

Abstract

A catalytic enantioselective approach to the eudesmane sesquiterpenoids is reported. The strategic use of a palladium-catalyzed enantioselective alkylation of vinylogous ester substrates forged the C(10) all-carbon quaternary center. This key transformation enabled a diastereoselective olefin hydrogenation to create the syn stereochemistry at C(7). The devised synthetic strategy allowed for the preparation of the antibacterial agent (+)-carissone and a formal synthesis of the P/Q-type calcium channel blocker (–)-α-eudesmol.
The flowering plants of the family Asteraceae (Compositae) have many historical uses, including rubber, medicines, edible oils, vegetables, and pesticides.1 Among these flora are a large number of species that are abundant in structurally diverse sesquiterpenoids, particularly ones that contain the eudesmane skeleton (Figure 1). Over 1000 eudesmanes have been identified from these sources, with their structures diverging based on oxygenation and oxidation patterns within the carbon framework.
Figure 1.

Representative Eudesmane Sesquiterpenoids
This ever-growing2 class of important secondary metabolites possesses a wide range of biological properties, including plant growth inhibition, insect antifeedant, antibacterial, antifungal, and antitumor activities. Representative eudesmanes include antibacterial agents (+)-carissone (1)3 and (+)-3-oxocostusic acid (2),4 as well as P/Q-type calcium channel blocker (−)-α-eudesmol (3)5 (Figure 1). These examples typify common structural motifs within this class of sesquiterpenoids, including the C(10) all-carbon quaternary stereocenter and stereogenic C(7) substituent. The structural similarities and interesting biology associated with this class of molecules has stimulated several synthetic efforts,6,7,8 most of which employ semi-synthetic or chiral pool strategies. To date, no catalytic asymmetric approaches toward these eudesmanes have been developed. Herein we report an approach9 that incorporates our recent method for the catalytic asymmetric formation of enantioenriched all-carbon quaternary stereocenters into a general synthetic strategy for this class of sesquiterpenoids.
In devising a strategy for the total synthesis of the eudesmanes, we simplified our target to enone 5, which has been utilized in the preparation of structures such as 4,6d and itself embodies many features present in various family members (cf. 5 and 1, 2) (Figure 2). We envisioned that the stereochemistry of the C(7) substituent could arise by means of the diastereoselective hydrogenation of a substituted cyclohexene (i.e., 6), the stereochemical outcome of which would be controlled by the C(10) quaternary stereocenter. This cyclohexene could be obtainable from a ring-closing methathesis of triolefin 7, which would be derived from an appropriately substituted α-quaternary ketone (i.e., 8). Thus, the key control element in the design of this synthetic strategy is the C(10) quaternary stereocenter,10 and we therefore sought to develop an efficient and selective means for the preparation of this moiety.
The enantioselective alkylation of ketone enolates is an area of intense investigation in our laboratory.11 This method has resulted in the preparation of wide range of carbonyl compounds with adjacent quaternary stereocenters with high levels of selectivity and excellent yields, some of which have proved valuable in synthetic endeavors.12 The application of α-quaternary ketones such as 8 for the devised strategy would require a carbonyl transposition (i.e., 8 → 7), and we therefore chose to exploit the unique properties of vinylogous esters (i.e., 8 where R2 = OR) pioneered by Stork and Danheiser13 for this purpose.
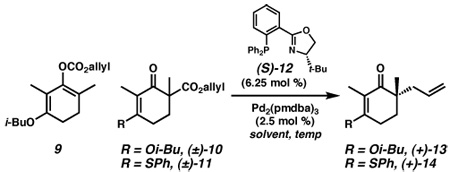
Our initial studies for the asymmetric generation of quaternary stereocenters utilitzing vinylgous ester derivatives focused on enol carbonates due to preliminary investigations12c,14 that have demonstrated successes for similar substrates. Exposure of allyl enol carbonate 9 to typical reaction conditions consisting of a palladium(0) catalyst and ligand (S)-1215 in toluene generated vinylogous ester (+)-13, albeit in variable yield and selectivity (Table 1, entry 1). Unfortunately, the instability of 9 impeded further studies as these results were highly dependent on the composition of this enol carbonate.16 Given the range of substrate possibilities for this transformation,11d we next focused on racemic β-ketoester (±)-10. Surprisingly, this substrate proved only modestly reactive at 50 °C, producing ketone (+)-13 in 19% yield and 79% ee (entry 2).17 Increasing the reaction temperature to 80 °C enabled complete conversion to ketone (+)-13, although with slightly reduced selectivity (entry 3). As the lack of reactivity seemed to be a major complication with this substrate, we considered vinylogous thioesters (i.e., (±)-11) for their reported activation properties.14 Indeed, racemic β-ketoester (±)-11 did prove more reactive and produced ketone (+)-14 at 50 °C in good yield and 92% ee (entry 4). A screen of solvents revealed that benzene (entry 5) and ethereal solvents (entries 6 and 7) provided similar selectivities to toluene.
Table 1.
Asymmetric Allylation of Vinylogous Ester Derivativesa
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate | solvent | temp (°C) |
product | yieldb (%) |
eec (%) |
| 1 | 9 | PhMe | 25 | 13 | 22–61 | 84–88 |
| 2 | 10 | PhMe | 50 | 13 | 19d | 79 |
| 3 | 10 | PhMe | 80 | 13 | 86 | 75 |
| 4 | 11 | PhMe | 50 | 14 | 86 | 92 |
| 5 | 11 | PhH | 50 | 14 | 61e | 92 |
| 6 | 11 | THF | 50 | 14 | 88 | 92 |
| 7 | 11 | dioxane | 50 | 14 | 90 | 91 |
pmdba = bis(4-methoxybenzylidene)acetone.
Isolated yields.
Enantiomeric excess determined by chiral HPLC or SFC.
β-ketoester (±)-10 was recovered in 69% yield.
β-ketoester (±)-11 was recovered in 26% yield.
With optimal conditions for the preparation of 14, we sought to demonstrate the feasibility of using this ketone for the total synthesis of (+)-carissone (1). Accordingly, racemic β-ketoester (±)-11 was converted to (−)-14 in 85% yield18 and 92% ee using ligand (R)-12, to correlate with the natural antipode of 1 (Scheme 2). Subsequent conversion of vinylogous thioester (−)-14 into vinylogous ester (−)-15 was achieved with sodium methoxide in refluxing methanol. Exposure of the resulting vinylogous ester to the substituted allylmagnesium bromide generated from 1619 and magnesium provided enone (−)-17 in 94% yield. The success of allylmagnesium bromide additions to vinylogous ester (−)-15 encouraged us to investigate similar reactions of various organometallic reagents with vinylogous thioester (−)-14, however, several conditions provided intractable mixtures with no desired products.20 Nonetheless, ring-closing metathesis of enone (−)-17 using Grubbs’ catalyst 1821 efficiently prepared the desired substrate (i.e., (−)-19) for the diastereoselective hydrogenation. Gratifyingly, heterogeneous hydrogenation utilizing Rh/Al2O3 catalyst22 in methanol, followed by TBS cleavage, provided alcohol (+)-20 in good overall yield with excellent diastereoselectivity.23 This notable transformation generates alcohol (+)-20 with the C(10) and C(7) stereocenters in the desired syn configuration required for 1. Conversion of alcohol (+)-20 to ester (+)-21 was achieved by a two-step process involving Dess–Martin oxidation,24 followed by chlorite oxidation25 with diazomethane workup.
Scheme 2.

Enantioselective Synthesis of the Eudesmane Core
The availability of ester (+)-21 in the desired configuration enabled preparation of (+)-carissone (1) in short order. Diastereoselective reduction of the enone carbonyl under Luche conditions,26 followed by treatment of the resulting alcohol with methylmagnesium bromide6m provided diol (+)-2227 in 71% yield (Scheme 3). The preparation of this diol intersects Aoyama’s synthesis (−)-α-eudesmol (3)6e and represents a formal total synthesis. Furthermore, facile allylic oxidation with manganese dioxide6a gave (+)-carissone (1), having spectroscopic data (1H NMR, 13C NMR, IR, HRMS, optical rotation) identical to that reported for natural 1.
Scheme 3.

End Game for (+)-Carissone (1)
In summary, we have described the palladium-catalyzed asymmetric alkylation of vinylogous ester substrates and demonstrated the utility of such products in a general synthetic approach for the total synthesis of the eudesmane sesquiterpenoids. Fundamental to this strategy is the use of the resulting C(10) quaternary stereocenter to control the C(7) stereochemistry via a diastereoselective hydrogenation, providing a highly selective and efficient route to the antibacterial agent (+)-carissone (1). Studies to understand the interplay between substrate reactivity and selectivity for the asymmetric alkylation of vinylogous ester derivatives, as well as the use of the resulting enantioenriched products in the synthesis of other bioactive natural substances28 is currently underway.
Scheme 1.

Retrosynthetic Analysis of the Eudesmanes
Acknowledgment
The authors thank Krastina V. Petrova and Justin T. Mohr (Caltech) for helpful discussions and experimental assistance. The authors gratefully acknowledge the NIH-NIGMS (RO1GM080269–01), Marcella R. Bonsall and the Dalton Fund (undergraduate fellowships to S.R.L.), Eli Lilly (predoctoral fellowship to M.R.K.), Amgen, Abbott Laboratories, Boehringer-Ingelheim, Materia, Merck, Bristol-Meyers Squibb, and the California Institute of Technology for financial support.
Footnotes
Supporting Information Available: Experimental procedures and NMR spectra for all intermediates. These materials are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a comprehensive review of the eudesmane sesquiterpenoids of the Asteraceae family, see: Wu Q-X, Shi Y-P, Jia Z-J. Nat. Prod. Rep. 2006;23:699–734. doi: 10.1039/b606168k.
- 2.Fraga BM. Nat. Prod. Rep. 2007;24:1350–1381. doi: 10.1039/b706427f. [DOI] [PubMed] [Google Scholar]
- 3.For the isolation and biological activity of (+)-carissone, see: Mohr K, Schindler O, Reichstein T. Helv. Chim. Acta. 1954;37:462–471. Joshi DV, Boyce SF. J. Org. Chem. 1957;22:95–97. Achenbach H, Waibel R, Addae-Mensah I. Phytochemistry. 1985;24:2325–2328. Lindsay EA, Berry Y, Jamie JF, Bremner JB. Phytochemistry. 2000;55:403–406. doi: 10.1016/s0031-9422(00)00343-5.
- 4.For the isolation and biological activity of (+)-3-oxocostusic acid, see: Bohlmann F, Jakupovic J, Lonitz M. Chem. Ber. 1977;110:301–314. Khanina MA, Kulyyasov AT, Bagryanskaya IY, Gatilov YV, Adekenov SM, Raldugin VA. Chem. Nat. Compd. 1998;34:145–147. Al-Dabbas MM, Hashinaga F, Abdelgaleil SAM, Suganuma T, Akiyama K, Hayashi H. J. Ethnopharmacol. 2005;97:237–240. doi: 10.1016/j.jep.2004.11.007. Mohamed AE-H, Ahmed AA, Wollenweber E, Bohm B, Asakawa Y. Chem. Pharm. Bull. 2006;54:152–155. doi: 10.1248/cpb.54.152.
- 5.For the isolation and biological activity of α-eudesmol, see: McQuillin FJ, Parrack JD. J. Chem Soc. 1956:2973–2978. Asakura K, Kanemasa T, Minagawa K, Kagawa K, Ninomiya M. Brain Res. 1999;823:169–176. doi: 10.1016/s0006-8993(99)01165-8. Toyota M, Yonehara Y, Horibe I, Minagawa K, Asakawa Y. Phytochemistry. 1999;52:689–694. Asakura K, Kanemasa T, Minagawa K, Kagawa K, Yagami T, Nakajima M, Ninomiya M. Brain Res. 2000;873:94–101. doi: 10.1016/s0006-8993(00)02527-0.
- 6.For syntheses of carissone, see: Pinder AR, Williams RA. J. Chem. Soc. 1963:2773–2778. Sathe VM, Rao AS. Indian J. Chem. 1971;9:95–97. Kutney JP, Singh AK. Can. J. Chem. 1982;60:1842–1846. Wang C-C, Kuoh C-S, Wu T-S. J. Nat. Prod. 1996;59:409–411. Aoyama Y, Araki Y, Konoike T. Synlett. 2001;9:1452–1454.
- 7.For syntheses of 3-oxocostusic acid, see: Ceccherelli P, Curini M, Marcotullio MC, Rosati O. Tetrahedron Lett. 1990;31:3071–3074. Xiong Z, Yang J, Li Y. Tetrahedron: Asymmetry. 1996;7:2607–2612.
- 8.For syntheses of α-eudesmol, see: Humber DC, Pinder AR, Williams RA. J. Chem. Soc. 1967:2335–2340. Taber DF, Saleh SA. Tetrahedron Lett. 1982;23:2361–2364. Schwartz MA, Willbrand AM. J. Org. Chem. 1985;50:1359–1365. Chou T-S, Lee S-J, Yao N-K. Tetrahedron. 1989;45:4113–4124. Frey B, Hünig S, Koch M, Reissig H-U. Synlett. 1991:854–856. Frey B, Schnaubelt J, Reiβig H-U. Eur. J. Org. Chem. 1999:1385–1393.
- 9.For a review of enantioselective methodologies inspired by target-directed synthesis, see: Mohr JT, Krout MR, Stoltz BM. Nature. 2008;455:323–332. doi: 10.1038/nature07370.
- 10.For reviews of the synthesis of quaternary stereocenters, see: Cozzi PG, Hilgraf R, Zimmermann N. Eur. J. Org. Chem. 2007;36:5969–5994. Trost BM, Jiang C. Synthesis. 2006:369–396. Christoffers J, Baro A, editors. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Weinheim: Wiley; 2005. Douglas CJ, Overman LE. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101. Denissova I, Barriault L. Tetrahedron. 2003;59:10105–10146.
- 11. Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15055. doi: 10.1021/ja044812x. Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem., Int. Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. Seto M, Roizen JL, Stoltz BM. Angew. Chem., Int. Ed. 2008;47:6873–6876. doi: 10.1002/anie.200801424. (d) For a review of the development of the enantioselective Tsuji allylation in our lab and others, see: Mohr JT, Stoltz BM. Chem.–Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183.
- 12.(a) McFadden RM, Stoltz BM. J. Am. Chem. Soc. 2006;128:7738–7739. doi: 10.1021/ja061853f. [DOI] [PubMed] [Google Scholar]; (b) Behenna DC, Stockdill JL, Stoltz BM. Angew. Chem., Int. Ed. 2007;46:4077–4080. doi: 10.1002/anie.200700430. [DOI] [PubMed] [Google Scholar]; (c) White DE, Stewart IC, Grubbs RH, Stoltz BM. J. Am. Chem. Soc. 2008;130:810–811. doi: 10.1021/ja710294k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Enquist JA, Jr, Stoltz BM. Nature. 2008;453:1228–1231. doi: 10.1038/nature07046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stork G, Danheiser RL. J. Org. Chem. 1973;38:1775–1776. [Google Scholar]
- 14.Trost BM, Bream RN, Xu J. Angew. Chem., Int. Ed. 2006;45:3109–3112. doi: 10.1002/anie.200504421. [DOI] [PubMed] [Google Scholar]
- 15.For the development of phosphinooxazoline (PHOX) ligands, see: Helmchen G, Pfaltz A. Acc. Chem. Res. 2000;33:336–345. doi: 10.1021/ar9900865. Williams JMJ. Synlett. 1996:705–710.
-
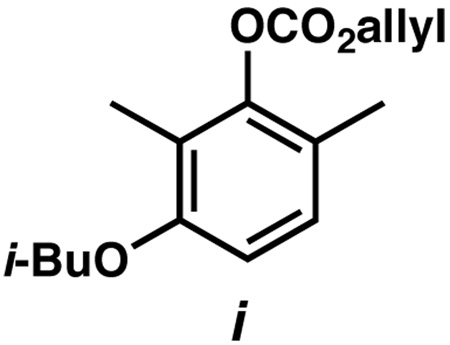
16.Enol carbonate 9 was unstable to air, forming complex reaction mixtures that substantially affected yields and selectivities for the alkylation. Aromatic carbonate i was identified from this mixture. See Supporting Information for more details.
-
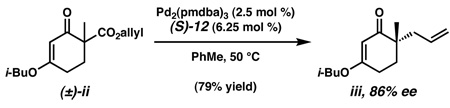
17.The reactivity of β-ketoester (±)-10 contrasts significantly with that of related derivative (±)-ii, which generates iii in 79% yield and 86% ee under identical conditions.
- 18.β-ketoester (±)-11 was recovered in 9% yield.
- 19.Breit B, Breuninger D. J. Am. Chem. Soc. 2004;126:10244–10245. doi: 10.1021/ja0467364. [DOI] [PubMed] [Google Scholar]
- 20.Studies employing either allylmagnesium bromide generated from 16, MeLi, or MeMgBr nucleophiles were unsuccessful. Difficulties with these types of additions into vinylogous thioesters have been noted (see Ref. 12).
- 21.Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 22.A variety of homogeneous and heterogeneous catalysts were screened for the hydrogenation of (−)-19. Of those that were reactive, most promoted undesired reactivity, including enone reduction and further decomposition. Rh/Al2O3 proved to be a mild catalyst at 1 atm H2 in MeOH for this substrate.
- 23.The stereochemistry of (+)-20 was initially verified using NOE correlations. See Supporting Information for details.
- 24.Dess DB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]
- 25.(a) Lindgren BO, Nilsson T. Acta Chem. Scand. 1973;27:888–890. [Google Scholar]; (b) Kraus GA, Roth B. J. Org. Chem. 1980;45:4825–4830. [Google Scholar]; (c) Bal BS, Childers WE, Pinnick HW. Tetrahedron. 1981;37:2091–2096. [Google Scholar]
- 26.Luche JL, Gemal AL. J. Am. Chem. Soc. 1979;101:5848–5849. [Google Scholar]
- 27.Pinder AR, Williams RA. J. Chem. Soc. 1963:2773–2778. [Google Scholar]
- 28.For a related study, see: Petrova KV, Mohr JT, Stoltz BM. Org. Lett. doi: 10.1021/ol802410t. in press.

