

Abstract
Refolding curves of the integral membrane protein Outer Membrane Protein A (OmpA) were measured to determine the conformational stabilities of this model system for membrane protein folding. Wild-type OmpA exhibits a free energy of unfolding ( ) of 10.5 kcal/mol. Mutants, containing a single tryptophan residue at the native positions 7, 15, 57, 102, or 143, are less stable than wild-type OmpA, with values of 6.7, 4.8, 2.4, 4.7, and 2.8 kcal/mol, respectively. The trend observed here is discussed in terms of non-covalent interactions, including aromatic interactions and hydrogen bonding. The effect of the soluble tail on the conformational stability of the transmembrane domain of OmpA was also investigated via truncated single-trp mutants; values for four of the five truncated mutants are greater by >2.7 kcal/mol relative to the full-length versions, suggesting that the absence of the soluble domain may destabilize the unfolded transmembrane domain. Finally, dynamic light scattering experiments were performed to measure the effects of urea and protein on vesicle size and stability. Urea concentrations greater than 1 M cause an increase in vesicle size, and these diameters are unaltered in the presence of protein. These dynamic light scattering results complement the fluorescence studies and illustrate the important effects of vesicle size on protein conformational stability.
Keywords: Outer Membrane Protein A, membrane protein folding, tryptophan fluorescence, conformational stability, dynamic light scattering, refolding curves
Membrane proteins adopt unique three-dimensional structures in cell membranes to be biologically active. Like soluble proteins, membrane proteins that form aberrant structures lead to a number of prevalent diseases, such as cystic fibrosis and type II diabetes (1, 2). A great deal of experimental and theoretical research has recently been devoted towards elucidating the molecular mechanisms, thermodynamics, and kinetics of protein folding in lipid bilayers (3–7). Despite these efforts, our knowledge of membrane protein folding remains inferior to our fundamental understanding of soluble protein folding. A number of factors contribute to the inherent difficulties in the study of membrane protein folding. First, in contrast to soluble proteins, a limited number of membrane protein systems are available to serve as models for membrane protein folding (5–8). An additional challenge is the membrane itself; lipid bilayers are complex systems, with a high degree of chemical heterogeneity (6, 9). Finally, the limited availability of membrane protein structures inhibits rapid expansion of experimental and theoretical work in this area.
A few α-helical and β-barrel membrane proteins and peptides have been studied to provide insight into the driving forces relevant to membrane protein folding. The energetically expensive process of dehydrating the peptide bond is balanced by two critical events: the formation of secondary-structure hydrogen bonds in the bilayer and favorable hydrophobic interactions. Peptide backbone stabilization in the form of extensive hydrogen bond networks dominates the formation of secondary structure; this large driving force may provide the physical basis for why unfolded proteins are not found within a bilayer (10). Hydrogen bonds involving side chains are also critical, and have been analyzed for soluble proteins (11, 12). Hydrophobic interactions have been quantified in the form of measured free energies of transfer from water to bilayer interface or to n-octanol on White-Wimley hydrophobicity scales (6, 13, 14). Aromatic amino acids also play critical roles in the stability of proteins (15, 16) and are considered to be anchoring residues for membrane proteins. Hence, the aromatic residues trp, tyr, and phe are found predominantly at the bilayer-water interface (17–21).
Outer Membrane Protein A (OmpA) is a monomeric, 325-residue integral membrane protein found in the outer membrane of Gram-negative bacteria (Figure 1 inset). It is one of the most abundant proteins in the outer membrane, and is reported to have a variety of functions, such as acting as a nonspecific pore, providing membrane stability, and serving as a receptor (22, 23). OmpA consists of a transmembrane β-barrel constructed from eight anti-parallel β-sheets and a soluble C-terminal domain located in the periplasm (24). The relative ease with which wild-type and mutant OmpA can be overexpressed in E. coli combined with the success with which OmpA spontaneously folds into a membrane bilayer or micelles (25–27) make OmpA an ideal candidate for β-barrel membrane protein folding studies. The existence of five native tryptophan residues that are sensitive reporters of conformational state provides an additional advantage of working with OmpA for membrane protein folding studies: Steady-state fluorescence experiments may be used to directly determine relative populations of folded and unfolded states.
Figure 1.

Representative tryptophan fluorescence spectra of folded protein (dark solid curve, λmax ~330nm) and unfolded protein (light solid curve, λmax ~350nm). Inset, crystal structure of the transmembrane portion of wild-type OmpA (PDB 1QJP), with a cartoon representation of the soluble periplasmic domain. The five native tryptophan residues are highlighted.
The in vitro folding mechanism of OmpA has been studied by several groups and techniques. Electronic spectroscopy, including fluorescence and circular dichroism, as well as vibrational techniques have yielded structural and kinetic information (25–31). Effects of temperature, bilayer composition, and bilayer curvature on the folding of OmpA have been investigated (25, 29, 32). Conversion between narrow and large pore sizes of OmpA folded in planar bilayers has also been reported (33, 34). These and other previous studies have helped establish successful conditions for performing folding experiments and have expanded our general understanding of β-barrel membrane protein folding.
Here, we present fluorescence data to shed light on conformational stabilities of wild-type and mutant OmpA folded in small unilamellar vesicles (SUVs), and discuss the observed trends in terms of important non-covalent interactions. An important goal is to elucidate molecular interactions of interfacial tryptophan residues that are critical for membrane protein stability. In the current studies, mutations have been made on wild-type OmpA to produce single-trp containing mutants, where four of the five native tryptophan residues have been replaced with phenylalanine residues. A similar study was recently reported by the Tamm group in which one or two aromatic amino acids was replaced by alanine to determine the contribution of tryptophan, tyrosine, or phenylalanine on conformational stability (35). In addition to the contribution of amphiphilic tryptophan residues to membrane protein stability, we compare refolding curves in the presence and absence of the soluble C-terminus. Surprisingly, the free energy of unfolding is greater when the soluble domain is removed, highlighting the importance of the unfolded state in these measurements of stability. Finally, we report dynamic light scattering measurements to elucidate changes in vesicle size in the presence of urea and/or protein, and probe changes in protein conformational stability as a function of vesicle diameter.
Materials and Methods
Preparation of single-tryptophan mutants
Site-directed mutagenesis was performed on a plasmid encoding for the single-tryptophan mutant at position 7 of Outer Membrane Protein A (OmpA) to produce additional full-length mutants with single trp residues in the other native positions (26). In these single-trp mutants, the remaining four native trp residues were mutated to phenylalanine residues. These quadruple mutants are referred to throughout the text as the single-trp mutants W7, W15, W57, W102, and W143 for mutants with a trp residue at position 7, 15, 57, 102, or 143, respectively. Truncated versions of these OmpA single-trp mutants were generated by introducing a stop codon at position 177 to cleave the periplasmic domain (147 residues), resulting in single-trp, transmembrane-spanning, truncated mutants W7t, W15t, W57t, W102t, and W143t. PCR products were transformed into XL1-Blue supercompetent cells; the resulting DNA was extracted using a Qiagen Miniprep Kit. All sequences were verified before expression.
Expression, isolation, and purification of OmpA
Previously published procedures were adapted to isolate and purify wild-type OmpA and OmpA mutants (25, 28). Wild-type OmpA, containing five native tryptophan residues, was obtained from E. coli strain JF701 (Genetic Stock Center, Yale University). Cells were initially grown overnight in sterile media (1% bactotryptone, 0.5% yeast extract, and 25 μg/mL streptomycin) at 37 °C before transferring to fresh LB containing 25 μg/mL streptomycin. Single-trp mutants of OmpA were overexpressed in an OmpA− and OmpF− E. coli strain JF733 (Genetic Stock Center, Yale University). Cells containing the appropriate plasmid for the mutant OmpA protein were initially grown overnight at 37 °C in sterile LB media with 0.5% glucose and 50 μg/mL ampicillin and then transferred to fresh LB containing 50 μg/mL ampicillin. Overexpression of OmpA was induced with 1 mM isopropyl β-D-thiogalactopyranoside (IPTG).
Wild-type and mutant OmpA were isolated and purified by the following procedure. Defrosted cells were placed in an ice bath and resuspended in a cold solution of 0.75 M sucrose in 10 mM tris-HCl buffer (pH 7.8). A solution of cold 20 mM EDTA with lysozyme (0.5 mg/mL) was added over a period of one minute. Phenylmethanesulphonylfluoride (PMSF) was also added. The solution was stirred in an ice bath until cells dissolved, and sonicated in an ice bath with a standard horn tip. After intact cells and other particulates were removed, the cell lysis was centrifuged and the resulting red-brown pellets containing OmpA was redissolved in pre-extraction buffer (3.5 M urea, 20 mM tris-HCl, pH 9, 0.05% 2-mercaptoethanol) in a 50 °C water bath to solubilize peripheral membrane proteins. This solution was centrifuged and the pellets containing OmpA were dissolved in a 1:1 mixture of 2-propanol: extraction buffer (8 M urea, 20 mM tris-HCl, 0.05% 2-mercaptoethanol, pH 8.5). The solution was stirred in a 50 °C water bath until pellets dissolved, and solubilized OmpA was isolated via centrifugation from the lipid membrane. The supernatant containing crude OmpA was purified by anion exchange chromatography. OmpA was eluted using a linear concentration gradient of up to 200 mM NaCl in urea buffer (8M urea, 15 mM tris-HCl, 0.5% 2-mercaptoethanol, pH 8.5) over 20 column volumes. Fractions containing purified protein were combined, washed, and concentrated via ultrafiltration (Amicon, PM-10 for full-length protein, and YM-3 for truncated). Stock protein samples were stored as unfolded protein in 8 M urea, 20 mM KPi, pH 7.3 buffer at −80 °C. SDS-PAGE was used to determine a purity of ~90%.
Preparation of unilamellar vesicles
Small unilamellar vesicles (SUVs) were prepared using an adaptation of a previously published procedure (25). A stock solution (20 mg/mL) of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC, Avanti Polar Lipids) in chloroform was dried under a stream of argon and resuspended in phosphate buffer (20 mM KPi, pH 7.3) to a final lipid concentration of 5 mg/mL. SUVs (~50 nm diameter) were made by sonication of the lipid solution with a Branson ultrasonicator microtip. The sonicated solution was passed through a 0.22 μm filter to remove titanium dust and other particulates. The SUVs were further diluted to a final concentration of 1 mg/mL for all experiments. Vesicle solutions were equilibrated overnight at 37 ° C and used the following day.
Small and large unilamellar vesicles were also made by extrusion (36). Stock DMPC solutions were dried and resuspended in KPi buffer as discussed above. Resuspended lipid solutions were passed through an extruder with a membrane of appropriately sized pores to produce nominal vesicle diameters of 50, 100, 200, and 400 nm. A gravity-flow size-exclusion column (Bio-Rad, 10DG) was used to separate the vesicles based on size. Dynamic light scattering experiments were performed to measure the average vesicle size for each fraction. Appropriately combined fractions with DLS-determined diameters of ~70, ~120, ~200, and ~300 nm were diluted to a final concentration of 1 mg/mL. The population of the nominally 400 nm vesicles was ~50% (300 nm) and ~50% >1 μm. Vesicle solutions were equilibrated overnight at 37 ° C and used the following day.
Steady-state fluorescence and absorption spectroscopy
OmpA refolding was monitored by tryptophan fluorescence. Steady-state fluorescence measurements with 290 nm excitation were performed with a Jobin Yvon-SPEX Fluorolog FL3-11 spectrofluorometer. Samples were maintained at 30 °C, which is above the phase transition temperature for DMPC. Absorbance measurements were recorded on an Agilent UV-visible spectrophotometer. Protein concentrations were calculated using ε280 values of 54 400 M−1cm−1, 32 300 M−1cm−1, and 26 000 M−1cm−1 for wild-type, single-trp, and truncated single-trp mutants of OmpA, respectively.
Equilibrium refolding and unfolding of OmpA
Stock solutions of 20 mM KPi buffer (pH 7.3), 10 M urea in 20 mM KPi buffer (pH 7.3), and appropriately sized vesicles were used to prepare 1 mg/mL vesicle samples with urea concentrations ranging from 0–8 M. Stock solution of unfolded protein was added to these vesicle solutions to a final OmpA concentration of ~ 4 μM for fluorescence experiments and ~12 uM for gel electrophoresis (SDS-PAGE) measurements. Blank samples (urea, KPi, and vesicles) containing no protein were also prepared. Fluorescence spectra of blank samples were subtracted from spectra of protein samples to remove contributions from buffer. Urea concentrations were determined from refractive index measurements using an Abbe 3L Bausch & Lomb refractometer (37). A time-dependence study was performed and determined that a 12 hour incubation period was sufficient to reach equilibrium conditions. All protein and blank samples for fluorescence and SDS-PAGE experiments were equilibrated overnight at 37 °C. SDS-PAGE experiments were performed to confirm that the folding of OmpA is reversible using an adaptation of a previously published method (32). The differential mobility of folded (30 kDa) and unfolded (35 kDa) protein was utilized for these gel-shift assays. For unfolding studies, unfolded stock protein was diluted into SUVs and incubated at 37 °C for refolding to occur. The solution was diluted 2x in appropriate urea solutions to a final protein concentration of ~12 uM. For refolding studies, the stock unfolded protein was diluted into solutions with SUV and varying urea concentrations. All refolding and unfolding samples were loaded onto 12.5% gels (full-length protein) or 15% gels (truncated protein). Unfolding gels of the truncated protein indicated a minor population (<15%) of the protein remained unfolded in low urea concentrations and folded at high urea concentrations. These gel-shift assays are presented in Figure S1 in Supplementary Information.
Determination of the effective free energy of unfolding for OmpA mutants
Gel-shift assays indicated fully reversible folding/unfolding conditions for full-length OmpA, and >85% reversibility for truncated OmpA under our experimental conditions. Due to the reversibility of refolding, all calculated free energies determined from the refolding curves are the effective free energies of unfolding. Emission maxima were determined for all protein samples, and the resulting plot of fraction unfolded, f, vs urea concentration were fit to a reversible, two-state model for protein unfolding (38, 39).
Dynamic light scattering measurements
Dynamic light scattering (DLS) was used to determine vesicle diameters. The apparatus has been described in detail elsewhere (40). Measurements were made using the 514.5-nm laser line from an argon ion laser with the samples held at a constant temperature of 30 °C. Ten autocorrelation traces were collected and averaged for each sample. Autocorrelation functions, g(τ), determined from DLS were calculated using the relationship g(τ) = exp(−t/τ) (41), where t is the correlation time, and τ is the characteristic lifetime of a particle diffusing within a defined volume. The translational diffusion constant, DT, can be calculated from the following relationship: τ−1 = 2DTq2. The scattering vector, q, is defined by q = (4πns/λ)sin(θ/2), where λ is the excitation wavelength, ns is the solvent refractive index, and θ is the scattering angle (θ = 90 °). The diffusion constant, DT, is defined by DT = kT/6πηRh, where η is the viscosity of the medium and Rh is the hydrodynamic radius of the particle. Particles were assumed to be spherical. The value of η was assumed to be 0.798×10−2 poise for water and scaled appropriately for changes in viscosity with increasing urea (42). The average characteristic lifetime, τ, of each sample obtained from experimental measurements was used to calculate Rh of the vesicles. Two populations with corresponding average diameters were obtained for each sample. The fraction of reported population was > 80% unless specified. Vesicle size was measured as a function of vesicle concentration from 0.1 to 1 mg/ml to ensure appropriate conditions for our measurements; these control experiments indicated variation of <20% in vesicle diameter across this concentration range and this error has been included in Figure 3.
Figure 3.

Representative vesicle diameters of samples used in refolding curve studies. Open symbols indicate vesicle samples containing 4 μM of protein; solid symbols indicate vesicle samples without protein (blanks).
Results
Refolding curves
Tryptophan fluorescence spectroscopy was used to determine conformational stabilities of single-trp mutants of OmpA in synthetic lipid bilayers. The single-trp residue in W15, W57, W102, and W143, is located on the β-barrel near the extracellular (intra-vesicle) region, while the trp residue in W7 is located closer to the periplasmic (extra-vesicle) side. Each trp is located along a different strand, except for W7 and W15 which are located along the same strand. Representative trp emission spectra of OmpA folded in vesicle (λmax = ~330 nm) and unfolded in 8 M urea (λmax = ~350 nm) are illustrated in Figure 1. This shift in emission maximum upon addition of denaturant was used to monitor the relative populations of folded and unfolded protein.
Refolding curves of wild-type and single-trp mutants of OmpA are displayed in Figure 2A. Wild-type OmpA was determined to be the most stable, with a value of 10.5 kcal/mol. Full-length mutants had lower free energies of unfolding compared to wild-type OmpA, with values ranging between 2.4 and 6.7 kcal/mol. W15 and W102 mutants had values of 4.8 and 4.7 kcal/mol, respectively, while W57 and W143 mutants had values of 2.4 and 2.8 kcal/mol, respectively. The most stable mutant appears to be the single-trp mutant W7, with a value of 6.7 kcal/mol. Despite the fact that W7 has a similar Cm value (~3 M urea) as W15 and W102, W7 has a greater overall conformational stability due to its larger m-value. Tabulated fitting parameters, m and Cm, and resulting values are summarized in Table 1.
Figure 2.

Denaturant-induced refolding curves of wild-type and full-length single-trp mutants (A) and single-trp truncated mutants (B) of OmpA along with fits (solid curves).
Table 1.
Fitting parameters and thermodynamic values determined from refolding curves of wild-type and single-trp OmpA mutants.
| Unfolding Curve Parameters |
|||
|---|---|---|---|
|
m kcal/mol M−1 |
Cm M |
kcal/mol |
|
| W7 | 2.18 | 3.07 | 6.7 +/− 1.0 |
| W7t | 2.98 | 3.16 | 9.4 +/− 1.4 |
| W15 | 1.53 | 3.17 | 4.8 +/− 0.7 |
| W15t | 1.70 | 2.28 | 3.9 +/− 0.6 |
| W57 | 1.36 | 1.78 | 2.4 +/− 0.4 |
| W57t | 2.01 | 2.64 | 5.3 +/− 0.8 |
| W102 | 1.51 | 3.13 | 4.7 +/− 0.7 |
| W102t | 3.00 | 3.69 | 11.1 +/− 1.7 |
| W143 | 1.22 | 2.30 | 2.8+/− 0.4 |
| W143t | 2.28 | 3.17 | 7.2 +/− 1.1 |
| WT | 2.36 | 4.43 | 10.5 +/− 1.6 |
Refolding curves were also determined for the truncated single-trp mutants that lack the soluble domain. These curves are shown in Figure 2B. Truncated mutants had free energy of unfolding values that ranged from 3.9 to 11.1 kcal/mol. The most stable mutants were W102t and W7t, with values of 11.1 and 9.4 kcal/mol, respectively. W143t was also stable, with of 7.2 kcal/mol. Even though W7t and W143t have almost identical Cm values of ~3.1 M urea, the higher stability for W7t can be traced to its larger m-value. However, given the difficulty of these experiments, our estimates of error (see below) prevent us from making definitive assessments of the relative stabilities of W7t and W143t at this time. values for W57t and W15t were determined to be 5.3, and 4.8 kcal/mol, respectively. Fitting parameters and values for the truncated mutants are summarized in Table 1. In general, the truncated mutants were more stable than their full-length counterparts: values for W7 and W57 increased by ~3 kcal/mol, for W143 increased by ~4 kcal/mol, and for W102 increased by ~6.5 kcal/mol upon removal of the soluble tail. In contrast to these mutants, for W15 decreased only ~ 1 kcal/mol when the periplasmic tail was eliminated; this difference for W15 vs. W15t falls within our experimental uncertainty and is therefore negligible.
Determination of vesicle size
DLS measurements determined vesicle size and stability to ensure reproducibility for the current protein unfolding studies. Fluorescence and DLS measurements were performed on the same day using solutions from the refolding studies for six mutants W57, W57t, W102, W102t, W143, and W143t. A representative data set for W143 OmpA is shown in Figure 3.
Diameters of vesicle-only solutions were measured with increasing urea concentration (0–8 M). The initial stock vesicle solution contained ~50 nm SUVs. Vesicle diameters remained ~50 nm in low concentrations of urea (< 1 M), and increased to ~100–300 nm at greater concentrations of urea (> 1 M). This trend was observed for all six vesicle-only (blank) solutions. Vesicle diameters were also measured in the presence of OmpA with increasing concentrations of urea. Vesicle diameters in protein-containing solutions followed the same trend as blank solutions. Diameters remained ~50 nm in low urea concentrations (< 1 M), and increased to ~80–300 nm at higher concentrations of urea (> 1 M). Similar trends of increased vesicle size with increasing urea concentration were also found for the W57, W57t, W102, and W102t mutants (data not shown).
The effects of small and large unilamellar vesicles on thermodynamic stability of OmpA
Refolding curves were measured for the single-trp mutant, W15t, using small unilamellar vesicles (SUVs) and large unilamellar vesicles (LUVs) to test the effects of vesicle size on thermodynamic stability. Figure 4 shows refolding curves of W15t OmpA using nominally 50, 100, 200, and 400 nm diameter vesicles. Actual vesicle diameters were ~70, ~120, ~200, and ~300 nm, respectively. For the latter sample, ~ 50% of the population exhibited an average diameter of ~ 300 nm and the remaining population had a very large average diameter (> 1μm). Measurements were taken 6, 12, and 24 hours after the addition of protein to determine equilibrium conditions. The data shown were measured after a 24-hour incubation period. The greatest stability was measured for solutions containing 50 nm vesicles, with of 3.9 kcal/mol, followed by solutions using ~100 and ~200 nm diameter vesicles which yielded the same value of 3.4 kcal/mol and is <15% lower than the value for 50 nm vesicles. Folding into nominally 400 nm diameter vesicles resulted in the lowest measured value of 2.2 kcal/mol. These measurements of conformational stabilities as a function of vesicle size combined with the range of urea concentrations over which OmpA unfolds indicates that the largest source of systematic error is the change in vesicle size over the urea concentrations of interest; therefore, we estimate an error of <15% in our reported values in Table 1.
Figure 4.
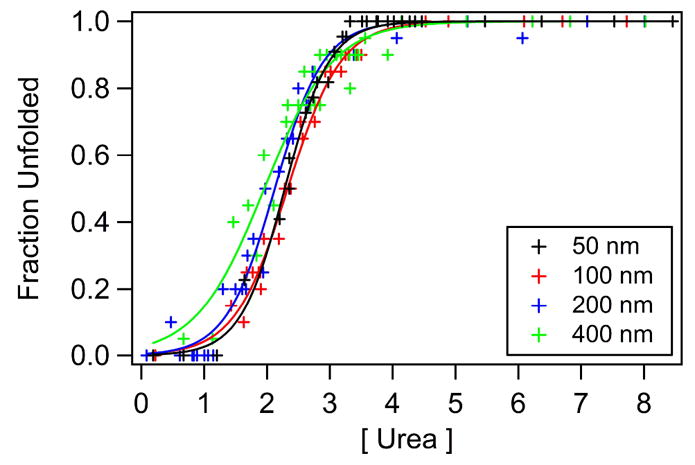
Refolding curves of W15t OmpA using nominally 50, 100, 200, and 400 nm diameter vesicles. Fits are solid curves.
Discussion
Full-length mutants
Non-covalent interactions are crucial to the stability of membrane proteins. Important forces that drive a peptide or protein towards a membrane include non-polar (hydrophobic) interactions, electrostatic interactions between residues and charged lipid head groups, formation of secondary structure, and electrostatic effects because of differences in the dielectric constants of water and the membrane (6, 43). A large variety of critical side-chain and backbone hydrogen bonds have been reported (44–46). The current study probes the thermodynamic stabilities associated with the five native tryptophan residues in wild-type and single-trp mutants of OmpA. Five native trp residues give rise to the refolding curve of wild-type OmpA, and hence provide an estimate of the global stability of the protein. In contrast, single-trp OmpA mutants in which four native trp residues are replaced with phe residues provide a unique opportunity to probe the effects of the trp environment on the overall stability of the protein.
The aromatic residues tryptophan, tyrosine, and phenylalanine contribute significantly to the thermodynamic stability of membrane proteins because of strong interactions with neighboring aromatic residues and favorable water-to-bilayer partition energies (13, 14, 16, 35). Individual trp residues provide greater contribution to membrane protein stability relative to phe residues; however both are energetically favored in the membrane and are able to participate in pairwise interactions with aromatic residues (35). Previous studies of OmpA showed similar secondary structure and trp environments when comparing wild-type and the single-trp mutants; therefore, it is likely that the range of conformational stabilities reported here for the single-trp mutants can be attributed to differences in local environment of trp residues as opposed to modification in global protein structure (26, 28).
As seen in Figure 2 and Table 1, the full-length single-trp mutants of OmpA were found to be less stable than wild-type OmpA. This result is consistent with removal of four of the five native anchoring trp residues. The change in free energy upon transfer of an individual trp residue from water to lipid bilayer is −1.85 kcal/mol; the analogous value for a phe residue is −1.13 kcal/mol (13). Trp and phe energy contributions determined from previous thermodynamic stability experiments on OmpA mutants showed an average contribution of −2.0 and −1.0 kcal/mol, respectively (35). Our results indicate that OmpA is destabilized 3.8 to 8.1 kcal/mol upon substitution of four trp residues for phe residues and are consistent with these previous reports; trp residues provide a greater contribution to the stability of OmpA relative to phe residues.
Three important molecular interactions involving trp residues are considered in the current study of single-trp mutants. First, the role of pairwise aromatic interactions has been shown to be critical to membrane protein stability. While it was previously suggested that the exact identity of aromatic amino acid involved in these interactions is not crucial (35), our results support a picture in which a trp residue is optimum for the aromatic pocket near Trp7, but may not be critical in the pairwise interactions involving Trp143 and Trp57. Second, hydrogen-bonding of the indole heteroatom provides stability in the transmembrane domain; this hydrogen bond is important in Trp15. Finally, the unique amphiphilic nature of trp plays an important role for interfacial stabilization of Trp102 relative to phe and tyr residues. These topics are discussed in detail below.
Stability of W7: Pairwise aromatic interactions
Neighboring aromatic residues may interact in pairwise fashion to contribute to the stability of proteins. Typical interaction distances of 4.5 to 7.0 Å between aromatic residue centroids in soluble proteins contribute up to 2 kcal/mol towards the stability of a protein (16). These pairwise interactions were recently investigated in OmpA and were estimated to be on the same order as those found for soluble proteins (35). The region near Trp7 has the greatest number of neighboring aromatic residues, Tyr8, Tyr43, Tyr168, and Phe170 (~ 7 Å between aromatic centroids). Our results support a picture in which a trp (and not phe) residue at position 7 is ideally suited for this aromatic pocket in the protein; mutant W7 is the most stable of all full-length single-trp species. This result modifies the previous report that suggested that these important pairwise interactions may not depend on the type of aromatic residue involved (35, 46); here, our results illustrate that maintaining a trp residue at position 7 while replacing the remaining four native trp residues with phe residues decreases the the least amount (by 3.8 kcal/mol) relative to the wild-type protein. This destabilization of W7 relative to WT can be attributed to removal of the hydrogen bond at position 15 and removal of the amphiphilic trp residue at position 102 (see below).
Other pairwise interactions exist in OmpA. W143 has two neighboring aromatic residues, Phe132 and Tyr141, and W57 has one neighboring aromatic residue, Tyr55, within ~ 7 Å of the indole centroid. These pairwise aromatic interactions contribute to the stability for OmpA. However, it is not clear that that the trp-to-phe mutation in positions 143 or 57 causes significant destabilization of the protein as was observed in the case of position 7. W143 and W57 are the least stable of all full-length single-trp mutants; this enhanced instability can be attributed to the combined effects of the loss of Trp7 in the aromatic pocket, loss of the hydrogen bond at position 15, and removal of the amphiphilic trp residue at position 102. In contrast, W15 and W102 have no neighboring aromatic residues; these observations are summarized in Table 2.
Table 2.
Summary of energies ( and ) and non-covalent interactions for the single-trp mutants of OmpA. values are from current experiments; are calculated from reference 15; number of aromatic residues is based on a ~ 7 Å radius centered on the aromatic centroid (PDB 1QJP); number of hydrogen bonds is based on a 3.5 Å radius centered on the indole nitrogen atom.
| Energy (kcal/mol) | Non-Covalent Interactions | |||||
|---|---|---|---|---|---|---|
| Mutants | # of Aromatic Residues | # of H-Bonds | ||||
| W7 | 6.7 | 3.2 | 4 | 0 | ||
| W15 | 4.8 | 2.0 | 0 | 1 | ||
| W102 | 4.7 | −1.4 | 0 | 0 | ||
| W143 | 2.8 | 2.2 | 2 | 0 | ||
| W57 | 2.4 | 0.7 | 1 | 0 | ||
Stability of W15: Hydrogen bonding
The mutant W15 is one of the next highest in stability following W7. A variety of side-chain hydrogen bonds in proteins have been described, including bonding between N-H, O-H, and C-H donor groups and N-, O-, and π– acceptor groups (11, 12). A range of hydrogen bonding energies has been reported, and the typical value for biological systems is on the order of a few kcal/mol. The crystal structure of OmpA indicates a likely hydrogen bond between Trp15 and the side chain of Asn33. Specifically, the distance between the heteroatom in indole and the carbonyl oxygen of Asn33 is less than 2.9 Å. The presence of this hydrogen bond likely contributes to the stability of W15, and we estimate the hydrogen-bond strength of Trp15 to be ~ 2 kcal/mol. The measured decrease in stability of W15 relative to WT is attributable to loss of Trp7 in the aromatic pocket and loss of amphiphilic stability at position 102. The observation that W15 is less stable than W7 supports a picture in which stabilization due to Trp7 in the aromatic pocket (< 4 kcal/mol, see below) is more significant than stabilization due to Trp15 hydrogen bond (~2 kcal/mol). These values are consistent with Hong et al (35), which determined that the trp residue at position 7 contributes 3.6 kcal/mol, and the trp residue at position 15 contributes 2.0 kcal/mol to the stability of the protein.
Stability of W102: Amphiphilic nature of tryptophan
In addition to contributions from interacting aromatic pairs and hydrogen-bonding partners, contributions from neighboring residues to the stability of tryptophan residues in OmpA were also investigated. Local partition energies for transfer from the bilayer to water to mimic protein unfolding were calculated using the White-Wimley interfacial hydrophobicity scale, which includes side-chain and backbone contributions (13). For hydrophobic residues, this partition energy is a positive value, and for hydrophilic residues, this value is typically negative. These calculations were performed to provide an estimate of the partition energy of the region surrounding each trp residue. All neighboring residues within a 7 Å radius of the trp were included in the determination of the local partition energies, and are tabulated in Table 2 under ΔGpart. This radius typically included 12 or 13 residues surrounding a given trp and included residues located on different strands of the β-barrel relative to the location of the trp.
The region with the largest partition energy is Trp7 (3.23 kcal/mol), and the region near Trp102 has the smallest partition energy (−1.38 kcal/mol) (13). The large positive partition energy for W7 is consistent with Trp7 being located in a highly hydrophobic environment: W7 displays the most blue-shifted emission maximum as well as a large m-value, suggesting that Trp7 undergoes a large increase in solvent exposure upon unfolding relative to the other trp positions (47). The region around Trp102 is the only region with a calculated negative partition energy, indicating a thermodynamic preference for this region to be in aqueous environment. This calculated negative partitioning energy for W102 is due to the presence of Gln75, Arg103, and Asp116.
Trp102 is found on a large loop that is likely in the interior of the vesicle due to the known directional insertion of OmpA (25). However, there is no evidence that Trp102 is solvent-exposed in the folded form despite it being the only trp residue located on a loop, that faces the interior of the barrel, and has a calculated negative partition energy. In fact, W102 remains in a hydrophobic region, evidenced by fluorescence experiments. The emission maximum of W102 folded in vesicle is 332 nm, which is identical to the emission maxima of the other trp residues located on the same side of the barrel, W15, W57, and W143. This similarity in emission maxima indicates a similar degree of hydrophobicity surrounding these trp residues. For W102, the local hydrophobicity can be attributed to neighboring nonpolar residues including Ala74, Met100, Val101, and Ala104, as opposed to the hydrophobic environment of the lipid bilayer. In addition to similar steady-state emission, the kinetics of refolding and the fluorescence lifetime of W102 were found to be similar to those of W15, W57, and W143 (26, 28).
The presence of both hydrophobic and hydrophilic residues in the local vicinity of Trp102 is consistent with the amphiphilic nature of tryptophan residues, and the substitution of trp for phe in this region may destabilize OmpA. The value of W102 is similar to that of W15; in the case of W102, loss of the hydrogen bond at position 15 and loss of Trp7 in the aromatic pocket destabilizes this mutant relative to WT. However, the observation that W102 and W15 have similar values for indicates that the energetic contributions from the amphiphilic nature of Trp102 and hydrogen-bonding ability of Trp 15 are similar, and each may contribute ~2 kcal/mol to overall protein stability.
Stability of W143 and W57
The least stable full-length mutants, W143 and W57, lack all three stabilizing effects discussed above. Namely, these mutants do not have Trp7 in the aromatic pocket, lack a hydrogen-bonded side chain at position 15, and do not possess the optimized amphiphilic trp residue in the unique loop region near Trp102. The absence of these effects results in the greatest destabilization of ~8 kcal/mol relative to WT. The observation that the values are similar for W143 and W57 despite the presence of two or one neighboring aromatic residues, respectively, suggests that the trp-to-phe mutation has not perturbed the energetics of these pairwise interactions significantly. This implication helps provide an upper estimate of ~4 kcal/mol to the stabilization effect of Trp7.
Truncated mutants
OmpA contains two distinct regions, the transmembrane domain located in the membrane which contains all five native trp residues, and the soluble periplasmic domain located in the exterior portion of the vesicle. There is no published structure of the C-terminal periplasmic tail of OmpA. A previous circular dichroism study of full-length and truncated mutants of OmpA showed a decrease in molar ellipticity and red-shift of the CD peak to ~216 nm when the soluble tail was removed (28). The origin of this variation in CD spectra is not known. While it was previously suggested that the soluble C-terminus may possess β-sheet structure, the observed CD changes are also consistent with a random coil soluble domain. Other studies demonstrated that OmpA forms two pores in planar bilayers: a small pore that involves the N-terminus domain only, and a large pore that requires both the N- and C-terminal domains (35, 36). We are unable to determine whether the soluble domain possesses secondary structure or whether an entirely new folded structure is formed in SUVs in the presence of the C-terminal domain; we are applying UV resonance Raman spectroscopy to probe changes in vibrational structures between full-length and truncated mutants.
Results from the current study indicate that removal of the soluble domain results in a greater free energy of unfolding for the transmembrane domain. The values for four of the five full- length mutants increased by >2.7 kcal/mol upon removal of the soluble tail. The trend in values of the full-length mutants was not identical to the trend for the values of the truncated mutants. This variation may indicate structural differences of the transmembrane domain upon removal of the soluble tail as well as reflect our experimental uncertainty. Previous studies have probed the differential stabilities of the transmembrane and soluble domains of a membrane protein (48, 49), but to our knowledge, this is the first report of direct measurements of the effect of a soluble domain on the conformational stability of a transmembrane domain.
Our result that the values increase upon removal of a significant (soluble) portion of the protein is surprising, and may be explained by one of the following scenarios in a simplified two-state picture consisting of folded and unfolded states of the transmembrane region. First, it is possible that the covalently bound soluble domain destabilizes the folded transmembrane domain such that removal of this energetically expensive soluble domain overall stabilizes the folded transmembrane portion. We have no experimental evidence or examples in literature to support this case and therefore, believe that this is an unlikely scenario. Second, the soluble domain may stabilize the unfolded transmembrane domain by, for example, providing favorable hydrophobic interactions in solution. Removal of this energetically favorable soluble domain destabilizes the unfolded transmembrane portion, resulting in the observed increase in . Other studies on the effects of the denatured state on the free energies of unfolding for soluble proteins have been reported (50–52). Currently, tryptophan emission studies on OmpA mutants do not show significant variations in fluorescence maxima of the unfolded transmembrane domain of the full-length and truncated protein; however stabilization provided by the soluble domain may not necessarily impact the emission properties of the native tryptophan residues in the unfolded state. Finally, the structure of the transmembrane domain may be different in the absence of the soluble C-terminus. For example, the transmembrane domain may be imbedded deeper into the bilayer when the soluble tail is removed. We do not have evidence to support a significant change in global protein structure upon truncation of the C-terminus, but do not dismiss the possibility. Based on our preliminary findings, we hypothesize that the second scenario is the most likely explanation for the increase in free energy of unfolding upon removal of the soluble tail; however, it is clear that more in-depth studies must be pursued.
Effects of denaturant on SUVs
To complement these studies of OmpA thermodynamics, we investigated the effects of denaturant and protein on SUVs. Previous studies have shown that factors such as bilayer composition, chain-length, and bilayer curvature affect protein folding and assembly mechanisms in the membrane (29, 32, 53, 54). In fact, conformational stabilities of OmpA ranged from ~2 to 6 kcal/mol depending on lipid composition, head-group size, and chain-length (29). From these studies, it is apparent that the properties of the bilayer can greatly affect the stability of the protein. Some important factors to consider are the following: Does the presence of a denaturant and/or folded protein affect the properties of the vesicles? How do the macroscopic properties of vesicles affect measurements of protein stability?
Urea has been shown to traverse the bilayer through a solubility-diffusion controlled mechanism, indicating equilibrium urea concentrations on the inside and outside of the vesicle in our experimental time-scale (55). Previous studies illustrated no change in vesicle size over a twelve hour period in the presence of ~100 mM urea (56) and little to no change in vesicle structure up to 3 M urea (57, 58); our results are consistent with these previous works in that low concentrations of urea (< 1M) do not affect vesicle size. However, at urea concentrations typical of unfolding studies, there is an increase in vesicle size; in blank solutions, the presence of > 1 M urea caused an increase in vesicle diameter with a high degree of variability in size (Figure 3). It appears that the presence of urea enhances vesicle fusion. A similar increase in vesicle diameter was observed in vesicle solutions containing protein and urea. This result suggests the protein itself does not cause vesicle fusion. Instead, the observed change in size is due to denaturant. It is not clear from our results that the presence of protein directly affects vesicle size as has been previously reported (59).
Effect of SUV size on
Figure 4 illustrates that values vary by less than 15% when determined with nominally 50, 100, or 200 nm diameter vesicles. Measurements of with 400 nm vesicles, however, resulted in a >40% decrease in observed value. It is not clear if this decrease is significant since the scattering became more pronounced and we have not thoroughly characterized these larger vesicles. Nonetheless, these results illustrate the importance of characterizing the effect of denaturant and protein on vesicle properties in these measurements of membrane protein conformational stabilities; the largest source of experimental error may be associated with the change in vesicle properties in the presence of denaturant.
Summary
The conformational stabilities of single-trp mutants of OmpA were measured and found to be less than that of wild-type OmpA: The mutants are destabilized ~ 4 kcal/mol (W7), ~6 kcal/mol (W15 and W102), and ~8 kcal/mol (W43 and W57) relative to WT. The trends in free energies of unfolding for the different mutants are likely due to a combination of non-covalent interactions, summarized in Table 2. Neighboring aromatic residues interact favorably to contribute to the stability of a protein. The presence of a specific trp residue is especially important in the aromatic pocket surrounding Trp7, and we estimate an upper limit of ~4 kcal/mol for the stabilizing effect of Trp7. The stability of W15 may be explained in terms of a hydrogen bond, which may provide ~2 kcal/mol of stabilization. The relatively high stability of W102 cannot be explained in terms of energetically favorable pairwise aromatic interactions or hydrogen-bonding partners, of which it has neither. The calculated bilayer-to-water partition energy near Trp102 is negative, indicating the local prevalence of hydrophilic residues, but the experimentally observed fluorescence maximum suggests a hydrophobic pocket. A likely explanation for the relatively high stability of W102 is the unique amphiphilic nature of the trp residue; substitution of trp for phe in this region reduces the stability of the protein by ~2 kcal/mol. Removal of the soluble domain appears to destabilize the unfolded transmembrane domain of the protein, resulting in the enhanced values for for the truncated proteins. This surprising result highlights the importance of the energetics of the unfolded state in measurements of overall protein conformational stability. DLS results presented here indicate that vesicle diameters increase in the presence of urea. In contrast, the presence of protein does not greatly affect vesicle size. values changed < 15% when vesicles in the size range between 50 and 200 were used. These and other findings may have important implications for future studies of membrane protein folding.
Supplementary Material
Acknowledgments
We thank Tiffany J. Neary for assistance with protein expression, Dustin Huard and Professor Akif Tezcan for help with gel-shift assays, and Professor Doug Madge for use of his DLS apparatus.
This work has been supported by the following funding agencies: DOE Graduate Assistance in Areas of National Need Fellowship (KMS), UCSD NIH Molecular Biophysics Training Grant GM 08326 (DES), and the National Science Foundation CHE-0645720.
Abbreviations
- OmpA
Outer Membrane Protein A
- WT
wild-type
- trp or W
tryptophan
- phe
phenylalanine
- tyr
tyrosine
- DLS
dynamic light scattering
- SUVs
small unilamellar vesicles
- LUVs
large unilamellar vesicles
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electropheoresis
Footnotes
Supporting Information Available: A supporting figure of gel-shift refolding/unfolding curves.
References
- 1.Sanders CR, Myers JK. Disease-related misassembly of membrane proteins. Annu Rev Biophys Biomol Struct. 2004;33:25–51. doi: 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- 2.Aridor M, Balch WE. Integration of endoplasmic reticulum signaling in health and disease. Nature Medic. 1999;5:745–751. doi: 10.1038/10466. [DOI] [PubMed] [Google Scholar]
- 3.Im W, Brooks CL., III De novo folding of membrane proteins: An exploration of the structure and NMR properties of the fd coat protein. J Mol Biol. 2004;337:513–519. doi: 10.1016/j.jmb.2004.01.045. [DOI] [PubMed] [Google Scholar]
- 4.Bond PJ, Cuthbertson JM, Deol SS, Sansom MSP. MD simulations of spontaneous membrane protein/detergent micelle formation. J Am Chem Soc. 2004;126:15948–15959. doi: 10.1021/ja044819e. [DOI] [PubMed] [Google Scholar]
- 5.Booth PJ, Templer RH, Meijberg W, Allen SJ, Curran AR, Lorch M. In vitro studies of membrane protein folding. Crit Rev in Biochem and Mol Biol. 2001;36:501–603. doi: 10.1080/20014091074246. [DOI] [PubMed] [Google Scholar]
- 6.White SH, Wimley WC. Membrane Protein Folding and Stability: Physical Principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 7.MacKenzie KR. Folding and stability of a-helical integral membrane proteins. Chem Rev. 2006;106:1931–1977. doi: 10.1021/cr0404388. [DOI] [PubMed] [Google Scholar]
- 8.Tamm LK, Hong H, Liang B. Folding and assembly of beta-barrel membrane proteins. Biochim Biophys Acta. 2004;1666:250–263. doi: 10.1016/j.bbamem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 9.Lee AG. Lipid-protein interactions in biological membranes: a structural perspective. Biochim Biophys Acta. 2003;1612:1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- 10.White SH. How hydrogen bonds shape membrane protein structure. Adv Protein Chem. 2006;72:157–172. doi: 10.1016/S0065-3233(05)72006-4. [DOI] [PubMed] [Google Scholar]
- 11.Weiss MS, Brand L, Suhnel J, Pal D, Hilgenfeld R. More hydrogen bonds for the (structural) biologist. Trends in Biochem Sci. 2001;26:521–523. doi: 10.1016/s0968-0004(01)01935-1. [DOI] [PubMed] [Google Scholar]
- 12.Steiner T, Koellner G. Hydrogen bonds with pi-acceptors in proteins: Frequencies and role in stabilizing local 3D structures. J Mol Biol. 2001;305:535–557. doi: 10.1006/jmbi.2000.4301. [DOI] [PubMed] [Google Scholar]
- 13.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 14.Wimley WC, Creamer TP, White SH. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry. 1996;35:5109–5124. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- 15.Burley SK, Petsko GA. Amino-aromatic interactions in proteins. FEBS Lett. 1986;203:139–143. doi: 10.1016/0014-5793(86)80730-x. [DOI] [PubMed] [Google Scholar]
- 16.Burley SK, Petsko GA. Aromatic-aromatic interaction: A mechanism of protein structural stabilization. Science. 1985;229:23–28. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 17.Domene C, Bond PJ, Deol SS, Sansom MSP. Lipid/protein interactions and the membrane/water interfacial region. J Am Chem Soc. 2003;125:14966–14967. doi: 10.1021/ja0364874. [DOI] [PubMed] [Google Scholar]
- 18.Babakhani A, Gorfe AA, Gullingsrud J, Kim JE, McCammon JA. Peptide insertion, positioning, and stabilization in a membrane: Insight from an all-atom molecular dynamics simulation. Biopolymers. 2007;85:490–497. doi: 10.1002/bip.20698. [DOI] [PubMed] [Google Scholar]
- 19.Yau W-M, Wimley WC, Gawrisch K, White SH. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 20.Reithmeier RAF. Characterization and modeling of membrane proteins using sequence analysis. Curr Op Struct Biol. 1995;5:491–500. doi: 10.1016/0959-440x(95)80034-4. [DOI] [PubMed] [Google Scholar]
- 21.Killian JA, von Heijne G. How proteins adapt to a membrane-water interface. Trends Biochem Sci. 2000 doi: 10.1016/s0968-0004(00)01626-1. [DOI] [PubMed] [Google Scholar]
- 22.Sugawara E, Nikaido H. Pore-forming activity of OmpA protein of Escherichia coli. J Biol Chem. 1992;267:2507–2511. [PubMed] [Google Scholar]
- 23.Wang Y. The Function of OmpA in Eschericia coli. Biochem Biophys Res Comm. 2002;292:396–401. doi: 10.1006/bbrc.2002.6657. [DOI] [PubMed] [Google Scholar]
- 24.Koebnik R, Locher KP, Van Gelder P. Structure and function of outer membrane proteins: Barrels in a nutshell. Molec Microbiol. 2000;37:239–253. doi: 10.1046/j.1365-2958.2000.01983.x. [DOI] [PubMed] [Google Scholar]
- 25.Surrey T, Jahnig F. Refolding and oriented insertion of a membrane protein into a lipid bilayer. Proc Natl Acad Sci USA. 1992;89:7457–7461. doi: 10.1073/pnas.89.16.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinschmidt JH, den Blaauwen T, Driessen AJM, Tamm LK. Outer membrane protein A of Escherichia coli inserts and folds into lipid bilayers by a concerted mechanism. Biochemistry. 1999;38:5006–5016. doi: 10.1021/bi982465w. [DOI] [PubMed] [Google Scholar]
- 27.Dornmair K, Kiefer H, Jahnig F. Refolding of an integral membrane protein. J Biol Chem. 1990;265:18907–18911. [PubMed] [Google Scholar]
- 28.Kim JE, Arjara G, Richards JH, Gray HB, Winkler JR. Probing folded and unfolded states of outer membrane protein A using steady-state and time-resolved tryptophan fluorescence. J Phys Chem B. 2006;110:17676–17662. doi: 10.1021/jp061991r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong H, Tamm LK. Elastic coupling of intergral membrane protein stability to lipid bilayer forces. Proc Natl Acad Sci USA. 2004;101:4065–4070. doi: 10.1073/pnas.0400358101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramakrishnan M, Qu J, Pocanschi CL, Kleinschmidt JH, Marsh D. Orientation of beta-barrel proteins OmpA and FhuA in lipid membranes. Chain length dependence from infrared dichroism. Biochemistry. 2005;44:3515–3523. doi: 10.1021/bi047603y. [DOI] [PubMed] [Google Scholar]
- 31.Sanchez KM, Neary TJ, Kim JE. Ultraviolet resonance Raman spectroscopy of folded and unfolded states of an integral membrane protein. J Phys Chem B. 2008;112:9507–9511. doi: 10.1021/jp800772j. [DOI] [PubMed] [Google Scholar]
- 32.Pocanschi CL, Patel GJ, Marsh D, Kleinschmidt JH. Curvature elasticity and refolding of OmpA in large unilamellar vesicles. Biophys J. 2006;91:L75–L77. doi: 10.1529/biophysj.106.091439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arora A, Rinehart D, Szabo G, Tamm LK. Refolded outer membrane protein A of Escherichia coli forms ion channels with two conductance states in planar lipid bilayers. J Biol Chem. 2000;275:1594–1600. doi: 10.1074/jbc.275.3.1594. [DOI] [PubMed] [Google Scholar]
- 34.Zakharian E, Reusch RN. Kinetics of folding of Escherichia coli OmpA from narrow to large pore conformation in a planar bilayer. Biochemistry. 2005;44:6701–6707. doi: 10.1021/bi047278e. [DOI] [PubMed] [Google Scholar]
- 35.Hong H, Park S, Flores Jimenez RH, Rinehart D, Tamm LK. Role of aromatic side chains in the folding and thermodynamic stability of integral membrane proteins. J Am Chem Soc. 2007;129:8320–8327. doi: 10.1021/ja068849o. [DOI] [PubMed] [Google Scholar]
- 36.Hope MJ, Bally MB, Webb G, Cullis PR. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim Biophys Acta. 1985;812:55–65. doi: 10.1016/0005-2736(85)90521-8. [DOI] [PubMed] [Google Scholar]
- 37.Shirley BA. Urea and guanidine hydrochloride denaturation curves. In: Shirley BA, editor. Protein Stability and Folding. Human Press Inc.; Totowa: 1995. pp. 177–190. [DOI] [PubMed] [Google Scholar]
- 38.Schellman JA. Solvent denaturation. Biopolymers. 1978;17:1305–1321. doi: 10.1002/bip.360260408. [DOI] [PubMed] [Google Scholar]
- 39.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 40.Toal SJ, Jones KA, Magde D, Trogler WC. Luminescent silole nanoparticles as chemoselective sensors for Cr(VI) J Am Chem Soc. 2005;127:11661–11665. doi: 10.1021/ja052582w. [DOI] [PubMed] [Google Scholar]
- 41.Berne BJ, Pecora R. Dynamic Light Scattering. Dover Publications, Inc.; Mineola, NY: 1976. [Google Scholar]
- 42.Kawahara K, Tanford C. Viscosity and density of aqueous solutions of urea and guanidinine hydrochloride. J Biol Chem. 1966;941:3228–3232. [PubMed] [Google Scholar]
- 43.Ben-Tal N, Ben-Shaul A, Honig B. Free-energy determinants of alpha-helix insertion into lipid bilayers. Biophys J. 1996;70:1803–1812. doi: 10.1016/S0006-3495(96)79744-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheiner S, Kar T, Pattanayak J. Comparison of various types of hydrogen bonds involving aromatic amino acids. J Am Chem Soc. 2002;124:13257–13264. doi: 10.1021/ja027200q. [DOI] [PubMed] [Google Scholar]
- 45.Brandl M, Weiss MS, Jabs A, Suhnel J, Hilgenfeld R. C-H..pi-interactions in proteins. J Mol Biol. 2001;307:357–377. doi: 10.1006/jmbi.2000.4473. [DOI] [PubMed] [Google Scholar]
- 46.Serrano L, Bycroft M, Fersht AR. Aromatic-aromatic interactions and protein stability. J Mol Biol. 1991;218:465–475. doi: 10.1016/0022-2836(91)90725-l. [DOI] [PubMed] [Google Scholar]
- 47.Wrabl J, Shortle D. A model of the changes in denatured state structure underlying m value effects in staphylococcal nuclease. Nature. 1999;6:876–883. doi: 10.1038/12338. [DOI] [PubMed] [Google Scholar]
- 48.Oikawa K, Lieberman DM, Reithmeier AF. Conformation and stability of the anion transport protein of human erythrocyte membranes. Biochemistry. 1985;24:2843–2848. doi: 10.1021/bi00333a005. [DOI] [PubMed] [Google Scholar]
- 49.Lau FW, Bowie JU. A method for assessing the stability of a membrane protein. Biochemistry. 1997;36:5884–5892. doi: 10.1021/bi963095j. [DOI] [PubMed] [Google Scholar]
- 50.Shortle D. The denatured state (the other half of the folding equation) and its role in protein stability. FASEB J. 1996;10:27–34. doi: 10.1096/fasebj.10.1.8566543. [DOI] [PubMed] [Google Scholar]
- 51.Arai M, Kataoka M, Kuwajima K, Matthews CR, Iwakura M. Effects of the difference in the unfolded-state ensemble on the folding of Escherichi coli dihydrofolate reductase. J Mol Biol. 2003;329:779–791. doi: 10.1016/s0022-2836(03)00511-4. [DOI] [PubMed] [Google Scholar]
- 52.Mok YK, Elisseeva EL, Davidson AR, Forman-Kay JD. Dramatic stabilization of an SH3 domain by a single substitution: Roles of the folded and unfolded states. J Mol Biol. 2001;307:913–928. doi: 10.1006/jmbi.2001.4521. [DOI] [PubMed] [Google Scholar]
- 53.Curran AR, Templer RH, Booth PJ. Modulation of folding and assembly of the membrane protein bacteriorhodopsin by intermolecular forces within the lipid bilayer. Biochemistry. 1999;38:9328–9336. doi: 10.1021/bi982322+. [DOI] [PubMed] [Google Scholar]
- 54.Kleinschmidt JH, Tamm LK. Secondary and tertiary structure formation of the beta-barrel membrane protein OmpA is syncrhonized and depends on membrane thickness. J Mol Biol. 2002;324:319–330. doi: 10.1016/s0022-2836(02)01071-9. [DOI] [PubMed] [Google Scholar]
- 55.Finkelstein A. Water and nonelectrolyte permeability of lipid bilayer membranes. J Gen Phys. 1976;68:127–135. doi: 10.1085/jgp.68.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pencer J, White GF, Hallett FR. Osmotically induced shape changes of large unilamellar vesicles measured by dynamic light scattering. Biophys J. 2001;81:2716–2728. doi: 10.1016/S0006-3495(01)75914-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kleinschmidt JH, Tamm LK. Structural transitions in short-chain lipid assemblies studied by 31P-NMR spectroscopy. Biophys J. 2002;83:994–1003. doi: 10.1016/S0006-3495(02)75225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng Y, Yu Z, Quinn PJ. Effect of urea, dimethylurea, and tetramethylurea on the phase behavior of dioleoylphosphatidylethanolamine. Chem Phys Lipids. 2002;114:149–157. doi: 10.1016/s0009-3084(01)00198-0. [DOI] [PubMed] [Google Scholar]
- 59.Dini L, Di Giulio A, Pavan A, Ravagnan G, Mossa G. Size and stability of dipalmitoylphosphatidylcholine/cholesterol unilamellar vesicles are affected by interaction with proteins. Biochim Biophys Acta. 1991;1062:108–112. doi: 10.1016/0005-2736(91)90341-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.