

Abstract
Mood disorders associated with dysfunction of the hypothalamic-pituitary-adrenal (HPA) axis are common psychiatric conditions. The glucocorticoid receptor (GR) is a steroid-activated nuclear receptor that, upon binding to cortisol, translocates to the nucleus where it targets genes related to neuronal metabolism and plasticity. In patients suffering from major depressive disorder (MDD), hypercortisolemia is a common finding. In the current study we investigated the molecular events associated with the FK506 binding proteins (FKBP) -52 and -51 response to cortisol exposure in neuronal cell cultures and their effect on GR translocation. We noted that FK506 altered nuclear localization of the GR and inhibited expression of GR-responsive genes. Furthermore, si-RNA knockdown of FKBP4 gene, coding for the immunophilin FKBP52, inhibited cortisol-activated GR nuclear translocation, while knockdown of FKBP5, coding for immunophilin FKBP51, was associated with increased baseline GR nuclear localization. We propose that immunophilins are modulators of the cortisol-HPA axis response to stress and related chronic brain disorders.
Keywords: Immunophilins, FKBP52, FKBP51, glucocorticoid receptor, fk506-binding proteins, nuclear translocation
1. Introduction
Psychiatric conditions associated with altered HPA axis function such as MDD and post-traumatic stress disorder (PTSD) are becoming increasingly recognized as significant causes of morbidity in the United States (Bao et al., 2007; Carroll et al., 2007) and there is a need to identify the underlying pathophysiological processes. In response to physical, emotional, and cognitive stress, corticotrophin releasing hormone (CRH) is secreted by the hypothalamus, signaling to the pituitary to release adrenocorticotropic hormone (ACTH). ACTH then stimulates the adrenal cortex to increase the production of corticosteroids, mainly glucorticoids such as cortisol. Glucocorticoids have an array of physiological effects to prepare the body for fight-or-flight, including effects on brain neurons and glia (Miyakawa et al., 1974; Tombaugh et al., 1992).
The type II corticosteroid receptor, GR, is expressed in the hypothalamus, hippocampus, and frontal cortex, and mediates feedback inhibition of the HPA axis (Chao et al., 1989). It is the low-affinity for cortisol receptor, its activity becomes prominent when the cortisol levels exceed the physiologic peaks of the circadian rhythm production as part of the physiologic response to stress. GR is a steroid-activated nuclear receptor and upon cortisol-binding it translocates to the nucleus where it binds target DNA sequences as a positive or negative transcription factor to mediate mRNA production of genes related to neuronal plasticity, neuronal activation, and neuronal metabolism (Maccari et al., 1992; Pavlides et al., 1995). In individuals with MDD, the negative feedback loop is deficient at reducing elevated cortisol levels, which is the basis of the dexamethasone suppression test for evaluation of HPA function (Sapolsky et al., 1986). In HPA axis dysregulation, for example Cushing's Disease and MDD, cortisol and ACTH are not suppressed and patients are called “non-suppressors” (Gold et al., 1988). The glucocorticoid cascade hypothesis proposes that deficiency of the HPA axis to regulate circulating cortisol results in a feed-forward mechanism of increased cortisol levels during stress and a decreased ability to return to resting conditions, consequently all cells, including neurons, are subject to prolonged GR activation and its downstream effects (Sapolsky et al., 1986).
This study analyzes the role of two adapter proteins, putative chaperones to the GR that are of the immunophilin protein class. FK506 binding protein of molecular weight 51 (FKBP51) and the related protein FKBP52 are thought to be involved in the regulation of GR signaling (Davies et al., 2002). FKBP51 acts as a competitive inhibitor to its genetic relative FKBP52 (gene ID FKBP4) (Davies et al., 2002). The protein products of FKBP4 were originally identified by co-precipitation of the activated GR/HSP90 complex as “HSP56” (Sanchez, 1990). Molecular studies have since clarified that these are two proteins of 52 and 59 kilodaltons, owing to two poly-adenylation sites of the FKBP4 gene producing two mRNA's (Callebaut et al., 1992). There is no known functional difference between the long or short form; for all purposes, FKBP52 and FKBP59 are synonymous and we will refer to the active protein as FKBP52.
FKBP52 colocalizes with microtubules, binds specifically to dynein by its N-terminal immunophilin (also known as prolyl isomerase) domain, and its cis/trans prolyl isomerase activity is required for this interaction (Harrell et al., 2004). The FKBP52 knockout male mouse displays decreased prostate development, malformed seminal vesicles, and reproductive abnormalities that correlates to decreased androgen levels or inadequate androgen receptor response (Cheung-Flynn et al., 2005). The female knockout has no uterine receptivity for embryo implantation, a progesterone receptor-mediated event (Tranguch et al., 2005). The effects of FKBP52 knockout on the developing or adult male and female mouse brain have not yet been studied. FKBP52, through its TPR domains, links GR to the molecular motor protein dynein (Davies et al., 2005). Dynein is a retrograde motor protein and the interaction between GR-FKBP52-dynein may be of particular importance in cells such as neurons where the distance between the receptor and the nucleus may be extensive.
There are data indicating that GR plays a role in MDD and mood disorders such as PTSD (Pariante and Miller, 2001). The administration of reserpine, a drug inducing depressive symptoms, causes altered GR signaling and glucocorticoid resistance (Lowy, 1990). In fact, transgenic mice that under- or over-express GR have been used as models of MDD or PTSD (Alexandre Urani, 2003; Chourbaji et al., 2008; Gass et al., 2001). Pariante et al showed that antidepressants exert effects on GR function, not expression (Pariante et al., 1999). It is thought that prolonged high glucocorticoid levels cause GR to be saturated with ligand leading to overburdening the recycling capacity of GR, and finally diminishing the function of GR (Pariante and Miller, 2001). Decreased GR mRNA was found in the frontal cortex of postmortem tissue of individuals with MDD, bipolar disorder and schizophrenia (Webster et al., 2002). In a rat model of PTSD, which involves subjecting the mice to a single-prolonged stress, long-term potentiation in the CA1 of the hippocampus was impaired, and administration of the GR antagonist RU40555 prevented this effect (Kohda et al., 2007). In this model, the rats exhibited enhanced contextual fear conditioning one week following the single-prolonged stress induction, and this effect was prevented by concomitant administration of the GR antagonist (Kohda et al., 2007). Interestingly, the abnormal glucocorticoid feedback regulation in HPA axis dysfunction was shown to occur at the level of the brain (Young et al., 1991), indicating that GR signaling in the brain would be a potentially important novel target of study for diseases like MDD.
Species of New World primates have general resistance to glucocorticoids (Brown et al., 1970), and as they do express the GR, that binds to cortisol, the mechanism of resistance was puzzling (Chrousos et al., 1982; Reynolds et al., 1999). Later, it was found that a soluble factor, FKBP51, inhibits the binding of cortisol to GR. Expression of FKBP51 is 13-fold higher in these primates, and FKBP52 expression less than one-half compared to humans; effectively rendering the monkeys resistant to the effects of cortisol mediated by GR (Denny et al., 2000). Structural differences between FKBP51 and FKBP52 are thought to cause FKBP51 to act as a competitive inhibitor of FKBP52 in the context of GR signaling (Sinars et al., 2003; Wu et al., 2004). These data, along with indications that brain GR function is dysregulated in MDD, indicate that FKBP52 / FKBP51 kinetics may be important in GR-mediated pathologies such as depression.
There may be a genetic component in variants of FKBP5 that could affect its function. Single nucleotide polymorphisms (SNP's) in FKBP5 were recently found to be linked to MDD. The functional significance of these SNP's is unknown (Binder et al., 2004; van Rossum et al., 2006). A study by Binder et al. linked polymorphisms in the FKBP5 gene to increased recurrence of depressive episodes and rapid response to drugs. Recently, they linked the FKBP5 polymorphisms with risk of PTSD symptoms in adults who suffered from abuse during childhood (Binder et al., 2008). The same alleles, rs3800373 and rs1360780, were associated with increased occurrence of peri-traumatic dissociation in children after physical trauma requiring medical attention (Koenen et al., 2005), which is a risk factor for developing adult PTSD (Ozer et al., 2003).
Past studies have clearly shown that FKBP52 and FKBP51 differentially regulate the function of the GR through the interaction of FKBP52 with dynein (Davies et al., 2002; Galigniana et al., 2002; Harrell et al., 2004; Pratt et al., 2004; Wochnik et al., 2005). On the other hand the evidence whether FK506 would enhance or inhibit nuclear translocation of the GR is still conflicting (Edinger et al., 2002). In the current study we hypothesized that inhibition or knockdown of FKBP4 would inhibit the effects of cortisol in neurons. We investigated the molecular functions of FKBP52 and FKBP51 in the context of cortisol exposure in neuronal cell cultures. Immunophilin ligand FK506, which is commonly used as an immunosuppressant in organ transplant patients, inhibited nuclear translocation of the GR and altered nuclear distribution of the FKBP51 chaperone in neurons. Knockdown by si-RNA of FKBP4 slowed cortisol-activated GR nuclear translocation. Knockdown of FKBP5 caused increased baseline localization of GR to the nucleus.
2. Results
2.1. FK506 Inhibits Cortisol-Induced Redistribution of GR and FKBP51 in Neurons
We sought to determine whether there was a cellular consequence of the presence of immunophilin ligand FK506 to GR and FKBP51 localization. We imaged GR and FKBP51 in neurons exposed to cortisol with and without 2 hr pre-incubation with FK506. We used a primary human brain cell culture system composed mainly of neurons and occasional glia; illustrated in Figure S2 showing Microtubule Associated Protein-2 (MAP2) and Glial Fibrilary Acidic Protein (GFAP) immunopositivity using fluorescent labeled antibodies. The GR was present in astrocytes (Figure S2a; GFAP positive cells), most abundant in neurons (neurofilament and MAP2 positive cells, Figure S2c).
Neuronal immunostaining for GR was apparent in nuclei without 100 nM cortisol, (Figure 1a). There was diffuse staining in the soma and dendrites with and without FK506 as well as diffusely in the nucleus (Figure 1a). Figure 1c shows intensity of staining for Hoechst and GR, outlining the location of the neuronal nuclei, indicating with and without FK506 were similar. After treatment with 100 nM cortisol, abundant punctate staining for GR was apparent in the nucleus (Figure 1b). The Hoechst stain indicated that the DNA became more diffuse as well, particularly in locations of GR foci (Figure 1c, white arrows). In the presence of FK506, however, the GR staining remains diffuse in the soma, dendrites, and nucleus, failing to form the GR foci, remaining relatively condensed when compared to the non-FK506 controls (Figure 1c).
Figure 1. Cortisol-induced nuclear translocation of GR slowed in primary human neuronal cultures with preincubation with 10 μM FK506.
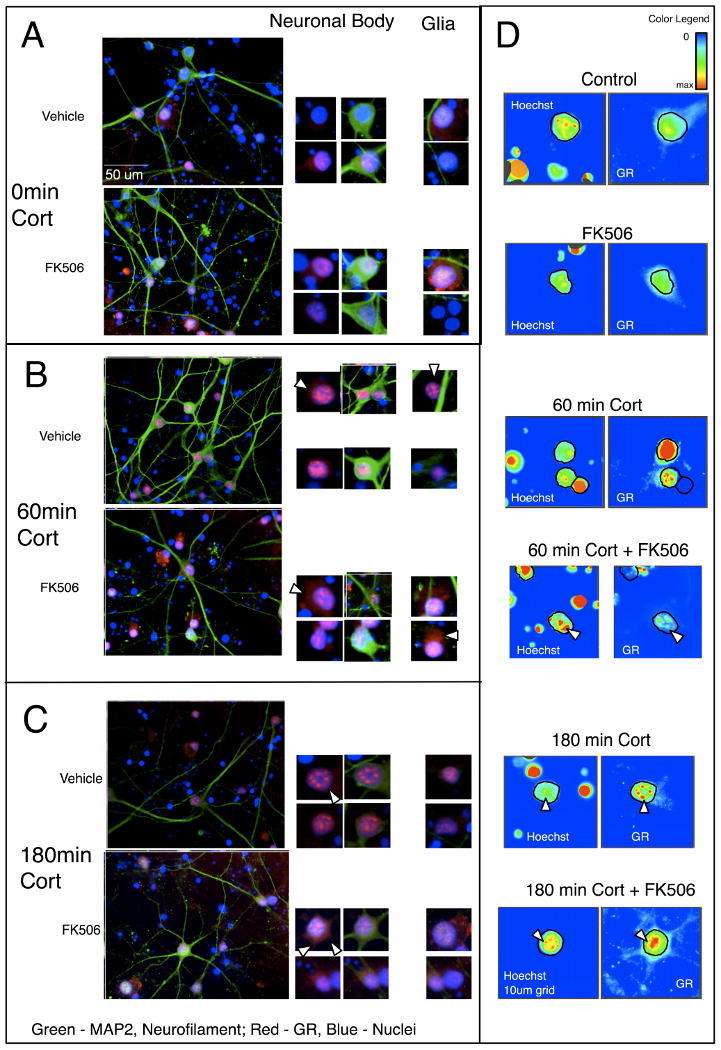
Primary human neuroglial cultures were pretreated with vehicle (1:500 ethanol) or 10 μM FK506 for 2 hr and subsequently treated with 100 nM Cortisol for 0 (a), 60 (b), 180 (c) min. The cell cultures were immunolabeled for neuronal markers (MAP2 and NF; green), and GR (red), and nuclei counterstained with Hoechst (blue); arrows indicate GR localization. More focal nuclear staining of GR was apparent in a time-dependent manner with cortisol. Increased cytoplasmic GR is noted at 180 min in cultures pretreated with FK506, as shown by fluorescence intensity plots; arrows indicate nuclear GR (d).
With regard to FKPB51, we found, as shown by others (Zhang et al., 2008), that it stained diffusely throughout the nucleus and cytoplasm of cells, shown in Figure 2a. In the presence of cortisol, the distribution of FKBP51 is not changed (Figure 2b). However, with FK506 treatment, FKB51 stains perinuclearly and in the soma of neurons, possibly in a polar fashion, congregating toward the axonal side of the neuronal soma (Figures 2b and 2c). The phenomenon of perinuclear and soma staining occurred in the absence of cortisol as well, as shown in Figure 2a and 2c. It is most apparent at the 60 min timepoint shown in Figure 2b.
Figure 2. Localization of FKBP51 in primary human neuronal cultures is altered in the presence of 10 μM FK506.
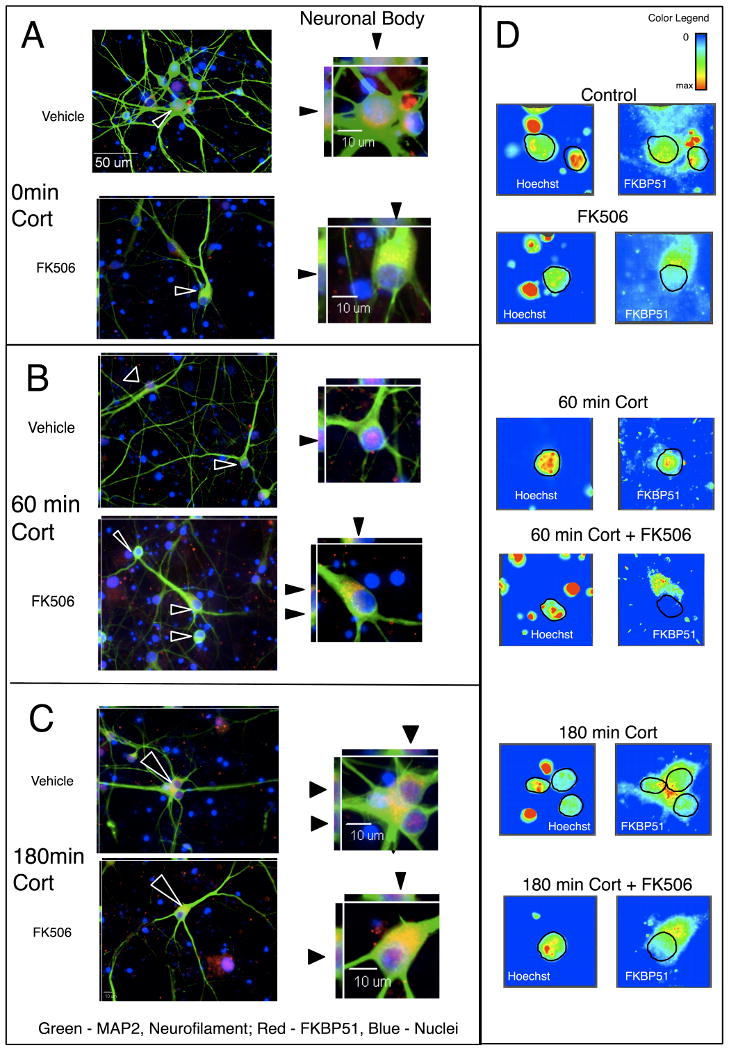
Primary human neuroglial cultures were pre-treated with vehicle (1:500 ethanol) or 10 μM FK506 for 2 hr and subsequently treated with 100 nM Cortisol for 60 (a), 60 (b) and 180 (c) min. The cell cultures were immunolabeled for neuronal markers (MAP2 and neurofilment; green), and FKBP51 (red), and nuclei counterstained with Hoechst (blue). Three-dimensional reconstruction showed diffused nuclear staining of FKBP51 with and without cortisol present as indicated in top panels and ▲ showing FKBP51 staining in z-axis imaging in and intensity plots (c) are illustrated. Pretreatment with FK506 caused exclusion of FKBP51 from the nucleus.
As FK506 apparently altered the cortisol-mediated distribution of GR in neuronal cells, we hypothesized that FK506 may alter gene transcription of a GR-responsive gene. For reproducibility and quantitation, we utilized the SH-SY5Y cell line for qPCR of these genes after exposure to cortisol, in the presence or absence of FK506. Two genes, FKBP5 and Bcl2L1, are induced by cortisol and GR-mediated gene transcription (Hubler and Scammell, 2004; Wang et al., 2004). The FKBP5 gene encodes the FKBP51 protein, contains glucocorticoid-responsive elements to which GR binds directly (Meijsing et al., 2009), and has polymorphisms that are associated with mood disorders (Binder et al., 2004; Gawlik et al., 2006; Hubler and Scammell, 2004; Koenen et al., 2005). The Bcl2-L1 gene encodes the protein Bcl-xL, a mitochondrial member of the Bcl2 family, thought to link mitochondria function, metabolism, and synaptic transmission (Jonas, 2006). We found that FKBP5 and Bcl2L1 mRNA were both elevated by 30% at 30 min in response to exposure to cortisol, as shown in Figure 3. FKBP5 expression returned to baseline at 24 hr (Figure 3a), while Bcl2L1 remained elevated for up to 48 hr (Figure 3b). In SH-SY5Y cells pretreated with FK506, the FKBP5 gene expression response was delayed and the Bcl2L1 genes expression remained below or close to baseline. In comparing the 2 hr FK506 pre-treatment with vehicle, at time-zero cortisol, gene transcription wasn't changed. Others found that FK506 pre-treatment, not coincubation, inhibited GR-induced transcription (Edinger et al., 2002), our findings seem to indicate that the effect may be due to the large-molecular FKBP repertoire of the cell-type; FKBP52 and FKBP51 being expressed in neurons, and the effect is on induction. These data are consistent with the model that pretreatment with FK506, in neurons, having altered immunophilin proteins present in the GR heterocomplex, slows GR-responsive gene induction.
Figure 3. FK506 Inhibits Cortisol - Induced Gene Expression of (a) FKBP5 and (b) Bcl2-l1 in Differentiated SH-SY5Y cells.
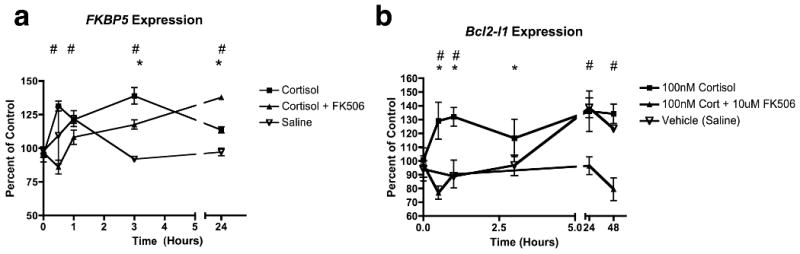
Differentiated SH-SY5Y cells were exposed to100 nM cortisol for up to 48 hr with (▲) or without (■) pretreatment with 10 μM FK506. Saline controls are indicated by ∇, mean and standard deviation of 3 measurements are plotted. RNA was isolated and qRT-PCR was performed as described in Methods, percent of control comparing to Time 0 for each treatment and time point for the (a) FKBP5 and (b) Bcl2-l1 transcripts are plotted. One-way ANOVA and Dunn's multiple comparison tests compared the treatments (p < 0.05 for significance). * Indicate significant difference at a particular time point of 100 nM Cortisol compared to Vehicle Control, and # indicate significant difference between 100 nM Cortisol compared to the FK506 pre-treated group. There is low induction of FKBP5 (A) with cortisol treatment (■) that is slowed but not abrogated by FK506 (▲). Induction of Bcl2-l1 is abrogated by FK506. The vehicle control (∇) elevated Bcl2-l1 (B) expression after 24 hr compared to Time point 0, which was not observed in the Cortisol + FK506 group (▲).
2.2. siRNA Knockdown of FKBP4 and FKBP5
Since FKBP52 protein distribution is predominantly neuronal (Tatro, 2008) and acts to promote GR trafficking, we sought to determine whether FKBP4 siRNA would inhibit the nuclear translocation of GR in SH-SY5Y differentiated cells (Figure S5). Exposure to 250 ng siRNA for 24 hr achieved 70% reduction in FKBP52 protein expression as determined by densitometry of western blot bands (Figure 4a). Figure 4b shows a western blot for FKBP51, indicating reduction after treatment with siRNA to the FKBP5 gene. Since our read-out is GR immunoreactivity, we wanted to ensure that any difference observed in GR localization was not due to changes in GR expression. Western blot analysis showed no changes in GR levels in the si-Control and si-FKBP4, along with si-Control and si-FKBP5 conditions (Figure 4c); the two siRNA experiments were performed independently and had specific controls: siRNA generated to the GFP gene supplied by the manufacturer.
Figure 4. Confirmation of siRNA knockdown of FKBP52 and FKBP51 in SH SY5Y cells.
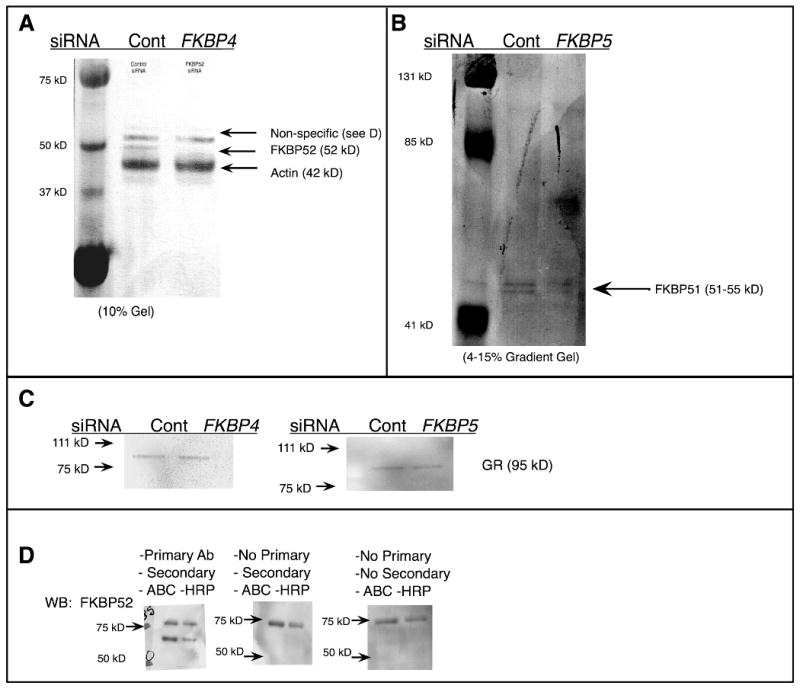
Differentiated SH-SY5Y cells on glass coverslips in 24 well plates were exposed to 250 ng siRNA generated against the FKBP4 (a) gene or against FKBP5 gene (b) and a control gene, encoding GFP provided by the manufacturer, for 24 hr; protein was isolated for Western blot. Concurrent to probing for FKBP52 a western blot for Actin was performed for the loading control (a). To ensure that a difference in GR immunofluorescence was not due to altered GR protein content, GR was assessed by western analysis (c). To discern non-specific bands in the western blots, membranes were developed with primary antibody (d-left), without primary antibody (d-middle) and without either secondary or primary antibody (d-right center), and it was determined that the non-specific band originates from the avidin-biotin amplification system in (a).
The si-RNA exposures were performed in 24 well plates. To compensate for the low protein yield we utilized the avidin-biotin-complex (ABC) system for amplification of western blot signal as described in Materials and Methods. It is for this reason also that Actin and FKBP52 were probed simultaneously. Since a non-specific band occurs above 53 kD we wanted to confirm that we are measuring the correct band. To do so, whole cell lysates from SH-SY5Y cells was separated by PAGE and transferred to a PVDF membrane, which was cut into three 2-lane strips, of 20 μg and 10 μg protein. Western blotting was performed as described in Materials and Methods except that the strips were incubated separately: the first was considered “normal” control (Figure 4d, left), the second strip was incubated without primary antibody (Figure 4d, center), and the third was incubated without primary or secondary antibody (Figure 4d, right).
2.3. Immunofluorescence Imaging and Quantification of Nuclear and Cytoplasmic GR
After exposure to either si-FKBP4, si-FKBP5 or siRNA control, cells were with saline or 100 nM cortisol for 15, 30, and 60 minutes. Immunofluorescent labeling for GR showed nuclear translocation of the GR with addition of cortisol (Figure 5a). With si-FKBP4, nuclear GR did not increase after addition of cortisol (Figure 5b and quantified in Figure 5d). With si-FKBP5, GR was elevated in the absence of cortisol compared to si-control. We quantified the intensity of cytoplasmic and nuclear signals and determined fluorescence intensity of the immunolabled GR per μm3, as illustrated in Figure 5d. The values were compared by two-way ANOVA with p < 0.01 set as statistically significant. Post-test comparing intensity between si-control and si-FKBP4 showed differences due to siRNA at 15 and 30 mins. Intensity was increased in the Control group due to cortisol, as expected, and was inhibited in the si-FKBP4 group (Figure 5d, left). Fluorescence intensity was increased in the absence of cortisol in si-FKBP5 compared to si-control (Figure 5d, right). GR translocation in the absence of FKBP51 is the same at the various timepoints and baseline (0 min cortisol) where GR is elevated (Figure 5d, right).
Figure 5. Quantification of nuclear and cytoplasmic immunofluorescent staining of GR after treatment with 100 nM cortisol and siRNA to FKBP4 and FKBP5.
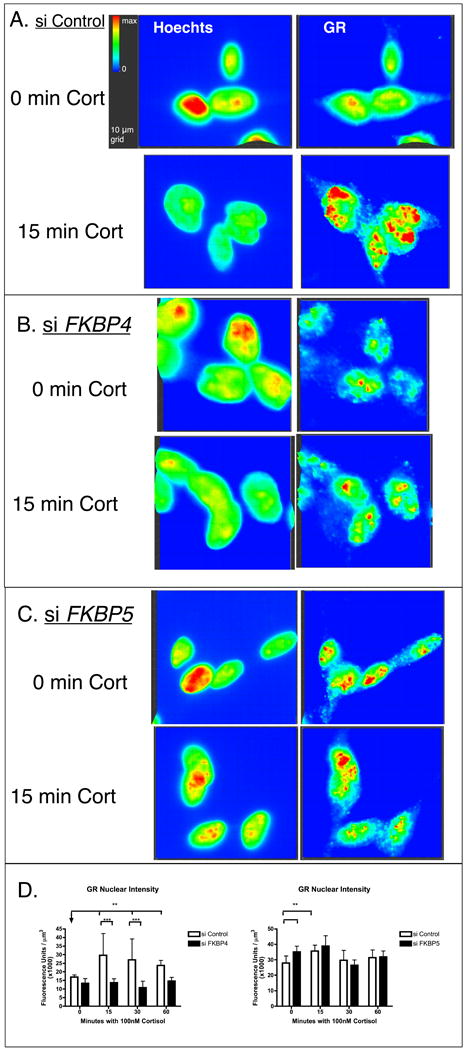
Differentiated SH-SY5Y cells were grown on glass coverslips and exposed to 250 ng control (a), FKBP4 (b), or FKBP5 (c) siRNA for 24 hr and then to saline or 100 nM cortisol 15, 30, or 60 min. Following fixation, cells were fluorescently labeled for GR (green) and nuclei (blue). Using Slidebook image analysis software, nuclear GR staining was partitioned according to the Methods and quantified in 10 3-D fields for each treatment, mean and standard deviation of intensity per μm3 is plotted for the treatment groups. Two-way ANOVA compared the effects of siRNA or cortisol on nuclear fluorescence intensity. Significance is represented by * (p < 0.01) and ** (p<0.001) between the treatment groups in (d). Si-FKBP4 contributed to lower nuclear staining intensity, and si-FKBP5 contributed to elevated nuclear staining intensity. The experiments were performed independently with separate controls, therefore, the scale of the intensity (arbitrary units) per μm3 is different.
3. Discussion
The GR is a highly dynamic protein both in the nucleus and in the cytoplasm: it is involved in the formation of various DNA and protein complexes and also undergoes proteolysis inside the nucleus (Defranco, 2000; Deroo et al., 2002; Elbi et al., 2004; Wang et al., 2002). Since some of the GR molecules may have already been involved in DNA-transcription factor complexes, prior to any cortisol exposure, our images seem to indicate that FK506 interrupts an interaction that would cause GR to be maintained in the nucleus or become involved in focal protein complexes; it may potentially interrupt the interaction with the chaperone complex that includes an immunophilin protein like FKBP51. While nuclear translocation occurs quickly, we examined the GR distribution at relatively late timepoints, as it would be relevant to a chronically stressful environment or hypercortisolemia that is a physiologic feature of MDD and PTSD. At 180 minutes of cortisol exposure, fewer aggregates are formed in the nucleus and the distribution is more diffuse. Furthermore, there is increased cytoplasmic FKBP51 staining noted in the presence of FK506. This would support the notion that perhaps FKBP51 promotes stability of the GR and the formation of foci, potentially protecting it from proteolysis; in this case it would be acting as a chaperone protein associated with GR in the nucleus.
It is not known whether FKBP51 binds in heterocomplexes with the DNA-attached GR. While the staining of GR in the nucleus showed specific foci, which became more prominent over time with cortisol, the FKBP51 nuclear staining remained diffuse. GR likely is in heterocomplex with FKBP51 in the nucleus when it is not DNA-bound or in foci, but rather soluble in the nucleus; potentially, the involvement of FKBP51 in the HSP90 complex protects non-DNA-bound-GR from proteolysis in the nucleus. Others have shown diffuse nuclear staining of FKBP51 in trabecular meshwork cells, (GTM-5 and NTM-5 cells), mouse embryonic fibroblasts (MEF), and rat intestinal epithelia (RIE) cells (Limited, 2008; Zhang et al., 2008). After treatment with FK506, FKBP51 was excluded from the nucleus and showed cytoplasmic, perinuclear staining in a similar pattern previously described in osteosarcoma cells (U2OS cell line) and in megakaryocytes (MK cells) (Giraudier et al., 2002; Limited, 2008). The differential localization of FKBP51, nuclear versus perinuclear, seems to be cell-type dependent. This suggests that nuclear localization of FKBP51 is dependent on some component present in MEF cells, neurons, GTM-5, NTM-5, REI cells that is not present in MK cells or U2OS.
In the cytoplasm, FKBP52 is hypothesized to be an accessory protein for GR trafficking to the nucleus. To test this hypothesis in neuronal cells, we knocked down FKBP52 via siRNA in differentiated SH-SY5Y cells. We treated the FKBP52-deficient cells with cortisol and stained for GR using immunofluorescence. FKBP52-deficiency did not totally abrogate nuclear translocation of GR. As shown in Figure 5, in the si-Control group, the nuclear GR fluorescence intensity increased with cortisol present. The FKBP52-deficient cells formed GR aggregates.
We hypothesized that the nuclear translocation of GR would be abrogated by the absence of FKBP52. We observed a significant reduction in the nuclear immunofluorescence signal for GR in the FKBP52 knockdown (Figure 5a), as expected from recent work using GFP-tagged GR (Banerjee et al., 2008). We assessed the effect of FKBP52 on the kinetics of relatively early cortisol-induced GR trafficking. However, if a spike in cortisol levels due to a stressful event caused ligand-induced GR signaling in neurons, and the physiologic cortisol levels dissipated quickly, FKBP52 may play a role in these transient stress-induced short cortisol spikes.
Si-FKBP5 contributed to increased nuclear GR translocation in the SH-SY5Y cells in the absence of cortisol, but not after cortisol-induction; which indicates that FKBP51 may play a role in chronic, baseline glucocorticoid signaling in the brain. Since in the absence of FKBP51 there was higher nuclear GR distribution without cortisol (Figure 5d), either GR import is increased or GR-turnover is decreased in the absence of FKBP51. There are SNPs in the FKBP5 gene that associate with MDD, PTSD, peritraumatic dissociative disorder, and impaired recovery from psychosocial stress in normal adults (Binder et al., 2004; Binder et al., 2008; Ising et al., 2008; Koenen et al., 2005). A microtubule-associated protein called Double-cortin-like was recently shown to regulate the transport of GR in neuroprogenitor cells (Fitzsimons et al., 2008). If SH-SY5Y cells express Double-cortin-like, then this would be a factor independent of FKBP52 and FKBP51 regulating GR translocation. Since SH-SY5Y is a cell line and it may express markers of “immature” neuronal cells our standard practice is to “differentiate” the cells by incubation for at least three days with retinoic acid prior to experiments; at that point they develop neuritic networks and the proliferation is stopped (Supplementary Information) (PÂhlman et al., 1984).
Treatment with FK506 did not alter the ability of the GR to form nuclear aggregates, but the aggregates were fewer and more diffuse. Also, there was appreciable GR present in the cytoplasm after 180 minutes if FK506 was present compared to cells that were not treated with FK506. This is evidence that association of GR with immunophilins supports GR stability in the nucleus. Based on this, changing the GR-immunophilin interaction, using FK506, seems to affect the formation and stability of GR in neuronal nuclei. Finally, knockdown of FKBP52 seemed to have the greatest effect, inhibiting GR nuclear translocation on cortisol-induction. However, no genetic association of its gene has been made with HPA-axis disorders, while the related gene, coding for FKBP51 has been associated multiple times with these disorders. Our findings show that knockdown of FKBP51 caused increased baseline nuclear localization of GR; and it may be the chronic, long-term, or baseline GR signaling that is relevant. The results suggest that FKBP51 is pertinent to inhibiting chronic, baseline signaling and FKBP52 is primarily involved in promoting acute, fast GR response. In conclusion, we propose that immunophilins may be modulators of the cortisol-HPA axis response to stress and related chronic brain disorders. Altering neuronal gene expression through controlling GR-mediated signaling pathways may represent therapeutic intervention for central nervous system diseases marked by HPA axis dysregulation such as MDD and PTSD.
4. Experimental Procedure
4.1. Cell culture
Human forebrain fetal tissue was acquired from Advanced Bioscience Resources (Alameda, CA) according to the University of California San Diego Internal Review Board guidelines. The tissue was processed as previously described (Avramut et al., 2001; White et al., 1999). Cells were seeded at 106 cells per well on glass coverslips coated with polyornithine and laminin in 24-well plates and maintained in DMEM-F12 media (Gibco 12634-010) supplemented with 22 mM glutamine, 5% human serum, and 10 μg/mL gentamycin sulphate (Gibco 15750) for four weeks with media-change every 3 days. The primary human neuroglial cultures were used in qualitative microscopic analyses.
The neuronal cell line SH-SY5Y was used for transfection with siRNA to knockdown the FKBP51 and FKBP52 translation. SH-SY5Y cells were plated at 5×105 cells/well on glass coverslips in 24-well plates which had been coated with poly-L-ornithine and laminin by sequentially incubating overnight at 37°C first with 0.1 mg/mL poly-L-ornithine hydrobromide in H2O solution (Sigma, St. Louis, MO, P5666) and then 2 μg/mL laminin in H2O (Sigma L2020). SH-SY5Y cells were grown in media composed of 1:1 mixture of Ham's F-12 Media (Gibco, Carlsbad, CA, 31765-035) and Dulbelco's Modified Eagle Media (DMEM) (Gibco 11960-044) supplemented with 2 mM glutamine (Gibco), sodium bicarbonate (Gibco 25080-094), sodium pyruvate (Gibco P333-1000), and 10% fetal bovine serum (Gibco 16140-071), and 50 μg/mL penicillin and streptomycin (Invitrogen, Carlsbad, CA, 15640-055). After three days in culture, the media were supplemented with 1 μg/mL retinoic acid (Fisher Scientific, Waltham, MA, AC41897) for differentiation, media were changed daily with the retinoic acid supplement until cells showed neuritic networks, typically 3-5 days.
4.2. Western Blotting
Primary antibodies used for Western Blotting: rabbit anti FKBP51 (Abcam, Cambridge, MA, 46002), Mouse anti FKBP52 (Stressgen, Ann Arbor, MI, SRA1400), Mouse anti GR (Abcam ab2768), Rabbit anti Actin (Sigma A3853). Biotin-conjugated secondary antibodies were used from Jackson Immunoresearch (Westgrove, PA, anti mouse 715-065-151, anti rabbit 711-065-152, both at 1:1,000, 2 hr at room temperature, and vector ABC kit was used for signal amplification (Vector Laboratories, Orton Southgate, England, PK-4002). Chemiluminescence was performed following manufacturer's instructions (Pierce Biologicals, Rockford, IL, 32106). As a control for secondary detection, 3 groups of 20 μg and 10 μg of total protein from SH SY5Y cells were run in a 10% polyacrylamide gel and transferred to a PVDF membrane and the membrane cut into 3 strips. For the first strip, the normal western blotting procedure was performed for FKBP52. For the second strip, the primary antibody was excluded. For the third strip both primary antibody and secondary antibody was excluded.
4.3. PCR and siRNA Synthesis
The Dicer siRNA Generation Kit (Gene Therapy Systems, San Diego, CA, T510001) was used to generate siRNA to FKBP52 following manufacturer's protocols. The mRNA sequences for human FKBP4 and human FKBP5 (Genebank accession numbers NM002014)were used to design primers to amplify 650 base-pair regions of the mRNAs spanning both Exons 2 and 3, coding for the linker region between the two immunophilin domains, a region of high variation between individuals and very low homology with FKBP5. The primers are: FKBP52-T7-F gcg-taatacgactcactatagggag-agcggtgaaggctatgctaag, FKBP52-T7-R gcg-taatacgactcactatagggag-acttctcgttgttgctgtcca, FKBP51-T7-F gcg-taatacgactcatatagggag-atccctcgaatgcaactctct, FKBP51-T7-R gcgtaatacgactcactatagggag-aaaggcagcaaggagaatga (leader-T7 RNA polymerase promoter- target sequence). Differentiated SH-SY5Y cells were exposed to siRNA (250 ng siRNA, 3.5μL GeneSilencer reagent per well) for 24 hr prior to cortisol exposure. Control siRNA, specific to green fluorescent protein (GFP), was generated using the template plasmid and primers for GFP supplied by the manufacturer.
4.4. Cortisol and FK506 Exposure
Differentiated SH-SY5Y cells or primary human neuroglia cultures were exposed to vehicle or 10 μM FK506 for 2 hr; followed by 100 nM cortisol for various time points. Each treatment (vehicle, FK506, cortisol 30, 60, 180 min, and cortisol pre-treated with FK506) was performed in triplicate and cells were fixed in 4% paraformaldehyde for immunofluorescent labeling of GR and Hoechst 33342 (Molecular Probes, Carlsbad, CA, H3570). Primary neuronal cultures were labeled for GR (mouse Abcam ab9568, 1:50), or FKBP51 (rabbit - Abcam ab54992, 1:40). To date, we have been unable to satisfactorily image FKBP52 in primary neuronal cultures, though the PCR and Western blotting products were detected (Supplementary Figure S1). To determine cell types in culture, counter-labeling was performed. To identify neuronal dendrites and soma we used antibodies to MAP2 (mouse - Sigma M9942, 1:500; or rabbit - Cell Signaling Technologies, Danvers, MA, #4542, 1:1000). To identify neuronal axons we used antibodies to neurofilament (rabbit – Covance, San Diego, CA, PRB574C, 1:500; or mouse - Cell Signaling Technologies, #2835, 1:1000). To identify astrocytes we used the mouse monoclonal antibody to GFAP (Cell Signaling, #3670, 1:400). Cells were permeabilized in 1% Triton × 100 for 30 min at room temp, and washed 3 times with phosphate buffered saline with 0.2% TWEEN-20 (PBS-T) prior to blocking for 1 hr with 10% normal donkey serum in PBS-T. After fixation, washing, and blocking, cells were incubated with primary antibody overnight at 4°C. To reduce non-specific antibody binding to extracellular debris previously observed using rabbit antibodies, the wash buffer was supplemented with 100 mM NaCl for primary antibodies generated in rabbit. Donkey anti-mouse Alexafluor 488 (Invitrogen, A21203) and Alexafluor 546 (Invitrogen, A10036); and donkey anti-rabbit Alexafluor 488 (Invitrogen 21206) and Alexafluor 546 (Alexafluor A10042); antibodies were used at 1:2000, and cells were incubated for 2 hr, at room temperature.
4.5. Quantitative PCR
To assess whether GR-responsive genes, FKBP5 and BCL21A are altered due to the presence of FK506, qPCR was performed. RNA was isolated from differentiated cells pretreated with or without 10 μM FK506 followed by 100 nM cortisol for various lengths of time. For qPCR, 20 ng cDNA per reaction was used and 6-carbosyfluorocein (FAM) -labeled 20× prevalidated probes were purchased from Applied Biosystems (Foster City, CA). For FKBP5, assay Hs00188025_m1; for BCL21A, assay Hs00236329_m1; and for endogenous control we used the actin gene, ACTB, assay 4352935E. TaqMan master mix (2×), purchased from Applied Biosystems was used in 20 μL reactions on 96 well-plates and assays were performed at the University of San Diego Center for AIDS Research Genomics Core. Gene expression is reported as fold-control versus the median of the control-group using the ΔΔ-CT method comparing to housekeeping gene ACTB whereby ΔCT=CTACTB–CTGene, ΔΔCT=ΔCTControl-ΔCTTreatment, and Fold-Control = 2 −ΔΔCT. Plotted are mean of percent-changes and standard deviation of triplicate.
4.6. Imaging
Images were acquired utilizing a 40× objective on a Carl Zeiss (Thornwood, NY) Axiovert 40 inverted fluorescent microscope with deconvolution capabilities. For 3D imaging, 10 planes were imaged at 0.5 μm steps and deconvolved using the Nearest Neighbor algorithm in the image analysis software Slidebook 4.2 (Intelligent Imaging, Santa Monica, CA) (Leffler, 1997). All the images for a particular experiment were acquired at the same time under identical parameters such as exposure time, intensity, 2× binning, and Normalization settings following the conventions outlined in Slidebook 4.2 (Leffler, 1997).
4.7. Quantitative Imaging of Glucocorticoid Receptor Nuclear Translocation in SH-SY5Y Cells
In order to quantify the nuclear staining of the GR in the SH-SY5Y cells, fluorescence intensity from the green channel was measured in the nuclei and expressed as fluorescence units per μm3. Ten fields from each treatment were acquired with a 40× objective on a Carl Zeiss Axiovert 40 inverted microscope. Fields were selected randomly and to reduce bias, the user focused only using the blue channel. For each field, 10 planes were imaged in 0.5 μm steps in the z-plane with 2× binning, green channel 1.75 sec, and blue channel 0.650 sec.
A mask was created in all planes and all images according to the Slidebook 4.2 (Leffler, 1997) procedures that encompassed the regions of blue intensity, designating the nuclei and mask statistics were exported to Excel for data analysis. Fluorescence intensity in the green channel accounts for intensity of GR staining, and intensity units per μm3 were calculated for each field, 3D imaging allowed for volume determination. Two-way ANOVA and Bonferroni's multiple comparison using a one-tailed test (we hypothesized greater intensity after treatment with cortisol), and significance is reported if p < 0.01.
Supplementary Material

Acknowledgments
Grants from the United States National Institutes of Health: MH076681 to CLA; R01MH079881, R41MH079728, R25MH074508, R25MH081482 and P30 MH62512 to IPE; R01 NS050621 to M.K.; T32 EB001026 to ETT. This work was performed in part with the support of the Genomics Core at the UCSD Center for AIDS Research (AI36214) and the San Diego Veterans Medical Research Foundation.
Abbreviations
- FKBP51
FK506 Binding Protein of 51 kilodaltons
- FKBP52
FK506 Binding Protein of 52 kilodaltons
- GR
glucocorticoid receptor
- HPA
hypothalamic-pituitary-adrenal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandre Urani PG. Corticosteroid Receptor Transgenic Mice. Ann N Y Acad Sci. 2003;1007:379–393. doi: 10.1196/annals.1286.037. [DOI] [PubMed] [Google Scholar]
- Avramut M, Zeevi A, Achim CL. The immunosuppressant drug FK506 is a potent trophic agent for human fetal neurons. Brain Res Dev Brain Res. 2001;132:151–7. doi: 10.1016/s0165-3806(01)00307-8. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Periyasamy S, Wolf IM, Hinds TD, Jr, Yong W, Shou W, Sanchez ER. Control of glucocorticoid and progesterone receptor subcellular localization by the ligand-binding domain is mediated by distinct interactions with tetratricopeptide repeat proteins. Biochemistry. 2008;47:10471–80. doi: 10.1021/bi8011862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao AM, Meynen G, Swaab DF. The stress system in depression and neurodegeneration: Focus on the human hypothalamus. Brain Res Rev. 2007 doi: 10.1016/j.brainresrev.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B, Papiol S, Seaman S, Lucae S, Kohli MA, Nickel T, Kunzel HE, Fuchs B, Majer M, Pfennig A, Kern N, Brunner J, Modell S, Baghai T, Deiml T, Zill P, Bondy B, Rupprecht R, Messer T, Kohnlein O, Dabitz H, Bruckl T, Muller N, Pfister H, Lieb R, Mueller JC, Lohmussaar E, Strom TM, Bettecken T, Meitinger T, Uhr M, Rein T, Holsboer F, Muller-Myhsok B. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. 2004;36:1319–25. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, Schwartz AC, Cubells JF, Ressler KJ. Association of FKBP5 Polymorphisms and Childhood Abuse With Risk of Posttraumatic Stress Disorder Symptoms in Adults. JAMA. 2008;299:1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GM, Grota LJ, Penney DP, Reichlin S. Pituitary-adrenal function in the squirrel monkey. Endocrinology. 1970;86:519–29. doi: 10.1210/endo-86-3-519. [DOI] [PubMed] [Google Scholar]
- Callebaut I, Renoir JM, Lebeau MC, Massol N, Burny A, Baulieu EE, Mornon JP. An immunophilin that binds M(r) 90,000 heat shock protein: main structural features of a mammalian p59 protein. Proc Natl Acad Sci U S A. 1992;89:6270–4. doi: 10.1073/pnas.89.14.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll BJ, Cassidy F, Naftolowitz D, Tatham NE, Wilson WH, Iranmanesh A, Liu PY, Veldhuis JD. Pathophysiology of hypercortisolism in depression. Acta Psychiatr Scand Suppl. 2007:90–103. doi: 10.1111/j.1600-0447.2007.00967.x. [DOI] [PubMed] [Google Scholar]
- Chao HM, Choo PH, McEwen BS. Glucocorticoid and mineralocorticoid receptor mRNA expression in rat brain. Neuroendocrinology. 1989;50:365–71. doi: 10.1159/000125250. [DOI] [PubMed] [Google Scholar]
- Cheung-Flynn J, Prapapanich V, Cox MB, Riggs DL, Suarez-Quian C, Smith DF. Physiological role for the cochaperone FKBP52 in androgen receptor signaling. Mol Endocrinol. 2005;19:1654–66. doi: 10.1210/me.2005-0071. [DOI] [PubMed] [Google Scholar]
- Chourbaji S, Vogt MA, Gass P. Mice that under- or overexpress glucocorticoid receptors as models for depression or posttraumatic stress disorder. Prog Brain Res. 2008;167:65–77. doi: 10.1016/S0079-6123(07)67005-8. [DOI] [PubMed] [Google Scholar]
- Chrousos GP, Renquist D, Brandon D, Eil C, Pugeat M, Vigersky R, Cutler GB, Jr, Loriaux DL, Lipsett MB. Glucocorticoid hormone resistance during primate evolution: receptor-mediated mechanisms. Proc Natl Acad Sci U S A. 1982;79:2036–40. doi: 10.1073/pnas.79.6.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies TH, Ning YM, Sanchez ER. A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem. 2002;277:4597–600. doi: 10.1074/jbc.C100531200. [DOI] [PubMed] [Google Scholar]
- Davies TH, Ning YM, Sanchez ER. Differential control of glucocorticoid receptor hormone-binding function by tetratricopeptide repeat (TPR) proteins and the immunosuppressive ligand FK506. Biochemistry. 2005;44:2030–8. doi: 10.1021/bi048503v. [DOI] [PubMed] [Google Scholar]
- Defranco DB. Role of molecular chaperones in subnuclear trafficking of glucocorticoid receptors. Kidney Int. 2000;57:1241–9. doi: 10.1046/j.1523-1755.2000.00957.x. [DOI] [PubMed] [Google Scholar]
- Denny WB, Valentine DL, Reynolds PD, Smith DF, Scammell JG. Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology. 2000;141:4107–13. doi: 10.1210/endo.141.11.7785. [DOI] [PubMed] [Google Scholar]
- Deroo BJ, Rentsch C, Sampath S, Young J, DeFranco DB, Archer TK. Proteasomal inhibition enhances glucocorticoid receptor transactivation and alters its subnuclear trafficking. Mol Cell Biol. 2002;22:4113–23. doi: 10.1128/MCB.22.12.4113-4123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edinger RS, Watkins SC, Pearce D, Johnson JP. Effect of immunosuppressive agents on glucocorticoid receptor function in A6 cells. Am J Physiol Renal Physiol. 2002;283:F254–61. doi: 10.1152/ajprenal.00337.2001. [DOI] [PubMed] [Google Scholar]
- Elbi C, Walker DA, Romero G, Sullivan WP, Toft DO, Hager GL, DeFranco DB. Molecular chaperones function as steroid receptor nuclear mobility factors. Proc Natl Acad Sci U S A. 2004;101:2876–81. doi: 10.1073/pnas.0400116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons CP, Ahmed S, Wittevrongel CF, Schouten TG, Dijkmans TF, Scheenen WJ, Schaaf MJ, de Kloet ER, Vreugdenhil E. The microtubule-associated protein doublecortin-like regulates the transport of the glucocorticoid receptor in neuronal progenitor cells. Mol Endocrinol. 2008;22:248–62. doi: 10.1210/me.2007-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galigniana MD, Harrell JM, Murphy PJM, Chinkers M, Radanyi C, Renoir JM, Zhang M, Pratt WB. Binding of hsp90-Associated Immunophilins to Cytoplasmic Dynein: Direct Binding and in Vivo Evidence that the Peptidylprolyl Isomerase Domain Is a Dynein Interaction Domain. Biochemistry. 2002;41:13602–13610. doi: 10.1021/bi020399z. [DOI] [PubMed] [Google Scholar]
- Gass P, Reichardt HM, Strekalova T, Henn F, Tronche F. Mice with targeted mutations of glucocorticoid and mineralocorticoid receptors: models for depression and anxiety? Physiol Behav. 2001;73:811–25. doi: 10.1016/s0031-9384(01)00518-2. [DOI] [PubMed] [Google Scholar]
- Gawlik M, Moller-Ehrlich K, Mende M, Jovnerovski M, Jung S, Jabs B, Knapp M, Stoeber G. Is FKBP5 a genetic marker of affective psychosis? A case control study and analysis of disease related traits. BMC Psychiatry. 2006;6:52. doi: 10.1186/1471-244X-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraudier S, Chagraoui H, Komura E, Barnache S, Blanchet B, LeCouedic JP, Smith DF, Larbret F, Taksin AL, Moreau-Gachelin F, Casadevall N, Tulliez M, Hulin A, Debili N, Vainchenker W. Overexpression of FKBP51 in idiopathic myelofibrosis regulates the growth factor independence of megakaryocyte progenitors. Blood. 2002;100:2932–2940. doi: 10.1182/blood-2002-02-0485. [DOI] [PubMed] [Google Scholar]
- Gold PW, Goodwin FK, Chrousos GP. Clinical and biochemical manifestations of depression. Relation to the neurobiology of stress (2) N Engl J Med. 1988;319:413–20. doi: 10.1056/NEJM198808183190706. [DOI] [PubMed] [Google Scholar]
- Harrell JM, Murphy PJ, Morishima Y, Chen H, Mansfield JF, Galigniana MD, Pratt WB. Evidence for glucocorticoid receptor transport on microtubules by dynein. J Biol Chem. 2004;279:54647–54. doi: 10.1074/jbc.M406863200. [DOI] [PubMed] [Google Scholar]
- Hubler TR, Scammell JG. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones. 2004;9:243–52. doi: 10.1379/CSC-32R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising M, Depping AM, Siebertz A, Lucae S, Unschuld PG, Kloiber S, Horstmann S, Uhr M, Müller-Myhsok B, Holsboer F. Polymorphisms in the FKBP5 gene region modulate recovery from psychosocial stress in healthy controls. European Journal of Neuroscience. 2008;28:389–398. doi: 10.1111/j.1460-9568.2008.06332.x. [DOI] [PubMed] [Google Scholar]
- Jonas E. BCL-xL Regulates Synaptic Plasticity. Mol Interv. 2006;6:208–222. doi: 10.1124/mi.6.4.7. [DOI] [PubMed] [Google Scholar]
- Koenen KC, Saxe G, Purcell S, Smoller JW, Bartholomew D, Miller A, Hall E, Kaplow J, Bosquet M, Moulton S, Baldwin C. Polymorphisms in FKBP5 are associated with peritraumatic dissociation in medically injured children. Mol Psychiatry. 2005;10:1058–9. doi: 10.1038/sj.mp.4001727. [DOI] [PubMed] [Google Scholar]
- Kohda K, Harada K, Kato K, Hoshino A, Motohashi J, Yamaji T, Morinobu S, Matsuoka N, Kato N. Glucocorticoid receptor activation is involved in producing abnormal phenotypes of single-prolonged stress rats: a putative post-traumatic stress disorder model. Neuroscience. 2007;148:22–33. doi: 10.1016/j.neuroscience.2007.05.041. [DOI] [PubMed] [Google Scholar]
- Leffler S. SlideBook. Silicon Graphics, Inc; 1997. Vol., ed.∧eds. [Google Scholar]
- Limited AS. Antibody Review to Mouse Anti FKBP51 ab46002. AbCam Incorporated; Cambridge, MA: 2008. Vol., ed.∧eds. [Google Scholar]
- Lowy MT. Reserpine-induced decrease in type I and II corticosteroid receptors in neuronal and lymphoid tissues of adrenalectomized rats. Neuroendocrinology. 1990;51:190–6. doi: 10.1159/000125336. [DOI] [PubMed] [Google Scholar]
- Maccari S, Mormede P, Piazza PV, Simon H, Angelucci L, Le Moal M. Hippocampal type I and type II corticosteroid receptors are modulated by central noradrenergic systems. Psychoneuroendocrinology. 1992;17:103–12. doi: 10.1016/0306-4530(92)90049-d. [DOI] [PubMed] [Google Scholar]
- Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA Binding Site Sequence Directs Glucocorticoid Receptor Structure and Activity. Science. 2009;324:4. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa T, Sumiyoshi S, Deshimaru M. Changes induced in the central nervous system of rats by steroid drug. Acta Neuropathol. 1974;30:85–9. doi: 10.1007/BF00685325. [DOI] [PubMed] [Google Scholar]
- Ozer EJ, Best SR, Lipsey TL, Weiss DS. Predictors of posttraumatic stress disorder and symptoms in adults: a meta-analysis. Psychol Bull. 2003;129:52–73. doi: 10.1037/0033-2909.129.1.52. [DOI] [PubMed] [Google Scholar]
- PÂhlman S, Ruusala AI, Abrahamsson L, Mattsson MEK, Esscher T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: a comparison with phorbolester-induced differentiation. Cell Differentiation. 1984;14:135–144. doi: 10.1016/0045-6039(84)90038-1. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Pearce BD, Pisell TL, Sanchez CI, Po C, Su C, Miller AH. The Proinflammatory Cytokine, Interleukin-1{alpha}, Reduces Glucocorticoid Receptor Translocation and Function. Endocrinology. 1999;140:4359–4366. doi: 10.1210/endo.140.9.6986. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biological Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Watanabe Y, Magarinos AM, McEwen BS. Opposing roles of type I and type II adrenal steroid receptors in hippocampal long-term potentiation. Neuroscience. 1995;68:387–94. doi: 10.1016/0306-4522(95)00151-8. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Galigniana MD, Harrell JM, DeFranco DB. Role of hsp90 and the hsp90-binding immunophilins in signalling protein movement. Cell Signal. 2004;16:857–72. doi: 10.1016/j.cellsig.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Reynolds PD, Ruan Y, Smith DF, Scammell JG. Glucocorticoid resistance in the squirrel monkey is associated with overexpression of the immunophilin FKBP51. J Clin Endocrinol Metab. 1999;84:663–9. doi: 10.1210/jcem.84.2.5429. [DOI] [PubMed] [Google Scholar]
- Sanchez ER. Hsp56: a novel heat shock protein associated with untransformed steroid receptor complexes. J Biol Chem. 1990;265:22067–70. [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- Sinars CR, Cheung-Flynn J, Rimerman RA, Scammell JG, Smith DF, Clardy J. Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc Natl Acad Sci U S A. 2003;100:868–73. doi: 10.1073/pnas.0231020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatro ET. [Dissertation] Cellular and Molecular Pathology. University of Pittsburgh School of Medicine; Pittsburgh, PA: 2008. Immunophilins FKBP52 and FKBP51 Modulate Glucocorticoid Receptor Distribution in Neurons and are Altered in HIV and Major Depressive Disorder; p. 210. [Google Scholar]
- Tombaugh GC, Yang SH, Swanson RA, Sapolsky RM. Glucocorticoids exacerbate hypoxic and hypoglycemic hippocampal injury in vitro: biochemical correlates and a role for astrocytes. J Neurochem. 1992;59:137–46. doi: 10.1111/j.1471-4159.1992.tb08884.x. [DOI] [PubMed] [Google Scholar]
- Tranguch S, Cheung-Flynn J, Daikoku T, Prapapanich V, Cox MB, Xie H, Wang H, Das SK, Smith DF, Dey SK. Cochaperone immunophilin FKBP52 is critical to uterine receptivity for embryo implantation. Proc Natl Acad Sci U S A. 2005;102:14326–31. doi: 10.1073/pnas.0505775102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rossum EFC, Binder EB, Majer M, Koper JW, Ising M, Modell S, Salyakina D, Lamberts SWJ, Holsboer F. Polymorphisms of the Glucocorticoid Receptor Gene and Major Depression. Biological Psychiatry. 2006;59:681–688. doi: 10.1016/j.biopsych.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR. From The Cover: Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proceedings of the National Academy of Sciences. 2004;101:15603–15608. doi: 10.1073/pnas.0407008101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Pongrac JL, DeFranco DB. Glucocorticoid receptors in hippocampal neurons that do not engage proteasomes escape from hormone-dependent down-regulation but maintain transactivation activity. Mol Endocrinol. 2002;16:1987–98. doi: 10.1210/me.2001-0287. [DOI] [PubMed] [Google Scholar]
- Webster MJ, Knable MB, O'Grady J, Orthmann J, Weickert CS. Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol Psychiatry. 2002;7:985–94. 924. doi: 10.1038/sj.mp.4001139. [DOI] [PubMed] [Google Scholar]
- White MG, Hammond RR, Sanders VJ, Bonaroti EA, Mehta AP, Wang G, Wiley CA, Achim CL. Neuron-enriched second trimester human cultures: growth factor response and in vivo graft survival. Cell Transplant. 1999;8:59–73. doi: 10.1177/096368979900800115. [DOI] [PubMed] [Google Scholar]
- Wochnik GM, Ruegg J, Abel GA, Schmidt U, Holsboer F, Rein T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem. 2005;280:4609–16. doi: 10.1074/jbc.M407498200. [DOI] [PubMed] [Google Scholar]
- Wu B, Li P, Liu Y, Lou Z, Ding Y, Shu C, Ye S, Bartlam M, Shen B, Rao Z. 3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc Natl Acad Sci U S A. 2004;101:8348–53. doi: 10.1073/pnas.0305969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young EA, Haskett RF, Murphy-Weinberg V, Watson SJ, Akil H. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry. 1991;48:693–9. doi: 10.1001/archpsyc.1991.01810320017003. [DOI] [PubMed] [Google Scholar]
- Zhang X, Clark AF, Yorio T. FK506-Binding Protein 51 Regulates Nuclear Transport of the Glucocorticoid Receptor {beta} and Glucocorticoid Responsiveness. Invest Ophthalmol Vis Sci. 2008;49:1037–1047. doi: 10.1167/iovs.07-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.