


Abstract
Replication of the promiscuous plasmid pMV158 requires expression of the initiator repB gene, which is controlled by the repressor CopG. Genes repB and copG are co-transcribed from promoter Pcr. We have studied the interactions between RNA polymerase, CopG and the promoter to elucidate the mechanism of repression by CopG. Complexes formed at 0°C and at 37°C between RNA polymerase and Pcr differed from each other in stability and in the extent of the DNA contacted. The 37°C complex was very stable (half-life of about 3 h), and shared features with typical open complexes generated at a variety of promoters. CopG protein repressed transcription from Pcr at two different stages in the process leading to the initiation complex. First, CopG hindered binding of RNA polymerase to the promoter. Second, CopG was able to displace RNA polymerase once the enzyme has formed a stable complex with Pcr. A model for the CopG-mediated disassembly of the stable RNA polymerase–Pcr promoter complex is presented.
INTRODUCTION
Inhibition of transcription initiation in bacteria can take place at any of the steps in the process leading to the formation of the elongation complex (1,2). Although some of the best-characterized transcriptional repressors seem to act by hindering the access of the RNA polymerase (RNAP) to the promoter, an increasing number of regulatory proteins have been reported that inhibit transcription initiation at a step subsequent to RNAP binding. Thus, the repressors λcI (3), LexA (4), LacI (5,6) or MalR (7) occlude binding of RNAP to their respective promoters (PR, PuvrA, Plac, or PM), while GalR bound to operator OE inhibits isomerization from the ternary closed complex to the open complex at promoter galP1 (8,9), and phage ϕ29 protein p4 prevents RNAP from leaving the A2c promoter (10). Inhibition of transcription at a step subsequent to promoter melting has also been shown for some of the reported regulatory activities of the architectural proteins H-NS and FIS. Thus, binding of FIS to two sites centred at positions −62 and −109 with respect to the transcription start point of the gyrB gene does not interfere with generation of an open complex at the gyrB promoter, but seems to prevent the NTP-driven isomerization of open complexes to initiation complexes (11). The presence of H-NS, while stimulating open complex formation at the rRNA rrnB P1 promoter of Escherichia coli, dramatically reduces the generation of transcription products longer than 3 nt from this promoter (12). An interesting example of growth phase-dependent regulation of gene expression by H-NS is the selective repression of the transcription initiated by the RNAP σ70 holoenzyme at promoter hdeAB (13). RNAP holoenzymes carrying either the house-keeping σ subunit (σ70) or the stationary phase σ (σ38) can form an open complex at the hdeAB promoter in the presence of H-NS, although the RNAP σ70 holoenzyme is unable to initiate transcription. This selective repression is attributed to differences in the degree of DNA wrapping around the two holoenzymes: the tighter wrapping around RNAP σ70 holoenzyme allows extension of H-NS by cooperative binding from its AT-tract nucleation site centred at position −118 to promoter-downstream sequences, which results in effective sealing of the DNA loop and trapping of RNAP (13).
The mechanism by which a given repressor acts can condition the relative location of its DNA target with respect to the promoter, and vice versa. Hence, the step at which transcription initiation is inhibited differs between the closely-related repressors of coliphages λ and 434 (14). These two proteins share both a common structure and the ability to bind specifically to a set of three operator sites (OR1, OR2 and OR3) located between two divergently oriented promoters, PR and PRM. In both phages, the region of the DNA containing OR1 and OR2 (to which the repressors bind with the highest affinity) overlaps the region containing the –35 and –10 boxes of PR, although the relative arrangement of the operator sites with respect to the promoter elements differs in λ and 434 (14). This distinct architecture has been invoked to argue the different mechanism of repression of the PR activity found in each of these phages. In λ, where OR1 and OR2 partially overlap the –10 and –35 elements of PR respectively, the binding of the repressor to either operator site blocks the access of RNAP to the promoter. In 434, where OR1 and OR2 are immediately downstream from the –10 element and the –35 element of PR respectively, binding of the repressor to OR2 is necessary and sufficient to inhibit transcription from PR through a mechanism that does not exclude the subsequent binding of RNAP. In this case, a stable (heparin-resistant) ternary complex is generated, which cannot progress to an open complex (14). From these latter examples, it becomes clear that prediction of the inhibitory mechanism of transcription initiation should be based on the relative arrangement and topology of RNAP and repressor on the DNA double helix, rather than in sequence or structural similarities to a reference protein acting at a known step of the initiation process.
Transcriptional repressor CopG, encoded by the promiscuous plasmid pMV158, is a small protein (45 amino-acid polypeptide chains) which, by inhibiting synthesis of the cop-rep mRNA, regulates both the expression of its own gene and that of the initiator of replication (repB) gene. Contacts of CopG to its target DNA span about 50 bp, through a region that includes the –35 and –10 boxes of the regulated Pcr promoter (15) (Figure 1A). In the centre of the contacted DNA, and overlapping the –35 box, there is a 13-bp pseudo-symmetric element (SE; Figure 1A) which constitutes the primary target of CopG. Although the structure solved from the co-crystals of CopG bound to either a 19- or a 22-bp double-stranded DNA shows two protein dimers, each interacting with a half of the SE, the working model for the whole DNA specifically contacted by CopG assumes the cooperative binding of four dimers of the protein (16,17). CopG belongs to the ribbon-helix-helix (RHH) class of DNA-binding proteins, which interact specifically with the bases of the DNA through residues located in the two-stranded antiparallel β-sheet (18,19). Members of the RHH class exhibit cooperativity based on protein–protein interactions, generating at least a dimer of protein dimers upon binding to their target DNA (20–22). CopG, which is no more than the RHH motif, is the smallest among these proteins and represents the minimal DNA-binding structure within this class of proteins (17). The peptide-backbone structure of the CopG dimer is almost identical to that of the RHH motif of Arc repressor from Salmonella bacteriophage P22, although this latter protein is slightly bigger (53 residues polypeptide chains) and contains an additional N-terminal region which also interacts with the target DNA (17,19). The similarity between the Arc- and CopG-mediated transcriptional regulatory systems also extends to the DNA moiety. In both systems, the operator to which the repressor binds overlaps the promoter region and contains two inversely-repeated copies of a 4-bp sequence (5′-TAGA-3′ for Arc operator, and the self-palindrome 5′-TGCA-3′ for CopG operator) which are 5 bp apart. This arrangement results in 13-bp SEs, whose dyad axes pass in both operators through a central G:C base pair. A dimer of either Arc or CopG interacts with each half of the respective SE establishing direct contacts with at least one base of each of the base pairs constituting the 4-bp inversely-repeated boxes, and with two further bases outside the 13-bp palindromic element (one at each side of the SE) (16,19). With respect to the mechanism of transcription inhibition, some detailed information has been published for the Arc-regulated system. Binding of Arc to its operator represses transcription from two divergent and overlapping promoters, Pant and Pmnt (23). It has been shown that binding of Arc does not preclude RNAP binding to Pant, but prevents the polymerase from forming a heparin-resistant stable complex at this promoter, so that the rate of generation of a transcriptionally-competent open complex is reduced (23). Although CopG-mediated repression of transcription from Pcr has been reported both in vivo and in vitro (15), the mechanisms of this repression have not been investigated previously. Here we characterize the complexes formed by binding of RNAP to the Pcr promoter at 0°C and 37°C. In addition, we present results showing that CopG does not only prevent binding of RNAP to the promoter, but is also able to dislodge the polymerase once it has formed a stable heparin-resistant complex.
Figure 1.

The Pcr promoter of pMV158. (A) Conserved elements of Pcr. The copG start codon, the +1 site, and the −35 and extended −10 elements are in capital letters; distances between them are shown. The SE of the CopG operator is boxed. (B) Footprinting pattern of the 0°C and 37°C RNAP–Pcr complexes. The scheme summarizes the results shown in Supplementary Figures S1 and S2. Promoter positions are numbered relative to the transcription start site. DNase I protections are denoted by numbered brackets. Thin dotted-line brackets indicate weak protections. Enhancements to HO• attack are shown by arrows.
MATERIALS AND METHODS
DNA, proteins and chemicals
A 239-bp DNA fragment containing Pcr and the overlapping CopG target was generated by PCR using Pfu DNA polymerase (Stratagene), a pair of synthetic primers (5′-CGCCTTTAGCCTTAGAG-3′ and 5′-CCATCTCTCTTGCCAT-3′), and pMV158 DNA as template. To label the DNA fragment at only one 5′-end, either of these primers was treated with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (Amersham) before performing the amplification reaction. The amplified fragment spanned from −144 to +95 relative to the Pcr transcription start site (15). This fragment was used in all the experiments described below. DNase I and E. coli RNAP holoenzyme were from Boehringer Mannheim. When indicated, RNAP holoenzyme from USB was used. Promoter binding activity of the commercial preparations of RNAP was determined by EMSA, using DNA concentrations 10, 20 or 40 times the total concentration of RNAP (4 nM). RNAP holoenzymes from Boehringer and USB were ∼15–20% and ∼65% active, respectively; concentrations refer to active holoenzyme. CopG was purified as described (24), and dialyzed against binding buffer (25 mM Tris–HCl pH 8.0, 1.5 mM EDTA, 6 mM DTT, 170 mM KCl, 5 mM MgCl2, 2.5 mM CaCl2, 7.5% glycerol and 5% ethylene glycol) to avoid changing the salt concentration when the protein is added to a binding mixture. Concentrations of CopG given throughout this article refer to total protomers. All other reagents were of the highest commercially available grade.
Formation and detection of protein–DNA complexes
Reactions were performed in 5 µl of the BB buffer (binding buffer plus 500 µg/ml bovine serum albumin). RNAP–DNA or CopG–DNA complexes were formed by mixing 2 nM (labelled) or 10 nM (unlabelled) DNA fragment with varying concentrations of either protein. RNAP–DNA-binding reactions were incubated either at 37°C for 30 min or at 0°C for 60 min. When indicated, RNAP–DNA complexes were treated with heparin (150 μg/ml final concentration, unless stated otherwise) for 5 min at the same temperature, to eliminate short-lived complexes. Treatment with the polyanion was omitted when binding was performed at 0°C, to avoid removal of the unstable complexes that usually result at this temperature. CopG–DNA mixtures were incubated at 37°C or 0°C for 10 min. Under the above conditions, binding mixtures reached equilibrium. Samples were loaded on running 5% PAA gels. The same temperature was maintained throughout the whole EMSA. Labelled DNA bands were detected by autoradiography and quantified with the storage phosphor technology, with the aid of a FLA-3000 (FUJIFILM) imaging system and the Quantity One software (Bio-Rad).
DNase I footprinting
Binding reactions (50 μl) contained 2 nM labelled DNA in BB buffer. Proteins CopG and/or RNAP were added at the concentrations indicated. Binding of RNAP was carried out at 37°C for 30 min, or at 0°C for 60 min. CopG binding was performed at 0°C or 37°C, for 10 min. Probing with DNase I was carried out at the same temperature as the binding of CopG and/or RNAP. Digestion (3 min) of the DNA was started by adding 2 × 10−5 (37°C) or 7 × 10−3 (0°C) units of DNase I. Samples were processed as described (15).
HO• footprinting
RNAP–DNA or CopG–DNA complexes were generated at 37°C in 100 μl of BB buffer. Samples were dialyzed against 8 mM Tris–HCl pH 8.0, for 60 min at 37°C to remove the glycerol, and then treated with heparin for 5 min at the same temperature. Although dialysis against low-salt buffer has been reported to stabilize the RNAP–promoter complexes (25), no differences in complex stability or in the footprinting pattern were observed when samples were dialyzed against 25 mM Tris–HCl pH 8.0 and 170 mM KCl. To cleave the DNA backbone, reagents for HO• production [Fe(EDTA)2−, H2O2 and sodium ascorbate] were added in two alternative ways: method 1, a freshly prepared 6×-concentrated mixture that gives final concentrations of 0.1 mM Fe (II), 0.2 mM Na2 EDTA, 0.3% H2O2 and 1 mM sodium ascorbate; or method 2, freshly made solutions which were added separately in the following order: sodium ascorbate to 1 mM, H2O2 to 0.03% and Fe (EDTA)2− [final concentrations of 9 mM for Fe (II) and 18 mM for Na2 EDTA]. Cleavage with HO• was allowed for 2 min at 37°C and stopped by addition of a thiourea/EDTA solution (final concentrations 9.5 mM thiourea and 1.7 mM EDTA). The treatment described as method 1, while not destabilizing the CopG–DNA complexes, disrupts the RNAP bound to the DNA so that only ∼30% of the initial fraction of complexes was observed after the 2 min HO• treatment. To obtain a clear RNAP-mediated footprinting pattern, RNAP–DNA complexes were next separated from free DNA by electrophoresis on native 5% PAA gels. The DNA of the complex was eluted and precipitated before loading onto the sequencing gel. When the RNAP binding mixtures were probed with HO• under the conditions described as method 2, more than 90% of the total DNA remained complexed to RNAP after the 2 min treatment, making isolation of RNAP–DNA complexes unnecessary, as was the case with the CopG binding mixtures treated as in method 1. DNA was analyzed on 8% PAA sequencing gels. G+A sequencing reactions were performed as described by (26).
Stability of the RNAP–DNA and CopG–DNA complexes
Equilibrium mixtures were prepared by incubating 2 nM labelled DNA fragment and varying concentrations of protein (RNAP or CopG) in BB buffer, at either 37°C or 0°C. Dissociation of complexes between labelled DNA and either protein was made irreversible by addition of the indicated molar excess of competing unlabelled promoter fragment. Samples were taken at intervals and applied directly to running gels. Fractions of complexed- and free-labelled DNA were directly quantified as above. Data were analyzed according to the equation for a first-order kinetic process:
| 1 |
where [Protein-DNA] represents the concentration of the complex between either RNAP or CopG and the labelled DNA at time t, and [Protein-DNA]0 is its value at t = 0. Dissociation rate constants of the complexes (kd ± 2 SE) were estimated from least-squares analysis of the data. The half-life (t1/2) of the complexes was calculated according to the following equation:
| 2 |
RESULTS
Analysis of the DNA backbone regions of promoter Pcr contacted by RNAP at 0°C and 37°C
Isomerization of a short-lived closed complex to a stable open complex is usually accompanied by extension of the enzyme contacts to regions downstream of the transcription start site (position +1) (25,27). In addition, kinetic studies have evidenced that unstable closed complexes accumulate at binding temperatures below 10°C, whereas the open complex predominates above 21°C (25,28,29). However, the characteristic DNA-protection pattern of the different complexes that can be ‘trapped’ and analyzed has to be investigated for each given promoter, as several examples differing from the above general scheme have been reported. Thus, a short-lived closed complex that displays RNAP contacts spanning ∼30 bases downstream of the +1 site is generated at 0°C between σ70 RNAP and the λPR promoter (30). Also, the downstream boundary of the DNase I footprint of the complex formed at 4°C between σ70 RNAP and the phage λ prmup-1 Δ265 promoter is around position +10 (31).
The contact pattern on both DNA strands of the binary complexes generated upon binding of RNAP to the pMV158 Pcr promoter at 0°C and at 37°C were analyzed by chemical (hydroxyl radical, HO•) and enzymatic (DNase I) probing of the DNA backbone reactivity. At 0°C, DNase I footprinting revealed strong RNAP-mediated protections extending from –43 to –25 and from –18 to –6 on the non-template strand, and, on the template strand, from –45 to –27 and from –25 to –8. In addition to these main footprints, a weak protection against DNase I cleavage was observed downstream of the transcription start site (from +16 to +22 on the non-template strand, and from +13 to +17 on the template strand; Figure 1B and Supplementary Figure S1). The different degree of protection observed between the upstream and downstream footprints suggests that the binding equilibrium at 0°C is heterogeneously populated. In the main complex, the overall protected area would only span upstream of the start site (between positions –45 and –6), yielding a footprint pattern similar to those observed for low-temperature complexes at a variety of promoters (typically from positions –55 to –5). The weakly protected area could arise from a small fraction of complexes in which the RNAP contacts extend further downstream, reaching around +20. The presence of a mixed population of RNAP–promoter complexes at equilibrium at a temperature as low as 0°C seems to be very uncommon, and has only been reported for the E. coli σ32 RNAP–groE promoter binary complex (32). At 37°C, the same average degree of protection was observed upstream and downstream of the +1 site, with strong footprints spanning from –45 to –25 and from –23 to +22 on the non-template strand, and from –45 to –27 and from –25 to +17 on the template strand (Figure 1B and Supplementary Figure S1). Thus, at 37°C the overall protected area extended further downstream by about 25 nt relative to that in the predominant complex generated at 0°C.
Additional information on the conformation of the RNAP–Pcr binary complex generated at 37°C was achieved by HO• probing of the reactivity of the DNA backbone of the template strand around the +1 site. High reactivity of the sequence between positions –7 and +2 of only the template strand has been observed in the E. coli RNAP–T7 A1 promoter complex generated at 37°C, whereas the same region is protected in the 20°C complex and, to a lesser extent, in the complex resulting at 30°C (25). This strand-specific, temperature-dependent sensitivity of the +1 region towards HO• has been proposed to characterize the open complexes by distinguishing them from intermediate closed complexes that otherwise have the same upstream and downstream boundaries of the overall protected region (25). After binding of RNAP to Pcr at 37°C and selection of the stable binary complexes by a brief treatment with heparin, the DNA was cleaved with HO•. Hypersensitive sites, surrounded by protected regions, were clearly visualized between positions –4 and +2 of the template strand (Figure 1B and Supplementary Figure S2), suggesting that at least a significant fraction of the complexes generated at 37°C are in an open conformation.
Stability of the binary RNAP–Pcr complexes generated at 0°C and 37°C
We analyzed the kinetics of dissociation of the short and extended RNAP–Pcr complexes generated at 0°C and 37°C, respectively. Equilibrium mixtures containing 2 nM-labelled promoter fragment and 150 nM RNAP were prepared at each temperature (under these conditions, essentially all Pcr promoters are bound by the polymerase). Dissociation was initiated by addition of a 1500-fold excess (3 μM) of unlabelled promoter fragment at t = 0, and samples were analyzed at intervals.
We were unable to measure the half-life of the 0°C complexes, since immediately after the addition of the competing unlabelled DNA only ∼10% of the labelled promoter DNA remained bound to RNAP, and this percentage barely decreased for the whole 30-min period of competition (not shown). These results reinforce the idea of the heterogeneity of the population of binary 0°C complexes, suggesting that it is mostly composed of very short-lived complexes which are in a rapid equilibrium with free RNAP and promoter. Only a minor fraction of the complexes populated at binding equilibrium at this temperature would be stable, dissociating very slowly into their components. These unstable and stable complexes might correspond, respectively, to the short-footprint and to the extended-footprint complexes that were assumed to coexist at binding equilibrium at 0°C from DNase I footprinting experiments (see above).
On the other hand, most of the binary complexes generated at 37°C resisted a brief exposition to the competitor, as ∼80% of the labelled promoter DNA remained bound to RNAP just after the addition of the unlabelled DNA (t = 0) (Figure 2A). In addition, dissociation of these RNAP–Pcr complexes was very slow (Figure 2B) and appeared to follow a first order kinetic which yielded a dissociation rate constant kd = (6.3 ± 0.7) × 10−5 s−1 (t1/2 ∼180 min). To discard this low kd being due to the stoichiometric excess of RNAP (150 nM) over the labelled promoter fragment (2 nM), experiments of dissociation of RNAP–Pcr complexes generated at 37°C were also performed by equilibrating 2 nM 32P-labelled DNA and 8 nM RNAP and, at t = 0, adding a 100-fold molar excess (200 nM) of unlabelled promoter fragment. In this case (Figure 2B), a kd = (6.0 ± 0.8) × 10−5 s−1 was estimated from the data, which was not significantly different from that obtained when 150 nM RNAP was used. Similar kd values were obtained when either 50 μg/ml or 150 μg/ml of heparin were added, instead of the unlabelled promoter fragment, to sequester free RNAP from pre-equilibrated binding mixtures containing 2 nM 32P-labelled DNA and 8 nM RNAP (data not shown). These results indicate that the apparent rate of dissociation of RNAP from Pcr is not affected by the nature or concentration of the RNAP-quenching agent, which would be thus unable to actively displace RNAP from the complexes, and would only sequester the free enzyme. Estimated kd values show that the major RNAP–Pcr complexes generated at 37°C are very stable, with a half-life of ∼3 h. The stability of these complexes is well within the range reported for the RNAP–promoter complexes generated at 37°C (6,33).
Figure 2.

Kinetic of dissociation of RNAP from Pcr at 37°C. (A) EMSA analysis of the stability of the RNAP–Pcr complexes. Equilibrium mixtures contained 150 nM RNAP and 2 nM labelled DNA. Samples were analyzed at the indicated times following addition of 3 μM unlabelled DNA. Bands corresponding to free DNA (fDNA) and to specific RNAP–DNA complexes are indicated. On addition of competitor (t = 0), the complex fraction was estimated to be 0.8. In the absence of competitor, slower-migrating complexes (C*) that contained several RNAP molecules were seen. All the lanes displayed came from the same gel. (B) Time course of RNAP–Pcr complex dissociation. Linear fits of data from each of two experimental conditions are shown.
CopG prevents binding of RNAP to Pcr promoter
To disclose the mechanism of CopG-mediated repression of Pcr we first investigated the step at which the repressor prevents transcription from this promoter. To this end, the promoter fragment (2 nM) was equilibrated, at 0°C or 37°C, with excess CopG (120 nM), so that virtual saturation of the operators was obtained. Subsequently, RNAP was added, also at a molar excess (150 nM) relative to the promoter DNA, and incubation continued for 60 more minutes at 0°C or for 30 more minutes at 37°C, prior to DNase I probing of the protein footprints (Figure 3A and B). At either temperature, the footprint pattern of the sample containing both proteins was almost identical to that of the DNA incubated with CopG alone. By decreasing the repressor concentration, the RNAP-specific footprint pattern was increasingly apparent (not shown). These results indicated that CopG was able to prevent subsequent binding of RNAP to Pcr. To verify that addition of RNAP to a preformed CopG–DNA complex does not result in the formation of a ternary repressor–DNA–RNAP complex, we analyzed the electrophoretic mobility of DNA samples that were incubated in the absence of CopG or in the presence of increasing concentrations of the repressor prior to the addition of RNAP (Figure 3C). The samples were treated with heparin before loading onto the gel, so that unspecific complexes resulting from the binding of additional molecules of RNAP to non-promoter regions were removed and could not interfere with identification of any existing ternary complex generated by simultaneous binding of both proteins to the operator/promoter region of the DNA. As heparin treatment also removes unstable specific closed complexes, electrophoretic mobility shift assay (EMSA) was only performed with binding mixtures prepared at 37°C. Bands that migrate as the specific binary CopG–DNA or RNAP–DNA complexes, but no super-shifted bands indicative of stable ternary complexes, were seen at all CopG concentrations. Increasing concentrations of the repressor resulted in a decrease of the fraction of DNA bound to RNAP and a concomitant increase of the fraction of CopG–DNA complexes (Figure 3C). Similarly, in experiments in which increasing concentrations of RNAP were added to a CopG–DNA equilibrium mixture, the fraction of RNAP–DNA complex was seen to increase while that of the CopG–DNA complexes decreased (not shown). Thus, the success of CopG in preventing formation of a stable RNAP–Pcr complex depends on its own concentration and that of RNAP. These results, together with those of the DNase I footprinting assays performed at 0°C or 37°C in the presence of both proteins, show that binding of CopG and RNAP are mutually exclusive, and that CopG competes with RNAP for binding to the target DNA. RNAP was able to displace DNA-bound CopG, as revealed by the appearance of RNAP–DNA complexes upon addition of the enzyme to binding mixtures in which virtually all the operator sites were saturated with CopG (Figure 3C, compare the samples containing 80 nM CopG, with and without RNAP). A passive displacement of the repressor from its complexes with the operator, resulting from the binding of RNAP to the free DNA present in the CopG equilibrium binding mixture, could account for this observation, provided that CopG–DNA complexes dissociate rapidly on the time scale of the incubation with RNAP.
Figure 3.

CopG and RNAP compete for binding to the Pcr region. (A, B) DNase I footprinting of binding mixtures containing CopG and/or RNAP. Labelled DNA was incubated at 0°C (A) or 37°C (B) in the presence (+) or in the absence (−) of CopG and RNAP prior to digestion with DNase I. When both proteins were included, CopG was added before RNAP. Cleavage products on the non-template DNA strand are shown, with regions protected by CopG or RNAP indicated by thin or thick brackets, respectively. RNAP footprints are named as in Figure 1B. Enhancements due to CopG binding are indicated by arrows. Dideoxy sequencing reactions on the same DNA are included. (C) EMSA analysis of the complexes formed at 37°C in the presence of CopG and/or RNAP. Unlabelled DNA (10 nM) was incubated with the indicated concentrations of CopG prior to addition of RNAP. Samples were treated with heparin before loading onto the gel. Bands corresponding to free DNA (fDNA), and to CopG–DNA and RNAP–DNA complexes are indicated.
CopG-operator complexes are unstable
The kinetics of dissociation of CopG-operator complexes at 37°C could only be measured by employing a moderate (50-fold) excess of competing unlabelled DNA (Figure 4). When a higher excess (500- or 1500-fold) was used, no DNA complexed to CopG could be detected immediately on addition of the unlabelled DNA (not shown). Thus, the lifetime of CopG-operator complexes seems to depend strongly on the concentration of competing DNA, which suggests that ‘direct transfer’ of CopG to another DNA molecule contributes to the dissociation mechanism of these complexes. DNA concentration-dependent dissociation has been shown for the lac repressor and CAP proteins complexed to their target DNAs (34,35). Data for dissociation kinetic arose from three independent experiments, each performed at a different CopG concentration and thus displaying a distinct distribution of free and complexed DNA in the equilibrium mixture prior to the addition of the competing DNA. All of them gave similarly high dissociation rate constants (Figure 4). When data of all three experiments were analyzed conjointly according to Equation (1) for a pseudo first-order process, the apparent dissociation rate constant was estimated to be (5.0 ± 0.3) × 10−2 s−1 (Figure 4), which corresponded to a t1/2 ∼14 s. The low stability of the CopG-operator complexes at 37°C could account for the observed displacement of DNA-bound CopG by RNAP (Figure 3C), as the addition of the polymerase to a previously equilibrated CopG–DNA binding mixture would passively displace the equilibrium in the direction of dissociation of the CopG–DNA complexes, by sequestering free DNA.
Figure 4.

Kinetic of dissociation of CopG from its operator at 37°C. (A) EMSA analysis of the stability of the CopG–DNA complexes. Dissociation of complexes between CopG (40 nM) and the labelled DNA was initiated by addition of a 50-fold excess of unlabelled DNA (t = 0), and samples were analyzed at the indicated times. The sum of the various CopG–DNA complexes (in brackets) was used to analyze the time course of the fraction of labelled DNA complexed to CopG. Samples of free DNA (fDNA) and of the equilibrium mixture without competitor were also loaded at t = 120. All the lanes displayed came from the same gel. (B) Time course of CopG–DNA complex dissociation. Data from three independent experiments, each performed at the indicated CopG concentration, are included. The kd values estimated were (5.5 ± 0.1) × 10−2 s−1 (16 nM CopG), (4.8 ± 0.2) × 10−2 s−1 (40 nM CopG), and (5.3 ± 0.7) × 10−2 s−1 (80 nM CopG). The solid line is the linear fit of all data.
CopG dislodges RNAP stably bound to Pcr promoter
Evidences of the ability of CopG to displace RNAP stably bound to the Pcr promoter were obtained by inverting the order in which the repressor and the polymerase were added to the target DNA. In the absence of CopG, RNAP formed very stable complexes with Pcr at 37°C (Figure 2). When preformed RNAP–Pcr complexes were challenged for 10 min with increasing concentrations of CopG, a decline of the RNAP-specific complexes was observed which was paralleled by an increase in the amount of CopG–DNA complexes (Figure 5A and B). The CopG–DNA complexes generated in the presence of RNAP exhibited the same electrophoretic mobility as those produced in the absence of the polymerase at identical repressor concentrations (Figure 5A and B). CopG-dependent decrease of the RNAP–Pcr complexes was observed irrespective of whether the concentration of RNAP used in the assays was 150 nM (Figure 5A) or 8 nM (Figure 5B). Almost complete removal of the RNAP–Pcr complexes was obtained after treatment at the highest CopG concentrations (Figure 5A and B), even though the 10-min incubation time is quite short relative to the half-life of these complexes (∼180 min). Therefore, it appeared of interest to analyze the time course of the CopG-mediated displacement of RNAP from Pcr by challenging the specific RNAP–Pcr complexes with CopG for different times. Several independent experiments were performed over a range of concentrations of CopG, RNAP and heparin (four of these experiments are shown in panels A, B, C and D of Figure 6). Each experiment included control samples in which only heparin (instead of heparin followed by CopG) was added at t = 0. In every experiment, the CopG-containing samples exhibited repressor-concentration-dependent decreases in the fraction of RNAP–Pcr complexes, compared with controls (Figure 6 and Supplementary Tables 1, 2, 3 and 4). Maximal extent of the CopG-mediated RNAP displacement was reached within 1 min of incubation with the repressor, while longer incubations did not result in any further significant decrease in the fraction of RNAP–Pcr complexes relative to the corresponding control samples (Figure 6 and Supplementary Tables 1, 2, 3 and 4). It is worth noting that the experiment in Figure 6D was carried out employing the E. coli RNAP holoenzyme from USB and two different heparin concentrations. As can be seen, the use of a heparin concentration more than three times greater than that required to compete unstable RNAP–DNA complexes did not significantly affect either the stability of the RNAP–Pcr specific complexes in the absence of CopG or the level and time course of the CopG-mediated displacement of RNAP from the promoter (Figure 6D and Supplementary Table 4). The time-course experiments also showed that CopG-mediated dissociation of the RNAP–Pcr complexes occurs regardless the concentration and the source of RNAP employed, which allows us to rule out any artefacts due to a particular commercial holoenzyme preparation.
Figure 5.

CopG-concentration-dependent dissociation of the RNAP–Pcr complexes at 37°C. RNAP (at the concentrations indicated in A and in B) and DNA (2 nM) were equilibrated at 37°C and then treated with heparin (150 μg/ml in A, and 10 μg/ml in B) before adding different amounts of CopG. Incubation of the mixtures continued for 10 more minutes before loading onto the gel. CopG–DNA complexes formed in the absence of RNAP were also analyzed. Bands corresponding to free DNA (fDNA) and to RNAP–DNA and CopG–DNA complexes are indicated. At high CopG concentrations (>200 nM), complexes migrating slower than the specific complex generated by binding of four CopG dimers to the operator DNA (17) were observed. These slowly-migrating complexes contain additional repressor molecules nucleated from the operator (39). All the lanes displayed came either from the same gel or from gels prepared and run in parallel. Only one of two independent experiments yielding identical results is shown in each panel. Note that in the absence of heparin, slowly migrating RNAP–DNA complexes that contained several RNAP molecules were seen (C* in panel B).
Figure 8.

Schematic representation of the contacts of CopG and RNAP with the DNA backbone of the Pcr promoter region at 37°C. RNAP contacts are denoted by red letters. Red arrows indicate positions in the RNAP–DNA complex with enhanced backbone cleavage. Contacts by CopG are shadowed in green. The SE is boxed. The binding sites of CopG dimers are indicated with brackets. The right half of the SE (RSE) is the CopG primary site. The −35 and extended −10 sequences of Pcr are denoted by lines and encircled letters in blue colour.
Figure 6.

CopG-mediated rapid dissociation of the RNAP–Pcr complexes at 37°C. (A, B, C, D) EMSA from four independent time-course experiments performed over a range of CopG and RNAP concentrations. RNAP holoenzyme was purchased from Boehringer (panels A, B and C) or from USB (panel D). DNA (2 nM) and RNAP at the indicated concentrations were equilibrated at 37°C and then treated at t = 0 with heparin (150 μg/ml in A and B, 10 μg/ml in C, and either 150 μg/ml or 500 μg/ml in D) followed by different concentrations of CopG. Incubation of the mixtures continued for several times before loading onto the gel. CopG–DNA complexes formed in the absence of RNAP were also analyzed. Bands corresponding to free DNA (fDNA) and to RNAP–DNA and CopG–DNA complexes are indicated. At high CopG concentrations (>200 nM), CopG–DNA complexes migrating slower than the specific complex generated by binding of four repressor dimers were observed. All the lanes shown in each experiment came either from the same gel or from gels prepared and run in parallel. Note that in the absence of heparin, slowly migrating RNAP–DNA complexes that contained several RNAP molecules were observed (C* in panel D). The percentages of RNAP–DNA and CopG–DNA complexes quantified from the EMSA displayed in panels A, B, C and D, are shown in Supplementary Tables 1, 2, 3 and 4, respectively.
Taken together, the above results show that CopG actively replaces the polymerase bound to the promoter/operator region. The CopG-mediated displacement of RNAP from Pcr depended on the repressor concentration and constituted a rapid process that took place in less than 1 min after addition of the repressor (Figures 5 and 6, and Supplementary Tables 1, 2, 3 and 4).
CopG and RNAP contact the promoter/operator region by different, though overlapping, sides of the DNA double helix
To analyze the mechanism by which CopG displaces RNAP from Pcr we determined the precise contacts of both proteins with the DNA backbone of the promoter/operator region (Figure 7 and Supplementary Figures S3 and S4).
Figure 7.

Summary of HO• footprints of the RNAP–Pcr complexes at 37°C. The schematized protection pattern corresponds to the footprinting experiment in Supplementary Figure S4. Promoter positions relative to the transcription start site are indicated. Protections are shown by brackets. Protections denoted by the same letter in both strands lie across the minor groove of the DNA. Enhancements are shown by arrows.
HO• footprinting of the CopG–DNA specific complexes generated at 37°C revealed five protected regions on each strand (a, b, c, d and e; Supplementary Figure S3), which basically matched the footprints observed upon binding of the protein at 20°C (15). CopG footprints showed that the protein binds to one face of the DNA helix (15), and delimit four regions of the major groove (Figure 8) whose bases are specifically contacted by CopG dimers [(17); unpublished data]. The two central CopG-binding sites correspond to the left (LSE) and right (RSE) halves of the SE, whose atomic structure in complex with CopG has been solved (16). The two additional binding sites, hereafter named Left Arm (LA) and Right Arm (RA), are located one helical turn apart from the LSE and the RSE, respectively (Figure 8).
HO• probing of the heparin-resistant RNAP–Pcr complexes generated at 37°C gave a footprint pattern that corresponded to extended binary complexes (Figure 7 and Supplementary Figure S4) and was similar to those observed for open complexes in other RNAP/promoter systems (25,31,36). Upstream of the −10 region of Pcr the periodicity of the RNAP footprints indicated that the polymerase contacted the DNA backbone by one face of the double helix, whereas downstream from this region the contacts involved the entire DNA helix (Figure 8). The large downstream footprint interrupted only around position +1 on the template strand at a few sugar-phosphate bonds which were sensitive or even hypersensitive towards HO• (Supplementary Figures S2 and S4). This strand-specific unprotected region around the transcription start site could be considered as a hallmark of the open complexes (25,27,36), which most likely populate the RNAP–Pcr complexes generated at 37°C. Upstream from the −10 region of Pcr, contacts of CopG with the DNA backbone were shifted on the double helix by some 80° (about 0.23 helix turns on average) relative to those of RNAP, although some sugar-phosphate bonds were protected by both proteins (Figure 8). Hence, CopG and RNAP interact with the promoter/operator region on different faces of the DNA.
The RSE of the operator DNA is the primary binding site of CopG
To know the relevance of the different elements constituting the operator of CopG, we analyzed the effect of changing the nucleotide sequence of each of the four CopG-binding sites on the affinity of the protein. To this end, we measured the binding affinity of CopG for mutant operators LSE−, RSE−, LA− and RA− relative to the wild-type operator (see the scheme of the mutants in Table 1 and the experimental design in Supplementary Materials and Methods section and Figure S5). Substitution of the RSE by an unspecific sequence affected binding of CopG most severely, resulting in a ∼300-fold reduction in the affinity of the protein for the RSE− mutant operator (Table 1). In contrast, a much lower reduction (by only ∼6-fold) in binding affinity was obtained when the LSE was replaced by the unspecific sequence (Table 1). Changing the sequence of either the LA or the RA also caused differential effects on the affinity of CopG binding: whereas replacement of the LA reduced the affinity by ∼2.5-fold, substitution of the RA had the same effect (a 6–7-fold reduction in the affinity) as changing the LSE (Table 1). These results indicated that the RSE contributed most to the high affinity of binding of CopG to its operator, while the LA had the lowest contribution. To verify whether the RSE was the preferred CopG-binding site, the relative affinity of the protein for mutant operators containing a single binding site was also measured (Table 1 and Supplementary Figure S5). CopG exhibited detectable specific binding affinity exclusively for the RSE+ operator, which only harboured the wild-type RSE. The relative affinity of CopG for the other single-binding-site operator mutants could not be measured, as was also the case for the non-specific (NS) oligonucleotide lacking the four CopG-binding sites (Table 1 and Supplementary Figure S5). These results show that the affinity of binding of CopG to the LSE+, LA+ and RA+ operator mutants is very low and most likely approaches non-specific levels. Remarkably, the CopG affinity for the RSE+ operator was ∼2.5-fold greater than the affinity of the protein for the RSE− mutant operator (Table 1), therefore showing that the presence of the sole RSE is more important for the binding affinity of the protein than the presence of the other three sites together. The overall results obtained from the analysis of the affinity of CopG for operator variants allow us to conclude that the RSE is the binding site preferred by CopG within the operator, that is to say, the RSE is the primary binding site of the protein. Moreover, the RSE is the only binding site whose presence guarantees specific binding of CopG. Since the RSE is required for specific and high-affinity complex formation by CopG, it appears that accessibility of this binding site is an essential factor for the binding of this protein to its operator.
Table 1.
CopG binding affinity to different operator variants
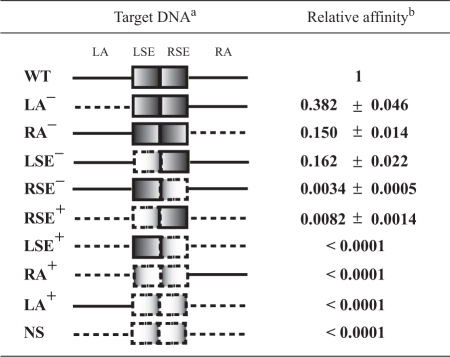 |
The four binding sites of the operator (LA, LSE, RSE and RA) are indicated on the schematic representation of the wild-type target DNA (WT). Continuous lines denote wild-type sequence regions. Dashed lines represent DNA regions with substituted sequence. The names of the mutant operators are indicated on the left.
The affinities of binding of CopG to the 55-bp operator variants relative to the 239 wild-type DNA fragment are shown. The number of samples independently analyzed to calculate each relative binding affinity varied between 17 and 33. The calculated relative affinities were normalized for the value of the ratio between the affinity of CopG for the 55-bp wild-type oligonucleotide and the affinity of the protein for the 239-bp wild-type operator DNA fragment (0.85 ± 0.04).
DISCUSSION
Plasmid pMV158 possesses a very promiscuous replicon that functions in E. coli (37). This implies that Pcr promoter, which directs transcription of the repB gene essential for plasmid replication, is recognized by RNAP from E. coli. In fact, transcription from Pcr by E. coli RNAP as well as CopG-mediated repression of it have been reported both in vivo and in vitro (15), although the bases underlying these processes had not been investigated. Here, we have characterized the complexes formed by RNAP at Pcr and analyzed the mechanism by which CopG represses transcription from this promoter.
As deduced from the contradictory story with Lac repressor (6), elucidation of the precise step at which a given repressor protein inhibits the reversible process of formation of a transcriptionally active open complex requires the previous knowledge of the dissociation rate constants of the operator/repressor and promoter/RNAP complexes. At promoter Pcr, typical unstable short closed and stable extended open complexes were generated upon binding of RNAP at 0°C and 37°C, respectively (Figures 1 and 2). The stability of the RNAP–Pcr complexes at 37°C (t1/2 ∼3 h; Figure 2) contrasted with the short half-life (t1/2 ∼14 s) of the complexes generated by binding of CopG to the promoter/operator region at this temperature (Figure 4). Hence, on the time scale of our experiments at 37°C, CopG–DNA complexes dissociated rapidly whereas RNAP–Pcr complexes hardly dissociated.
Clues to the mechanism of CopG-mediated repression of transcription from Pcr were provided from competition assays in which the order of addition of polymerase and repressor was inverted: (i) no ternary specific complexes were observed in any case (Figure 3C), which evidences mutually exclusive binding of CopG and RNAP to the promoter/operator region; (ii) CopG induced disassembly of preformed stable RNAP–Pcr complexes (Figures 5 and 6, and Supplementary Tables 1, 2, 3 and 4); and (iii) in the presence of CopG and RNAP, the apparent equilibrium extent of formation of RNAP–Pcr complexes depended on the concentration of both proteins (Figures 3 and 5). Therefore, CopG seems to repress transcription initiation from Pcr by acting at two different steps in the process of formation of an open complex. First, CopG competes with RNAP for binding to the same region of DNA, where promoter and operator overlap. At this stage, CopG bound to its target DNA would hinder the access of RNAP to the promoter region by a steric exclusion mechanism, which constitutes a classic model of repressor action. The strong DNA bend induced by CopG toward the repressor–DNA interface (16) might play a role in this mechanism as well. Secondly, CopG is able to displace RNAP stably bound to Pcr, which constitutes an unexpected ability with no obvious explanation. We propose a model for the CopG-induced disassembly of the stable RNAP–Pcr complexes which takes into account the small size of CopG (a dimeric protein of 45 amino acids per subunit), the sequential and cooperative binding of CopG to the four sites that constitute the operator, and the accessibility of the CopG primary binding site in the RNAP–Pcr complexes. Specific CopG–operator complexes result from cooperative binding of four CopG dimers on the same face of the DNA helix (Figure 8). The repressor interacts specifically through the major groove of the DNA with four sites separated one helical turn from each other (17). The two central sites correspond to the LSE and RSE elements of the operator (Figure 8). Each of the two outer sites (LA or RA) is located one helical turn apart from either central site. By employment of operator mutants that either lack or harbour only one of these binding sites, we have shown that the RSE is the primary binding site of CopG (Table 1 and Supplementary Figure S5) and hence it is, most likely, the first site occupied during the sequential and cooperative binding of the repressor. This primary CopG-binding site could be accessible to the repressor even in the stable RNAP–Pcr complexes generated at 37°C since at the RSE region there are only two phosphate groups contacted by both proteins (Figure 8), and there are no expected contacts of RNAP with specific bases of the DNA. This situation is unique for the primary binding site of CopG, because the LSE and RA sites where subsequent repressor molecules would bind contain, respectively, the −35 and the −10 hexamers of Pcr, as well as a higher number of phosphate groups contacted by CopG and RNAP (Figure 8). Thus, the proposed sequence of events in the model for CopG-induced disassembly of the stable RNAP–Pcr complexes would be as follows: a repressor dimer reaches its primary binding site at the right half of the SE and establishes contacts with specific bases of the DNA through the major groove. This preliminary CopG–operator interaction might change the conformation of the DNA, thus weakening the contacts between RNAP and the nucleic acid. If repressor concentration is high enough, binding of CopG to the primary site may promote rapid successive binding to the other sites in the operator, so that CopG would displace RNAP from the promoter (Figure 8). By acting at two different steps during open complex formation, repression by CopG does not consist in a simple mechanism of competition between free molecules of RNAP and repressor for binding to the same target DNA, since CopG can also establish interactions with its operator when RNAP is already bound to the Pcr promoter. Repression of transcription initiation at two different steps might allow a fine-tuning of the expression from Pcr in response to the intracellular concentration of CopG. Hence, the proposed dual repression mechanism could be appropriate for a control system involved in regulation of plasmid replication, as variations in the intracellular conditions would be rapidly sensed due to the short half-life of the CopG/operator complexes and to the ability of CopG to dislodge the bound RNAP.
Repression of transcription initiation by disassembly of the open complex seems to be an uncommon mechanism, which has only been reported previously for the E. coli repressor IclR (38). IclR represses expression of the aceBAK operon (involved in the glyoxylate bypass) through two different mechanisms, each acting at a distinct step of the open complex formation process. First, IclR binds to its primary site (IclR box II), located between −52 and −19 of the aceB promoter, thus interfering with binding of RNAP to this promoter. Second, at higher repressor concentrations, IclR binds to its secondary site (IclR box I), located between −125 and −99 of the aceB promoter, even after formation of an open complex on this promoter, and induces disassembly of RNAP from the promoter. This latter mechanism of transcription repression is based on the establishment of direct interactions between the C-terminal domain of RNAP α subunit and IclR bound to its secondary binding site (38).
CopG- and IclR-mediated repression of transcription from Pcr and aceB promoters, respectively, displays both similarities and differences. CopG and IclR can act at two distinct steps of the open complex formation, namely by competing with RNAP for binding to the DNA or by disassembling the open complex at the respective promoter. However, whereas repression by IclR involves two different mechanisms (steric exclusion of, and interaction with, RNAP) and two different binding sites (boxes II and I), cooperative binding of four CopG molecules to its single operator would be responsible for the dual repression mechanism reported here. From this, a picture emerges of CopG being the minimal protein element able to bind specifically and cooperatively to the operator in such a way that it not only prevents the binding of the RNAP, but also displaces efficiently the polymerase bound to the regulated promoter.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministerio de Ciencia e Innovación [grants BFU2004-00687/BMC and BFU2007-63575 to G.d.S.]; [grant INTERMODS-CSD2008-00013 to M.E.]; A.M.H.A. by [grant BFU2005-03911 to Prof. R. Díaz-Orejas]. Funding for open access charge: Grant BFU2007-63575 to G.d.S. from Ministerio de Ciencia e Innovación.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Thanks are due to M.T. Alda for purification of CopG; to members of our lab for helpful discussions and to A.M.L. Jones for correcting the English throughout the manuscript.
REFERENCES
- 1.Rojo F. Repression of transcription initiation in bacteria. J. Bacteriol. 1999;181:2987–2991. doi: 10.1128/jb.181.10.2987-2991.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rojo F. Mechanisms of transcriptional repression. Curr. Opin. Microbiol. 2001;4:145–151. doi: 10.1016/s1369-5274(00)00180-6. [DOI] [PubMed] [Google Scholar]
- 3.Hawley DK, Johnson AD, McClure WR. Functional and physical characterization of transcription initiation complexes in the bacteriophage λ OR region. J. Biol. Chem. 1985;260:8618–8626. [PubMed] [Google Scholar]
- 4.Bertrand-Burggraf E, Hurstel S, Daune M, Schnarr M. Promoter properties and negative regulation of the uvrA gene by the LexA repressor and its amino-terminal DNA binding domain. J. Mol. Biol. 1987;193:293–302. doi: 10.1016/0022-2836(87)90220-8. [DOI] [PubMed] [Google Scholar]
- 5.Majors J. Initiation of in vitro mRNA synthesis from the wild-type lac promoter. Proc. Natl Acad. Sci. USA. 1975;72:4394–4398. doi: 10.1073/pnas.72.11.4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlax PJ, Capp MW, Record MT. Inhibition of transcription initiation by lac repressor. J. Mol. Biol. 1995;245:331–350. doi: 10.1006/jmbi.1994.0028. [DOI] [PubMed] [Google Scholar]
- 7.Nieto C, Puyet A, Espinosa M. MalR-mediated Regulation of the Streptococcus pneumoniae malMP Operon at Promoter PM. Influence of a proximal divergent promoter region and competition between MalR and RNA polymerase proteins. J. Biol. Chem. 2001;276:14946–14954. doi: 10.1074/jbc.M010911200. [DOI] [PubMed] [Google Scholar]
- 8.Choy HE, Park S-W, Aki T, Parrack P, Fujita N, Ishihama A, Adhya S. Repression and activation of transcription by Gal and Lac repressors: involvement of alpha subunit of RNA polymerase. EMBO J. 1995;14:4523–4529. doi: 10.1002/j.1460-2075.1995.tb00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuhnke G, Theres C, Fritz HJ, Ehring R. RNA polymerase and gal repressor bind simultaneously and with DNA bending to the control region of the Escherichia coli galactose operon. EMBO J. 1989;8:1247–1255. doi: 10.1002/j.1460-2075.1989.tb03498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monsalve M, Mencía M, Rojo F, Salas M. Activation and repression of transcription at two different phage ϕ29 promoters are mediated by interaction of the same residues of regulatory protein p4 with RNA polymerase. EMBO J. 1996;15:383–391. [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider R, Travers A, Kutateladze T, Muskhelishvili G. A DNA architectural protein couples cellular physiology and DNA topology in Escherichia coli. Mol. Microbiol. 1999;34:953–964. doi: 10.1046/j.1365-2958.1999.01656.x. [DOI] [PubMed] [Google Scholar]
- 12.Schröder O, Wagner R. The bacterial DNA-binding protein H-NS represses ribosomal RNA transcription by trapping RNA polymerase in the initiation complex. J. Mol. Biol. 2000;298:737–748. doi: 10.1006/jmbi.2000.3708. [DOI] [PubMed] [Google Scholar]
- 13.Shin M, Song M, Rhee JH, Hong Y, Kim Y-J, Seok Y-J, Ha K-S, Jung S-H, Choy HE. DNA looping-mediated repression by histone-like protein H-NS: specific requirement of Eσ70 as a cofactor for looping. Genes Dev. 2005;19:2388–2398. doi: 10.1101/gad.1316305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J, Koudelka GB. Repression of transcription initiation at 434 PR by 434 repressor: effects on transition of a closed to an open promoter complex. J. Mol. Biol. 2001;309:573–587. doi: 10.1006/jmbi.2001.4702. [DOI] [PubMed] [Google Scholar]
- 15.del Solar G, Pérez-Martín J, Espinosa M. Plasmid pLS1-encoded RepA protein regulates transcription from repAB promoter by binding to a DNA sequence containing a 13-base pair symmetric element. J. Biol. Chem. 1990;265:12569–12575. [PubMed] [Google Scholar]
- 16.Gomis-Rüth FX, Solá M, Acebo P, Párraga A, Guasch A, Eritja R, González A, Espinosa M, del Solar G, Coll M. The structure of plasmid-encoded transcriptional repressor CopG unliganded and bound to its operator. EMBO J. 1998;17:7404–7415. doi: 10.1093/emboj/17.24.7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.del Solar G, Hernández-Arriaga AM, Gomis-Rüth FX, Coll M, Espinosa M. A genetically economical family of plasmid-encoded transcriptional repressors involved in control of plasmid copy number. J. Bacteriol. 2002;184:4943–4951. doi: 10.1128/JB.184.18.4943-4951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips SEV. Specific β-sheet interactions. Curr. Opin. Struct. Biol. 1991;1:89–98. [Google Scholar]
- 19.Raumann BE, Rould MA, Pabo CO, Sauer RT. DNA recognition by beta-sheets in the Arc repressor-operator crystal structure. Nature. 1994;367:754–757. doi: 10.1038/367754a0. [DOI] [PubMed] [Google Scholar]
- 20.Berggrun A, Sauer RT. Contributions of distinct quaternary contacts to cooperative operator binding by Mnt repressor. Proc. Natl Acad. Sci. USA. 2001;98:2301–2305. doi: 10.1073/pnas.041612198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Somers WS, Phillips SEV. Crystal structure of the met repressor-operator complex at 2.8 Å resolution reveals DNA recognition by β-strands. Nature. 1992;359:387–393. doi: 10.1038/359387a0. [DOI] [PubMed] [Google Scholar]
- 22.Smith TL, Sauer RT. P22 Arc repressor: role of cooperativity in repression and binding to operators with altered half-site spacing. J. Mol. Biol. 1995;249:729–742. doi: 10.1006/jmbi.1995.0332. [DOI] [PubMed] [Google Scholar]
- 23.Smith TL, Sauer RT. Role of operator subsites in Arc repression. J. Mol. Biol. 1996;264:233–242. doi: 10.1006/jmbi.1996.0637. [DOI] [PubMed] [Google Scholar]
- 24.Gomis-Rüth FX, Solá M, Pérez-Luque R, Acebo P, Alda MT, González A, Espinosa M, del Solar G, Coll M. Overexpression, purification, crystallization and preliminary X-ray diffraction analysis of the pMV158-encoded plasmid transcriptional repressor protein CopG. FEBS Lett. 1998;425:161–165. doi: 10.1016/s0014-5793(98)00219-1. [DOI] [PubMed] [Google Scholar]
- 25.Schickor P, Metzger W, Werel W, Lederer H, Heumann H. Topography of intermediates in transcription initiation of E. coli. EMBO J. 1990;9:2215–2220. doi: 10.1002/j.1460-2075.1990.tb07391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maxam AM, Gilbert W. Sequencing end-labelled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–559. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 27.Record MT, Reznikoff WS, Craig ML, McQuade KL, Schlaz PJ. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. 2nd. Neidhart FC, Curtiss III R, Ingraham JL, Lin ECC, Low KR, Magasanik B, Reznikoff WS, Riley M, Schaetchter M, Umbarger HE, editors. Washington, DC: ASM Press; 1996. pp. 792–820. [Google Scholar]
- 28.Kirkegaard K, Buc H, Spassky A, Wang JC. Mapping of single-stranded regions in duplex DNA at the sequence level: single-strand-specific cytosine methylation in RNA polymerase-promoter complexes. Proc. Natl Acad. Sci. USA. 1983;80:2544–2548. doi: 10.1073/pnas.80.9.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Straney DC, Crothers DM. Intermediates in transcription initiation from the E. coli lac UV5. Cell. 1985;43:449–459. doi: 10.1016/0092-8674(85)90175-8. [DOI] [PubMed] [Google Scholar]
- 30.Craig ML, Tsodikov OV, McQuade KL, Schlax PE, Capp MW, Saecker RM, Record MT. DNA footprints of the two kinetically significant intermediates in formation of an RNA polymerase-promoter open complex: evidence that interactions with start site and downstream DNA induce sequential conformational changes in polymerase and DNA. J. Mol. Biol. 1998;283:741–756. doi: 10.1006/jmbi.1998.2129. [DOI] [PubMed] [Google Scholar]
- 31.Li XY, McClure RW. Characterization of the closed complex intermediate formed during transcription initiation by Escherichia coli RNA polymerase. J. Biol. Chem. 1998;273:23549–23557. doi: 10.1074/jbc.273.36.23549. [DOI] [PubMed] [Google Scholar]
- 32.Cowing DW, Mecsas J, Record MT, Jr., Gross CA. Intermediates in the formation of the open complex by RNA polymerase holoenzyme containing the sigma factor σ32 at the groE promoter. J. Mol. Biol. 1989;210:521–530. doi: 10.1016/0022-2836(89)90128-9. [DOI] [PubMed] [Google Scholar]
- 33.Roe JH, Burgess RR, Record MT., Jr. Kinetics and mechanism of the interaction of Escherichia coli RNA polymerase with the λPR promoter. J. Mol. Biol. 1984;176:495–521. doi: 10.1016/0022-2836(84)90174-8. [DOI] [PubMed] [Google Scholar]
- 34.Fried MG, Liu G. Molecular sequestration stabilizes CAP-DNA complexes during polyacrylamide gel electrophoresis. Nucleic Acids Res. 1994;22:5054–5059. doi: 10.1093/nar/22.23.5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vossen KM, Fried MG. Sequestration stabilizes lac repressor-DNA complexes during gel electrophoresis. Anal. Biochem. 1997;245:85–92. doi: 10.1006/abio.1996.9944. [DOI] [PubMed] [Google Scholar]
- 36.Craig ML, Suh W-C, Record MT., Jr. HO• and DNase I probing of Eσ70 RNA polymerase-λPR promoter open complexes: Mg2+ binding and its structural consequences at the transcription start site. Biochemistry. 1995;34:15624–15632. doi: 10.1021/bi00048a004. [DOI] [PubMed] [Google Scholar]
- 37.Lacks SA, López P, Greenberg B, Espinosa M. Identification and analysis of genes for tetracycline resistance and replication functions in the broad-host-range plasmid pLS1. J. Mol. Biol. 1986;192:753–765. doi: 10.1016/0022-2836(86)90026-4. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto K, Ishihama A. Two different modes of transcription repression of the Escherichia coli acetate operon by IclR. Mol. Microbiol. 2003;47:183–194. doi: 10.1046/j.1365-2958.2003.03287.x. [DOI] [PubMed] [Google Scholar]
- 39.Costa M, Solá M, del Solar G, Eritja R, Hernández-Arriaga AM, Espinosa M, Gomis-Rüth FX, Coll M. Plasmid transcriptional repressor CopG oligomerises to render helical superstructures unbound and in complexes with oligonucleotides. J. Mol. Biol. 2001;310:403–417. doi: 10.1006/jmbi.2001.4760. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.