was also a gift from Dr. Parkinson. Verification of cDNA constructs was by ABI automated sequencing (Applied Biosystems, Warrington, UK).
Antibodies
Anti-Ha tag (mouse IgG) and rabbit polyclonals anti-14-3-3θ, anti-PKC-ζ, anti-RelA, anti-WT1 (C-19), p-JNK, Par4, p38, and Bcl2 were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-GFP (rabbit polyclonal) was from Abcam (Cambridge, UK). Anti-acetylated tubulin (mouse) was from Sigma-Aldrich (Dorset, UK). IκB-α and phospho-PKCζ/λ(Thr410/403) rabbit polyclonals were from Cell Signaling Technology (Danvers, MA). Anti–Alexa Fluor 488 antibody (rabbit) and anti–Alexa Fluor 496 (mouse) secondaries were from Invitrogen (Paisley, UK).
Generation of Par4−/− Mice
Mice genetically deficient for Par4 were produced by homologous recombination on a CD1 outbred background as described by Garcia-Cao et al.36
Histologic Analysis of Murine Kidney Sections
The kidneys of killed mice were fixed in 10% neutral buffered formalin, dehydrated, and subsequently wax embedded in paraffin. Sections were cut at 4-μm thickness and stained with hematoxylin and eosin using a standard method.
Immunohistochemistry
Sections were dewaxed in Histoclear (National Diagnostics, Atlanta, GA) and rehydrated through graded ethanols. Antigen retrieval was performed by boiling in citrate buffer (10 mM citric acid and 0.05% Tween 20 [pH 6.0]) for 10 min. Nonspecific binding was blocked by incubation in blocking medium. Two-hour incubations with primary antibody at dilutions between 1:50 and 1:200 and secondary antibody at a dilution of 1:200 were performed in blocking medium at room temperature. For double labeling, cells were additionally incubated with acetylated-tubulin antibody (1:50) and an anti–Alexa Fluor 496 (red) secondary. Sections were mounted under coverslips in Citifluor (London, UK). Fluorescence was visualized on a confocal laser scanning microscope (Leica Aristoplan, Heidelberg, Germany).
Electron Microscopy
Kidney tissue was fixed in 3% gluteraldehyde, 0.1 M sodium cacodylate, and 5 mM calcium chloride (pH 7.2) at room temperature. Postfixation was carried out with 1% osmium tetroxide for 1 h, followed by dehydration of tissue in graded ethanols and infiltration with 1:1 propylene oxide/epoxy resin for 3 h at room temperature. Samples were re-infiltrated with pure epoxy resin overnight at 4°C, embedded, and polymerized at 60°C for 48 to 72 h. Ultrathin sections (60 nm) were collected onto copper grids. Sections were stained with 25% uranyl acetate, washed in methanol, and contrasted with lead citrate. Electron micrographs were taken with a Zeiss 910 Advanced Transmission Electron Microscope (Zeiss, Oberkochen, Germany).
Cell Culture
MDCK cells were cultured in DMEM (Glutamax I) and 10% FBS (Life Technologies, Grand Island, NY) using standard techniques. Conditionally immortalized WT and mutant human podocytes were cultured according to methods described by Saleem et al.40
Protein Extraction from Cultured Cells
Ice-cold NETN buffer (20 mM Tris-HCl [pH 8], 100 mM NaCl, 1 mM EDTA, 0.5% NP40, and protease inhibitor cocktail [Boehringer Mannheim, Manheim, Germany]) was used for protein extraction for Western blotting and glutathione precipitation. Phospho-proteins were extracted using ice-cold HEPES buffer (50 mM HEPES [pH 7.5], 10% glycerol, 1% Triton X-100, 1.5 mM MgCl, 150 mM NaCl, 1 mM EGTA, and 1 mM PMSF, with protease inhibitors and phosphatase inhibitor cocktail [Roche Applied Science, Burgess Hill, UK]). Lysates were centrifuged at 4°C, and supernatants were stored at −70°C.
Immunocytochemistry
MDCK cells and human podocytes were propagated to 70% confluence on glass microscope slides using the Lab-Tek Chamber Slide system (Nunc, Fisher Scientific Loughborough, UK). Cells were fixed with either methanol at −20°C or 2% paraformaldehyde for 10 min. Permeabilization with 0.3% Triton X was used for WT1, nephrin, and Par4. Cells were blocked for 1 h in 10% FCS in PBS and 1% BSA and incubated with primary and anti–Alexa Fluor 488 (green, rabbit) secondary antibody for 1 h, respectively, at room temperature. Cells were mounted under coverslips in Citifluor. In double-labeling experiments, cells were also incubated with acetylated-tubulin antibody for 1 h as before. Fluorescence was visualized using a confocal laser scanning microscope (Leica Aristoplan).
Luciferase Assays in MDCK and Human Podocyte Cell Lines
Cells (1 × 105) seeded in six-well culture plates were grown to 50 to 70% confluence. Lipofectamine 2000 reagent (Invitrogen) was used for transient transfections according to the manufacturer's instructions. A total of 5 μg of DNA was transfected in total, 1 μg of pNF-κB-TA-Luc reporter (Mercury Pathway Profiling System; Clontech, Mountain View, CA), 1 μg of pcDNA3(CMV)-β-galactosidase vector (transfection efficiency), and 1 μg of combinations of 1 to 2 μg of expression vectors encoding WT full-length nephrin, W1201X or R1160X nephrin mutants, podocin, IκBα, dominant negative IκBα (Clontech), 14-3-371 and aPKCζ,72 or empty vector. Cells were incubated for 48 h before harvesting. Extracts were prepared in 1× Reporter Lysis buffer (RLB; Promega, Southhampton, UK). Standardized luciferase activity was determined using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Experiments were in quadruplicate, and statistical analysis was by paired t test.
Glutathione Precipitation of Endogenous 14-3-3θ from MDCK Cells by GST-Nephrin (1057 to 1241)
A total of 50 μl of glutathione-Sepharose beads (Amersham Biosciences, Little Chalfont, UK) preblocked with 1% BSA was washed with NETN buffer containing protease inhibitor cocktail (Boehringer Mannheim) and stored as a 50% slurry. MDCK cells were transfected with either pcDNA3-GST empty vector or pCDNA3-GST-nephrin (1057 to 1241) using Lipofectamine 2000. Cells were lysed on ice in NETN 48 h after transfection. A total of 400 μg of cell lysate and 50 μl of beads were used per precipitation at 4°C for 3 h. Pelleted glutathione precipitates were washed with NETN and resuspended in 2× Laemmli Buffer (20 μl). Samples were boiled for 5 min before SDS-PAGE. Precipitated native 14-3-3θ was detected using a rabbit polyclonal 14-3-3θ antibody (Santa Cruz Biotechnology).
Disclosures
None.
Acknowledgments
This work was funded by the Wellcome Trust (A.K. and S.H.); the Sir Jules Thorn Trust (A.K. and L.R.); and a Charlotte Eyke & Elizabeth Wherry Award, British Medical Association (A.K.).
We thank Prof. K Trygvasson and Dr. S. Parker for gifts of antibodies and plasmids.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “A Novel Role for Nephrin in the Maintenance of Glomerular Structure,” on pages 1661–1663.
References
- 1.Hayden MS, Ghosh S: Signaling to NF-kappaB. Genes Dev 18: 2195–2224, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Saccani S, Pantano S, Natoli G: Modulation of NF-kappaB activity by exchange of dimers. Mol Cell 11: 1563–1574, 2003 [DOI] [PubMed] [Google Scholar]
- 3.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M: The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91: 243–252, 1997 [DOI] [PubMed] [Google Scholar]
- 4.Pahl HL: Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18: 6853–6866, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Ballard DW, Dixon EP, Peffer NJ, Bogerd H, Doerre S, Stein B, Greene WC: The 65-kDa subunit of human NF-kappa B functions as a potent transcriptional activator and a target for v-Rel-mediated repression. Proc Natl Acad Sci U S A 89: 1875–1879, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanz L, Sanchez P, Lallena MJ, az-Meco MT, Moscat J: The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J 18: 3044–3053, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z: MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity 7: 837–847, 1997 [DOI] [PubMed] [Google Scholar]
- 8.Moscat J, Diaz-Meco MT, Rennert P: NF-kappaB activation by protein kinase C isoforms and B-cell function. EMBO Rep 4: 31–36, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lallena MJ, az-Meco MT, Bren G, Paya CV, Moscat J: Activation of IkappaB kinase beta by protein kinase C isoforms. Mol Cell Biol 19: 2180–2188, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duran A, az-Meco MT, Moscat J: Essential role of RelA Ser311 phosphorylation by zetaPKC in NF-kappaB transcriptional activation. EMBO J 22: 3910–3918, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leitges M, Sanz L, Martin P, Duran A, Braun U, Garcia JF, Camacho F, az-Meco MT, Rennert PD, Moscat J: Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol Cell 8: 771–780, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Diaz-Meco MT, Municio MM, Frutos S, Sanchez P, Lozano J, Sanz L, Moscat J: The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell 86: 777–786, 1996 [DOI] [PubMed] [Google Scholar]
- 13.Diaz-Meco MT, Lallena MJ, Monjas A, Frutos S, Moscat J: Inactivation of the inhibitory kappaB protein kinase/nuclear factor kappaB pathway by Par-4 expression potentiates tumor necrosis factor alpha-induced apoptosis. J Biol Chem 274: 19606–19612, 1999 [DOI] [PubMed] [Google Scholar]
- 14.Kriz W, Lemley KV: The role of the podocyte in glomerulosclerosis. Curr Opin Nephrol Hypertens 8: 489–497, 1999 [DOI] [PubMed] [Google Scholar]
- 15.Mundel P, Shankland SJ: Podocyte biology and response to injury. J Am Soc Nephrol 13: 3005–3015, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K: Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998 [DOI] [PubMed] [Google Scholar]
- 17.Wartiovaara J, Ofverstedt LG, Khoshnoodi J, Zhang J, Makela E, Sandin S, Ruotsalainen V, Cheng RH, Jalanko H, Skoglund U, Tryggvason K: Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest 114: 1475–1483, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doublier S, Ruotsalainen V, Salvidio G, Lupia E, Biancone L, Conaldi PG, Reponen P, Tryggvason K, Camussi G: Nephrin redistribution on podocytes is a potential mechanism for proteinuria in patients with primary acquired nephrotic syndrome. Am J Pathol 158: 1723–1731, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saran AM, Yuan H, Takeuchi E, McLaughlin M, Salant DJ: Complement mediates nephrin redistribution and actin dissociation in experimental membranous nephropathy. Kidney Int 64: 2072–2078, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Lenkkeri U, Mannikko M, McCready P, Lamerdin J, Gribouval O, Niaudet PM, Antignac CK, Kashtan CE, Homberg C, Olsen A, Kestila M, Tryggvason K: Structure of the gene for congenital nephrotic syndrome of the Finnish type (NPHS1) and characterization of mutations. Am J Hum Genet 64: 51–61, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koziell A, Grech V, Hussain S, Lee G, Lenkkeri U, Tryggvason K, Scambler P: Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration. Hum Mol Genet 11: 379–388, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C: NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24: 349–354, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, Miner JH, Shaw AS: Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 286: 312–315, 1999 [DOI] [PubMed] [Google Scholar]
- 24.Lehtonen S, Ryan JJ, Kudlicka K, Iino N, Zhou H, Farquhar MG: Cell junction-associated proteins IQGAP1, MAGI-2, CASK, spectrins, and alpha-actinin are components of the nephrin multiprotein complex. Proc Natl Acad Sci U S A 102: 9814–9819, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quack I, Rump LC, Gerke P, Walther I, Vinke T, Vonend O, Grunwald T, Sellin L: beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity. Proc Natl Acad Sci U S A 103: 14110–14115, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verma R, Wharram B, Kovari I, Kunkel R, Nihalani D, Wary KK, Wiggins RC, Killen P, Holzman LB: Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J Biol Chem 278: 20716–20723, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Lahdenpera J, Kilpelainen P, Liu XL, Pikkarainen T, Reponen P, Ruotsalainen V, Tryggvason K: Clustering-induced tyrosine phosphorylation of nephrin by Src family kinases. Kidney Int 64: 404–413, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, Huang H, Larose L, Li SS, Takano T, Quaggin SE, Pawson T: Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 440: 818–823, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB: Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest 116: 1346–1359, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng L, Sinniah R, Hsu SI: In situ glomerular expression of activated NF-kappaB in human lupus nephritis and other non-proliferative proteinuric glomerulopathy. Virchows Arch 448: 172–183, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Fujihara CK, Antunes GR, Mattar AL, Malheiros DM, Vieira JM, Jr., Zatz R: Chronic inhibition of nuclear factor-kappaB attenuates renal injury in the 5/6 renal ablation model. Am J Physiol Renal Physiol 292: F92–F99, 2007 [DOI] [PubMed] [Google Scholar]
- 32.Takano T, Cybulsky AV, Yang X, Aoudjit L: Complement C5b-9 induces cyclooxygenase-2 gene transcription in glomerular epithelial cells. Am J Physiol Renal Physiol 281: F841–F850, 2001 [DOI] [PubMed] [Google Scholar]
- 33.Choi J, Krushel LA, Crossin KL: NF-kappaB activation by N-CAM and cytokines in astrocytes is regulated by multiple protein kinases and redox modulation. Glia 33: 45–56, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Reidy M, Zihlmann P, Hubbell JA, Hall H: Activation of cell-survival transcription factor NFkappaB in L1Ig6-stimulated endothelial cells. J Biomed Mater Res A 77: 542–550, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Pasparakis M, Luedde T, Schmidt-Supprian M: Dissection of the NF-kappaB signalling cascade in transgenic and knockout mice. Cell Death Differ 13: 861–872, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Cao I, Lafuente MJ, Criado LM, az-Meco MT, Serrano M, Moscat J: Genetic inactivation of Par4 results in hyperactivation of NF-kappaB and impairment of JNK and p38. EMBO Rep 4: 307–312, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Jeanpierre C, Dressler GR, Lacoste M, Niaudet P, Gubler MC: WT1 and PAX-2 podocyte expression in Denys-Drash syndrome and isolated diffuse mesangial sclerosis. Am J Pathol 154: 181–192, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohtaka A, Ootaka T, Sato H, Soma J, Sato T, Saito T, Ito S: Significance of early phenotypic change of glomerular podocytes detected by Pax2 in primary focal segmental glomerulosclerosis. Am J Kidney Dis 39: 475–485, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Wagner KD, Wagner N, Guo JK, Elger M, Dallman MJ, Bugeon L, Schedl A: An inducible mouse model for PAX2-dependent glomerular disease: insights into a complex pathogenesis. Curr Biol 16: 793–800, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Saleem MA, O'Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P: A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 13: 630–638, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Martinka S, Bruggeman LA: Persistent NF-kappaB activation in renal epithelial cells in a mouse model of HIV-associated nephropathy. Am J Physiol Renal Physiol 290: F657–F665, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coward RJ, Welsh GI, Koziell A, Hussain S, Lennon R, Ni L, Tavare JM, Mathieson PW, Saleem MA: Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes 56: 1127–1135, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Huber TB, Kottgen M, Schilling B, Walz G, Benzing T: Interaction with podocin facilitates nephrin signaling. J Biol Chem 276: 41543–41546, 2001 [DOI] [PubMed] [Google Scholar]
- 44.Li H, Lemay S, Aoudjit L, Kawachi H, Takano T: SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J Am Soc Nephrol 15: 3006–3015, 2004 [DOI] [PubMed] [Google Scholar]
- 45.Blasutig IM, New LA, Thanabalasuriar A, Dayarathna TK, Goudreault M, Quaggin SE, Li SS, Gruenheid S, Jones N, Pawson T: Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol Cell Biol 28: 2035–2046, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sellin L, Huber TB, Gerke P, Quack I, Pavenstadt H, Walz G: NEPH1 defines a novel family of podocin interacting proteins. FASEB J 17: 115–117, 2003 [DOI] [PubMed] [Google Scholar]
- 47.Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, Shaw AS, Holzman LB, Mundel P: Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest 108: 1621–1629, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aguilera C, Fernandez-Majada V, Ingles-Esteve J, Rodilla V, Bigas A, Espinosa L: Efficient nuclear export of p65-IkappaBalpha complexes requires 14-3-3 proteins. J Cell Sci 119: 3695–3704, 2006 [DOI] [PubMed] [Google Scholar]
- 49.Huber TB, Hartleben B, Kim J, Schmidts M, Schermer B, Keil A, Egger L, Lecha RL, Borner C, Pavenstadt H, Shaw AS, Walz G, Benzing T: Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol 23: 4917–4928, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P: The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med 14: 931–938, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartleben B, Schweizer H, Lubben P, Bartram MP, Moller CC, Herr R, Wei C, Neumann-Haefelin E, Schermer B, Zentgraf H, Kerjaschki D, Reiser J, Walz G, Benzing T, Huber TB: Neph-Nephrin proteins bind the Par3-Par6-atypical protein kinase C (aPKC) complex to regulate podocyte cell polarity. J Biol Chem 283: 23033–23038, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnstone RW, See RH, Sells SF, Wang J, Muthukkumar S, Englert C, Haber DA, Licht JD, Sugrue SP, Roberts T, Rangnekar VM, Shi Y: A novel repressor, par-4, modulates transcription and growth suppression functions of the Wilms' tumor suppressor WT1. Mol Cell Biol 16: 6945–6956, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie J, Guo Q: Par-4 is a novel mediator of renal tubule cell death in models of ischemia-reperfusion injury. Am J Physiol Renal Physiol 292: F107–F115, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Lafuente MJ, Martin P, Garcia-Cao I, az-Meco MT, Serrano M, Moscat J: Regulation of mature T lymphocyte proliferation and differentiation by Par-4. EMBO J 22: 4689–4698, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moscat J, Rennert P, Diaz-Meco MT: PKCzeta at the crossroad of NF-kappaB and Jak1/Stat6 signaling pathways. Cell Death Differ 13: 702–711, 2006 [DOI] [PubMed] [Google Scholar]
- 56.Lallena MJ, az-Meco MT, Bren G, Paya CV, Moscat J: Activation of IkappaB kinase beta by protein kinase C isoforms. Mol Cell Biol 19: 2180–2188, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drumm K, Gassner B, Silbernagl S, Gekle M: Albumin in the mg/l-range activates NF-kappaB in renal proximal tubule-derived cell lines via tyrosine kinases and protein kinase C. Eur J Med Res 6: 247–258, 2001 [PubMed] [Google Scholar]
- 58.Harris TJ, Peifer M: aPKC controls microtubule organization to balance adherens junction symmetry and planar polarity during development. Dev Cell 12: 727–738, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uberall F, Hellbert K, Kampfer S, Maly K, Villunger A, Spitaler M, Mwanjewe J, Baier-Bitterlich G, Baier G, Grunicke HH: Evidence that atypical protein kinase C-lambda and atypical protein kinase C-zeta participate in Ras-mediated reorganization of the F-actin cytoskeleton. J Cell Biol 144: 413–425, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nemeth ZH, Deitch EA, Davidson MT, Szabo C, Vizi ES, Hasko G: Disruption of the actin cytoskeleton results in nuclear factor-kappaB activation and inflammatory mediator production in cultured human intestinal epithelial cells. J Cell Physiol 200: 71–81, 2004 [DOI] [PubMed] [Google Scholar]
- 61.O'Dea EL, Barken D, Peralta RQ, Tran KT, Werner SL, Kearns JD, Levchenko A, Hoffmann A: A homeostatic model of IkappaB metabolism to control constitutive NF-kappaB activity. Mol Syst Biol 3: 111, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoffmann A, Baltimore D: Circuitry of nuclear factor kappaB signaling. Immunol Rev 210: 171–186, 2006 [DOI] [PubMed] [Google Scholar]
- 63.Burt D, Salvidio G, Tarabra E, Barutta F, Pinach S, Dentelli P, Camussi G, Perin PC, Gruden G: The monocyte chemoattractant protein-1/cognate CC chemokine receptor 2 system affects cell motility in cultured human podocytes. Am J Pathol 171: 1789–1799, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dallot E, Mehats C, Oger S, Leroy MJ, Breuiller-Fouche M: A role for PKCzeta in the LPS-induced translocation NF-kappaB p65 subunit in cultured myometrial cells. Biochimie 87: 513–521, 2005 [DOI] [PubMed] [Google Scholar]
- 65.Haas C, Car B, Ryffel B, Le HM: Lipopolysaccharide-induced glomerulonephritis develops in the absence of interferon-gamma signaling. Exp Nephrol 4: 222–230, 1996 [PubMed] [Google Scholar]
- 66.Reiser J, von GG, Loos M, Oh J, Asanuma K, Giardino L, Rastaldi MP, Calvaresi N, Watanabe H, Schwarz K, Faul C, Kretzler M, Davidson A, Sugimoto H, Kalluri R, Sharpe AH, Kreidberg JA, Mundel P: Induction of B7–1 in podocytes is associated with nephrotic syndrome. J Clin Invest 113: 1390–1397, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bondeva T, Roger T, Wolf G: Differential regulation of Toll-like receptor 4 gene expression in renal cells by angiotensin II: dependency on AP1 and PU.1 transcriptional sites. Am J Nephrol 27: 308–314, 2007 [DOI] [PubMed] [Google Scholar]
- 68.Banas MC, Banas B, Hudkins KL, Wietecha TA, Iyoda M, Bock E, Hauser P, Pippin JW, Shankland SJ, Smith KD, Stoelcker B, Liu G, Grone HJ, Kramer BK, Alpers CE: TLR4 links podocytes with the innate immune system to mediate glomerular injury. J Am Soc Nephrol 19: 704–713, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Esteban V, Ruperez M, Vita JR, Lopez ES, Mezzano S, Plaza JJ, Egido J, Ruiz-Ortega M: Effect of simultaneous blockade of AT1 and AT2 receptors on the NFkappaB pathway and renal inflammatory response. Kidney Int Suppl 86: S33–S38, 2003 [DOI] [PubMed] [Google Scholar]
- 70.de Haij S, Daha MR, van KC: Mechanism of steroid action in renal epithelial cells. Kidney Int 65: 1577–1588, 2004 [DOI] [PubMed] [Google Scholar]
- 71.Thorson JA, Yu LW, Hsu AL, Shih NY, Graves PR, Tanner JW, Allen PM, Piwnica-Worms H, Shaw AS: 14-3-3 proteins are required for maintenance of Raf-1 phosphorylation and kinase activity. Mol Cell Biol 18: 5229–5238, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Diaz-Meco MT, Berra E, Municio MM, Sanz L, Lozano J, Dominguez I, az-Golpe V, Lain de Lera MT, Alcami J, Paya CV, Arenzana-Seisdedos F, Virelizier J-L, Moscat J: A dominant negative protein kinase C zeta subspecies blocks NF-kappa B activation. Mol Cell Biol 13: 4770–4775, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]