


Abstract
The transcription factor Hypoxia-inducible factor 1 (HIF-1) plays a central role in the transcriptional response to oxygen flux. To gain insight into the molecular pathways regulated by HIF-1, it is essential to identify the downstream-target genes. We report here a strategy to identify HIF-1-target genes based on an integrative genomic approach combining computational strategies and experimental validation. To identify HIF-1-target genes microarrays data sets were used to rank genes based on their differential response to hypoxia. The proximal promoters of these genes were then analyzed for the presence of conserved HIF-1-binding sites. Genes were scored and ranked based on their response to hypoxia and their HIF-binding site score. Using this strategy we recovered 41% of the previously confirmed HIF-1-target genes that responded to hypoxia in the microarrays and provide a catalogue of predicted HIF-1 targets. We present experimental validation for ANKRD37 as a novel HIF-1-target gene. Together these analyses demonstrate the potential to recover novel HIF-1-target genes and the discovery of mammalian-regulatory elements operative in the context of microarray data sets.
INTRODUCTION
Maintenance of oxygen homeostasis in mammalian cells is fundamental to the survival of the organism. One of the pivotal mediators of the cellular response to hypoxia is hypoxia-inducible factor (HIF), a transcription factor that contains a basic helix-loop-helix motif as well as PAS domain. There are three known members of the HIF family (HIF-1, HIF-2 and HIF-3) and all are α/β heterodimeric proteins. HIF-1 was the first factor to be cloned and is the best understood isoform (1). HIF-3 is a distant relative of HIF-1 and little is currently known about its function and involvement in oxygen homeostasis. HIF-2, however, is closely related to HIF-1 and both activate hypoxia-dependent gene transcription. In all three isoforms the α-subunit senses oxygen levels (2–5). In normoxia the α-subunit is ubiquitously expressed but is rapidly degraded through interaction with the VHL ubiquitin ligase complex (6). In hypoxic conditions, it is stabilized through multiple mechanisms that are transcriptional, post-transcriptional, as well as post-translational (6–9). The β-subunit is widely expressed and generally not regulated by hypoxia. Once stabilized HIF-1 and HIF-2 interact with co-activators such as CREBBP/EP300 and bind to a consensus sequence element (5′-RCGTG-3′) in the proximal promoters of hypoxia-regulated genes. Together these genes control cellular process including a switch from oxidative to glycolytic metabolism, inhibition of cellular proliferation, and stimulation of oxygen delivery through erythropoiesis and angiogenesis (10). Metabolic deregulation is the final outcome if the adaptive response is inadequate.
Hypoxia can arise in a variety of developmental, physiological and pathological states. Solid tumors, such as breast cancer, quickly outgrow their blood supply and manipulate the hypoxia pathways to promote their own survival. The role of HIF-1 in tumor onset, progression, invasion and metastasis has been demonstrated in several solid tumors (11,12). In addition, cancer specific-regulatory roles for HIF-2 have been demonstrated in an expanding list of cancers (13). For instance, in clear renal cell carcinoma the normal predominance of HIF1A expression is altered in favor of EPAS1 (HIF2α) expression and promotes prosurvival rather than proapoptotic factors (14,15). As a result HIF-target genes have emerged as potential targets for cancer specific therapy (16,17).
HIF-1 is an important metabolic regulator that allows rapid adaptation to oxygen availability. This effect is mediated through key regulators of bioenergetics and growth. For instance, PDK1 is directly induced by HIF-1 resulting in attenuation of mitochondrial function and oxygen consumption (18) and CDKN1A is induced leading to growth arrest at the G1 phase (19). Identifying such HIF-target genes is central to our understanding of the HIF network and the mechanism of cellular pathways modulation during hypoxia. In addition, identifying additional binding sites in HIF-regulated genes might contribute to the paradigm of inferring function from sequence.
Currently, studies attempting to identify novel HIF-target genes rely on microarray expression profiles to detect genes that respond to changes in HIF protein levels. Typically a gene of interest is selected and experimentally validated for HIF binding to its promoter. Kim et al. identified 25 transcripts that increase by at least 4-fold during hypoxia in a B cell line. PDK1 was selected for further validation due to its role in regulating the TCA cycle. Consequently they were able to show that HIF-1 binds to the PDK1 promoter (18). Shen et al. (20) used a more rationale microarray design where tissues from Caenorhabditis elegans were profiled in normoxia and in hypoxia in a wild-type, HIF-1 knockout and a VHL mutant strain. This study resulted in 63 HIF-1 dependent genes that respond to hypoxia but only 12 of those had a human homolog. Elvidge et al. (21) compared expression profiles of human breast cancer cells in normoxia, hypoxia and in normoxia treated with a 2-OG-dependent dioxygenase inhibitor, dimethyloxalylglycine (DMOG) which activates HIF-1. In addition, they compared expression profiles in hypoxia and in hypoxia treated with siRNAs to HIF1A and EPAS1 (HIF-2α). Manalo et al. (22) profiled arterial endothelial cells in normoxia and in hypoxia as well as in normoxia in the presence of a HIF-1α constitutively active form. Only 45 genes were identified in both studies as HIF-1 dependent. Although these approaches are more likely to filter out genes that are not dependent on HIF-1, it remains challenging to determine whether the affected transcripts were directly or indirectly modulated by HIF.
Several methods for in-silico identification of transcription factor-binding sites (TFBS) have been proposed (23–29). These methods fall into two categories. First, identification of TFBS based on predefined position weight matrices (PWM). Second, de novo prediction of sequence motifs over-represented in proximal promoters compared to control sequences [for a review, see (30,31)]. Examples for the former include STORM in CREAD (27), CORE_TF (28) and GeneAct (29) and for the latter DME (32) and the genomic identification of motifs described by Xie et al. (26). PWMs of HIF-1 have been previously used to identify genome-wide target genes. For instance, the Gene Set Enrichment Analysis program contains a set of HIF-1-target genes as part of its Molecular Signature Database (26,33). However, these approaches do not take into consideration promoter availability. In addition, the consensus HIF-binding sequence is very short, has low information content and is ubiquitously present in the human genome making in-silico genome-wide prediction unreliable. To generate a reliable prediction we propose to reduce the search space to a subset of genes expressed in specific tissues under specific conditions. This can be achieved by microarray expression profiles under the assumption that promoters of genes that respond to a stimulus are available for TF binding. Several groups previously demonstrated that microarray expression profiles can be used to improve TFBS prediction (32,34–36) and that cis-regulatory modules in proximal promoters correlate with differential expression of downstream-target genes (27).
In this study we present an integrative genomic approach combining both computational and experimental strategies to identify HIF-target genes. Using this approach we recovered 41% of the known HIF-1-target genes that were differentially expressed in hypoxia, defined a ranked list of HIF-target genes and experimentally validated ANKRD37 as a novel HIF-1 target.
MATERIALS AND METHODS
Microarrays
Raw microarray data was obtained from GEO (37) and ArrayExpress (38). Microarray studies were filtered to identify those that profiled the same tissue in normoxia and hypoxia on Affymetrix platform and used at least two replicates and provided raw data. Affymetrix-based data sets were normalized using the GCRMA module (39) of bioconductor (40) and present–absent calls were calculated for each probe using the MAS5 module. All probes with no present calls were removed and from the remaining at least one sample was required to have an expression value larger than log2 (100), as recommended by Affymetrix. In all experiments, only pairwise comparisons were made with respect to normoxia. After the initial filtering was applied, the expression values were centered to the mean for each probe with replicates. Only probes for which the expression values of the replicates were all positive or all negative were further tested for differential expression. The Limma module (41) of biocoductor was used to fit a linear model for each probe and P-values were corrected using the false discovery rate method (42).
Promoters
Promoter sequences of human genome build 18 were obtained from UCSC genome browser (43). For each protein-coding gene, the proximal promoter was defined as 700 bases upstream and 300 bases downstream of the transcription start site. In cases where multiple transcripts were annotated all alternative promoters were obtained and analyzed individually. Due to restriction of conservation analysis programs, for all genes the forward strand sequence of the genome was used. Background promoters for enrichment analysis were selected randomly from the promoter collection to match the CpG content and base composition of the foreground promoters. We obtained CpG sequence location from UCSC genome browser (43) and matched their location to the promoters defined. Each promoter was classified as CpG or non-CpG and for the background set the same proportion of CpG and non CpG promoters were selected randomly according to the ratio in the foreground set. Next we verified that the base composition matched within ±2% for each nucleotide with the foreground data set. For all enrichment purposes a set of 1000 promoters was used as a background.
Enrichment of binding sites
The program MOTIFCLASS v1.40 (27) was employed to identify binding sites over-represented in promoters of a foreground set (genes differentially expressed in microarrays) compared to a background set of 1000 randomly selected promoters. The relative error-rate was optimized for each motif and a P-value was calculated using 10 000 permutations. Finally, only motifs with an enrichment specificity of at least 0.5 were reported.
De-novo prediction of motifs
The program DME (v2.0) (27,32) was employed using the ZOOPS model to identify motifs of length 5–12 bases compared to a background set. Only the top 50 enriched motifs were reported and compared to known motifs using the program MATCOMPARE in CREAD (27).
Binding site prediction
The program STORM in CREAD (27,44) was used to match PWMs to genomic DNA and was employed using a P-value cutoff of 1e-4. This cutoff was found to be optimal since a cutoff of P < 1e–3 was not stringent enough predicting nearly every promoter in the genome as having a HIF-1 site while P < 1e–5 was too stringent predicting only a handful of promoters in the genome with a HIF-1 site. Conservation information of 28 species genome alignments (43) was used in two approaches. First, the software multiSTORM [also in CREAD (27)] was employed to identify the number of genomes in which the PWM could still be matched with the same P-value cutoff as used in STORM. Second, the software site-cons was employed to calculate the level of conservation at each binding site compared to the flanking 100 bases (27). The site-cons P-value was obtained by extracting 100 bases upstream and downstream of the binding site and sampling randomly a number of columns equal to the length of the predicted binding site. A conservation score was then calculated for these columns and the process was iterated 10 000 times to obtain a distribution. Finally, the P-value was calculated by comparing the original score for the binding site to the distribution. Conservation information was obtained from the 28-genome alignments available at UCSC genome browser (43). Namely, human (hg18), chimpanzee (panTro2), rhesus (rheMac2), bushbaby (otoGar1), tree shrew (tupBel1), mouse (mm8), rat (rn4), guinea pig (cavPor2), rabbit (oryCun1), shrew (sorAra1), hedgehog (eriEur1), dog (canFam2), cat (felCat3), horse (equCab1), cow (bosTau3), armadillo (dasNov1), elephant (loxAfr1), tenrec (echTel1), opossum (monDom4), platypus (ornAna1), lizard (anoCar1), chicken (galGal3), frog (xenTro2), fugu (fr2), tetraodon (tetNig1), stickleback (gasAcu1), medaka (oryLat1) and zebrafish (danRer4).
Transcription network
For each predicted HIF-target gene, the single best scoring HIF site was selected and other binding sites were identified within ±25, ±50, ±75, ±100, ±125 and ±150 bases of that site as well as in the entire promoter. To obtain a P-value the frequency of each site within that window was compared to a randomly selected background set of 1000 promoters from genes that respond to hypoxia but had no detectable HIF-binding site. Sequence composition for the foreground and background set were matched as described above. In each of the background promoters a single binding site was randomly selected and binding sites were identified within the window size as selected for the HIF site. A chi-square test was performed for each TF, comparing the occurrence of binding sites in the neighborhood of HIF to their occurrence in the neighborhood of the randomly selected binding site in the background set. This process was iterated 100 times and the average P-value for each TFBS was used as the final P-value. No correction for P-values was performed.
Functional pathway analysis
The KEGG database (release 47) (45) was obtained and used to place predicted HIF-target genes into molecular pathways. Statistical analysis was performed using a hypergeometric distribution as described in the GOhyperGAll module of Bioconductor for gene ontology terms (40). The R program was employed (v2.6.2) with the following command: phyper(x − 1, m, n–m, k and lower.tail = FALSE), where x is the number of predicted target gene within a specific pathway; m, the number of genes in that pathway; n total number of unique genes in KEGG; and k, the number of predicted HIF-target genes that have an entry in any of the KEGG pathways. P-values were not corrected for multiple testing. This statistical analysis was repeated using the Reactome pathway database (46), the curated gene sets of Gene Set Enrichment Molecular Signature database (33) and the microRNA predicted targets database TargetScan (47). Enrichment for protein interactions was performed by first compiling a list of interactors for each human protein (Data obtained from NCBI Gene database). The HomoloGene database (48) was used to identify interaction in other species that can be mapped to human proteins. Finally, enrichment was calculated as described for KEGG by considering all interactors of each protein. Protein domains were obtained from Interpro (49) and enrichment was calculated for each domain.
Selection of mitochondrial genes
Genes that localize to the mitochondria were selected by identifying (i) all human Swiss-Prot proteins annotated as localized to the mitochondria (50); (ii) genes annotated with Gene Ontology terms that include the keywords: mitochondria, mitochondrion or mitochondrial (51); and (iii) A predicted catalog of genes localized to the mitochondria (52). Genes from the predicted list of mitochondrial genes that conflicted with existing knowledge and annotation were removed.
Cell culture
DLD-1, HCT116, SW480, Lovo, Panc-1, HeLa and MCF7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Hypoxic conditions were achieved by culturing cell lines in a sealed hypoxia chamber (Billups-Rothenberg) for 18 h after flushing with a mixture of 1% O2, 5% CO2 and 94% N2 (Compressed pre-mixed gas, Airgas) (53). To minimize the effect of serum growth factors, the culture medium was switched to serum-free UltraCulture (Lonza) before the cells were subjected to hypoxia.
Quantitative polymerase chain reaction analysis (qPCR)
Total RNA was isolated from cultured cells using the RNeasy kit (Qiagen) and reverse transcription with random hexanucleotide primers and SuperScript Reverse Transcriptase III (Invitrogen) was performed. The resulting cDNA was amplified by real-time polymerase chain reaction using the iQ5 Real-Time PCR Detection System (BioRad) and Power SYBR Green PCR Master Mix (Applied Biosystems). Primer sequences were 5′-GTCGCCTGTCCACTTAGCC-3′ and 5′-GCTGTTTGCCCGTTCTTATTACA-3′ for ANKRD37, 5′-AGGCCAGCACATAGGAGAGA-3′ and 5′-TTTCTTGCGCTTTCGTTTTT-3′ for VEGFA and 5′-CGGCTACCACATCCAAGGAA-3′ and 5′-GCTGGAATTACCGCGGCT-3′ for 18S rRNA. Transcript levels of ANKRD37 were normalized to 18S rRNA.
Western blotting
Cells were lysed in chilled lysis buffer (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM leupeptin) supplemented with the Complete protease inhibitor cocktail (Roche). Total 50 µg of protein extracts were resolved on a 3–8% NuPAGE Tris–acetate SDS polyacrylamide gel (Invitrogen) and transferred onto a polyvinylidene difluoride membrane (Millipore). The blots were probed with anti-HIF1A (Transduction Laboratories; 1:500) and anti-β actin (Sigma; 1:100 000) antibodies. Immunoreactive proteins were visualized using the Western Lighting Chemiluminescence Reagent Plus (PerkinElmer Life Sciences).
Plasmid constructions
Fragments from −325 bp to +230 bp (promoter 1) −166 bp to +230 bp (promoter 2) or −78 bp to +230 bp (promoter 3) relative to the transcriptional start site of the human ANKRD37 promoter were amplified by polymerase chain reaction (PCR) using 1 µg of human genomic DNA extracted from DLD-1 cells. The PCR primers were as follows: −325 bp sense primer; 5′-AACCAAACGCGTCCAGTTTCCTGGTTACGTG-3′, −166 bp sense primer; 5′-AACCAAACGCGTTCTCACGCCCACTGACTTA-3′, −78 bp sense primer; 5′-AACCAAACGCGTATCCACAGCTGGCCAATCG-3′ and +230 bp antisense primer; 5′-GTGGTTGAGATCTTAAGGCAGATGACAAGAGTCC-3′. The PCR-amplified products were digested with MluI and BglII and subcloned into the pGL3-basic vector (Promega). Site directed mutations were introduced at three potential HIF-binding sites (1, 2 and 4). To prepare site 1 and site 4 mutants, the −325 ANKRD37-luc pGL3 plasmid was digested with MluI/PstI or PmlI/Tth111I, respectively, and ligated with a double-stranded oligonucleotide in which the consensus HIF-binding site was disrupted. The oligonucleotide sequences were as follows: 5′-CGCGTCCAGTTTCCTGGTTAAAAGCGCGCCGGCAGAGCCAAAACCTGCA-3′ and 5′-GGTTTTGGCTCTGCCGGCGCGCTTTTAACCAGGAAACTGGA-3′ for site 1 at −310 bp and 5′-GTGCCAGTGTTTGTGTAAAAGCGGCCGTGGCGGGGCTGGACA-3′ and 5′-CTGTCCAGCCCCGCCACGGCCGCTTTTACACAAACACTGGCAC-3′ for site 4 at +14 bp. To prepare the site 2 mutant, PCR-based site-directed mutagenesis was performed using primers 5′-TACCGGGAGAAAAGTCAAACTCA-3′ and 5′-TGAGTTTGACTTTTCTCCCGGTA-3′. The nucleotide sequences of all constructs were confirmed by sequencing.
Transfections and reporter assays
Transient transfections were performed using Fugene 6 (Roche) in 24-well tissue culture plates according to the manufacturer's specifications. Total 150 ng of ANKRD37-luciferase pGL3 reporter constructs were co-transfected with 2 ng of pRL-CMV (Promega) as a control. Ten hours after transfection, the culture medium was switched to UltraCulture and cells were grown in normoxic or hypoxic conditions for 18 h. Luciferase activity was measured with a dual luciferase reporter assay system (Promega). The experiments were performed in triplicate wells a minimum of three times.
RNA interference
HCT116 cells were transfected with 20 nM siRNA duplex oligos directed to HIF1A or a non-specific control siRNA using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Nucleotide sequences of siRNAs were as follows: HIF1A, 5′-r(AUCCAGAGUCACUGGAACU)d(TT)-3′, and control, 5′-r(GCGCGCUUUGUAGGAUUCG)d(TT)-3′. The silencing effect was confirmed by Western blotting 48 h after transfection. ANKRD37 promoter reporter constructs were transfected 12 h after siRNA transfection.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from MCF7 cells cultured in either normoxic or hypoxic conditions for 12 h utilizing NE-PER nuclear extraction reagent (Pierce). The ANKRD37 promoter sequence utilized as an oligonucleotide probe was 5′-CTACCGGGAGACGTGTCAAACTCAG-3′. The 5′-end of the oligonucleotide was labeled with biotin, and a complementary oligonucleotide was annealed to generate double-stranded fragments. EMSA was performed using LightShift chemiluminescent kit (Pierce) as previously described (54). Specificity of shifts was confirmed by utilizing 200-fold molar excess of unbiotinylated oligonucleotides as a specific competitor. Mutagenesis was performed to further define the elements responsible for the specific shifts obtained. The sequence of the mutant oligonucleotide probe that includes point mutations in the HIF consensus-binding site was: 5′-CTACCGGGAGAAAAGTCAAACTCAG-3′.
Chromatin immunoprecipitation (ChIP)
MCF7 cells were cultured in either normoxic or hypoxic conditions for 6 h and then fixed with 1% formaldehyde at 37°C for 10 min. The ChIP assays were performed using a Chromatin Immunoprecipitation Assay Kit (Upstate) according to the manufacturer's protocol. Briefly, cells were lysed and sonicated to achieve DNA sizes ranging from 200 to 1000 bp. Diluted supernatants were pre-cleared by incubating with salmon sperm DNA/protein-A agarose, 50% slurry at 4°C for 30 min. Supernatants were then incubated with anti-HIF-1α antibody (Novus Biologicals number 100–105) or normal mouse IgG (Santa Cruz) at 4°C overnight, or directly processed to the next step without antibody. Immunocomplexes were collected with salmon sperm DNA/protein A-agarose, 50% slurry, washed, eluted and cross-linkage was reversed by heating at 65°C for 4 h. The eluates were then digested with proteinase K at 45°C for 1 h. The precipitated DNAs were recovered using PCR Purification Kit (Qiagen) and amplified by PCR using PrimeSTAR HS DNA Polymerase (Takara Bio). The PCR primers used for amplification of site 2 (primers A) and a non-HIF region upstream of site 2 (primers B) were as follows: sense primer A; 5′-CCAGTTTCCTGGTTACGTGC-3′, anti-sense primer A; 5′-TAAGTCAGTGGGCGTGAGAG-3′, sense primer B; 5′-TGGAGGCACTTTCACCACAC-3′ and anti-sense primer B; 5′-TGTGAGGTGAGCACACGTTG-3′.
RESULTS
HIF-1 and HIF-2 and their downstream-target genes are activated in response to hypoxia. To identify HIF-target genes we first identified genes that responded to hypoxia using publicly available microarray data sets. NCBI GEO and EBI ArrayExpress were searched for experiments where the same tissue was profiled comparing normoxia and hypoxia. Experiments that used replicates, were profiled on Affymetrix platform and provided raw data were accepted for further analysis (Supplementary Table S1). The same identical analysis protocol was applied to all experiments (see ‘Materials and Methods’ section). Although all microarrays were profiled on Affymetrix arrays, the number of genes differentially expressed in each cell type varied significantly and ranged from 486 genes in monocytes to 2119 genes in HeLa, with a surprisingly small overlap. The differentially expressed genes in each cell type are the potential HIF-target genes and were subject to in-silico analysis of HIF-binding sites.
In-silico optimization of HIF-binding site prediction
Two databases of PWMs were obtained and searched for HIF-1 PWM. The Jaspar collection (v3.0) contained no HIF-1 PWM while the TRANSFAC collection (v8.2) contained 2 HIF-1 PWMs (M00466, M00797). These PWMs are 12 and 14 bases long and were compiled based on only 12 and 23 validated binding sites, respectively. We compiled an additional HIF-1 PWM based on 104 validated binding sites in human, mouse and rat collected by Wenger et al. (10) (see Supplementary Figure S1; submitted to Jaspar). Despite the apparent variability in PWM length, it is clear from the alignment in Supplementary Figure S1 that most of the information content is within the HIF consensus binding site: RCGTG. The composition preference in the region flanking the core RCGTG observed in the Transfac matrices is a consequence of using a small number of validated HIF-1-binding sites.
Proximal promoters were defined from 700 bases upstream to 300 bases downstream of the transcription start site (TSS) of each transcript. Initially, HIF-1 PWMs were used to identify binding sites; however, we observed a too small overlap between the three HIF-1 PWMs for any given P-value and conservation filtering criteria. Differences in PWM length (12–18 bp) and in the sequences flanking the consensus binding site were found to introduce a bias in HIF-1-binding-site prediction since calculation of P-values and conservation consider the entire length of the matrix and all bases defined within. To avoid this bias all HIF consensus-binding sites sequences (RCGTG) were extracted from each promoter and tested for conservation in other species (see ‘Materials and Methods’ section). A shift from A to G and vice versa in the first position was allowed for the purpose of conservation. Next, the software site-cons in CREAD was employed to calculate the level of conservation at each binding site compared to the flanking 100 bases. A site was predicted as a HIF-binding site if it was conserved in at least 10 species (including human) or if conserved in four species with a site-cons P < 0.05. When the HIF consensus site is preceded by a C, it may form the E-box-binding site CACGTG which is a known binding site for several bHLH transcription factors including HIF-1. In 43% of the E-box sites other TFs were predicted to bind at the same location as HIF-1, compared to only 2% of the non-Ebox sites. The transcription factors that are most commonly predicted at the same location as HIF are ARNT (HIF-1β; as homodimer), AHR:ARNT, MYC, MAX, MYCN, CLOCK:ARNTL and USF1 (Supplementary Figure S2). We only accepted genes with at least one non-Ebox HIF site as HIF-target genes. Throughout this strategy our filtering criteria were very stringent in order to obtain a catalogue of high confidence at the expense of not recovering some known HIF targets. Each gene was scored as follows:
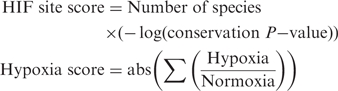 |
The hypoxia/normoxia ratio was inverted and assigned a negative value for genes that were downregulated in hypoxia. Each gene was assigned two ranks, one based on the highest scoring HIF site and one on the hypoxia score. Finally, the sum of both ranks was calculated and all genes were ranked based on that value. For instance, the gene BNIP3, a bona fide HIF-1 target, was ranked 515 by HIF site score and 15 by hypoxia score. Using the sum of ranks BNIP3 was ranked 56 among all genes. This scoring scheme is biased towards genes that respond to hypoxia in multiple cells. While some genes could be HIF targets in only one cell, we reason that a gene that responds to hypoxia in more than one cell is more likely to be a HIF target. This strategy is summarized in Figure 1. Despite the stringent selection criteria of this approach we were able to identify a HIF-binding site in 35/86 promoters of known target genes. A list of previously identified known HIF-1-target genes is shown in Supplementary Table S2.
Figure 1.

Prediction strategy for identifying HIF-1-target genes. Candidate genes that respond to hypoxia were first identified by microarrays. Each data set was subjected to a computational analysis in which HIF-binding sites were detected in proximal promoters. Each gene was scored for its response to hypoxia and for the best HIF-binding site. No cutoff was set for determining HIF-target genes. Finally, all genes were ranked.
The majority of hypoxia responsive genes lack a HIF-binding site
HIF-target genes were predicted for each data set separately with the criteria outlined in ‘Materials and Methods’ section. Of the known and validated HIF-target genes that responded to hypoxia in each data set, we recovered 37–55% (Table 1). On average 70% of the hypoxia responsive genes did not contain a detectable HIF-binding site in their proximal promoter (Table 1). To verify this observation and to test that no bias was introduced by our stringent conservation filtering above, genes that responded to hypoxia in each data set were divided to two groups. Those with a predicted HIF-binding site (A) and those with no detectable HIF-binding site (B). Each group was tested for over-representation of any PWM compared to a random set of promoters with similar base composition and CpG content. This analysis was performed using the program MOTIFCLASS in CREAD and is independent from the prediction analysis and selection criteria outlined above. In addition, MOTIFCLASS does not consider conservation and is based on sequence analysis alone. As expected HIF PWMs were identified in the top 20 over-represented binding sites in group A and none of the HIF PWMs were detected in the top 20 PWMs from group B. The results for the top 20 enriched motifs in each group in MCF7 breast cancer cells are shown in Figure 2A and B. The highest scoring PWM was a HIF-1 matrix that had a classification error of 0.407. In each of the six data sets we observed HIF-1 enrichment in group A while group B varied significantly between cell types (Supplementary Table S3). We did not observe any significant HIF enrichment within the top 20 PWMs in group B throughout all data sets.
Table 1.
Transcriptional response to hypoxia across six different cell types as determined by microarrays
| MCF7 | U251 | Astrocytes | Monocytes | B cells | HeLA | |
|---|---|---|---|---|---|---|
| Genes that respond to hypoxia | 830 | 1702 | 1371 | 486 | 1920 | 2119 |
| Genes that respond to hypoxia in which a HIF binding site was identified | 278 (33%) | 546 (32%) | 380 (28%) | 159 (33%) | 534 (28%) | 555 (26%) |
| Known target genes that respond to hypoxia | 35 | 31 | 20 | 22 | 32 | 27 |
| Known target genes recovery | 19 (54%) | 17 (55%) | 8 (40%) | 11 (50%) | 13 (41%) | 10 (37%) |
Figure 2.

(A and B) Binding site enrichment analysis. The program MOTIFCLASS was employed to identify binding sites significantly over-represented in the promoters of genes that respond to hypoxia in B cells. (A) Totally 770 promoters of genes that responded to hypoxia in which a detectable HIF-1-binding site was identified. HIF-1 PWMs were identified as the most enriched. (B) Totally 1456 promoters of genes that responded to hypoxia but do not contain a detectable HIF-1-binding site. No HIF-1 PWM was identified in the top 20 matrices. Sn, sensitivity; Sp, specificity; Error, classification error rate; Pval, P-value. (C) Correlation between the number of HIF-target genes and the number of cells in which they responded to hypoxia. Only 31% of the genes that responded to hypoxia in one of six cells was predicted as a HIF-target gene, compared to 71% of the genes that responded to hypoxia in six of six cell lines.
The general response to hypoxia is dependent on a small number of genes
Previous studies have suggested that the response to hypoxia varies by cell type (55–57). However, we expected a core set of genes to be shared, e.g. genes that control the shift from oxidative to glycolytic metabolism and inhibition of cellular proliferation. Yet, the cellular response to hypoxia was surprisingly selective between cells. Only 17 genes responded to hypoxia in all studied cell types. These genes include HIF targets such as ENO1, BHLHB2 and BNIP3. We observed a positive correlation between the number of cells in which a gene responded to hypoxia and the identification of HIF-binding sites in its promoter (Figure 2C). Only 23% of the genes that responded to hypoxia in a single cell type had a predicted HIF-binding site in their promoter compared to 59% of the genes that responded in six cell types. The top scoring gene was TMEM45A which responded to hypoxia in all studied cell types with an average of 21.6-fold induction and a HIF-binding site conserved in 13 species with a conservation P-value <0.0001 compared to the flanking 100 bp. At least 5 of the top 10 genes have been previously identified as HIF-1-target genes, namely DDIT4 (58,59), STC2 (60), P4HA1 (61), JMJD1A (62) and ALDOC (63). We did not find any correlation between the response to hypoxia and the HIF-binding site score for the top 100 predicted HIF-target genes. To obtain a cutoff of high confidence, the top 500 ranked genes were plotted versus the cumulative number of previously validated HIF-1 targets (Supplementary Figure S3). The resulting curve showed a logarithmic increase in validated targets for genes ranking below 200 and approached saturation between 200 and 500. Therefore, the 200th gene rank was used as a cutoff of high confidence. Supplementary Table S4 summarizes our prediction results for the top 500 HIF-target genes.
In the top 200 predicted HIF-target genes, 81 responded to hypoxia in at least three cell types (Figure 3). We hypothesize that these genes are likely to be part of the core response to hypoxia. In addition, these genes have a higher probability of being HIF-1 targets since their response to hypoxia was detected in multiple experiments. Of the 81 genes, 22 genes were previously reported to be HIF-target genes and 60 genes (74%) were upregulated in hypoxia. In addition, 80 (99%) of the genes contained a CpG island in their proximal promoter, significantly higher than the 51% CpG containing promoters in our entire promoter database. CpG islands are thought to be non-tissue specific (64), further supporting the selection of the 81 genes as a shared response to hypoxia. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to identify over-represented pathways and processes within the 81 genes. In Figure 4A a metabolic map of HIF-1-target genes was constructed from the resulting analysis (Supplementary Table S5). The metabolic map contained several known HIF-1-target genes, such as LDHA, ALDOC and GAPDH and also several novel targets such as GYS1 in starch and sucrose metabolism (65) and fumarate hydratase (FH) in the TCA cycle (66). One of the key cellular responses to HIF-1 in hypoxia is a metabolic shift from oxidative phosphorylation to glycolytic production of ATP. To highlight direct targets that may be involved in HIF-1 regulation of mitochondria function the predicted HIF targets that localize to the mitochondria (see ‘Materials and Methods’ section) were used to create a mitochondrial map (Figure 4B). Eight of eleven genes were downregulated in hypoxia and are involved in critical steps of mitochondrial function including two ribosomal subunits, cytochrome c, the iron transporter SLC25A28 and the iron–sulfur assembly and repair protein ISCU.
Figure 3.
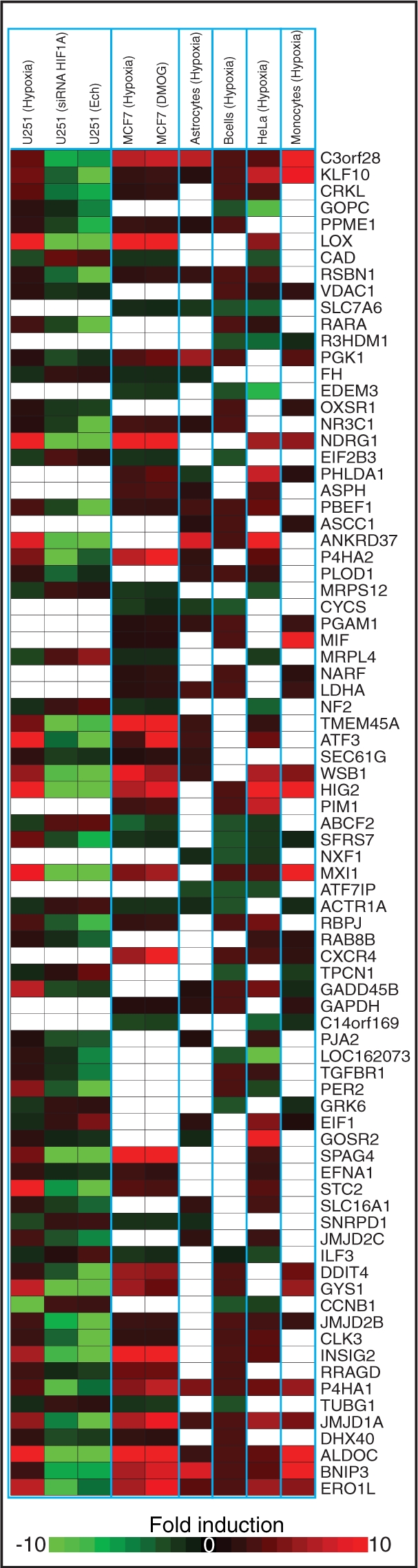
HIF dependent response to hypoxia across six cell types for 81 selected genes that respond to hypoxia in at least three cell types and were ranked within the top 200 HIF-target genes. The heatmap shows a fold induction compared to normoxia within each cell type. White blocks indicate the gene did not respond to hypoxia in that cell type.
Figure 4.

(A) Metabolic map of HIF-target genes. The KEGG pathway database was employed to map 81 HIF-target genes defined as the core response to hypoxia to pathways. Metabolism related processes are shown here (black objects) and those over-represented with a P < 0.05 are shown with a dotted white frame. Upregulated genes are shown as red nodes while downregulated genes as green nodes. Previously identified HIF-1-target genes are indicated by a diamond shape. (B) Mitochondrial map. Genes within the top 200 predicted HIF targets that localize to the mitochondria were mapped according to their functional annotation. Upregulated genes are shown as red nodes and downregulated as green nodes. Genes indicated in bold responded to hypoxia in at least three cell types. Dotted lines from iron–sulfur protein assembly and repair illustrate a few of the iron–sulfur proteins that are key to mitochondrial function, such as NADH dehydrogenase. (C) Graphical representation of protein-protein interaction network and Reactome pathways (D) enrichment for previously validated HIF-1 targets (red) and novel predicted targets (yellow). Only proteins/pathways that were significantly enriched (P < 0.05) are shown. See Supplementary Table S6 for analysis details.
Classification of novel HIF targets
All previously validated HIF-1 targets (Supplementary Tables S2 and S4) were tested for functional enrichment in KEGG pathway databases, Reactome database, Gene Set Enrichment Molecular Database (GSEA), protein interactions, InterPro protein domains and microRNA-target prediction database TargetScan (see ‘Materials and Methods’ section). This analysis allows identification of pathways or attributes that are highly represented by the HIF targets compared to an equivalent random list of genes. We next mapped novel HIF targets to these significantly enriched attributes as demonstrated by selected examples in Table 2 (full analysis shown in Supplementary Table S6). Analysis of protein domains identified 2-oxoglutarate and Fe(II)-dependent oxygenase family as highly enriched in validated HIF targets (5 of 19 proteins, P < 2e–7). This family includes the prolyl hydroxylases known to regulate HIF-1α as part of the oxygen sensing mechanism. PLOD1, is a novel predicted HIF target that is part of the same family. Analysis of microRNA targets identified five microRNAs that target validated HIF targets. miR-1/206, for instance, is predicted to target 583 human genes, of which nine are known to be HIF-1 targets and 14 are predicted HIF targets. Analysis of gene sets from GSEA highlighted a group of 104 genes that predict a poor breast cancer prognosis and included nine validated HIF targets (P = 1e–7) and four predicted ones. A graphical representation of protein interactions and Reactome pathways is shown in Figure 4C and D and highlights genes such as PCAF. PCAF interacts with four validated HIF targets and six predicted targets.
Table 2.
Selected examples of functional enrichment of 101 previously validated HIF-1 targets and mapping of novel targets to these categories
| Source | Description | Number of genes | Previously validated HIF-1 targets |
Novel HIF-1 targets | |
|---|---|---|---|---|---|
| P-value | Gene symbols | ||||
| Protein domains | IPR005123; 2-oxoglutarate (2OG) and Fe(II)-dependent oxogenase | 6/19 | 2.E-07 | P4HA2, EGLN3, EGLN1, P4HA1, PH-4 | PLOD1 |
| GSEA | Poor progonosis marker genes in Breast Cancer | 13/104 | 1.E-07 | CA9, CP, PGK1, EGLN1, TFRC, VEGFA, NDRG1, ADM, BNIP3 | TMEFF1, IVNS1ABP, TMEM45A, RRAGD |
| KEGG | Arginine and proline metabolism | 6/35 | 4.E-05 | P4HA2, NOS3, P4HA1, NOS2A | EPRS, ALDH4A1 |
| Reactome | Platelet activation | 7/86 | 1.E-02 | PDGFA, TGFB3, FN1, VEGFA, ALDOA, SERPINE1 | ARHGEF1 |
| TargetScan | MiR-1/206 | 23/583 | 3.E-03 | PDGFA, NAMPT, HSP90B1, MET, ETS1, VEGFA, EDN1, CITED2, STC2 | RNF165, GRK6, SLC7A6, PTPLAD1, ASPH, MYLK, RSBN1, SLC31A1, EVI1, PGAM1, SOX6, BTAF1, MXD1, GLCCI1 |
| Protein interactions | PCAF | 10/106 | 9.E-05 | ENO1, NR4A1, PFKL, PGK1 | RARA, CCNB1, SSRP1, RBPJ, EVI1, BTAF1 |
The number of genes reflects the total number of known and predicted targets within the functional group. P-values for enrichment of validated HIF-1 targets were obtained using the hypergeometric distribution as described in ‘Materials and Methods’ section.
ANKRD37 is a novel HIF-target gene
To demonstrate the utility of our method in identifying novel HIF-1-target genes, we selected five genes that were previously unknown to be HIF-1 targets, ANKRD37 (ranked 8), KLF10 (ranked 24) WSB1 (ranked 32), GYS1 (ranked 34) and ELL2 (ranked 465). Using quantitative PCR, ANKRD37 was induced up to 35-fold in hypoxia compared to normoxia, KLF10 up to 2.5-fold, WSB1 up to 4.9-fold, GYS1 up to 7.8-fold, ELL2 was not induced in hypoxia and VEGFA that was used as a positive control was induced up to 8.3-fold (Figure 5 and Supplementary Figure S4A). The gene ANKRD37 was studied further due to its extreme induction in hypoxia and its high ranking within the predicted targets. ANKRD37 is a short (158 amino acids, 17 kDa) ankyrin repeat protein with unknown function that is conserved in mammals and zebrafish. ANKRD37 responded to hypoxia in four of six cell types and four HIF-1-binding sites were predicted within the proximal promoter. The response to hypoxia was validated under hypoxic conditions across seven cell lines. We observed a strong induction of ANKRD37 in all cell lines but most significantly in the colon cancer cell lines DLD-1, HCT116 and SW480 (Figure 5A). ANKRD37 hypoxic induction was measured after 4, 8 and 12 h of incubation in hypoxic conditions in HCT116 cells and compared to VEGFA. HIF-1 protein levels were determined by western blot and showed a significant induction after 4 and 8 h compared to normoxia and a slight decrease after 12 h (Figure 5B). ANKRD37 and VEGFA mRNA levels showed a similar profile that matched the HIF-1 induction; however, ANKRD37 was induced 3-fold over the level of VEGFA induction (Figure 5C).
Figure 5.

ANKRD37 response to hypoxia. (A) ANKRD37 mRNA levels were monitored by qPCR across seven cell lines and normalized to 18S rRNA. (B, C) ANKRD37 hypoxic induction over time during hypoxia. (B) Endogenous HIF1A protein levels determined after 0, 4, 8 and 12 h of hypoxic incubation by western blot. (C) mRNA levels of ANKRD37 and VEGFA measured by qPCR after 0, 4, 8 and 12 h of hypoxic incubation.
We next investigated the HIF-binding sites in the ANKRD37 promoter. We identified four HIF-1-binding sites that matched our filtering criteria and were located at −311 to −306 bp (site 1), −228 to −223 bp (site 2), −142 to −137 bp (site 3) and +12 to +17 bp (site 4). As shown in Figure 6A, three ANKRD37 promoters encompassing these different HIF-binding sites were cloned upstream of a luciferase reporter. Promoter 1 was induced 4.1, 7.4 and 10.7-fold in hypoxia compared to normoxia in DLD-1, HCT116 and MCF7 cells, respectively. In contrast the induction observed with promoters 2 and 3 was in the range of 1.5–3-fold (Figure 6B). To determine whether the hypoxia induction was HIF-1 dependent, luciferase activity of ANKRD37 promoter 1 was measured in HCT116 cells in the presence of HIF1A siRNA. Endogenous HIF1A knockdown was confirmed by western blot (Supplementary Figure S4A). The 5-fold induction of promoter 1 in hypoxia was reduced to 1.3-fold in the presence of HIF1A siRNA (Supplementary Figure S4B). These results suggested that sites 1 and 2 are the physiologically relevant HIF-1-binding sites. Therefore, sites 1, 2 and 4 in promoter 1 were selectively mutated at the HIF consensus-binding site (ACGTG > AAAAG). As shown in Figure 6C, the hypoxic induction of promoter 1 was maximally inhibited by a mutation in site 2 while the other mutations had a minimal effect.
Figure 6.

Mapping of HIF-1 site in ANKRD37 promoter. (A) Genome browser view of the four HIF-binding sites identified in the ANKRD37 promoters and the three promoter sequences that were cloned to identify the dominant HIF-1 site. The site score is shown in parenthesis for each site, higher is better. The larger box of the ANKRD37 transcript (green) indicates the coding sequence. Note that the conservation shown on the plot is computed by PhastCons which scores conservation based on the evolutionary distance and does not necessarily reflect the number of species (95). (B) DLD1, HCT116 and MCF7 cells were transiently transfected and incubated under normoxic or hypoxic conditions. Luciferase activity of the three promoters was measured and normalized to promoter 1 in normoxic conditions. All studies were performed in triplicate and data represent the mean ± SD for three different wells. (C) Luciferase activity of promoter 1 in normoxia versus hypoxia with mutations in sites 1, 2 and 4 in DLD1, HCT116 and MCF7 cells. Experimental procedure and data analysis was performed as described for panel B. (D) In vitro binding of HIF-1 protein to site 2 in the ANKRD37 promoter. EMSAs were performed using biotinylated probe corresponding to HIF-binding site 2. Nuclear extracts (NE) from MCF7 cells cultured in either normoxia (N) or hypoxia (H) were utilized. The asterisk indicates the specific shift obtained in hypoxia. 200× M excess of non-labeled probe as a specific competitor or probe with a specific mutation in the HIF-binding site2 (ACGTG > AAAAG) was utilized to confirm specificity. WT = wild-type probe. MT = mutant probe. (E) Nuclear extracts from MCF7 cells grown in hypoxia were incubated with antibody specific to HIF-1α or normal mouse IgG. The double asterisk indicates the super-shift obtained only with a HIF-1α antibody. N.S.; nonspecific band. (F) In-vivo binding of HIF-1α protein to site 2 in the ANKRD37 promoter. ChIP assays were performed using primer sets corresponding to HIF-binding site 2 (primer A) or a region upstream of site 2 (primer B). DNA–protein complexes from MCF7 cells cultured in either normoxia (N) or hypoxia (H) were immunoprecipitated with or without antibody specific to HIF-1α or normal mouse IgG. Precipitated DNA or input samples were amplified by PCR.
To establish whether endogenous HIF-1 protein binds to site 2 in the ANKRD37 promoter, EMSA assays were performed using nuclear extracts from MCF7 cells incubated in either normoxia or hypoxia. A unique band shift was obtained only when nuclear extracts from cells grown in hypoxic conditions were incubated with the probe corresponding to site 2 (Figure 6D, lane 3). When non-biotinylated probe was co-incubated as a competitor, or a probe with specific mutations in the HIF consensus-binding site (ACGTG > AAAAG) was utilized, the band obtained in hypoxic conditions was lost (Figure 6D, lanes 4 and 5). Nuclear extracts from cells grown in hypoxia were then incubated with an antibody specific to HIF-1α or control mouse IgG prior to performing EMSA assays. Incubation with an antibody against HIF-1α resulted in a supershift of this specific band (Figure 6E). To further address whether HIF-1α physiologically binds to site 2, chromatin immunoprecipitation assays were performed using MCF7 cells incubated in either normoxia or hypoxia. Cross-linked cell lysates were immunoprecipitated with or without anti- HIF-1α antibody or control mouse IgG and PCR was performed to amplify a 178 bp or 220 bp fragment of the ANKRD37 promoter using primers corresponding to site 2 (primers A) and a region upstream of site 2 (primers B), respectively. DNA bound to HIF-1α was detected when DNA–protein complexes from hypoxic cells were precipitated with an anti-HIF-1α antibody and amplified with primers A, whereas no binding was observed at an upstream promoter region with primers B (Figure 6F). These data indicate that site 2, the most conserved and highest scoring HIF site, is indeed a HIF-1-binding site in DLD-1, HCT116 and MCF7 cells.
The HIF-transcriptional network
To identify transcription factors that are part of the HIF network we analyzed the presence of other binding sites in the 81 genes that ranked in top 200 and responded to hypoxia in at least three cell types. Matrices for validated transcription factors from the TRANSFAC and Jaspar collections were used to identify binding sites within 150 bases away from the highest scoring HIF-binding site in each of the promoters as well as within the entire promoter. For this analysis we used STORM with P < 1e–4 and accepted only binding sites predicted in at least four species and significantly (P < 0.05) more conserved compared to the flanking 100 bases (see ‘Materials and Methods’ section). To obtain a P-value for co-occurring binding sites, a chi-square analysis was performed compared to TFBS occurrence in a background set of 1000 promoters of genes that responded to hypoxia in at least one cell type but had no detectable HIF binding (see ‘Materials and Methods’ section). The analysis was performed in increments of 25 bases from the highest scoring HIF site and the most significant P-value for each PWM was selected. Only binding sites that were not predicted at the HIF consensus binding site were considered. We identified 10 TFBS that were significantly enriched within a specific distance from the highest scoring HIF site including ATF2, CREB1, SP1, WT1 and JUN which have been previously linked to hypoxia and HIF-1 (Supplementary Table S6). When considering the entire promoter, we observed enrichment for P53:P73-binding sites and multiple members of the forkhead family, including FOXO4, FOXD1 and FOXF2. To test for potential mechanism in up or down regulation of HIF-target genes, a similar analysis was employed to upregulated and downregulated transcripts separately. Most of the sites co-occurring for all HIF targets above were also enriched for the upregulated genes, albeit with more significant P-values. SP-1 and NF-Y were the only TFs significantly co-occurring within a specific distance from the HIF site in downregulated genes. Surprisingly, four of five predicted NF-Y targets (ABCF2, CYCS, FH, MRPL4) were genes localized to mitochondria (Supplementary Table S7).
HIF-1 motifs are identified de-novo in predicted HIF-1-target genes
To identify de-novo motifs over-represented in the promoters of the 81 HIF-target genes selected above, DME2 was employed. For each motif length in the range of 5–12 bases we identified the top 50 over-represented motifs compared to a suitable background set (see ‘Materials and Methods’ section). These motifs were compared to the TRANSFAC and Jaspar collections of PWMs using the CREAD program MATCOMPARE. Comparison was estimated using a divergence score in the range of 0 to 1, where a divergence of 0 indicates the motif predicted de-novo is identical to a PWM. HIF-1 PWMs were matched best to six bases long de-novo motifs. For motifs longer than 7 bases, no HIF-1 PWMs were ranked within the top 50 (Supplementary Table S8). Comparing all available PWMs to de-novo predicted motifs with a divergence of 0.25 or lower identified ARNT (HIF-1β), ATF2, NF-Y and ATF6 as the most enriched in the data set (Supplementary Table S9). Other motifs that were predicted de-novo include CREB1, AP-1 and EVI1 and MIZF (Supplementary Table S9). Some of these are also identified above as part of the HIF-transcriptional network.
DISCUSSION
In this study we presented a computational strategy to identify HIF-1-target genes. This strategy integrates gene expression profiles and computational-binding site analysis of proximal promoters. The utility of our strategy in detecting novel HIF-target genes was demonstrated by recovering known HIF-1-target genes and experimentally validating ANKRD37. ANKRD37 is a previously uncharacterized protein that contains four ankyrin repeats. Ankyrin repeats are thought to mediate protein–protein interactions (67) and are known to be involved in regulation of key transcription factors such as NFκB and TP53 (68–70). ANKRD37 induction was 3-fold higher than VEGFA (Figure 5E), the validated HIF-1-binding site was conserved in 10 species and was significantly induced in hypoxia in multiple cell types analyzed here. Taken together, these data suggest ANKRD37 is likely to have an important role in the hypoxia response.
The cellular response to hypoxia was different between cell types, consistent with previous reports (55,56). The union of all differentially expressed genes in the data sets presented here contained over 6000 unique genes. Even when comparing studies that attempted to detect a HIF dependent response to hypoxia (21,22,71), only 45 genes were differentially expressed in all three. Importantly the analysis presented here indicates that 60% of the transcripts that responded to hypoxia did not have a detectable conserved HIF-binding site in their proximal promoter. This observation was also supported by MOTIFCLASS enrichment, which is an independent analysis that does not consider conservation and did not detect any enrichment for HIF-binding sites in those promoters. In addition, transcripts with no detectable HIF site were enriched for other TFBS that were previously shown to participate in the cellular response to hypoxia including, DEAF1, ELK1, POU2F1 and IRF1 (72,73). Moreover, genes that responded to hypoxia and did not have a detectable HIF-binding site were used as a background for the HIF-1-transcriptional network. One possible mechanism that may explain the different hypoxic response between cells is that genes directly regulated by HIF are involved in key processes and their regulation mediates a robust effect. As illustrated in the mitochondrial map, where several predicted downregulated genes affect key steps in the mitochondria. The predicted HIF-target genes SLC25A28, an iron transporter in the mitochondria and ISCU, an iron–sulfur scaffold protein are both downregulated in hypoxia. These two genes could affect several of the mitochondria iron–sulfur proteins including, NADH dehydrogenease, Coenzyme Q—cytochrome c reductase and Succinate—coenzyme Q reductase are critical to the function of the electron transport chain and the TCA cycle. In addition, the predicted HIF targets MRPL4 and MRPS12 are components of the mitochondrial ribosome that are required for mitochondrial protein translation. Additional examples include cell growth arrest by inhibition of CAD, a component of pyrimidine synthesis (74); RRP9, a component of snoRNP predicted to participate in processing and modification of pre-ribosomal RNA (75); EIF2B3, a translation initiation factor subunit; and CCNB1, a regulatory protein involved in mitosis (74). One novel predicted HIF target that is highlighted by the HIF-1 KEGG metabolic pathways map is GYS1, a gene involved in glycogen biosynthesis (65,76) that was validated as responsive to hypoxia by quantitative PCR in multiple cell types.
To highlight predicted HIF targets of interest and to suggest potential functional roles for these targets, a bioinformatics analysis of validated HIF targets was performed and predicted targets that were not previously validated were mapped to the significantly enriched functional categories. This analysis is summarized in Supplementary Table S6 and identifies several attractive predicted HIF targets. The family of 2-oxoglutarate and Fe(II)-dependent oxygenase has 19 members in the human genome of which five are bona fide HIF targets, including the HIF-1α regulators EGLN1 and EGLN3. The predicted HIF target PLOD1, is a member of that family and a lysyl hydroxylase involved in collagen synthesis and localized to the endoplasmic reticulum (77). Mutations in PLOD1 have been associated with type VI Ehlers–Danlos syndrome, a genetic disorder caused by defects in collagen synthesis (78). In another example, microRNA-target predictions from TargetScan were analyzed and validated HIF targets were enriched for five microRNAs including miR-1/206 and miR-130/301. miR-1/206 were shown to be upregulated in a rat model of myocardial infarction (79) and miR-130 was shown to regulate angiogenesis (80). Both are relevant to the HIF pathway and would be attractive candidate microRNAs to study further with respect to the validated and predicted targets. Finally, protein interaction data presented in Figure 4C can be used to identify potential regulators, co-factors or adapter proteins that play a role in the HIF pathway. Finally, P300/CBP-associated factor (PCAF) interacts with four validated HIF targets and six predicted targets. PCAF was previously shown to be a HIF-1α co-factor that regulates p53-transcriptional activity in hypoxia (81). The specific HIF targets that interact with PCAF are attractive candidates for functional studies.
The method presented here is not without limitation. First, the microarray itself presents a limitation since only genes represented on the microarray are considered. However, this could be improved by using multiple platforms that offer complete coverage of annotated genes. Of the 86 previously validated HIF-1 targets (Supplementary Table S2), only 56 responded to hypoxia in at least one cell type. One example of a bona fide HIF-target gene that was not recovered by our analysis is EPO (erythropoietin). A close examination of the data for EPO revealed that the probe did not respond to hypoxia in any of the cells that were profiled. EPO was previously reported to be highly induced in the kidney (82). Second, HIF-binding sites may be located outside the promoter region we defined or may not be conserved in other species. The binding site for the well-characterized HIF-1 target VEGFA is located at −985 bp (83) and therefore was not recovered in the present analysis. HIF-1-binding sites have been identified as far as 12-kb downstream (PHD3, (84) and 5-kb upstream [eNos; (85)] of the TSS. The defined promoter region was found optimal to capture most of the known sites (60% of known HIF-1-binding sites) while minimizing potential false positive prediction.
In this study we provide a ranked catalogue of genes predicted to be direct HIF-target genes. Given the different hypoxia response between cell types, it is currently difficult to estimate how many HIF-target genes exist. Our analysis across six cell types was stringent in an effort to obtain the highest specificity and the top 200 ranked genes are of high confidence. The ranking system by itself offers a level of confidence for the prediction and was shown as reliable by high ranking for previously validated targets (Supplementary Figure S3). Currently, almost all known HIF-target genes are upregulated in hypoxia. While the majority of predicted HIF targets were upregulated in hypoxia, nearly all downregulated genes are novel. The top ranking downregulated gene is EDEM3, an α1,2-mannosidase involved in quality control of misfolded proteins (86). In addition, a subset of downregulated genes highlighted in the mitochondrial map is consistent with the observed attenuation of oxidative phosphorylation by HIF-1 (87). We also identified NF-Y as significantly co-occurring adjacent to the highest scoring HIF-1 site in four of these downregulated mitochondrial genes. It is currently unclear whether NF-Y is part of the hypoxic regulation of those genes. However, it was previously shown in yeast that the NF-Y homolog regulates the mitochondrial enzyme COX (88), which was also shown to be regulated by HIF-1 to optimize efficiency of mitochondrial respiration (89).
To describe the transcriptional network of HIF, we identified additional TFBS over-represented within ±150 bp from the highest scoring HIF site as well as TFBS enriched in the entire promoter. We identified only a few TFs within a specific distance from the HIF site, however, almost all were previously associated with HIF-1, including CREB1, ATF2 and SP1 (90–92). The recovery of these TFs increases the confidence in the genes predicted as HIF targets and also identifies specific gene targets in Supplementary Table S6 as predicted targets with higher confidence. WSB1, for instance, is a novel predicted HIF-1 target that contains binding sites for CREB1, ATF6, ATF2 and JUN:ATF2, a profile very similar to LDHA that is a bona fide HIF-1 target. WSB1 was previously shown to be involved in the ubiquitination and degradation of HIPK2 (93), a kinase that was shown to repress HIF-1α transcription (94). WSB1 was also confirmed to be hypoxia responsive by quantitative PCR (Supplementary Figure S4). Taken together these data suggest WSB1 is highly likely to be a direct HIF-1 target and to be part of a feedback loop that includes HIF-1α, WSB1 and HIPK2. TFs that were previously associated with HIF-1 appear to be enriched only in genes upregulated during hypoxia. YY-1 and NF-Y were detected as enriched in downregulated genes. Further studies will be necessary to validate these observations and to determine their functional relevance to HIF-1.
De-novo motif prediction suggested that no significant information flanking the HIF consensus-binding site could be found in the predicted HIF-target genes. This is consistent with the observation that enhancing the number of validated HIF-binding sites reduces the information content of the flanking nucleotides (Supplementary Figure S1). Conservation was not considered in the de-novo prediction process. Yet, several TFs identified as part of the HIF-transcriptional network have also been identified de-novo, including, CREB1, JUN:ATF2, ATF2, ATF6 and AP-1. The ability to recover these TFs de-novo confirms their significant over-representation and demonstrates the utility of this approach in recovering previously validated TFs and co-factor that are part of the HIF network. Furthermore, novel TFs that may be part of the HIF-transcriptional network are currently supported by sequence analysis data alone and additional studies are required to validate these findings.
HIF-1 and HIF-2 are both key transcription factors with that modulate metabolic pathways, bioenergetics and processes relevant to cancer onset and progression. This study not only gives insights into the genes potentially regulated by HIF but also serves as a guide to study direct targets of other transcription factors.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Harris Fellowship to Y.B.; National Institutes of Health [grant number DK043351, AI062773, DK83756] for work in RJX lab; National Institutes of Health [grant number HG001696] for work in MZQ lab; and National Institutes of Health [grant number CA92594] for work in DCC lab. Funding for open access charge: [grant number HG001696, CA45508] for MZQ lab.
Conflict of interest statement. None declared.
Supplementary Material
Footnotes
Present address: Andrew D. Smith, Department of Biology, University of Southern California, Los Angeles, CA 90089, USA
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
REFERENCES
- 1.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 2.Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000;275:25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 3.Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, Gavin BJ, Kley N, Kaelin WG, Iliopoulos O. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol. Cell. 1998;1:959–968. doi: 10.1016/s1097-2765(00)80096-9. [DOI] [PubMed] [Google Scholar]
- 4.Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc. Natl Acad. Sci. USA. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19:4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 7.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 9.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 10.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci. STKE. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 11.Krishnamachary B, Semenza GL. Analysis of hypoxia-inducible factor 1alpha expression and its effects on invasion and metastasis. Methods Enzymol. 2007;435:347–354. doi: 10.1016/S0076-6879(07)35017-9. [DOI] [PubMed] [Google Scholar]
- 12.Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007;67:563–572. doi: 10.1158/0008-5472.CAN-06-2701. [DOI] [PubMed] [Google Scholar]
- 13.Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15:678–685. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–246. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 16.Semenza GL. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 17.Patiar S, Harris AL. Role of hypoxia-inducible factor-1alpha as a cancer therapy target. Endocr. Relat. Cancer. 2006;13(Suppl. 1):S61–S75. doi: 10.1677/erc.1.01290. [DOI] [PubMed] [Google Scholar]
- 18.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Cho YS, Bae JM, Chun YS, Chung JH, Jeon YK, Kim IS, Kim MS, Park JW. HIF-1alpha controls keratinocyte proliferation by up-regulating p21(WAF1/Cip1) Biochim. Biophys. Acta. 2008;1783:323–333. doi: 10.1016/j.bbamcr.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 20.Shen C, Nettleton D, Jiang M, Kim SK, Powell-Coffman JA. Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J. Biol. Chem. 2005;280:20580–20588. doi: 10.1074/jbc.M501894200. [DOI] [PubMed] [Google Scholar]
- 21.Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1alpha, HIF-2alpha, and other pathways. J. Biol. Chem. 2006;281:15215–15226. doi: 10.1074/jbc.M511408200. [DOI] [PubMed] [Google Scholar]
- 22.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Q, Wong WH. CisModule: de novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc. Natl Acad. Sci. USA. 2004;101:12114–12119. doi: 10.1073/pnas.0402858101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta M, Liu JS. De novo cis-regulatory module elicitation for eukaryotic genomes. Proc. Natl Acad. Sci. USA. 2005;102:7079–7084. doi: 10.1073/pnas.0408743102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Z, Shendure J, Church GM. Discovering functional transcription-factor combinations in the human cell cycle. Genome Res. 2005;15:848–855. doi: 10.1101/gr.3394405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith AD, Sumazin P, Xuan Z, Zhang MQ. DNA motifs in human and mouse proximal promoters predict tissue-specific expression. Proc. Natl Acad. Sci. USA. 2006;103:6275–6280. doi: 10.1073/pnas.0508169103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hestand MS, van Galen M, Villerius MP, van Ommen GJ, den Dunnen JT, t Hoen PA. CORE_TF: a user-friendly interface to identify evolutionary conserved transcription factor binding sites in sets of co-regulated genes. BMC bioinformatics. 2008;9:495. doi: 10.1186/1471-2105-9-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheung TH, Kwan YL, Hamady M, Liu X. Unraveling transcriptional control and cis-regulatory codes using the software suite GeneACT. Genome Biol. 2006;7:R97. doi: 10.1186/gb-2006-7-10-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulyk ML. Computational prediction of transcription-factor binding site locations. Genome Biol. 2003;5:201. doi: 10.1186/gb-2003-5-1-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown CT. Computational approaches to finding and analyzing cis-regulatory elements. Methods Cell Biol. 2008;87:337–365. doi: 10.1016/S0091-679X(08)00218-5. [DOI] [PubMed] [Google Scholar]
- 32.Smith AD, Sumazin P, Zhang MQ. Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc. Natl Acad. Sci. USA. 2005;102:1560–1565. doi: 10.1073/pnas.0406123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bussemaker HJ, Li H, Siggia ED. Regulatory element detection using correlation with expression. Nature Genet. 2001;27:167–171. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- 35.Conlon EM, Liu XS, Lieb JD, Liu JS. Integrating regulatory motif discovery and genome-wide expression analysis. Proc. Natl Acad. Sci. USA. 2003;100:3339–3344. doi: 10.1073/pnas.0630591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das D, Banerjee N, Zhang MQ. Interacting models of cooperative gene regulation. Proc. Natl Acad. Sci. USA. 2004;101:16234–16239. doi: 10.1073/pnas.0407365101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrett T, Suzek TO, Troup DB, Wilhite SE, Ngau WC, Ledoux P, Rudnev D, Lash AE, Fujibuchi W, Edgar R. NCBI GEO: mining millions of expression profiles – database and tools. Nucleic Acids Res. 2005;33:D562–D566. doi: 10.1093/nar/gki022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parkinson H, Kapushesky M, Shojatalab M, Abeygunawardena N, Coulson R, Farne A, Holloway E, Kolesnykov N, Lilja P, Lukk M, et al. ArrayExpress – a public database of microarray experiments and gene expression profiles. Nucleic Acids Res. 2007;35:D747–D750. doi: 10.1093/nar/gkl995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Association. 2004;99:909–917. [Google Scholar]
- 40.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21:2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- 42.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995;57:289–300. [Google Scholar]
- 43.Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, Diekhans M, Giardine B, Harte RA, Hinrichs AS, Hsu F, et al. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36:D773–D779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schones DE, Smith AD, Zhang MQ. Statistical significance of cis-regulatory modules. BMC Bioinformatics. 2007;8:19. doi: 10.1186/1471-2105-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vastrik I, D’Eustachio P, Schmidt E, Joshi-Tope G, Gopinath G, Croft D, de Bono B, Gillespie M, Jassal B, Lewis S, et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol. 2007;8:R39. doi: 10.1186/gb-2007-8-3-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 48.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Church DM, DiCuccio M, Edgar R, Federhen S, Helmberg W, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2005;33:D39–D45. doi: 10.1093/nar/gki062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bradley P, Bork P, Bucher P, Cerutti L, et al. InterPro, progress and status in 2005. Nucleic Acids Res. 2005;33:D201–D205. doi: 10.1093/nar/gki106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, et al. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R. The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Res. 2004;32:D262–D266. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calvo S, Jain M, Xie X, Sheth SA, Chang B, Goldberger OA, Spinazzola A, Zeviani M, Carr SA, Mootha VK. Systematic identification of human mitochondrial disease genes through integrative genomics. Nature Genet. 2006;38:576–582. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 53.Mizukami Y, Li J, Zhang X, Zimmer MA, Iliopoulos O, Chung DC. Hypoxia-inducible factor-1-independent regulation of vascular endothelial growth factor by hypoxia in colon cancer. Cancer Res. 2004;64:1765–1772. doi: 10.1158/0008-5472.can-03-3017. [DOI] [PubMed] [Google Scholar]
- 54.Mizukami Y, Fujiki K, Duerr EM, Gala M, Jo WS, Zhang X, Chung DC. Hypoxic regulation of vascular endothelial growth factor through the induction of phosphatidylinositol 3-kinase/Rho/ROCK and c-Myc. J. Biol. Chem. 2006;281:13957–13963. doi: 10.1074/jbc.M511763200. [DOI] [PubMed] [Google Scholar]
- 55.Chi JT, Wang Z, Nuyten DS, Rodriguez EH, Schaner ME, Salim A, Wang Y, Kristensen GB, Helland A, Borresen-Dale AL, et al. Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med. 2006;3:e47. doi: 10.1371/journal.pmed.0030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Denko NC, Fontana LA, Hudson KM, Sutphin PD, Raychaudhuri S, Altman R, Giaccia AJ. Investigating hypoxic tumor physiology through gene expression patterns. Oncogene. 2003;22:5907–5914. doi: 10.1038/sj.onc.1206703. [DOI] [PubMed] [Google Scholar]
- 57.Gardner LB, Corn PG. Hypoxic regulation of mRNA expression. Cell Cycle. 2008;7:1916–1924. doi: 10.4161/cc.7.13.6203. [DOI] [PubMed] [Google Scholar]
- 58.Jin HO, An S, Lee HC, Woo SH, Seo SK, Choe TB, Yoo DH, Lee SB, Um HD, Lee SJ, et al. Hypoxic condition- and high cell density-induced expression of Redd1 is regulated by activation of hypoxia-inducible factor-1alpha and Sp1 through the phosphatidylinositol 3-kinase/Akt signaling pathway. Cell Signal. 2007;19:1393–1403. doi: 10.1016/j.cellsig.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 59.Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell Biol. 2002;22:2283–2293. doi: 10.1128/MCB.22.7.2283-2293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leonard MO, Cottell DC, Godson C, Brady HR, Taylor CT. The role of HIF-1 alpha in transcriptional regulation of the proximal tubular epithelial cell response to hypoxia. J. Biol. Chem. 2003;278:40296–40304. doi: 10.1074/jbc.M302560200. [DOI] [PubMed] [Google Scholar]
- 61.Takahashi Y, Takahashi S, Shiga Y, Yoshimi T, Miura T. Hypoxic induction of prolyl 4-hydroxylase alpha (I) in cultured cells. J. Biol. Chem. 2000;275:14139–14146. doi: 10.1074/jbc.275.19.14139. [DOI] [PubMed] [Google Scholar]
- 62.Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, Eltzschig HK, Buhrer C. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem. Biophys. Res. Commun. 2008;372:892–897. doi: 10.1016/j.bbrc.2008.05.150. [DOI] [PubMed] [Google Scholar]
- 63.Jean JC, Rich CB, Joyce-Brady M. Hypoxia results in an HIF-1-dependent induction of brain-specific aldolase C in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;291:L950–L956. doi: 10.1152/ajplung.00087.2006. [DOI] [PubMed] [Google Scholar]
- 64.Barrera LO, Li Z, Smith AD, Arden KC, Cavenee WK, Zhang MQ, Green RD, Ren B. Genome-wide mapping and analysis of active promoters in mouse embryonic stem cells and adult organs. Genome Res. 2008;18:46–59. doi: 10.1101/gr.6654808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cid E, Gomis RR, Geremia RA, Guinovart JJ, Ferrer JC. Identification of two essential glutamic acid residues in glycogen synthase. J. Biol. Chem. 2000;275:33614–33621. doi: 10.1074/jbc.M005358200. [DOI] [PubMed] [Google Scholar]
- 66.Bourgeron T, Chretien D, Poggi-Bach J, Doonan S, Rabier D, Letouze P, Munnich A, Rotig A, Landrieu P, Rustin P. Mutation of the fumarase gene in two siblings with progressive encephalopathy and fumarase deficiency. J. Clin. Invest. 1994;93:2514–2518. doi: 10.1172/JCI117261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li J, Mahajan A, Tsai MD. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry. 2006;45:15168–15178. doi: 10.1021/bi062188q. [DOI] [PubMed] [Google Scholar]
- 68.Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, Coles CH, Yu X, Hay RT, Ley SC, et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH) Proc. Natl Acad. Sci. USA. 2006;103:14767–14772. doi: 10.1073/pnas.0606877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 70.Dreyfus DH, Nagasawa M, Gelfand EW, Ghoda LY. Modulation of p53 activity by IkappaBalpha: evidence suggesting a common phylogeny between NF-kappaB and p53 transcription factors. BMC Immunol. 2005;6:12. doi: 10.1186/1471-2172-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nickols NG, Jacobs CS, Farkas ME, Dervan PB. Modulating hypoxia-inducible transcription by disrupting the HIF-1-DNA interface. ACS Chem. Biol. 2007;2:561–571. doi: 10.1021/cb700110z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choi KO, Lee T, Lee N, Kim JH, Yang EG, Yoon JM, Lee TG, Park H. Inhibition of the catalytic activity of hypoxia-inducible factor-1alpha-prolyl-hydroxylase 2 by a MYND-type zinc finger. Mol. Pharmacol. 2005;68:1803–1809. doi: 10.1124/mol.105.015271. [DOI] [PubMed] [Google Scholar]
- 73.Yan SF, Lu J, Zou YS, Soh-Won J, Cohen DM, Buttrick PM, Cooper DR, Steinberg SF, Mackman N, Pinsky DJ, et al. Hypoxia-associated induction of early growth response-1 gene expression. J. Biol. Chem. 1999;274:15030–15040. doi: 10.1074/jbc.274.21.15030. [DOI] [PubMed] [Google Scholar]
- 74.Sigoillot FD, Berkowski JA, Sigoillot SM, Kotsis DH, Guy HI. Cell cycle-dependent regulation of pyrimidine biosynthesis. J. Biol. Chem. 2003;278:3403–3409. doi: 10.1074/jbc.M211078200. [DOI] [PubMed] [Google Scholar]
- 75.Lukowiak AA, Granneman S, Mattox SA, Speckmann WA, Jones K, Pluk H, Venrooij WJ, Terns RM, Terns MP. Interaction of the U3-55k protein with U3 snoRNA is mediated by the box B/C motif of U3 and the WD repeats of U3-55k. Nucleic Acids Res. 2000;28:3462–3471. doi: 10.1093/nar/28.18.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kollberg G, Tulinius M, Gilljam T, Ostman-Smith I, Forsander G, Jotorp P, Oldfors A, Holme E. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N. Engl. J. Med. 2007;357:1507–1514. doi: 10.1056/NEJMoa066691. [DOI] [PubMed] [Google Scholar]
- 77.Takaluoma K, Hyry M, Lantto J, Sormunen R, Bank RA, Kivirikko KI, Myllyharju J, Soininen R. Tissue-specific changes in the hydroxylysine content and cross-links of collagens and alterations in fibril morphology in lysyl hydroxylase 1 knock-out mice. J. Biol. Chem. 2007;282:6588–6596. doi: 10.1074/jbc.M608830200. [DOI] [PubMed] [Google Scholar]
- 78.Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) Am. J. Med. Genet. 1998;77:31–37. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 79.Shan ZX, Lin QX, Fu YH, Deng CY, Zhou ZL, Zhu JN, Liu XY, Zhang YY, Li Y, Lin SG, et al. Upregulated expression of miR-1/miR-206 in a rat model of myocardial infarction. Biochem. Biophys. Res. Commun. 2009;381:597–601. doi: 10.1016/j.bbrc.2009.02.097. [DOI] [PubMed] [Google Scholar]
- 80.Chen Y, Gorski DH. Regulation of angiogenesis through a microRNA (miR-130a) that down-regulates antiangiogenic homeobox genes GAX and HOXA5. Blood. 2008;111:1217–1226. doi: 10.1182/blood-2007-07-104133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xenaki G, Ontikatze T, Rajendran R, Stratford IJ, Dive C, Krstic-Demonacos M, Demonacos C. PCAF is an HIF-1alpha cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene. 2008;27:5785–5796. doi: 10.1038/onc.2008.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Westenfelder C, Biddle DL, Baranowski RL. Human, rat, and mouse kidney cells express functional erythropoietin receptors. Kidney Int. 1999;55:808–820. doi: 10.1046/j.1523-1755.1999.055003808.x. [DOI] [PubMed] [Google Scholar]
- 83.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pescador N, Cuevas Y, Naranjo S, Alcaide M, Villar D, Landazuri MO, Del Peso L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem. J. 2005;390:189–197. doi: 10.1042/BJ20042121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Coulet F, Nadaud S, Agrapart M, Soubrier F. Identification of hypoxia-response element in the human endothelial nitric-oxide synthase gene promoter. J. Biol. Chem. 2003;278:46230–46240. doi: 10.1074/jbc.M305420200. [DOI] [PubMed] [Google Scholar]
- 86.Hirao K, Natsuka Y, Tamura T, Wada I, Morito D, Natsuka S, Romero P, Sleno B, Tremblay LO, Herscovics A, et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006;281:9650–9658. doi: 10.1074/jbc.M512191200. [DOI] [PubMed] [Google Scholar]
- 87.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 88.Fontanesi F, Jin C, Tzagoloff A, Barrientos A. Transcriptional activators HAP/NF-Y rescue a cytochrome c oxidase defect in yeast and human cells. Hum. Mol. Genet. 2008;17:775–788. doi: 10.1093/hmg/ddm349. [DOI] [PubMed] [Google Scholar]
- 89.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 90.Dimova EY, Jakubowska MM, Kietzmann T. CREB binding to the hypoxia-inducible factor-1 responsive elements in the plasminogen activator inhibitor-1 promoter mediates the glucagon effect. Thromb. Haemost. 2007;98:296–303. [PubMed] [Google Scholar]
- 91.Choi JH, Park MJ, Kim KW, Choi YH, Park SH, An WG, Yang US, Cheong J. Molecular mechanism of hypoxia-mediated hepatic gluconeogenesis by transcriptional regulation. FEBS Lett. 2005;579:2795–2801. doi: 10.1016/j.febslet.2005.03.097. [DOI] [PubMed] [Google Scholar]
- 92.Sanchez-Elsner T, Botella LM, Velasco B, Langa C, Bernabeu C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J. Biol. Chem. 2002;277:43799–43808. doi: 10.1074/jbc.M207160200. [DOI] [PubMed] [Google Scholar]
- 93.Choi DW, Seo YM, Kim EA, Sung KS, Ahn JW, Park SJ, Lee SR, Choi CY. Ubiquitination and degradation of homeodomain-interacting protein kinase 2 by WD40 repeat/SOCS box protein WSB-1. J. Biol. Chem. 2008;283:4682–4689. doi: 10.1074/jbc.M708873200. [DOI] [PubMed] [Google Scholar]
- 94.Nardinocchi L, Puca R, Guidolin D, Belloni AS, Bossi G, Michiels C, Sacchi A, Onisto M, D'Orazi G. Transcriptional regulation of hypoxia-inducible factor 1alpha by HIPK2 suggests a novel mechanism to restrain tumor growth. Biochim. Biophys. Acta. 2009;1793:368–377. doi: 10.1016/j.bbamcr.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 95.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.