


Abstract
Accurate and efficient recognition of splice sites during pre-mRNA splicing is essential for proper transcriptome expression. Splice site usage can be modulated by secondary structures, but it is unclear if this type of modulation is commonly used or occurs to a significant degree with secondary structures forming over long distances. Using phlyogenetic comparisons of intronic sequences among 12 Drosophila genomes, we elucidated a group of 202 highly conserved pairs of sequences, each at least nine nucleotides long, capable of forming stable stem structures. This set was highly enriched in alternatively spliced introns and introns with weak acceptor sites and long introns, and most occurred over long distances (>150 nucleotides). Experimentally, we analyzed the splicing of several of these introns using mini-genes in Drosophila S2 cells. Wild-type splicing patterns were changed by mutations that opened the stem structure, and restored by compensatory mutations that re-established the base-pairing potential, demonstrating that these secondary structures were indeed implicated in the splice site choice. Mechanistically, the RNA structures masked splice sites, brought together distant splice sites and/or looped out introns. Thus, base-pairing interactions within introns, even those occurring over long distances, are more frequent modulators of alternative splicing than is currently assumed.
INTRODUCTION
Pre-mRNA splicing provides an important window for post-transcriptional control of the transcriptome, with alternative splicing leading to a huge expansion in proteomic diversity (1,2). The large, multi-complex spliceosome is assembled de novo onto each intron, for which the precise recognition of the intron borders by the spliceosome is essential (3). Each intron is defined by a donor site and an acceptor site at its 5′ and 3′ ends, respectively. However, as these core splicing signals are highly degenerative, intron/exon definition requires a network of protein–protein and protein–RNA interactions to ensure that the correct splice sites are recognized and used (3). Much attention has been focused on the regulation of this process through RNA-binding proteins, which can mediate the effects of splicing enhancers or silencers at a specific site (4,5).
Splicing regulation can also be effected by the presence of secondary structure within the pre-mRNA (6). There is a general consensus that secondary structures within pre-mRNA will be formed locally, rather than over long distances, since folding occurs cotranscriptionally (7–9). Cotranscriptional folding of pre-RNA was suggested to occur mainly within a window of about 60 nucleotides downstream of the transcribing polymerase (8). Recently, many specific examples have been documented in which the presence of local secondary structure is shown to affect the splicing outcome (6). For instance, the efficiency of splicing of an intron in the Drosophila Adh gene was reduced when a hairpin structure within the intron was disrupted (10). In the human tau pre-mRNA, a stem structure that occurs locally masks the donor site of exon 10 (11). Silent mutations linked with neurogenerative diseases have been shown to destabilize the stem structure, thereby increasing the availability of the donor site with a concurrent increase in exon 10 inclusion (11,12).
A recent analysis of the human genome revealed a correlation between secondary structure encompassing a splice site and alternative splicing, suggesting that local secondary structures frequently modulate alternative splicing by masking splice sites (13). In another human genome-wide analysis, splicing enhancer and silencer signals were found more frequently in a single-stranded than in a double-stranded context (14). Correspondingly, the signals were less effective as splicing regulators if incorporated into a double-stranded context, suggesting that local RNA secondary structure is under evolutionary selection (14).
Long-range base-pairing within pre-mRNA has also been implicated in modulating pre-mRNA splicing in a few cases. One of the most dramatic examples is offered by the Drosophila Dscam pre-mRNA, where the formation of an intronic stem structure between a region downstream of the donor site, and one of the regions upstream of each of the 48 potential acceptor sites, appears to modulate the binding of splicing regulators and allow for mutually exclusive splicing to a single exon of the exon 6 cluster (15). Such interactions would occur over distances ranging from 1000 to 12 000 nucleotides. Similarly to short-range interactions, long-range interactions could mask splicing signals or create novel binding sites for protein binding to double-stranded RNA. They could also affect the context of splicing signals to a greater degree, for example, by looping out an exon, or by bringing distant splice sites in closer proximity to each other. For instance, GC-rich motifs surrounding alternatively spliced exons in humans were implicated in looping-out these exons and thereby leading to exon skipping, even though the interactions between these motifs would occur over long distances (16).
To determine the extent to which long-range interactions modulate pre-mRNA splicing, we took advantage of the availability of the 12 sequenced Drosophila genomes to perform phylogenetic searches for conserved intronic stem structures. Specifically, we first searched the D. melonogaster genome for complementary stretches of at least nine nucleotides (hereafter termed ‘boxes’) that could base-pair, with the requirement that each box be located near an intron boundary to maximize the potential for the stem structures to influence splicing. This set was then narrowed down to those pairs that were also phylogenetically conserved, resulting in 202 pairs of conserved boxes, of which approximately 50% were within alternatively spliced introns. Several pairs of boxes were experimentally tested within mini-genes to determine whether the stem structures predicted to form over long distances could influence the splicing outcome. Indeed, mutagenesis studies revealed that base-pairing of the boxes was critical in determining the resulting ratio of alternatively spliced mRNAs. We suggest that the formation of long-distance secondary structure plays a much greater role in modulating alternative splicing in Drosophila than previously assumed. This modulation provides a way of amplifying the alternative splicing repertoire as well as a platform for further regulation in trans.
MATERIALS AND METHODS
Mini-genes and splicing assays
Mini-genes containing the exons/introns of interest were amplified from the D. melanogaster genomic DNA using Taq Precision Plus polymerase (Stratagene) and inserted into the pRMHA5 plasmid under a copper-inducible metallothionein promoter. Schneider S2-L4 cells were transfected using the Effectene Transfection Reagent (Qiagen) as recommended. The promoter was induced 24 h following transfection by the addition of 10 µM copper to the medium, and cells were harvested 24 h later. RNA was purified using the RNeasy Mini Kit (Qiagen) as recommended. Reverse transcription was carried out on 1 µg of RNA with oligo-dT reverse primer, and semi-quantitative PCR was performed with a plasmid-specific forward primer and a reverse primer specific for either the vector or the gene, as indicated, on 1/40 of the RT (except where indicated). Semi-quantitative RT–PCR for the endogenous mRNAs were carried out with oligo-dT primer for the RT, using 1 µg of total S2 cell RNA, and primers that were located within the exons border the splicing event to be analyzed for the PCR, using 1/20 of the total RT for the PCR. Controls were performed without the addition of the reverse transcriptase enzyme, to differentiate between RNA and DNA amplification. Splicing was visualized on agarose gels, and bands were quantified with the NIH ImageJ program. Reverse cDNAs of the spliced products were cloned into the pGEM-T Easy vector (Promega) and identified by sequencing. Mutagenesis was performed using the QuikChange method (Stratagene) as recommended, and the resulting mutants were verified by sequencing.
Splicing database
The sequence data on D. melanogaster introns were obtained from the release 3.2 of the genome annotations available at FlyBase (17). The database comprised of 50 000 introns, of which 17% were alternative, and ∼2% contained putative polyadenylation events.
This database was extended to D. sechellia, D. simulans, D. erecta, D. yakuba, D. ananassae, D. pseudoobscura, D. persimillis, D. willistoni, D. mojavensis, D. virilis and D. grimshawi using pairwise nucleotide BLASTZ alignments (18). In each of the species, we identified possible orthologs of D. melanogaster splice sites using chain alignments (18). Approximately 95% of D. melanogaster splice sites were conserved in at least seven of the 12 species. The strength of a splice site consensus (w) was computed by using scoring matrices covering five nucleotides upstream and seven nucleotides downstream of the donor site, and nine nucleotides upstream and three nucleotides downstream of the acceptor site, respectively (19). Equilibrium free energies were computed at 37°C based on thermodynamic parameters for RNA folding (20). More information can be found in Supplementary Data.
Tests of significance
Tests of significance for proportions (e.g. the proportion of alternative introns in the box containing intron set versus that in the population of all D. melanogaster introns) were carried out using the one-sample z-test for np > 5, and using the Poisson approximation to the binomial distribution for n ≤ 5, where n is the sample size and p is the population proportion. The reference population was defined uniquely by the context in each test. It was not unreasonable to assume normal distribution for splice site strengths (see Supplementary Data). Average splice site strengths (Δw) were compared using the one-sample z-test, with the exception of the test for strong cryptic splice sites, in which case the matched two-sample procedure was used (see Supplementary Data). The number that follows the ± sign denotes the standard error. The standard deviation was multiplied by the square root of 2 when strengths of individual splice sites (Δw) were compared. Throughout the article, we report one-tailed P-values (P). Statistical analysis of gene functions was carried out by using the GOSTAT software with the Benjamini correction for multiple tests (21).
Randomization procedures
The rate of false positive predictions was estimated by random sampling (without replacement) of 8000 introns, each from a different gene. These introns were randomly matched in pairs, and sequences surrounding splice sites were rewired, i.e. the donor splice site of one intron was set in correspondence with the acceptor splice site of the other intron and vice versa. This yielded a set of non-cognate donor–acceptor pairs which did not correspond to any existing intron. The proportion of false positive predictions was computed from the number of boxes found in the original set and in the rewired set. This sampling procedure was repeated 100 times to estimate the average false positive rate. Since every splice site has equal chances to be paired with any other splice site, we expect that the potential confounding effects of CG content or intron length average out during repetitive sampling. Additionally, we scored each pair of boxes by computing an individual p-value, as explained in detail in the Supplementary Data.
RESULTS
To address the possibility that stable secondary structure is a common modulator of splicing, we searched D. melanogaster introns for pairs of sequences (boxes) that could potentially base-pair. We did not restrict the distance between the two complementary boxes but required that they were located intronically, within 150 nucleotides of the intron boundaries (i.e. a box near the donor splice site, and the complementary box near the acceptor splice site). Note that the definition of introns and exons is relative to particular splicing events, so that our boxes could also be located in intronic regions that can also be exonic. To ensure for stability, we required that the sequences contain a continuous stretch of nine complementary nucleotides, with at least two GC pairs and a maximum of one GU base-pair. When such a stretch of nine complementary nucleotides was detected, it was extended to the longest common secondary structure (see Supplementary Data). To obtain box pairs that are biologically relevant, we added the strong requirement that each sequence is evolutionarily conserved. Specifically, each set of sequences had to be phylogenetically conserved in at least seven of the 12 Drosophila genomes, and contain a maximum variation of three nucleotides across these genomes (see Supplementary Data for details).
We obtained a set of 202 intronic box pairs that met our criteria (Table 1 and Supplementary Table S1). The high level of conservation of these sequences is striking, given that these sequences occur intronically, and that intronic sequences are not conserved as strongly as exons. The average false positive rate, obtained by applying an identical search procedure to a dataset consisting of randomly matched donor and acceptor sites from unrelated genes, was determined to be 6% ± 4% (see ‘Materials and Methods’ section). The average length and equilibrium free energy (of the extended structures) were 11 ± 2 nt and 19 ± 5 kcal/mol, respectively (see Table 1 and the Supplementary Table S1 for the individual lengths and free energies for each pair).
Table 1.
Genes containing predicted intronic secondary structures
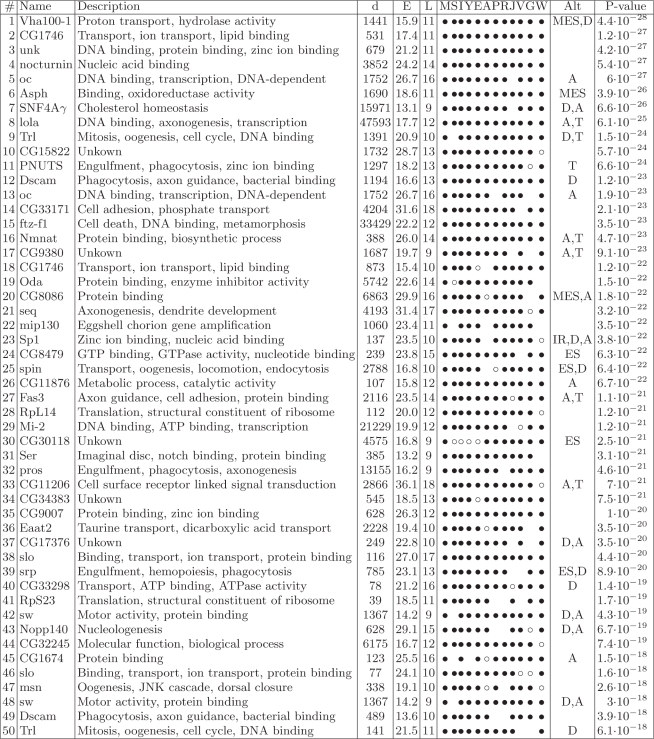 |
The 50 best-scoring predictions, determined by P-values, are shown here; the remaining are listed in Supplementary Table 1. The columns from left to right are: rank number (#); name of the gene (FlyBase); gene annotation (GO or FlyBase; description); distance between boxes (d); equilibrium free energy of the predicted stem (E); length of the stem (L); list of species (see abbreviations below); type of alternative splicing if present (see below; Alt); and P-value (see Supplementary Data). Species are abbreviated with an M for Drosophila melanogaster; S for D. sechellia; I for D. simulans; Y for D. yakuba; E for D. erecta; A for D. ananassae; P for D. pseudoobscura; R for D. persimillis; J for D. mojavensis; V for D. virilis; G for D. grimshawi; and W for D. willistoni. Bullets denote that a structure was found; open circles denote that an orthologous intron, but not structure, was found; and an empty space indicated that no orthologous intron was found. Splicing events are denoted as: D, alternative donor site; A, alternative acceptor site; T, putative polyadenylation site; SE, skipped exon; MES, multiple exon skipping; IR, intron retention; no sign, constitutive splicing (note that the alternative splicing categories are not necessarily mutually exclusive). Additional information about the positions and sequences of the boxes can be found in Supplementary Table 2.
As a cross-validation test, we compared our predictions to those obtained by RNAalifold (22), a program that predicts a consensus secondary structure in a set of aligned sequences. Since all RNA structure prediction programs are limited by sequence length but the majority of the box pairs (>80%) were more than 150 nt apart from each other, we narrowed the search space to the subset of short (less than 150 nt) introns (see ‘Methods’ section). Of our set, only five box pairs occur within such short introns, while only two were detected by RNAalifold. When the requirement of the minimal number of base pairs within the stem structures was reduced from nine to eight, our method retrieved 32 boxes compared to seven by RNAalifold. Thus, the predictions of RNAalifold constituted a proper subset of our predictions, indicating 100% sensitivity of our method, with respect to RNAalifold as a baseline.
Characteristics of introns with conserved complementary sequences
Analysis of the set of introns that contain the complementary boxes revealed several characteristics that set this group apart statistically. There was a significant enrichment in alternatively spliced introns, as compared to the general population (of 50%, compared to 17% overall; n = 202, P = 1 × 10–36) (Figure 1A). Alternatively spliced introns are better conserved overall than are constitutively spliced introns (23), as is reflected by the enrichment in alternatively spliced introns (of 30%, P = 2 × 10–7) observed in the control set when splice sites were rewired (alternative to alternative and constitutive to constitutive). Nonetheless, the enrichment within the set of introns that contain complementary boxes is still significant compared to that in the rewired control (50% versus 30%, P = 1 × 10–12). No significant difference in equilibrium free energies was found between structures located in alternative and constitutive introns (P = 0.16).
Figure 1.

Statistical properties of the box-containing intron set. (A) Percentage of alternative introns (alternative) and introns containing putative polyadenylation events (polyA) in the set of introns with predicted secondary structures (dark bars), as compared those in the population of all Drosophila introns (light bars). Error bars indicate standard errors; n = 202 (see Materials and Methods). (B) Percentage of alternatively-spliced introns with predicted secondary structure (dark bars) are compared to all alternatively-spliced introns (light bars) in the categories (left to right): introns with alternative donor sites, introns with alternative acceptor sites, introns containing skipped exons, retained introns and introns with both alternative acceptor sites and internal polyadenylation signals; n = 102. (C) Distribution of box positions relative to splice sites. Light grey bars indicate the position of the 5′-box relative to the donor site, while dark grey bars indicate the position of the 3′-box relative to the acceptor site. (D) and (E) Log distributions of intron lengths for the set of introns with predicted secondary structures (dark bars) as compared to that for the population of all introns (light bars) in D, and for the set of alternative introns with predicted secondary structures (dark bars) as compared to that for the population of alternative introns (light bars) in E.
Within the subgroup of alternatively spliced introns, we see an enrichment in introns that contain alternative acceptor sites (n = 102, P = 0.005), and especially those that contain both alternative acceptor sites and potential polyadenylation signals (n = 102, P = 0.0001) (Figure 1B). The predicted set is also enriched for weaker-than-average acceptor sites, with respect to all introns (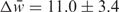 , n = 192, P = 0.0006), and even with respect to all alternatively spliced introns (
, n = 192, P = 0.0006), and even with respect to all alternatively spliced introns (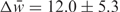 , n = 99, P = 0.01). In contrast, no discernable differences were observed for the donor site strength (
, n = 99, P = 0.01). In contrast, no discernable differences were observed for the donor site strength (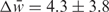 , n = 193, P = 0.13). Additionally, introns containing the complementary boxes were more likely to contain a strong cryptic acceptor site within 100 nucleotides of the annotated acceptor site (see Supplementary Data) than were alternatively spliced introns overall (P = 0.004). This suggests that there is a stronger modulation of alternative splicing of acceptor sites than donor sites through conserved secondary structures.
, n = 193, P = 0.13). Additionally, introns containing the complementary boxes were more likely to contain a strong cryptic acceptor site within 100 nucleotides of the annotated acceptor site (see Supplementary Data) than were alternatively spliced introns overall (P = 0.004). This suggests that there is a stronger modulation of alternative splicing of acceptor sites than donor sites through conserved secondary structures.
The presence of the intronic stem structures could influence splicing in highly complex ways, in addition to directly masking splice sites. Indeed, our search selected against sequences that covered splice sites, since the sequences were required to be intronic, to reduce the false positive rate (see below). Interestingly, the distribution of the sequences within the introns also revealed a minimal distance to the splice sites that differed between the 5′ ends and the 3′ ends: the 5′-box was located on average at 60 nt downstream of the donor site, while the 3′-box was located on average at 80 nt upstream of the acceptor site (Figure 1C). The same skew was observed when the requirement of having at least two GC pairs was eliminated (data not shown). This spatial arrangement suggests that the majority of the stems are located as not to interfere with the polypyrimidine tract and branch point.
Although Drosophila introns range in size between 40 bp to more than 70 kb, more than half of all introns have an average length of around 60 bp (19,24). Thus, it was interesting that we also observe an enrichment for longer introns within our set of box-containing introns, as compared to the length of introns overall, within the groups of introns ranging from 100 to 1000, or from 1000 to 10 000 nucleotides (Figure 1D). Since alternatively spliced introns in Drosophila are in general longer than those of the overall population, we also compared the alternatively spliced introns within our set to all alternatively spliced introns. In this case, while we lose the enrichment in the medium length intron class (e.g. 100–1000 nt), we still observe an enrichment in the long intron class of 1000–10 000 nt (of 52%, as compared to 37%, P = 0.0008) (Figure 1E). The presence of stems within long introns could bring together distant splice sites, since each box is within 150 nt of each splice site), thereby facilitating splicing of long introns.
The Gene Ontology (GO) analysis revealed a strong association between the occurrence of conserved secondary structures and gene function, with statistically detectable enrichment for genes related to morphogenesis and developmental processes, and especially in those associated with nervous system (P < 0.00001; data not shown). Although there is a potential confounding effect of alternative splicing in this association due to the overall high frequency of alternative splicing among developmental genes (25,26), only a few changes were observed in the list of over-represented GO terms when the reference set was narrowed to alternatively spliced genes.
Since our analysis used highly restrictive structure and conservation constraints, we explored how the number of predictions would change under different search conditions. We varied each of the parameter values and re-computed the number of predicted secondary structures along with the corresponding false positive rates. As expected, the number of predictions correlated with the noise level (Table 2). The number of predicted structures remained within the same order of magnitude when the maximum number of GU base pairs, the minimum number of GC base pairs, or the maximum Hamming distance (the number of nucleotides by which boxes differ between species) were varied. However, the predicted set increased dramatically (even compared to the noise level) when the seed length (e.g. the minimum stretch of complementary nucleotides) or the minimum number of species were decreased, and computation of the false discovery rate demonstrated that this increase is not due solely to the noise. Similarly, broader windows captured more structures, although also at the expense of increased noise.
Table 2.
The number of introns with conserved secondary structures predicted at different parameter values
| Parameter | Value | Predicted | FPR (%) |
|---|---|---|---|
| Seed length | 8 | 539 | 23 ± 7 |
| 9 | 202 | 6 ± 4 | |
| 10 | 101 | 4 ± 3 | |
| Max. number of GU | 0 | 105 | 4 ± 2 |
| 1 | 202 | 6 ± 4 | |
| 2 | 307 | 19 ± 7 | |
| Min. number of GC | 0 | 272 | 9 ± 6 |
| 1 | 242 | 7 ± 5 | |
| 2 | 202 | 6 ± 4 | |
| 3 | 154 | 6 ± 5 | |
| Max. hamming distance | 1 | 129 | 7 ± 4 |
| 3 | 202 | 6 ± 4 | |
| 5 | 321 | 10 ± 6 | |
| Min. number of species | 4 | 1599 | 37 ± 5 |
| 5 | 872 | 26 ± 5 | |
| 6 | 355 | 11 ± 6 | |
| 7 | 202 | 6 ± 4 | |
| 8 | 117 | 6 ± 5 | |
| 9 | 70 | 6 ± 5 | |
| 10 | 45 | 4 ± 4 | |
| 11 | 28 | 6 ± 6 | |
| 12 | 11 | N/A | |
| Nucleotides in exon | 10 | 243 | 11 ± 5 |
| 20 | 263 | 14 ± 6 | |
| 30 | 300 | 16 ± 6 | |
| Window length | 100 | 84 | 7 ± 6 |
| 150 | 202 | 6 ± 4 | |
| 200 | 298 | 11 ± 6 | |
| 250 | 414 | 14 ± 7 | |
| 300 | 560 | 16 ± 6 |
Columns from left to right are: parameter name (see text); parameter value; the number of predicted secondary structures; and the estimated false positive rate (see Materials and Methods section). Numbers that follow the ± sign are standard errors.
We also explored a possibility of including up to 30 nt of the exonic sequence into the search space. As a result, the number of predictions increases from 202 to 300. However, due to higher sequence conservations rate in coding regions, this increase was also accompanied by a substantial increase in the noise level. Although some of these predictions could represent interesting cases of secondary structures that are involved in masking splice sites, they are located in a conserved background and thus are less statistically significant compared to the intronic predictions.
On the basis of Table 2, we suggest that our set of 202 highly conserved secondary structures is an under-representation of secondary structures that could potentially influence splicing of pre-mRNAs.
Stem structures modulate alternatively spliced introns
To test our prediction that the conserved pairs of sequences can form stable secondary structures that influence splicing, we chose several of these to analyze experimentally. The choice of introns to test was made based on gene function, type of splicing, and whether it was feasible to clone the region into a mini-gene, and thus was relatively random with respect to box sequence and location. We constructed mini-genes for the regions of interest surrounding the introns, and analyzed the splicing following transfection into D. melanogaster S2 cells. To determine whether the boxes base-pair and form an RNA structure that can influence splicing, we mutated each box separately, to disrupt potential stem structures, or both boxes simultaneously with complementary mutations, to re-establish a stem structure with a novel sequence. In this way, we directly monitored splicing and could infer whether a stable secondary structure was formed only if this was critical for the splicing outcome.
We chose to test three alternatively spliced and three constitutively spliced introns (see Table 1, #16, 40 and 84, or #3, 57 and 69, respectively, for gene names). For the constitutively spliced introns, we did not observe any discernible changes when the single boxes were mutated (data not shown). In contrast, we observed changes in the splicing pattern of each of three alternatively spliced mini-genes tested, when either one or the other box was mutated (Figures 2–4, as discussed in detail below).
Figure 2.

Splicing to alternative donor sites in the CG33298 mini-gene is regulated by the stem structure formed by the conserved box sequences. (A) Top panel: Schematic representation of the CG33298 mini-gene, which contains the chromosomal region 2L:9519321–9519987. The box 1 sequence overlaps with the proximal donor splice site of exon 13. The location of the primers used for PCR amplification of the mini-gene mRNAs (in the vector) and the endogenous mRNAs (in the exons) are indicated. Bottom panel: Multiple sequence alignment for the intronic regions containing the boxes. 100% conserved positions are indicated by asterisks. No orthologous sequence was found for D. mojavensis. Complementary boxes (highlighted) are conserved in all but two positions; the conservation rate for the rest of the intron is less than 3%. (B) Secondary structure formed by the conserved boxes affects donor site usage. mRNA products from either the mini-gene (lanes 1–8) or from the endogenous gene (lanes 9–10) were analyzed by RT–PCR. Bands are labeled as D, distal donor; P, proximal donor; or IR, intron 13 retention. The addition (+) or absence (−) of the reverse transcriptase (RT) enzyme to the reaction is indicated. The results of three independent splicing assays are represented graphically in the bottom panel, as the ratio of the band intensity (P or D) to the total intensity (P + D). (C) Predicted base-pairing for the wild-type, box 1, box 2 and box 1/2 mutants (point mutations are shown in boldface), with the estimated equilibrium free energies (given in kcal/mol). The box 1 sequence is shown above the box 2 sequence.
Figure 3.

Alternative splicing of Atrophin (CG6964, also called Grunge and Gug) is regulated by a conserved secondary structure element. (A) Schematic representation of the Atrophin mini-gene, which encompasses exons 8–11 (chromosome 3L:8462561–8463475). A non-annotated proximal acceptor site was determined to be located in the box 2 sequence. The multiple sequence alignment of the intronic regions containing the boxes is shown in the bottom panel. The complementary boxes and the sequence downstream of box 2 are 100% conserved. Splicing to the proximal acceptor site is predicted to add 66 nucleotides to the exon, and the predicted amino acid insertion is shown below the sequence. The legend for (B) and (C) is the same as in Figure 1, except that P is the proximal acceptor site, and D, the distal acceptor site.
Figure 4.

A stem structure regulates alternative usage of acceptor splice sites in the Nmnat (CG13645) mini-gene. (A) Top panel: Representation of the Nmnat mini-gene (chromosome 3R:20771699–20772905). Exon 5 is an internal terminal exon that is cleaved when included (the poly-adenylation/3′-processing signals are indicated), while exon 6 is an internal exon. Box 2 is located upstream of the poly(A) signal in exon 5. Primers used in the PCR amplifications are indicated. Bottom panel: Multiple sequence alignment of the sequence between exons 4 and 6. The complementary boxes are almost 100% conserved, with only one change of GU to AU in the base pairs. (B) Splicing products from the mini-gene were amplified with a reverse primer to the vector to amplify the isoforms formed by splicing to the distal acceptor (D) or to the proximal acceptor (P) that had not been cleaved. The results of three independent splicing assays are represented graphically in the bottom panel for distal acceptor usage. Samples were normalized prior to loading against an independent PCR performed in parallel with a reverse primer to exon 4, to visualize the constitutively-spliced product of exon 3–exon 4 (data not shown). (C) As in (B), except that a reverse primer in exon 5 was used to amplify splice products to proximal acceptor (P) or with intron 4 retention (IR). Proximal acceptor usage is depicted graphically at the bottom. (D) Endogenous mRNA was amplified with reverse primers in exon 6 or exon 5. (E) Predicted base pairing for the wild-type, box 1, box 2 and box 1/2 mutants (point mutations are shown in boldface), and their estimated equilibrium free energies. Since the sequence was completely exchanged during mutagenesis, no base-pairing is predicted to occur for the single box mutations (box 1 and box 2).
The CG33 298 gene encodes an ATPase with phospholipid-translocating activity, and alternative donor usage during the splicing of its pre-mRNA is predicted to change the C-termini of the proteins (Figure 2A). Box 1 overlaps with a proximal donor site and is separated from box 2 by 185 nt. Both donor sites are predicted to be equally strong (P = 0.46). However, splicing of the endogenous pre-mRNA reveals almost exclusive splicing to the distal donor site, and this preference is also observed within the splicing of the pre-mRNA from the mini-gene (Figure 2B). Within the mini-gene construct, we introduced four point mutations to the sequence of either box 1 or box 2, to interfere with stem-structure formation but not with the donor site (of AGGU) in box 1 (Figure 2C). In both cases, the mutations led to a switch to almost exclusive use of the proximal donor site (Figure 2B). Importantly, when both boxes were mutated at the same time to re-establish the base-pairing potential, the distal donor was again the preferentially used donor site. We conclude that the boxes form a stem structure, the presence of which is necessary to modulate the donor site usage. Since the mutations in both single boxes had the same general effect on the splicing pattern, we conclude that it is the formation of the stem structure per se, rather than the conserved sequences, which is the major determinant of donor site usage.
Atrophin encodes a transcriptional co-repressor with histone deacetylase activity (27). Splicing analysis of the mini-gene products revealed the presence of an unannotated acceptor site within box 2, proximal to the annotated one (Figure 3A). RT–PCR analysis of the endogenous mRNA revealed that this proximal acceptor site is indeed used (Figure 3B), and correspondingly, the novel exonic region is completely conserved phylogenetically (Figure 3A). While the proximal acceptor is predicted to be stronger than the distal one (P = 0.09), both acceptor sites are used, with an approximate ratio of 1:1 for the mini-gene and endogenous mRNAs (Figure 3B). However, mutation of either box within the mini-gene construct resulted in splicing mainly to the proximal acceptor site located in box 2 (Figure 3). Re-establishing a stem with a novel sequence, and with a similar stability as the wild-type stem (Figure 3C), led to a switch in the splicing pattern to approximately that of the wild-type (Figure 3B). Thus, the stem structure suppresses the proximal acceptor site, thereby equalizing two splice sites of distinct strengths by incorporating the stronger site into a stem structure.
Within the nicotinamide mononucleotide adenylytransferase (Nmnat) pre-mRNA, the boxes surround the proximal of two alternative acceptor sites within the alternatively spliced intron 4 (Figure 4A). Splicing to the proximal acceptor site introduces an alternative terminal exon with a polyadenylation site, while splicing to the distal acceptor site introduces an internal exon, resulting in distinct C-termini of the Nmnat protein isoforms. Splicing of this intron was first analyzed for usage of the distal acceptor site (Figure 4B). Completely exchanging the sequence of either box 1 or box 2 to eliminate complementarity (Figure 4E) drastically reduced the level of splicing to the distal acceptor (Figure 4B). Re-establishing a stem structure with the novel sequence (box 1/2; Figure 4B) reversed this effect, demonstrating the role of the stem structure in modulating the distal acceptor site usage. In contrast, analysis of the use of the proximal acceptor site (by using a primer specific for exon 5) revealed that, although this site is used in the wild-type mini-gene, its usage increased with both the box 1 and the box 2 mutations (by about 1.5-fold) and again decreased with the novel stem formation (Figure 4C). Mechanistically, the actions of the stem structure could be explained in a dual manner. First, since the proximal acceptor site is the stronger of the two sites (P = 0.04), looping it out with the stem structure could make it less competitive. Second, the distal acceptor site is more than 400 nt downstream of the proximal one, making the intervening intron much longer than the average intron in Drosophila. Forming a stem by the two complementary sequences, which are separated by about 350 nt, could physically bring this distal site to the proximity of the donor site and thereby promote its usage.
DISCUSSION
The extent to which secondary structures influence splicing was analyzed in a genome-wide manner, using the strength of phylogenomic comparisons in Drosophila, for which 12 genomes are available (28). We uncovered a set of 202 intronic sequence pairs that could engage in thermodynamically stable stems, and that are highly conserved among fruit flies. Our search included base-pairing over relatively long RNA distances. Experimentally, we demonstrated for three cases that the predicted stem structures influence the outcome of alternative splicing. We propose that alternative splicing is often modulated by long-range RNA secondary structures, through a variety of mechanisms that promote specific splice site usage.
Alternative splicing modulation by stem structures
Taking advantage of phylogenetic comparisons, we identified a set of highly conserved complementary sequences that could form stem structures, which we predict could influence splicing. Testing several of these experimentally demonstrated that the stem structures indeed influenced splicing when they occurred in alternatively, but not constitutively, spliced introns. Consistently, there is an enrichment for alternatively spliced introns within our set. However, we cannot exclude the possibility that constitutive splicing is likewise affected by the presence of the stem structures, but that our over-expression system is technically not able to detect changes in these splicing events upon stem disruption. For example, a hairpin structure was found to influence the splicing of the Drosophila Adh pre-mRNA and alter the subsequent protein expression levels, although the changes observed in splicing in vivo when the hairpin was disrupted were only 6% (10). It is also possible that stem structures are important within constitutively spliced introns for sequestering and thereby silencing cryptic splice sites, thus allowing splicing to occur constitutively. Additionally, some of the introns in our set which are classified as constitutively spliced may actually contain undocumented alternative splicing events, such as those we observed for the Atrophin intron (which had an undocumented alternative acceptor site; Figure 3).
Why should secondary structures play such a frequent role in regulating alternative splicing? Modulation of alternative splicing by secondary structures provides a built-in mechanism for balancing the splicing output. Our results exemplify this principle. In the case of Atrophin alternative splicing, two alternative acceptor sites are used equally well only when a stem structure masks the stronger one of these sites (see Figure 3). The use of the stronger acceptor site adds 22 amino acids to the resulting protein, which could change its function. Thus, the balanced use of the two acceptor sites is ensured by the stem structure formation, without the prerequisite for additional trans-acting factors. In the second case, a stem structure also masks an alternative splice site in the CG33298 intron; however, the splicing outcome of this event differs from that of Atrophin, since the masked splice site is highly suppressed and only used to a small degree. When the stem is prevented from forming, there is an almost complete switch of splicing to the previously masked splice site (which is predicted to be the stronger one). Thus, several parameters determine how alternative splicing can be modulated by secondary structure formation, such as splice site strength, splice site competitiveness due to positioning, and regulation through the kinetics and thermodynamics of secondary structure formation. This complexity is evident for the Nmnat intron, in which the stem loop was required to approximate a distal splice site, and to reduce the competitiveness of a proximal splice site, in order to allow usage of both splice sites (see Figure 4). Thus, the formation of stem structures over long ranges of RNA greatly amplifies the potential for alternative splice site choices.
Modulation of alternative splicing by stem structures also opens the possibility for directed regulation. Regulation could come through the propensity of the secondary RNA structures themselves to change, in response to different cellular situations. For example, changes in transcription rate could change the kinetics of stem formation, leading to specific changes in the alternative splicing outcome. This would be somewhat similar to the bacterial attenuators which are regulated by ribosome pausing (29). Additional regulation could likewise come from the binding of trans-acting factors, such as proteins and small RNAs. Regulation through local RNA structures has been demonstrated for the yeast ribosomal L30 protein, which binds a structure in its own pre-mRNA that resembles its rRNA target. L30 binding prevents subsequent U2 snRNP association, thereby auto-regulating its own pre-mRNA splicing (30). Splicing regulation has also been demonstrated in plants and fungi to occur by riboswitches modulated by the binding of thiamine pyrophosphate (TPP) in some pre-mRNAs that encode proteins involved in TPP metabolism, thereby changing the alternative splicing outcome (31,32). The kinetics of stem structure formation could also be regulated by sequestering binding sites of single-stranded RNA-binding proteins (intronic splicing enhancers and silencers). Such regulation could then affect the ratio of splicing isoforms produced and would have a strong potential to fine-tune sensitive splicing events.
Conservation and frequency of secondary structures
The high degree of conservation of not only the RNA structure but also the complementary sequences (almost always 100%) in our dataset is remarkable. Indeed, although the search allowed for up to three mismatches in a 9 nt stretch, the boxes usually differed by at most one nucleotide between the species (Figures 2–4, and Supplementary Table S2). This is quite surprising since the Drosophila species analyzed here have been diverging for over 40 million years of evolution. One possible explanation for this extreme conservation is that sequence evolutionary rate is slower in base-paired regions because two simultaneous mutations are needed to maintain secondary structure. This effect has been reported previously in bacterial terminators and attenuators (33). However, it is also possible that the strong conservation we observe reflects further interactions with trans-acting factors for one or both of the sequences of each pair, in addition to a direct role of the stem structures on splicing. Indeed, sequence covariation over the evolution of ribosomal RNA structure was very strong and allowed these structures to be resolved through comparative modeling (34). Since our search would not have included stem structures that have been conserved structurally with covariation, we could predict that such a group would further expand our list of stem structures that could influence splicing.
By allowing the distance between the boxes to be determined by intron length, we have considered long-range as well as short-range interactions, despite the common belief that the long-range interactions are less likely to occur. One of the main arguments against considering long-range pre-RNA interactions is that they will not likely occur kinetically during transcription, which is believed to promote local RNA structure formation in the wake of the RNA polymerase (8). However, a study in yeast analyzing the ability of a sequence to base-pair with and disrupt the formation of a ribozyme revealed that the competitor sequence was more effective when it was transcribed before the ribozyme rather than after it in vitro, whereas there was no positional effect in vivo (e.g., the competitor sequence was equally effective at disrupting ribozyme formation when transcribed either before or after it) (35). This suggests that the formation of RNA secondary structure is more dependent on other factors, such as transiently binding proteins, which could allow a ‘delayed folding’ of the RNA (35,36). Since our set of stem structures are predicted to be thermodynamically highly stable, we propose that the kinetics of the stem formation is the main regulatory mechanism.
What is the probable frequency of secondary structures that influence splicing? Our search conditions were deliberately overly restrictive, to generate a smaller data set with a high potential for being relevant for splicing modulation. In fact, we did not find the few RNA structures known to influence alternative splicing in Drosophila, such as those involved in Dscam splicing (15,37), because these did not meet our search criteria (such as distance to splice sites, or phylogenetic conservation). Note that these structures can still be found by relaxing the search constraints, but with an unacceptable increase of the false discovery rate. Additionally, several of our restrictions do not reflect necessary conditions for secondary structures to influence splicing. For example, the extremely high phylogenetic conservation of secondary structures in our set is a strong indicator that these play an important role, for instance, in splicing regulation. However, structures that are not conserved could also be involved in splicing modulation and could be important for species-specific alternative splicing. Likewise, restricting the stem structures to introns allowed us to visualize the ‘islands’ of conservation of these sequences in the low-conservation intronic regions (as compared to the relatively high conservation of the exons). Nonetheless, stem structures that are partially or entirely present in exonic regions would also be able to efficiently modulate splicing.
Therefore, we propose that the modulation of alternative splicing by RNA stem structures in Drosophila is more common than it is currently believed. We predict that this type of modulation plays an important role in alternative splicing in other eukaryotic species as well.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Howard Hughes Medical Institute [grant number 55005610]; the Program ‘Molecular and Cellular Biology’ of the Russian Academy of Sciences; and Russian Foundation of Basic Research [grant number 09-04-92742]. V.A.R. is a Ramon y Cajal fellow and D.D.P. is an INTAS YS fellow.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Elisa Izaurralde, Britta Hartmann and Juan Valcárcel for invaluable discussions and support. Some of the computations were performed at Scientific Computing and Visualization facility at Boston University.
REFERENCES
- 1.Stolc V, Gauhar Z, Mason C, Halasz G, van Batenburg MF, Rifkin SA, Hua S, Herreman T, Tongprasit W, Barbano PE, et al. A gene expression map for the euchromatic genome of Drosophila melanogaster. Science. 2004;306:655–660. doi: 10.1126/science.1101312. [DOI] [PubMed] [Google Scholar]
- 2.Castle JC, Zhang C, Shah JK, Kulkarni AV, Kalsotra A, Cooper TA, Johnson JM. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat Genet. 2008;40:1416–1425. doi: 10.1038/ng.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Will CL, Lührmann L. Spliceosome structure and function. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA World. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2006. pp. 369–400. [Google Scholar]
- 4.Smith CW, Valcarcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem. Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- 5.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 6.Buratti E, Baralle FE. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol. Cell Biol. 2004;24:10505–10514. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schroeder R, Grossberger R, Pichler A, Waldsich C. RNA folding in vivo. Curr. Opin. Struct. Biol. 2002;12:296–300. doi: 10.1016/s0959-440x(02)00325-1. [DOI] [PubMed] [Google Scholar]
- 8.Eperon LP, Graham IR, Griffiths AD, Eperon IC. Effects of RNA secondary structure on alternative splicing of pre-mRNA: is folding limited to a region behind the transcribing RNA polymerase? Cell. 1988;54:393–401. doi: 10.1016/0092-8674(88)90202-4. [DOI] [PubMed] [Google Scholar]
- 9.Solnick D, Lee SI. Amount of RNA secondary structure required to induce an alternative splice. Mol. Cell Biol. 1987;7:3194–3198. doi: 10.1128/mcb.7.9.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Stephan W. Compensatory evolution of a precursor messenger RNA secondary structure in the Drosophila melanogaster Adh gene. Proc. Natl Acad. Sci. USA. 2003;100:11499–11504. doi: 10.1073/pnas.1932834100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 12.Donahue CP, Muratore C, Wu JY, Kosik KS, Wolfe MS. Stabilization of the tau exon 10 stem loop alters pre-mRNA splicing. J. Biol. Chem. 2006;281:23302–23306. doi: 10.1074/jbc.C600143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shepard PJ, Hertel KJ. Conserved RNA secondary structures promote alternative splicing. RNA. 2008;14:1463–1469. doi: 10.1261/rna.1069408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiller M, Zhang Z, Backofen R, Stamm S. Pre-mRNA secondary structures influence exon recognition. PLoS Genet. 2007;3:e204. doi: 10.1371/journal.pgen.0030204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graveley BR. Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell. 2005;123:65–73. doi: 10.1016/j.cell.2005.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miriami E, Margalit H, Sperling R. Conserved sequence elements associated with exon skipping. Nucleic Acids Res. 2003;31:1974–1983. doi: 10.1093/nar/gkg279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crosby MA, Goodman JL, Strelets VB, Zhang P, Gelbart WM. FlyBase: genomes by the dozen. Nucleic Acids Res. 2007;35:D486–D491. doi: 10.1093/nar/gkl827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mount SM, Burks C, Hertz G, Stormo GD, White O, Fields C. Splicing signals in Drosophila: intron size, information content, and consensus sequences. Nucleic Acids Res. 1992;20:4255–4262. doi: 10.1093/nar/20.16.4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 21.Beissbarth T, Speed TP. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20:1464–1465. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- 22.Hofacker IL, Stadler PF. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics. 2006;22:1172–1176. doi: 10.1093/bioinformatics/btl023. [DOI] [PubMed] [Google Scholar]
- 23.Sorek R, Shamir R, Ast G. How prevalent is functional alternative splicing in the human genome? Trends Genet. 2004;20:68–71. doi: 10.1016/j.tig.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 24.Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 25.Resch A, Xing Y, Alekseyenko A, Modrek B, Lee C. Evidence for a subpopulation of conserved alternative splicing events under selection pressure for protein reading frame preservation. Nucleic Acids Res. 2004;32:1261–1269. doi: 10.1093/nar/gkh284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D, Thanaraj TA, Soreq H. Function of alternative splicing. Gene. 2005;344:1–20. doi: 10.1016/j.gene.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Rajan H, Pitman JL, McKeown M, Tsai CC. Histone deacetylase-associating Atrophin proteins are nuclear receptor corepressors. Genes Dev. 2006;20:525–530. doi: 10.1101/gad.1393506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drosophila 12 Genome Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- 29.Yanofsky C. Attenuation in the control of expression of bacterial operons. Nature. 1981;289:751–758. doi: 10.1038/289751a0. [DOI] [PubMed] [Google Scholar]
- 30.Macias S, Bragulat M, Tardiff DF, Vilardell J. L30 binds the nascent RPL30 transcript to repress U2 snRNP recruitment. Mol. Cell. 2008;30:732–742. doi: 10.1016/j.molcel.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Bocobza S, Adato A, Mandel T, Shapira M, Nudler E, Aharoni A. Riboswitch-dependent gene regulation and its evolution in the plant kingdom. Genes Dev. 2007;21:2874–2879. doi: 10.1101/gad.443907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheah MT, Wachter A, Sudarsan N, Breaker RR. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature. 2007;447:497–500. doi: 10.1038/nature05769. [DOI] [PubMed] [Google Scholar]
- 33.Vitreschak AG, Rodionov DA, Mironov AA, Gelfand MS. Riboswitches: the oldest mechanism for the regulation of gene expression? Trends Genet. 2004;20:44–50. doi: 10.1016/j.tig.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 34.Gutell RR, Lee JC, Cannone JJ. The accuracy of ribosomal RNA comparative structure models. Curr. Opin. Struct. Biol. 2002;12:301–310. doi: 10.1016/s0959-440x(02)00339-1. [DOI] [PubMed] [Google Scholar]
- 35.Mahen EM, Harger JW, Calderon EM, Fedor MJ. Kinetics and thermodynamics make different contributions to RNA folding in vitro and in yeast. Mol. Cell. 2005;19:27–37. doi: 10.1016/j.molcel.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 36.Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 37.Kreahling JM, Graveley BR. The iStem, a long-range RNA secondary structure element required for efficient exon inclusion in the Drosophila Dscam pre-mRNA. Mol. Cell Biol. 2005;25:10251–10260. doi: 10.1128/MCB.25.23.10251-10260.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.