

Abstract
Phosphorylation of the myosin regulatory light chain (RLC) in skeletal muscle has been proposed to act as a molecular memory of recent activation by increasing the rate of force development, ATPase activity, and isometric force at submaximal activation in fibers. It has been proposed that these effects stem from phosphorylation-induced movement of myosin heads away from the thick filament backbone. In this study, we examined the molecular effects of skeletal muscle myosin RLC phosphorylation using in vitro motility assays. We showed that, independently of the thick filament backbone, the velocity of skeletal muscle myosin is decreased upon phosphorylation due to an increase in the myosin duty cycle. Furthermore, we did not observe a phosphorylation-dependent shift in calcium sensitivity in the absence of the myosin thick filament. These data suggest that phosphorylation-induced movement of myosin heads away from the thick filament backbone explains only part of the observed phosphorylation-induced changes in myosin mechanics. Last, we showed that the duty cycle of skeletal muscle myosin is strain dependent, consistent with the notion that strain slows the rate of ADP release in striated muscle.
Keywords: in vitro motility assay, mechanics
all known muscle systems share the characteristic that the regulation of actomyosin interaction is mediated by the gated release of calcium. The specific mechanism by which calcium regulates this interaction depends on the muscle type. In molluscan muscle, calcium binds directly to the myosin essential light chain (ELC) that, together with the regulatory light chain (RLC) and part of the myosin heavy chain (MHC), constitutes the myosin regulatory domain that switches on the myosin motor in response to calcium binding (86). In vertebrate cardiac and skeletal muscle, calcium binds to troponin C, causing a shift in the position of tropomyosin along actin and allowing myosin cross bridges to bind strongly to the thin filament and shorten the sarcomere (for review, see Ref. 22). Smooth muscle has yet another pathway for activation in which calcium binding to calmodulin activates myosin light chain kinase (MLCK), phosphorylating the RLC and activating smooth muscle contraction (for review, see Ref. 70).
It was shown by Perrie et al. (50) that vertebrate striated muscle RLC could also be phosphorylated by activated MLCK, and although this interaction is not necessary for activation of contraction, it appears to modulate skeletal and cardiac muscle contractility (40). Phosphorylation of the RLC in striated muscle fibers has been shown to enhance both the magnitude (13, 52, 66, 74, 79) and rate of tension development (42, 73) as well as to increase the ATPase rate (79) at submaximal calcium levels (for review, see Ref. 71). Furthermore, it has been shown that myosin phosphorylation can cause potentiation of posttetanic twitch (39, 40) and the rate of cross-bridge attachment (14, 42, 48, 73, 79). These effects have also been shown to be removed by knocking out skeletal muscle MLCK (89). The extent of RLC phosphorylation correlates with the frequency of activation of the muscle (39, 44), leading to the proposal that phosphorylation of striated muscle myosin RLC acts as a molecular memory, increasing actomyosin interactions after repeated muscle activation (60, 71). Phosphorylation of the RLC was also recently shown to be an important determinant of stretch activation in myocardium (13, 66).
Structural studies have suggested that phosphorylation of the RLC causes a disordering of the myosin heads in the thick filament by facilitating detachment of the myosin heads from the thick filament backbone and increasing their accessibility for actin binding, giving rise to the phosphorylation-induced changes in contractility (37). The myosin RLC is attached to the elongated α-helical neck region of the myosin molecule, which works as a lever arm amplifying small conformational changes at the active site of the myosin head into large movements required to generate force and motility (6, 55, 56, 61). Furthermore, the neck region also serves as a molecular strain sensor, transmitting load directly to the active site (for review, see Ref. 47). Consistent with this idea, single-molecule studies have suggested that straining the myosin lever arm can cause changes in the kinetics of ADP release, with a negative strain slowing the release rate and a positive strain accelerating the rate (30, 84).
It has been shown that under physiological conditions, the phosphorylation of striated muscle myosin results in a conformational change in the RLC (59). It is possible that phosphorylation-induced changes in the RLC could modulate myosin kinetics and mechanics in a way similar to how cation binding to the RLC modulates myosin conformation and function (1, 27, 57, 75, 76). To investigate the effects of RLC phosphorylation, we measured the velocity, duty cycle, and calcium sensitivity of velocity of phosphorylated and dephosphorylated myosins using the in vitro motility assay (34). We observed that phosphorylation of the RLC causes a decrease in myosin velocity, independent of the myosin thick filament backbone. We propose that the reduction in velocity is due to an increase in the myosin duty cycle resulting from phosphorylation-induced stiffening of the myosin lever arm. Also, contrary to studies with filamentous myosin (52, 71, 74, 79), we did not observe the phosphorylation-dependent increase in calcium sensitivity, suggesting that this shift might be due to interactions between myosin molecules within the thick filament. We have also shown that the duty cycle of skeletal muscle myosin is strain dependent, consistent with the notion that strain slows the rate of ADP release in striated muscle.
MATERIALS AND METHODS
Preparation of endogenously dephosphorylated rabbit skeletal muscle myosin.
Dephosphorylated rabbit skeletal muscle myosin was isolated from rabbit fast skeletal muscle taken from freshly euthanized New Zealand White rabbits. Briefly, muscle was minced and then extracted on ice for 20 min in 1.5 l/500 g of ice-cold Guba Straub solution of the following composition: 0.1 M potassium monophosphate, 0.05 M potassium diphosphate, and 0.3 M potassium chloride (pH 6.5). After extraction, the mince was centrifuged at 11,000 g for 30 min at 4°C. The resulting supernatant was then poured through glass wool to clarify it and precipitated with 14 volumes of cold 1 mM EDTA. The precipitate was collected by serial centrifugation at 7,000 g for 10 min with a final spin at 11,000 g. The pellet was then resuspended in 20 mM MOPS (pH 7), 1 mM DTT, and KCl to a final concentration of 0.5 M. The myosin was then ultracentrifuged at 200,000 g for 1.5 h at 4°C to remove myosin aggregates. After ultracentrifugation, the supernatant was precipitated on ice with 14 volumes of ice-cold double-distilled water. The precipitated myosin was again collected by centrifugation at 6,500 g for 10 min. To ensure that myosin was fully dephosphorylated, pellets of myosin were kept overnight on ice. The following day, myosin pellets were resuspended as described above and subjected to a second round of ultracentrifugation. The myosin supernatant was then analyzed by SDS-PAGE to determine myosin purity, mixed with an equal volume of glycerol, and stored at −20°C until use.
Preparation of endogenously phosphorylated rabbit skeletal muscle myosin.
Isolation of phosphorylated rabbit skeletal muscle myosin was performed according to Stepkowski et al. (67). In brief, the first steps of myosin extraction were the same as described above, but the myosin extract was precipitated with 5 mM potassium phosphate buffer (pH 7) instead of 1 mM EDTA. Pellets resulting from this first precipitation were resuspended in 20 mM potassium phosphate buffer (pH 8.0) and a final concentration of 0.5 M KCl. Before ultracentrifugation, myosin was incubated in the solution containing 20 mM phosphate buffer (pH 8), 5 mM ATP, 12.5 mM MgCl2, and 0.1 mM CaCl2 for 30 min at room temperature. This step was necessary to phosphorylate myosin by the endogenous MLCK. After incubation, myosin was cooled to 4°C, ultracentrifuged, and processed in the same way as described above. In addition, all steps to obtain fully phosphorylated rabbit skeletal muscle myosin were performed on the same day, and no overnight treatments were performed. Purity again determined by SDS-PAGE analysis and the phosphorylation status was examined on 8% polyacrylamide gel in the presence of 8 M urea (58).
Preparation of MLCK.
Isolation and purification of skeletal muscle MLCK were performed simultaneously with the preparation of phosphorylated myosin (67), allowing the myosin-bound MLCK to be copurified with phosphorylated myosin (see MLCK purification in the supplemental material in the online version of this article). However, after incubation of myosin in the solution containing 20 mM phosphate buffer (pH 8), 5 mM ATP, 12.5 mM MgCl2, and 0.1 mM CaCl2 for 30 min at room temperature and ultracentrifugation at 200,000 g for 1.5 h at 4°C, the myosin was subjected to an ammonium sulfate precipitation instead of precipitation with 5 mM potassium phosphate buffer (see above). The first ammonium sulfate cut 0–42.5% was to remove impurities of actin, whereas the second 42.5–55% contained the complex of phosphorylated myosin and MLCK. The separation of myosin from MLCK occurred during dialysis of the ammonium sulfate pellet containing myosin-MLCK complex. Pellet was resuspended in a minimal volume of 40 mM KCl, 2 mM MgCl2, 10 mM imidazole (pH 6.6), 10 mM Bis-Tris, 0.5 mM PMSF, and 0.5 mM DTT and then dialyzed against the same buffer diluted four times. Precipitated myosin was collected by centrifugation at 11,000 g for 10 min at 4°C, and the supernatant contained endogenous MLCK. The preparation of MLCK was then concentrated, mixed with protease inhibitor cocktail (Sigma, St. Lois, MO), aliquoted, and stored frozen at −80°C.
Phosphorylation of dephosphorylated myosin with endogenous MLCK.
Myosin (100 μg) was precipitated in 10 mM DTT for 1 h on ice before centrifugation at 16,000 g for 30 min at 4°C. The supernatant was removed, and the pellet was solubilized in 50 μl of buffer containing 20 mM potassium phosphate buffer (pH 8.0) and 30 mM KCl. Phosphorylation of myosin was performed in a phosphorylation buffer consisting of 20 mM potassium phosphate buffer (pH 8.0), 12 mM MgCl2, 0.1 mM CaCl2, and 5 mM ATP. The reaction was initiated by addition of 500 μM CaM (AG Scientific, San Diego, CA) and 100 μg of MLCK. The mixture was incubated overnight at 4°C. The next morning, the myosin-MLCK-CaM mixture was diluted with 1.5 ml of 10 mM DTT solution, incubated on ice for 1 h, and pelleted by centrifugation at 16,000 g for 30 min at 4°C. Phosphorylated myosin was then resuspended in the desired volume of myosin buffer (300 mM KCl, 25 mM imidazole, 1 mM EGTA, 4 mM MgCl2, and 10 mM DTT) and used in the experiments. Phosphorylation of MLCK-treated myosin was examined on 8% polyacrylamide gel in the presence of 8 M urea.
Dephosphorylation of endogenously phosphorylated myosin by calf intestinal alkaline phosphatase.
Myosin (100 μg) was precipitated in 10 mM DTT for 1 h on ice before being centrifuged at 16,000 g for 30 min at 4°C. The supernatant was removed, and the pellet was solubilized in 50 μl of 1× NEB buffer 3 (New England Biolabs, Ipswich, MA). Calf intestinal alkaline phosphatase (CIAP; 1 μl, 100 units; New England Biolabs) was added, and the solution was incubated for 1 h at 37°C and placed on ice overnight. The next morning, the myosin was precipitated by adding 1.5 ml of 10 mM DTT solution to the myosin-CIAP solution and allowing the mixture to incubate on ice for 1 h. The dephosphorylated myosin was then pelleted by centrifugation at 16,000 g for 30 min at 4°C and then resuspended in the desired volume of myosin buffer. Dephosphorylation of CIAP-treated myosin was examined on 8% polyacrylamide gel in the presence of 8 M urea.
Actin purification and labeling.
Unlabeled actin was prepared from chicken pectoralis muscle acetone powder using the method of Straub (69) with the modification of Drabikowski and Gergely (17). The actin was suspended in actin buffer (25 mM KCl, 1 μM EGTA, 10 μM DTT, 25 μM imidazole, and 4 μM MgCl2). TRITC phalloidin-labeled actin was prepared by reacting a 1:1 molar ratio mixture of TRITC phalloidin and actin in actin buffer overnight at 4°C.
Regulatory proteins.
Tropomyosin (Tm) and troponin (Tn) subunits (TnT, TnI, and TnC) were prepared according to standard methods (53). Formation of the Tn complex was carried out as described by Szczesna et al. (78) and Gomes et al. (21).
Unregulated motility assay.
The in vitro motility assays were performed as previously described (76) with some subtle modifications. Approximately 200 μg of myosin in 50% glycerol were suspended in 1 ml of 10 mM DTT and allowed to precipitate for 1 h on ice. Myosin competent to form filaments was collected by centrifugation at 16,000 g for 30 min at 4°C and resuspended in 200 μl of myosin buffer (composition defined above). Damaged myosin heads that were unable to bind and release from actin in an ATP-dependent manner were removed by adding 1 mM ATP and 1.1 μM actin and pelleting in an Airfuge for 30 min at 30 psi (135,000 g). The myosin concentration after centrifugation was determined using a Bradford assay (Bio-Rad, Hercules, CA) and diluted to the desired concentration in myosin buffer.
Flow cells were constructed by forming a channel between a nitrocellulose-coated coverslip and a standard glass slide with double-stick tape (3M, St. Paul, MN). Myosin was adsorbed to the coverslip surface by adding 30 μl of myosin (100 μg/ml) in myosin buffer to the flow cell and incubating for 1 min. Any remaining surface lacking myosin was blocked by adding 30 μl of 0.5 mg/ml bovine serum albumin (BSA) in myosin buffer followed by a 60-μl wash with actin buffer. As an additional measure to minimize the effects of damaged myosin heads, 30 μl of 1 μM unlabeled actin in actin buffer were vortexed and added to the flow cell. After incubation for 2 min, the flow cell was washed with 60 μl of actin buffer containing 1 mM ATP and then 120 μl of actin buffer without ATP. Thirty microliters of 5 nM TRITC-labeled actin was then added to the flow cell and allowed to incubate for 1 min. Motility was initiated by the addition of motility buffer (actin buffer with the addition of 0.5% methyl cellulose, 1 mM ATP, 2 mM dextrose, 160 units of glucose oxidase, and 2 μM catalase).
Duty cycle estimation.
The duty cycle of myosin was estimated by measuring actin filament sliding velocity vs. actin filament length using the method of Uyeda et al. (82). This method has been used successfully by several groups to calculate the duty cycle (24–26, 83). The procedure was identical to the standard motility assay, except that only 25 μg/ml myosin was incubated on the motility assay surface and actin filament velocity was recorded as a function of filament length. Since the number of heads available to interact with the actin filament is proportional to the actin filament length, the number of heads available to interact with the actin filament was calculated using the data of Harris and Warshaw (26), who measured surface concentration as a function of filament length. Plots of velocity as a function of the number of heads available to interact with a particular filament were obtained and fit by nonlinear regression analysis (SigmaPlot; Systat Software, San Jose, CA) to Eq. 2 with the duty cycle, f, as a fit parameter.
Strain-dependent duty cycle measurements.
The procedure for measuring the strain-dependent duty cycle was identical to the unstrained duty cycle measurements, except the myosin was mixed with 1.25 μg/ml of α-actinin (Cytoskeleton, Denver, CO) before being incubated in the flow cell.
Troponin-tropomyosin-regulated motility assays.
The procedure for regulated motility assays was the same as the unregulated motility assays with minor modifications. After the TRITC phalloidin-labeled actin had bound to the surface for 30 s, 30 μl of a mixture of 150 nM Tm-Tn complex in actin buffer was added to the flow cell. After 10 min of incubation allowing for the Tm and Tn to bind to the actin, 30 μl of appropriate pCa motility buffer containing balanced ions as determined using a Bathe program (19) were added with the inclusion of 75 nM Tm-Tn complex and an appropriate amount of calcium. In the high-temperature experiments, the temperature of the flow cell was regulated using a Bioptechs objective heater (Butler, PA).
Microscopy.
TRITC-phalloidin-labeled actin filaments were observed on a Nikon Eclipse TE2000-U microscope (Melville, NY) with standard epifluoresence illumination. The images were recorded using video microscopy and captured with a Scion frame grabber (model AG-5). Using Scion Image (Frederick, MD). Images were captured at an appropriate frame rate (ranging from 1 to 7 s per frame). The filament velocity was determined using the freeware motility software Retrac (http://mc11.mcri.ac.uk/Retrac). Fifteen to thirty filaments were analyzed from at least two different areas of the flow cell to ensure that there were no surface artifacts. Only moving filaments were counted in the average velocities. For the duty cycle measurements, the filament lengths were measured using ImageJ (http://rsb.info.nih.gov/ij/NIH).
Statistics.
For each flow cell, the velocity and standard errors were calculated for 15–30 sliding filaments over the course of 5–10 frames. A two-tailed t-test was used to examine the significance of the differences between velocities. The P value was calculated from the Student's t-test distribution and corrected for multiple comparisons using the Holm t-test criteria.
For the duty cycle measurements, a large volume of data needed to be collected due to the inherent noise in the assay. The sliding velocities of 145–204 different actin filaments over the course of 5–10 frames were measured for each condition for at least two different myosin preparations. Each point represents the average sliding velocity of a single actin filament over the course of 5–10 frames. For the regulation experiments, fewer data were required since the experimental noise was less. Each point represents the average sliding velocity of 15–30 actin filaments averaged over 5–10 frames with the error bars representing the standard error in the mean velocity of the 15–30 actin filaments. For all experiments where the data were fit to a model, a least-squares algorithm was employed (SigmaPlot). Parameter values (Vmax, f, pCa50, nH) and the errors in these parameters were determined from the errors in the least-squares fit. The regression lines were tested for coincidence by an analysis of the variance. When different conditions (i.e., the duty cycle of phosphorylated and dephosphorylated myosins in the absence of strain) were compared, the standard error in the estimate was calculated for both the pooled estimate of the variance about separate nonlinear regression curves and a common regression curve. The improvement gained by fitting to the two separate nonlinear regression curves instead of one common regression curve was quantified by calculating the F statistic. All of the conditions tested were found to be significantly different (P < 0.05) using the F statistic. Once it was established that the different regression lines were not coincident, a two tailed t-test was used to examine whether there were statistically significant changes to the myosin duty cycle based on the values obtained from the least-squares fit.
RESULTS
Myosin was purified from rabbit psoas muscle and endogenously phosphorylated or dephosphorylated during preparation. Phosphorylation of endogenously dephosphorylated myosin was also achieved by incubation with calmodulin, MLCK, ATP, calcium, and high magnesium, whereas dephosphorylation of endogenously phosphorylated myosin was also achieved by incubation with CIAP in the absence of ATP and calcium. The extent of endogenous or MLCK treatment-induced RLC phosphorylation was measured by urea gel electrophoresis and densitometry (Fig. 1A). The endogenously phosphorylated myosin showed 81% RLC phosphorylation (Fig. 1A, lane c), whereas endogenous dephosphorylated myosin showed no detectable RLC phosphorylation (Fig. 1A, lane a). Treatment of endogenously dephosphorylated myosin with MLCK yielded 80% RLC-phosphorylated myosin, whereas treatment of endogenously phosphorylated myosin with CIAP yielded nearly complete RLC dephosphorylation as determined by densitometry (Fig. 1A, lanes b and d).
Fig. 1.

A: 8% polyacrylamide 8 M urea gel showing the reversibility of the phosphorylation reaction. Solid arrows denote dephosphorylated regulatory light chain (RLC); shaded arrows denote phosphorylated RLC. Lane a, endogenously dephosphorylated rabbit skeletal muscle myosin; lane b, Ca2+/CaM myosin light chain kinase (MLCK)-treated rabbit skeletal muscle (in vitro) dephosphorylated myosin; lane c, endogenously phosphorylated rabbit skeletal muscle myosin; lane d, calf intestinal alkaline phosphatase (CIAP)-treated rabbit skeletal muscle (in vitro) phosphorylated myosin. LC1–LC3, light chains 1–3. B: The unloaded sliding velocities of RLC-dephosphorylated (Dephos) and RLC-phosphorylated (Phos) myosins before and after treatment with either MLCK or CIAP were measured in the in vitro motility assay.
Velocity.
To determine the effects of RLC phosphorylation on myosin's motion-generating capacity, we measured the sliding velocity of fluorescently labeled actin filaments propelled by a bed of either phosphorylated or dephosphorylated myosin using the in vitro motility assay. We observed that endogenously dephosphorylated myosin propelled actin filaments at a sliding velocity (V) of 1.05 ± 0.04 μm/s, whereas endogenously phosphorylated myosin moved actin at a significantly lower velocity of 0.84 ± 0.08 μm/s (P < 0.01) (Fig. 1B). The decrease in velocity with phosphorylation may be even greater with 100% phosphorylation.
To ensure that the observed change in velocity was due to RLC phosphorylation-induced changes in the properties of myosin and not a result of the purification procedures for endogenously phosphorylated and dephosphorylated myosin, we repeated the sliding velocity assay using MLCK-phosphorylated or CIAP-dephosphorylated myosin preparations (Fig. 1B). MLCK-treated dephosphorylated myosin (V = 0.83 ± 0.08 μm/s) showed a similar 20% reduction in velocity, giving a velocity that was indistinguishable from the endogenously phosphorylated myosin (P = 0.91). Likewise, CIAP treatment of phosphorylated myosin (V = 1.05 ± 0.02 μm/s) caused an increase in velocity that was indistinguishable from endogenously dephosphorylated myosin (P = 0.98). Therefore, the observed 20% reduction in velocity was due to phosphorylation-induced changes in the myosin molecule and not experimental artifact due to the purification procedure. Furthermore, since MLCK- and CIAP-treated myosins are indistinguishable from their endogenously phosphorylated and dephosphorylated counterparts, endogenously phosphorylated and dephosphorylated myosins were used for the remainder of the experiments.
Duty cycle.
The velocity, V, of myosin-induced actin filament translocation in an in vitro motility assay is given by (81)
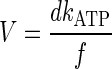 |
(1) |
where d is the unitary step size of myosin, kATP is the actin-activated ATPase rate, and f is the duty cycle of the myosin (i.e., the fraction the myosin biochemical cycle spent attached to actin). Changes in actin filament velocity in the in vitro motility assay are indicative of changes in at least one of these parameters. It has previously been shown that phosphorylation has no effect on the ATPase rate (49, 51); thus any observed changes in V must be due to changes in d or f. Therefore, we measured the myosin duty cycle to examine whether the phosphorylation-induced reduction in actin filament sliding velocity could be ascribed to changes in the duty cycle.
At a low density of surface myosin, the actin filament sliding velocity in the in vitro motility assay is given by the nonlinear relationship (82)
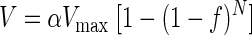 |
(2) |
where α relates to the efficiency of force transmission of the myosin, Vmax is the maximal velocity, f is the duty cycle, and N is the number of myosin heads available to interact with the actin filament. Since an actin filament in a motility assay will move at its maximal velocity if at least one myosin head is interacting with it at any given time, and the probability of a head interacting with an actin filament increases nonlinearly with myosin concentration, a myosin with a higher duty cycle will require fewer myosin heads to move the actin filament at maximal velocity. When we measured the duty cycle of both phosphorylated and dephosphorylated rabbit skeletal muscle myosin (Fig. 2), we found that there was a significant 45 ± 1% increase in duty cycle upon phosphorylation (P = 0.05). This significant increase in duty cycle is consistent with a decrease in velocity that exceeds the observed 20% decrease seen in RLC-phosphorylated myosin.
Fig. 2.

The duty cycles (f) of RLC-dephosphorylated and RLC-phosphorylated myosins were calculated by measuring the velocity of actin filament sliding vs. the number of myosin heads available to interact with the actin filament and fitting to Eq. 2. Each point represents the average sliding velocity of a single actin filament across 10 video frames; 176 actin filaments were followed for RLC-dephosphorylated myosin and 166 filaments were followed for RLC-phosphorylated myosin. The duty cycle for RLC-dephosphorylated myosin was 3.21 ± 0.45%, whereas the duty cycle for RLC-phosphorylated myosin was 4.64 ± 0.60%.
Strain dependence of the duty cycle.
It is possible that RLC phosphorylation might affect the RLC-MHC interaction and thus neck/lever domain stiffness. It is therefore conceivable that the duty cycle of myosin could change under an applied force since it has been shown that straining the myosin lever arm can change the myosin kinetics (84). We therefore measured the duty cycle under load using the same experimental procedure as the unloaded duty cycle measurements (Fig. 2), except that a small amount of α-actinin, a low-affinity actin-binding protein that can be used to exert a frictional drag on sliding actin filaments (5), was introduced into the flow cell with the myosin. To determine the loaded duty cycle, we added enough α-actinin to slow velocity but not stop it. In the presence of the frictional load (Fig. 3), we found that the duty cycle of both dephosphorylated (fdephos. unstrained = 3.21 ± 0.45%, fdephos. strained = 6.8 ± 1.6%; P < 0.05) and phosphorylated (fphos. unstrained = 4.64 ± 0.60%, fphos. strained = 9.2 ± 2.0%; P < 0.05) myosins increased. As was seen in the unloaded case, there was an increase (35 ± 1%) in duty cycle for RLC-phosphorylated myosin compared with the dephosphorylated myosin under load.
Fig. 3.

Comparison of the effect of applied strain on the duty cycle on RLC-dephosphorylated (A) and RLC-phosphorylated myosins (B). Each point represents the average sliding velocity of a single actin filament across 5–10 video frames; 145 actin filaments were followed for RLC-dephosphorylated myosin, and 204 actin filaments were followed for RLC-phosphorylated myosin. The duty cycle for strained dephosphorylated myosin was 6.8 ± 1.6%, and the duty cycle for strained phosphorylated myosin was 9.2 ± 2.0%. These measured duty cycles are ∼2-fold higher than the measured unstrained duty cycles.
Troponin-tropomyosin-regulated motility assays.
We also studied the effects of RLC phosphorylation on the sliding velocity of regulated thin filaments. In regulated motility assays, thin filaments are coated with tropomyosin and troponin and allowed to slide over a bed of randomly oriented surface myosin in the presence of different concentrations of calcium (4). The relationship between calcium concentration and the sliding velocity of thin filaments in a regulated motility assay is described by a Hill equation:
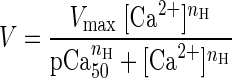 |
(3) |
where Vmax is the maximal sliding velocity, pCa50 is the calcium concentration at which half-maximal velocity is achieved, and nH is the Hill coefficient, a measure of the cooperativity of thin filament activation. We measured thin filament sliding velocity vs. calcium concentration at 24°C (Fig. 4). There is no significant change in calcium sensitivity (as shown by the pCa50 value; P = 0.6) or cooperativity of thin filament activation (as shown by the Hill coefficient; P = 0.85) upon phosphorylation. These results contrast with the results of fiber studies (52, 66, 74) and the actin-activated ATPase measurements performed with partially filamentous myosin (79), where a shift toward lower calcium concentrations of the force-pCa dependence was observed with phosphorylation of RLC.
Fig. 4.

A: regulated thin filament (reconstituted with tropomyosin and troponin) sliding velocity was measured as a function of calcium concentration at both 24 and 35°C. B: normalized plots of the regulated sliding velocities (V/Vmax). Each point represents the average sliding velocity of 15–30 actin filaments averaged over 10 frames. The raw data were fit to Hill plots (Eq. 3). The best fit curves are indicated as follows: black solid line, RLC-dephosphorylated myosin at 35°C; shaded dotted line, RLC-phosphorylated myosin at 35°C; black dashed-dotted line; RLC-dephosphorylated myosin at 24°C; and shaded dashed line, RLC-phosphorylated myosin at 24°C. Each point represents the average velocity (mean ± SE).
Since it has been reported that the phosphorylation-induced shift toward submaximal calcium activation becomes more pronounced with increased temperature (18, 74), we measured the in vitro regulation of RLC-phosphorylated and dephosphorylated myosin at 35°C as a function of calcium concentrations (Fig. 4). Once again, there was no change in calcium sensitivity of velocity (as evidenced by the unchanged pCa50 value; P = 0.35) or in the cooperativity of thin filament activation (no difference in the Hill coefficient; P = 0.93) between phosphorylated and dephosphorylated myosins. There was, however, a significant increase in the calcium sensitivity of velocity at 35 compared with 24°C (P < 0.001) for both RLC-phosphorylated and dephosphorylated myosin (P < 0.01) (Fig. 4A). The cooperativity of the velocity-pCa relationship remained unchanged for phosphorylated (P = 0.15) and dephosphorylated (P = 0.17) myosins.
DISCUSSION
Examining endogenously phosphorylated and dephosphorylated myosins in the in vitro motility assay, we observed that the unloaded sliding velocity of myosin was decreased 20% upon RLC phosphorylation. Fiber studies have shown conflicting data as to whether phosphorylation changes the unloaded sliding velocity of myosin, with some studies showing no differences in sliding velocity upon phosphorylation (7, 45, 52, 72) and others showing a depression in velocity (11, 12). However, the earlier experiments showing a depression in actin filament sliding velocity were performed under fatigue-like conditions (7, 74) and, as has been shown, fatigue-like conditions would cause a depression of phosphorylated myosin unloaded shortening velocity (7, 18, 31).
Since our result showing a reduction in unloaded sliding velocity with phosphorylation was somewhat unexpected, we exogenously phosphorylated endogenously dephosphorylated myosin using Ca2+/CaM-activated exogenous MLCK. We also dephosphorylated endogenously phosphorylated myosin using CIAP (Fig. 1A). We demonstrated that the 20% depression of the phosphorylation-induced unloaded sliding velocity was reversible (Fig. 1B). Thus the observed reduction in velocity is due to phosphorylation-based changes in myosin motor function and not due to purification-induced artifacts. Furthermore, this decrease in velocity with phosphorylation was seen in both unregulated (Figs. 1–3) and regulated thin filaments at high and low temperatures (Fig. 4A).
Previous studies have shown that there is no difference in the maximal rate of actin-activated ATPase with phosphorylated myosin in either the absence (49, 51) [using heavy meromyosin (HMM) and myosin filaments, respectively] or presence of the regulatory proteins (79) [using full-length myosin]. Fiber studies have shown that phosphorylated myosin has an increased rate of transition from the weakly to the strongly bound state (14, 16, 42, 48, 73, 79). Furthermore, it has been proposed that in fibers, the rate of transition from a strongly to weakly bound state is either unaffected (73) or decreased (16, 48) by phosphorylation. A phosphorylation-induced increase in the rate of transition from weak to strong binding with or without a decrease in the rate of transition from strong to weak binding would tend to populate the strongly bound states and thus increase the myosin duty cycle. This is consistent with our data suggesting that phosphorylation causes a 44% increase in the myosin duty cycle. In support of this, the binding of phosphorylated HMM to actin was shown to be stronger than the dephosphorylated HMM in the conditions when the Ca2+-Mg2+ binding site of the phosphorylated HMM was saturated with Mg2+ (77), similar to the conditions used in our motility assays.
Based on a detachment limited model of actomyosin interaction (3, 29, 64, 81), the velocity of myosin in the in vitro motility assay is inversely proportional to the myosin duty cycle (Eq. 1). On the basis of our observed increase in duty cycle, one would expect to see a decrease in the myosin unloaded shortening velocity in RLC-phosphorylated fibers as we do in the in vitro motility assay; however, such a reduction in velocity has only been seen in fibers under fatigue-like conditions (18, 31, 68). Under fatigue-like conditions, acidosis of the muscle causes an increase in the number of weakly bound cross bridges (2, 15, 18, 32, 43). Along these lines, phosphorylation of the RLC has been shown to reduce actin filament velocity in fibers treated with vanadate and blebbistatin, conditions that increase the population of weakly bound states (18, 68). It is possible that the geometry of the in vitro motility assay, in which the myosins are randomly oriented on the motility assay surface, would make the myosin more prone to populating fatigue-like, weakly bound states that would exert a drag force on sliding actin filaments, reducing the actin filament velocity. Although this may play a role in reducing the actin filament sliding velocity of phosphorylated myosin in our assays, we believe that the primary cause of the reduction in velocity stems from the observed increase in the duty cycle of phosphorylated myosin. Even if these weakly bound cross bridges were to cause a slight reduction in velocity, they do not generate force and do not contribute to the observed increase in duty cycle. According to Eq. 1, a change in duty cycle is expected to decrease myosin velocity, and thus it seems reasonable that the observed decrease in velocity is primarily due to the changes in the myosin duty cycle and not due to the population of weakly bound dragging cross bridges.
The observed increase in myosin duty cycle with phosphorylation should decrease the velocity by 44% (Eq. 1). However, we only observed a 20% reduction. Given that the ATPase rates of the phosphorylated and dephosphorylated myosin are unchanged (49, 51, 79), one explanation for this discrepancy could be that RLC phosphorylation causes an increase in the unitary step size of the myosin. A phosphorylation-induced change in the unitary step size of myosin would not be unreasonable, since the RLC supports the neck region of myosin, the structure that is believed to act as a lever arm, amplifying small conformational changes generated by the myosin head. This neck region also has been proposed to be one of the principal sources of compliance of the actomyosin cross bridge (28). Phosphorylation of the RLC adds negative charges to the neck region of the myosin, possibly stiffening the lever arm (37). In fact, such stiffening of the lever arm with RLC phosphorylation has been observed in smooth muscle myosin (33). Stiffening of the lever arm would allow for more efficient transmission of conformational changes at the active site of the myosin head down the lever arm, potentially increasing the myosin unitary step size. This increased step size would offset the depressive effect of the increase in duty cycle on velocity, explaining why we saw only a 20% decrease in velocity but a 44% increase in duty cycle. The hypothesis of phosphorylation-induced stiffening of the myosin lever arm is also supported by the results of Ritz-Gold et al. (59), who showed that phosphorylation of the myosin RLC alters the conformation of myosin, making myosin less susceptible to proteolysis. On the other hand, Franks-Skiba et al. (18) saw no difference in the stiffness of phosphorylated and dephosphorylated fibers. However, this result does not preclude the possibility of a stiffening of the myosin lever arm, since the increase in lever arm stiffness upon phosphorylation could be offset by a potential decrease in stiffness of the thick filaments due to release of the phosphorylated myosin heads from the thick filament backbone (35–37). Single-molecule studies of myosin step size and stiffness are needed to address this issue.
Since the myosin neck region has been proposed to act as a strain sensor, transmitting forces from the neck region to the myosin head (and visa versa), it is possible that changing the stiffness of the myosin cross bridge may cause changes in the strain sensitivity of myosin biochemistry (47). A phosphorylation-induced stiffening of the neck region would tend to enhance the propagation of forces down the myosin heavy chain neck region. This additional sensitivity to load, in turn, could slow the rate of ADP release, explaining the observed changes in detachment rate (14, 42, 73, 79) and duty cycle with RLC phosphorylation. This hypothesis is consistent with the proposal that phosphorylation of the RLC could cause changes in the rate of ADP release from myosin, reducing the myosin velocity and increasing the duty cycle (18, 48). This would not be unreasonable, since it has previously been shown that nucleotide binding in the head region causes an increase in the solvent exposure of the RLC (and that this effect was greater in RLC-phosphorylated myosin), suggesting that changes in the myosin head can affect the conformation of the RLC (41).
We also compared the duty cycle of myosin under both loaded and unloaded conditions. Introduction of an α-actinin-based frictional load reduced the actin sliding velocity by 50% and caused a substantial increase in duty cycle (112% increase for RLC-dephosphorylated myosin and 98% increase for RLC-phosphorylated myosin). This change in duty cycle would probably be even greater under isometric conditions. However, our method does not allow for the measurement of the strained duty cycle in the absence of actin filament sliding. The observed increase in duty cycle in the presence of α-actinin provides strong evidence for a strain dependence in one or more of the myosin biochemical transitions. An increase in duty cycle is indicative of an increase in the population of strongly bound myosin heads and would occur via changes in the rates of phosphate release, ADP release, or ATP binding (47). It has previously been shown using optical trapping techniques that strain significantly slows the rate of ADP release in nonmuscle myosins (9, 54) and smooth muscle myosin II (10, 30, 84), but such a slowing has not been observed in fast muscle myosin, where the rate of ADP release is too fast to be measured using optical traps. It has been suggested that the strain sensitivity of smooth muscle myosin to ADP release is associated with a rotation of the lever arm with ADP release (20, 47). Although such a rotation has been observed in optical trapping data of skeletal muscle (8), it has not been observed by either electron paramagnetic resonance or crystallographic studies (8, 47). There is data suggesting that the myosin biochemical state observed after loading the myosin with exogeously added ADP from the apo-state (as was done in these structural studies) might be different from the state that occurs with the release of ADP from ATP hydrolysis (3, 8, 63, 80), possibly explaining why no lever arm swing was detected in structural studies. Furthermore, it is important to note that although these data are consistent with a slowing of the rate of ADP release with strain in fast skeletal muscle, they do not preclude a different step in the biochemical cycle from being strain dependent.
Regulation by tropomyosin and troponin.
The shift toward submaximal calcium activation with phosphorylation has led to the suggestion that phosphorylation acts as a molecular memory (71), enabling the development of force in muscle fibers at lower levels of activation after repeated stimulation. Actin-Tm-Tn-activated ATPase measurements of phosphorylated and dephosphorylated filamentous myosin revealed a shift toward submaximal calcium activation of force development and ATPase for phosphorylated myosin (79). A similar shift toward submaximal calcium activation after phosphorylation has been observed for force development in skinned fibers (52, 66, 74). On the other hand, our in vitro motility data using randomly oriented (nonfilamentous) surface myosins show no phosphorylation-induced changes in either the pCa50 value or the Hill coefficient of activation.
The lack of effect on calcium sensitivity in our regulated motility assays could be explained by three different, nonexclusive mechanisms. First, sliding velocity and ATPase have different rate-limiting steps, whereas ATPase is limited by an isomerization of the phosphate-bound state (65), product release (38), or ATP hydrolysis (85), and sliding velocity is limited by ADP release (62, 64). It is possible that phosphorylation has a more pronounced effect on the rate of phosphate release than the ADP release rate, and thus the effect of phosphorylation on submaximal calcium activation was masked, consistent with the results of Davis et al. (14). Second, it is possible that the observed lack of change in the calcium sensitivity of phosphorylated myosin could be due to the geometric constraints of the motility assay. Yang et al. (87) suggested that the effects of phosphorylation on submaximal calcium activation require a separation of at least 10 nm between the thick and thin filaments. When the skinned muscle fiber was artificially compressed, the submaximal calcium activation induced by phosphorylation was removed. In the in vitro motility assay, actin lies on the surface of a bed of myosin, and thus the distance from the myosin to the actin filament could be less than 10 nm, possibly causing a masking of the effect of phosphorylation on submaximal calcium activation. However, we consider this possibility unlikely, since solution ATPase measurements using regulated thick filaments, where there are no imposed geometric constraints, also showed submaximal calcium activation (79). Finally, our motility assay utilized monomeric myosin, whereas the fiber studies and the ATPase experiments used filamentous or partially filamentous myosin, respectively. It is possible that the thick filament couples the myosin monomers, cooperatively changing the kinetics of the myosin.
Another interesting observation from the regulation experiments was that an increase in temperature caused a shift toward submaximal calcium activation for both phosphorylated and dephosphorylated myosins. This shift can be explained by the different temperature dependencies of the various steps in the myosin ATPase cycle. Activation of the thin filament occurs during the transition of myosin from a weakly bound to a strongly bound state (23). Myosin strong binding to the thin filament changes the equilibrium position of tropomyosin, lowering the energy barrier for successive myosins to bind to actin (for review, see Ref. 22). Therefore, a shift toward submaximal calcium activation at higher temperatures could be indicative of an increase in the number of strongly bound bridges attached to the thin filament, consistent with data suggesting that the transition to strong binding has the greatest sensitivity to temperature (as evidenced by the Q10) of any step in the biochemical cycle (88).
Perspectives and Significance
There are several differences observed in our in vitro motility studies compared with skinned fiber studies. First, our experiments, done with purified contractile proteins, have the distinct advantage in probing molecular changes in myosin contractility over skinned fiber experiments in that all of the phosphorylation-induced results can be attributed to changes in the myosin molecule and not broader cellular changes, changes in lattice structure, or changes in other sarcomeric proteins. Second, in muscle fibers, myosin is in a filamentous form, whereas our experiments utilize randomly oriented myosin molecules. As such, our results probe the molecular properties of monomeric myosin and not any possible structural changes or allosteric coupling of myosins in the thick filament. Interestingly, we observed the changes in velocity and duty cycle independently of the presence of the thick filament backbone, suggesting that the basis for these alterations originates at the molecular level. These changes are not accounted for by a model in which the primary effect of phosphorylation is to move the myosin heads away from the thick filament backbone (37, 87). Instead, it is more likely that these changes are due to changes in the stiffness of the myosin lever arm or actomyosin kinetics upon phosphorylation.
Finally, the effects of phosphorylation on calcium sensitivity are only observed with an intact thick filament (52, 66, 74, 79), suggesting that the thick filament backbone is required to see these phosphorylation-induced changes in thin filament regulation. This idea is consistent with the suggestion of Myburgh and Cooke (46), who proposed that the properties of phosphorylation could require an intact myofilament array. Phosphorylation-induced movement of myosin heads away from the thick filament backbone could exert its effects by increasing the probability of the formation of actomyosin cross bridges by decreasing the diffusional barrier for activation (37, 87). It also is possible that the thick filament couples the individual myosin force generators, altering the kinetics of the myosin through an allosteric mechanism. Our experiments using monomeric myosin would not be able to detect these changes. Future experiments using filamentous myosin motility assays are necessary to allow us to test these possibilities.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL077280 (to J. R. Moore) and HL071778 (to D. Szczesna-Cordary) and American Heart Association Grants 0435434T (to J. R. Moore) and 0815704D (to M. J. Greenberg).
Supplementary Material
Acknowledgments
We thank Raymond Stephens for helpful discussions and critical reading of the manuscript.
REFERENCES
- 1.Alexis MN, Gratzer WB. Interaction of skeletal myosin light chains with calcium ions. Biochemistry 17: 2319–2325, 1978. [DOI] [PubMed] [Google Scholar]
- 2.Allen DG, Lannergren J, Westerblad H. Muscle cell function during prolonged activity: cellular mechanisms of fatigue. Exp Physiol 80: 497–527, 1995. [DOI] [PubMed] [Google Scholar]
- 3.Baker JE, Brosseau C, Joel PB, Warshaw DM. The biochemical kinetics underlying actin movement generated by one and many skeletal muscle myosin molecules. Biophys J 82: 2134–2147, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bing W, Fraser ID, Marston SB. Troponin I and troponin T interact with troponin C to produce different Ca2+-dependent effects on actin-tropomyosin filament motility. Biochem J 327: 335–340, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bing W, Knott A, Marston SB. A simple method for measuring the relative force exerted by myosin on actin filaments in the in vitro motility assay: evidence that tropomyosin and troponin increase force in single thin filaments. Biochem J 350: 693–699, 2000. [PMC free article] [PubMed] [Google Scholar]
- 6.Brust-Mascher I, LaConte LE, Baker JE, Thomas DD. Myosin light-chain domain rotates upon muscle activation but not ATP hydrolysis. Biochemistry 38: 12607–12613, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Butler TM, Siegman MJ, Mooers SU, Barsotti RJ. Myosin light chain phosphorylation does not modulate cross-bridge cycling rate in mouse skeletal muscle. Science 220: 1167–1169, 1983. [DOI] [PubMed] [Google Scholar]
- 8.Capitanio M, Canepari M, Cacciafesta P, Lombardi V, Cicchi R, Maffei M, Pavone FS, Bottinelli R. Two independent mechanical events in the interaction cycle of skeletal muscle myosin with actin. Proc Natl Acad Sci USA 103: 87–92, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clemen AE, Vilfan M, Jaud J, Zhang J, Barmann M, Rief M. Force-dependent stepping kinetics of myosin-V. Biophys J 88: 4402–4410, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cremo CR, Geeves MA. Interaction of actin and ADP with the head domain of smooth muscle myosin: implications for strain-dependent ADP release in smooth muscle. Biochemistry 37: 1969–1978, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Crow MT, Kushmerick MJ. Myosin light chain phosphorylation is associated with a decrease in the energy cost for contraction in fast twitch mouse muscle. J Biol Chem 257: 2121–2124, 1982. [PubMed] [Google Scholar]
- 12.Crow MT, Kushmerick MJ. Phosphorylation of myosin light chains in mouse fast-twitch muscle associated with reduced actomyosin turnover rate. Science 217: 835–837, 1982. [DOI] [PubMed] [Google Scholar]
- 13.Davis JS, Hassanzadeh S, Winitsky S, Lin H, Satorius C, Vemuri R, Aletras AH, Wen H, Epstein ND. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell 107: 631–641, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Davis JS, Satorius CL, Epstein ND. Kinetic effects of myosin regulatory light chain phosphorylation on skeletal muscle contraction. Biophys J 83: 359–370, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Debold EP, Beck SE, Warshaw DM. Effect of low pH on single skeletal muscle myosin mechanics and kinetics. Am J Physiol Cell Physiol 295: C173–C179, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diffee GM, Patel JR, Reinach FC, Greaser ML, Moss RL. Altered kinetics of contraction in skeletal muscle fibers containing a mutant myosin regulatory light chain with reduced divalent cation binding. Biophys J 71: 341–350, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drabikowski W, Gergely J. The effect of the temperature of extraction and of tropomyosin on the viscosity of actin. In: Biochemistry of Muscle Contraction, edited by Gergely J. Boston, MA: Little, Brown, 1964, p. 125–131.
- 18.Franks-Skiba K, Lardelli R, Goh G, Cooke R. Myosin light chain phosphorylation inhibits muscle fiber shortening velocity in the presence of vanadate. Am J Physiol Regul Integr Comp Physiol 292: R1603–R1612, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J Gen Physiol 80: 279–297, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gollub J, Cremo CR, Cooke R. ADP release produces a rotation of the neck region of smooth myosin but not skeletal myosin. Nat Struct Biol 3: 796–802, 1996. [DOI] [PubMed] [Google Scholar]
- 21.Gomes AV, Liang J, Potter JD. Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J Biol Chem 280: 30909–30915, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev 80: 853–924, 2000. [DOI] [PubMed] [Google Scholar]
- 23.Gorga JA, Fishbaugher DE, VanBuren P. Activation of the calcium-regulated thin filament by myosin strong binding. Biophys J 85: 2484–2491, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo B, Guilford WH. The tail of myosin reduces actin filament velocity in the in vitro motility assay. Cell Motil Cytoskeleton 59: 264–272, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Harada Y, Sakurada K, Aoki T, Thomas DD, Yanagida T. Mechanochemical coupling in actomyosin energy transduction studied by in vitro movement assay. J Mol Biol 216: 49–68, 1990. [DOI] [PubMed] [Google Scholar]
- 26.Harris DE, Warshaw DM. Smooth and skeletal muscle myosin both exhibit low duty cycles at zero load in vitro. J Biol Chem 268: 14764–14768, 1993. [PubMed] [Google Scholar]
- 27.Holroyde MJ, Potter JD, Solaro RJ. The calcium binding properties of phosphorylated and unphosphorylated cardiac and skeletal myosins. J Biol Chem 254: 6478–6482, 1979. [PubMed] [Google Scholar]
- 28.Howard J, Spudich JA. Is the lever arm of myosin a molecular elastic element? Proc Natl Acad Sci USA 93: 4462–4464, 1996. [PubMed] [Google Scholar]
- 29.Huxley HE Sliding filaments and molecular motile systems. J Biol Chem 265: 8347–8350, 1990. [PubMed] [Google Scholar]
- 30.Kad NM, Patlak JB, Fagnant PM, Trybus KM, Warshaw DM. Mutation of a conserved glycine in the SH1-SH2 helix affects the load-dependent kinetics of myosin. Biophys J 92: 1623–1631, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karatzaferi C, Franks-Skiba K, Cooke R. Inhibition of shortening velocity of skinned skeletal muscle fibers in conditions that mimic fatigue. Am J Physiol Regul Integr Comp Physiol 294: R948–R955, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Kentish JC Combined inhibitory actions of acidosis and phosphate on maximum force production in rat skinned cardiac muscle. Pflügers Arch 419: 310–318, 1991. [DOI] [PubMed] [Google Scholar]
- 33.Khromov AS, Somlyo AV, Somlyo AP. Thiophosphorylation of myosin light chain increases rigor stiffness of rabbit smooth muscle. J Physiol 512: 345–350, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kron SJ, Spudich JA. Fluorescent actin filaments move on myosin fixed to a glass surface. Proc Natl Acad Sci USA 83: 6272–6276, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levine RJ, Kensler RW, Yang Z, Stull JT, Sweeney HL. Myosin light chain phosphorylation affects the structure of rabbit skeletal muscle thick filaments. Biophys J 71: 898–907, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levine RJ, Kensler RW, Yang Z, Sweeney HL. Myosin regulatory light chain phosphorylation and the production of functionally significant changes in myosin head arrangement on striated muscle thick filaments. Biophys J 68, Suppl 4: 224S, 1995. [PMC free article] [PubMed] [Google Scholar]
- 37.Levine RJ, Yang Z, Epstein ND, Fananapazir L, Stull JT, Sweeney HL. Structural and functional responses of mammalian thick filaments to alterations in myosin regulatory light chains. J Struct Biol 122: 149–161, 1998. [DOI] [PubMed] [Google Scholar]
- 38.Lymn RW, Taylor EW. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 10: 4617–4624, 1971. [DOI] [PubMed] [Google Scholar]
- 39.Manning DR, Stull JT. Myosin light chain phosphorylation-dephosphorylation in mammalian skeletal muscle. Am J Physiol Cell Physiol 242: C234–C241, 1982. [DOI] [PubMed] [Google Scholar]
- 40.Manning DR, Stull JT. Myosin light chain phosphorylation and phosphorylase A activity in rat extensor digitorum longus muscle. Biochem Biophys Res Commun 90: 164–170, 1979. [DOI] [PubMed] [Google Scholar]
- 41.Mazhari SM, Selser CT, Cremo CR. Novel sensors of the regulatory switch on the regulatory light chain of smooth muscle Myosin. J Biol Chem 279: 39905–39914, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Metzger JM, Greaser ML, Moss RL. Variations in cross-bridge attachment rate and tension with phosphorylation of myosin in mammalian skinned skeletal muscle fibers. Implications for twitch potentiation in intact muscle. J Gen Physiol 93: 855–883, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Metzger JM, Moss RL. Effects of tension and stiffness due to reduced pH in mammalian fast- and slow-twitch skinned skeletal muscle fibres. J Physiol 428: 737–750, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore RL, Stull JT. Myosin light chain phosphorylation in fast and slow skeletal muscles in situ. Am J Physiol Cell Physiol 247: C462–C471, 1984. [DOI] [PubMed] [Google Scholar]
- 45.Morano I, Arndt H, Bachle-Stolz C, Ruegg JC. Further studies on the effects of myosin P-light chain phosphorylation on contractile properties of skinned cardiac fibres. Basic Res Cardiol 81: 611–619, 1986. [DOI] [PubMed] [Google Scholar]
- 46.Myburgh KH, Cooke R. Response of compressed skinned skeletal muscle fibers to conditions that simulate fatigue. J Appl Physiol 82: 1297–1304, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Nyitrai M, Geeves MA. Adenosine diphosphate and strain sensitivity in myosin motors. Philos Trans R Soc Lond B Biol Sci 359: 1867–1877, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olsson MC, Patel JR, Fitzsimons DP, Walker JW, Moss RL. Basal myosin light chain phosphorylation is a determinant of Ca2+ sensitivity of force and activation dependence of the kinetics of myocardial force development. Am J Physiol Heart Circ Physiol 287: H2712–H2718, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Pemrick SM The phosphorylated L2 light chain of skeletal myosin is a modifier of the actomyosin ATPase. J Biol Chem 255: 8836–8841, 1980. [PubMed] [Google Scholar]
- 50.Perrie WT, Smillie LB, Perry SB. A phosphorylated light-chain component of myosin from skeletal muscle. Biochem J 135: 151–164, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Persechini A, Stull JT. Phosphorylation kinetics of skeletal muscle myosin and the effect of phosphorylation on actomyosin adenosinetriphosphatase activity. Biochemistry 23: 4144–4150, 1984. [DOI] [PubMed] [Google Scholar]
- 52.Persechini A, Stull JT, Cooke R. The effect of myosin phosphorylation on the contractile properties of skinned rabbit skeletal muscle fibers. J Biol Chem 260: 7951–7954, 1985. [PubMed] [Google Scholar]
- 53.Potter JD Preparation of troponin and its subunits. Methods Enzymol 85: 241–263, 1982. [DOI] [PubMed] [Google Scholar]
- 54.Purcell TJ, Sweeney HL, Spudich JA. A force-dependent state controls the coordination of processive myosin V. Proc Natl Acad Sci USA 102: 13873–13878, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quinlan ME, Forkey JN, Goldman YE. Orientation of the myosin light chain region by single molecule total internal reflection fluorescence polarization microscopy. Biophys J 89: 1132–1142, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 261: 50–58, 1993. [DOI] [PubMed] [Google Scholar]
- 57.Reinach FC, Nagai K, Kendrick-Jones J. Site-directed mutagenesis of the regulatory light-chain Ca2+/Mg2+ binding site and its role in hybrid myosins. Nature 322: 80–83, 1986. [DOI] [PubMed] [Google Scholar]
- 58.Reisfeld RA, Small PA Jr. Electrophoretic heterogeneity of polypeptide chains of specific antibodies. Science 152: 1253–1255, 1966. [DOI] [PubMed] [Google Scholar]
- 59.Ritz-Gold CJ, Cooke R, Blumenthal DK, Stull JT. Light chain phosphorylation alters the conformation of skeletal muscle myosin. Biochem Biophys Res Commun 93: 209–214, 1980. [DOI] [PubMed] [Google Scholar]
- 60.Ryder JW, Lau KS, Kamm KE, Stull JT. Enhanced skeletal muscle contraction with myosin light chain phosphorylation by a calmodulin-sensing kinase. J Biol Chem 282: 20447–20454, 2007. [DOI] [PubMed] [Google Scholar]
- 61.Shih WM, Gryczynski Z, Lakowicz JR, Spudich JA. A FRET-based sensor reveals large ATP hydrolysis-induced conformational changes and three distinct states of the molecular motor myosin. Cell 102: 683–694, 2000. [DOI] [PubMed] [Google Scholar]
- 62.Siemankowski RF, Wiseman MO, White HD. ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle. Proc Natl Acad Sci USA 82: 658–662, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sleep JA, Hutton RL. Exchange between inorganic phosphate and adenosine 5′-triphosphate in the medium by actomyosin subfragment 1. Biochemistry 19: 1276–1283, 1980. [DOI] [PubMed] [Google Scholar]
- 64.Spudich JA How molecular motors work. Nature 372: 515–518, 1994. [DOI] [PubMed] [Google Scholar]
- 65.Stein LA, Chock PB, Eisenberg E. The rate-limiting step in the actomyosin adenosinetriphosphatase cycle. Biochemistry 23: 1555–1563, 1984. [DOI] [PubMed] [Google Scholar]
- 66.Stelzer JE, Patel JR, Moss RL. Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. J Gen Physiol 128: 261–272, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stepkowski D, Szczesna D, Wrotek M, Kakol I. Factors influencing interaction of phosphorylated and dephosphorylated myosin with actin. Biochim Biophys Acta 831: 321–329, 1985. [DOI] [PubMed] [Google Scholar]
- 68.Stewart M, Franks-Skiba K, Cooke R. Myosin regulatory light chain phosphorylation inhibits shortening velocities of skeletal muscle fibers in the presence of the myosin inhibitor blebbistatin. J Muscle Res Cell Motil 30: 17–27, 2009. [DOI] [PubMed] [Google Scholar]
- 69.Straub FB Actin. In Stud Inst Med Chem Univ Szeged 2: 3–16, 1942. [Google Scholar]
- 70.Sweeney HL Regulation and tuning of smooth muscle myosin. Am J Respir Crit Care Med 158: S95–S99, 1998. [DOI] [PubMed] [Google Scholar]
- 71.Sweeney HL, Bowman BF, Stull JT. Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. Am J Physiol Cell Physiol 264: C1085–C1095, 1993. [DOI] [PubMed] [Google Scholar]
- 72.Sweeney HL, Kushmerick MJ. Myosin phosphorylation in permeabilized rabbit psoas fibers. Am J Physiol Cell Physiol 249: C362–C365, 1985. [DOI] [PubMed] [Google Scholar]
- 73.Sweeney HL, Stull JT. Alteration of cross-bridge kinetics by myosin light chain phosphorylation in rabbit skeletal muscle: implications for regulation of actin-myosin interaction. Proc Natl Acad Sci USA 87: 414–418, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sweeney HL, Stull JT. Phosphorylation of myosin in permeabilized mammalian cardiac and skeletal muscle cells. Am J Physiol Cell Physiol 250: C657–C660, 1986. [DOI] [PubMed] [Google Scholar]
- 75.Szczesna-Cordary D, Guzman G, Ng SS, Zhao J. Familial hypertrophic cardiomyopathy-linked alterations in Ca2+ binding of human cardiac myosin regulatory light chain affect cardiac muscle contraction. J Biol Chem 279: 3535–3542, 2004. [DOI] [PubMed] [Google Scholar]
- 76.Szczesna-Cordary D, Jones M, Moore JR, Watt J, Kerrick WG, Xu Y, Wang Y, Wagg C, Lopaschuk GD. Myosin regulatory light chain E22K mutation results in decreased cardiac intracellular calcium and force transients. FASEB J 21: 3974–3985, 2007. [DOI] [PubMed] [Google Scholar]
- 77.Szczesna D, Sobieszek A, Kakol I. Binding of phosphorylated and dephosphorylated heavy meromyosin to F-actin. FEBS Lett 210: 177–180, 1987. [DOI] [PubMed] [Google Scholar]
- 78.Szczesna D, Zhang R, Zhao J, Jones M, Guzman G, Potter JD. Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. J Biol Chem 275: 624–630, 2000. [DOI] [PubMed] [Google Scholar]
- 79.Szczesna D, Zhao J, Jones M, Zhi G, Stull J, Potter JD. Phosphorylation of the regulatory light chains of myosin affects Ca2+ sensitivity of skeletal muscle contraction. J Appl Physiol 92: 1661–1670, 2002. [DOI] [PubMed] [Google Scholar]
- 80.Trybus KM, Taylor EW. Transient kinetics of adenosine 5′-diphosphate and adenosine 5′-(beta, gamma-imidotriphosphate) binding to subfragment 1 and actosubfragment 1. Biochemistry 21: 1284–1294, 1982. [DOI] [PubMed] [Google Scholar]
- 81.Tyska MJ, Warshaw DM. The myosin power stroke. Cell Motil Cytoskeleton 51: 1–15, 2002. [DOI] [PubMed] [Google Scholar]
- 82.Uyeda TQ, Kron SJ, Spudich JA. Myosin step size. Estimation from slow sliding movement of actin over low densities of heavy meromyosin. J Mol Biol 214: 699–710, 1990. [DOI] [PubMed] [Google Scholar]
- 83.VanBuren P, Waller GS, Harris DE, Trybus KM, Warshaw DM, Lowey S. The essential light chain is required for full force production by skeletal muscle myosin. Proc Natl Acad Sci USA 91: 12403–12407, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Veigel C, Molloy JE, Schmitz S, Kendrick-Jones J. Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nat Cell Biol 5: 980–986, 2003. [DOI] [PubMed] [Google Scholar]
- 85.White HD, Belknap B, Webb MR. Kinetics of nucleoside triphosphate cleavage and phosphate release steps by associated rabbit skeletal actomyosin, measured using a novel fluorescent probe for phosphate. Biochemistry 36: 11828–11836, 1997. [DOI] [PubMed] [Google Scholar]
- 86.Xie X, Harrison DH, Schlichting I, Sweet RM, Kalabokis VN, Szent-Gyorgyi AG, Cohen C. Structure of the regulatory domain of scallop myosin at 2.8 A resolution. Nature 368: 306–312, 1994. [DOI] [PubMed] [Google Scholar]
- 87.Yang Z, Stull JT, Levine RJ, Sweeney HL. Changes in interfilament spacing mimic the effects of myosin regulatory light chain phosphorylation in rabbit psoas fibers. J Struct Biol 122: 139–148, 1998. [DOI] [PubMed] [Google Scholar]
- 88.Zhao Y, Kawai M. Kinetic and thermodynamic studies of the cross-bridge cycle in rabbit psoas muscle fibers. Biophys J 67: 1655–1668, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhi G, Ryder JW, Huang J, Ding P, Chen Y, Zhao Y, Kamm KE, Stull JT. Myosin light chain kinase and myosins phosphorylation effect frequency-dependent potentiation of skeletal muscle contraction. Proc Natl Acad Sci USA 102: 17,519–17,524, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.