

Abstract
The striatum is particularly sensitive to the irreversible inhibitor of succinate dehydrogenase 3-nitropropionic acid (3-NP). In the present study, we examined early changes in behavior and dopamine and glutamate synaptic physiology created by a single systemic injection of 3-NP in Fischer 344 rats. Hindlimb dystonia was seen 2 h after 3-NP injections, and rats performed poorly on balance beam and rotarod motor tests 24 h later. Systemic 3-NP increased NMDA receptor-dependent long-term potentiation (LTP) at corticostriatal synapses over the same time period. The 3-NP-induced corticostriatal LTP was not attributable to increased NMDA receptor number or function, because 3-NP did not change MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine] binding or NMDA/AMPA receptor current ratios. The LTP seen 24 h after 3-NP was D1 receptor dependent and reversed by exogenous addition of dopamine or a D2 receptor agonist to brain slices. HPLC and fast-scan cyclic voltammetry revealed a decrease in dopamine content and release in rats injected 24 h earlier with 3-NP, and much like the enhanced LTP, dopamine changes were reversed by 48 h. Tyrosine hydroxylase expression was not changed, and there was no evidence of striatal cell loss at 24–48 h after 3-NP exposure. Sprague Dawley rats showed similar physiological responses to systemic 3-NP, albeit with reduced sensitivity. Thus, 3-NP causes significant changes in motor behavior marked by parallel changes in striatal dopamine release and corticostriatal synaptic plasticity.
Keywords: LTP, LTD, voltammetry, NMDA, D1, D2
Introduction
The striatum is particularly sensitive to metabolic deficits created by stroke, cardiac arrest, and exposure to mitochondrial toxins (Nishino et al., 2000; Ste-Marie et al., 2000; Carmichael, 2005). These different forms of metabolic crisis share a unique profile of creating striatal lesions, which preferentially impact the ventrolateral striatum, and within the lesion site, a sparing of NADPH diaphorase and cholinergic interneurons (Ferriero et al., 1988; Burke et al., 1994; Ste-Marie et al., 2000; Blum et al., 2002). The striatal lesions created by stroke, cardiac arrest, and mitochondrial toxins are mediated at least in part by oxidative stress, and a critical pathological role is also implicated for striatal dopamine (DA) (Beal et al., 1993a,b; Hashimoto et al., 1994; Lancelot et al., 1995; Bogdanov et al., 1998; Reynolds et al., 1998; Toner and Stamford, 1999; Klivenyi et al., 2000; Fernagut et al., 2002; Kim and Chan, 2002; Pang et al., 2002; Callaway et al., 2003; Fancellu et al., 2003; Golden et al., 2003; Gu et al., 2004). This striatal blueprint created by metabolic challenge indicates that common mechanisms are likely triggered to create such a distinctive phenotype of pathology.
Most studies investigating striatal vulnerability to metabolic challenge focus on an endpoint of cell death and quantifying the size of lesions created by the challenge. Many questions still exist, however, about cellular and synaptic events that precede dehydrogenase 3-nitropropionic acid (3-NP)-induced cell death or those that may occur without cell death (Newcomb et al., 2005). We intentionally reduced the dose of 3-NP to begin to address changes in synaptic physiology, which may underlie the pathology created by larger doses or increased risk to other physiological challenges (Simpson and Isacson, 1993). This scenario is more likely to occur in the dorsomedial striatum, which is spared in models of stroke and mitochondrial poisoning. Electrophysiological studies on ischemia and 3-NP toxicity in the striatum have focused primarily on responses to bath delivery of the toxic insult to brain slices (Calabresi et al., 2001; Gubellini et al., 2004). This study differs in its analysis of electrophysiological changes created by systemic, whole-animal exposure to 3-NP and in the use of a normal divalent cation saline to bathe brain slices instead of Mg2+-free saline often used to examine 3-NP effects in brain slices (Centonze et al., 1999, 2001a; Calabresi et al., 2001, 2003). The 3-NP-induced increase in corticostriatal long-term potentiation (LTP) expression we observed is particularly noteworthy, because long-term synaptic depression (LTD) is most often reported for corticostriatal synapses (Calabresi et al., 1992; Partridge et al., 2000; Centonze et al., 2001a; Smith et al., 2001; Wang et al., 2006; Kreitzer and Malenka, 2007).
Dalbem et al. (2005) reported that systemic 3-NP caused a reduction in LTD of corticostriatal field potentials for 48–60 h after a 4 d regimen of 3-NP injections. In this study, we examined the potential mechanisms underlying changes in corticostriatal function created by systemic 3-NP and discovered that 3-NP created unique conditions for enhancing corticostriatal LTP expression produced by a change in dopamine modulation of corticostriatal synapses.
Materials and Methods
3-NP injections and behavioral testing.
Experiments were performed on 2-month-old Fischer 344 male rats obtained from a local vendor (Charles River Breeding Laboratories). An additional set of Sprague Dawley rats was also examined to study potential strain-specific differences in 3-NP sensitivity (see Fig. 8). The age of rats at the time of injection was held constant, because 3-NP sensitivity is age dependent (Beal et al., 1993a). All protocols were approved by the Animal Use and Care Committee at the University of Southern California and were in accordance with guidelines established by the United States Public Health Service Policy on Humane Care and Use of Laboratory Animals. Single intraperitoneal injections of 3-NP (15.0–16.5 mg/kg) (Sigma) using an injection volume of 0.5 cc/200 g were delivered to 2-month-old rats, and they were allowed to survive 24 or 48 h after the injection. 3-NP was dissolved in saline and neutralized to pH 7.4 using sodium hydroxide. The 15–16.5 mg/kg dose was chosen empirically by looking at maximal changes in synaptic plasticity, while still having good 48 h survival rates. A single dose of 20 mg/kg was consistently lethal in the Fischer 344 rat. Rats were ranked for early signs of 3-NP-induced motor problems 6–8 h after the 3-NP injection according to the following rating scale developed by Ouary et al. (2000): intermittent dystonia of one hindlimb, score = 1; intermittent dystonia of two hindlimbs, score = 2; permanent dystonia of hindlimbs, score = 3; uncoordinated and wobbling gate, score = 4; animal lying on one side but showing uncoordinated movement when stimulated, score = 5; near death recumbence, score = 6.
Figure 8.
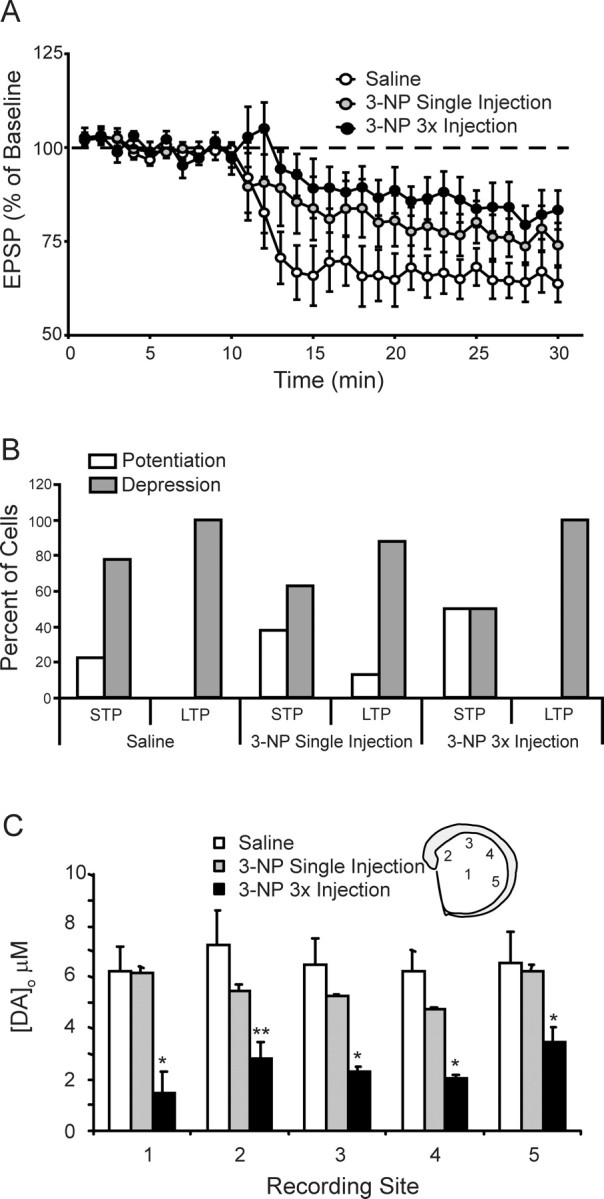
Sprague Dawley rats show reduced sensitivity to systemic 3-NP. A, Plots of tetanus-induced long-term synaptic plasticity at dorsomedial corticostriatal synapses are shown. No difference was found in the tetanus-induced plasticity between slices taken from saline-injected Sprague Dawley rats and Sprague Dawley rats injected 24 h earlier with 25–35 mg/kg 3-NP. A trend toward reduced LTD was seen in slices taken from rats injected once a day for 3 d with 3-NP (15, 10, and 10 mg/kg). B, Bar graphs showing distribution of cells expressing tetanus-induced potentiation (>100%) and depression (<100%) for STP and LTP under each experimental condition (saline, single injection, and 3× injection). C, Systemic injection of 3-NP creates regionally dependent decreases in dopamine release in the striatum. The amount of dopamine release was determined at 5 regions within the striatum including (1) midstriatum, (2) dorsomedial, (3) dorsal, (4) dorsolateral, and (5) ventrolateral (inset). Bar graph illustrates peak dopamine released by a single intrastriatal stimulus (200 μA, 0.1 ms) applied to striatal brain slices at each of the above indicated regions for saline-injected rats, rats injected 24 h earlier with 3-NP (25–35 mg/kg 3-NP), and rats injected once a day for 3 d with 3-NP (15, 10, and 10 mg/kg) and killed 24 h after the last injection. No differences were observed between saline-injected and single injected (25–35 mg/kg 3-NP) Sprague Dawley rats. A significant reduction in dopamine release was seen in rats injected once a day for 3 d with 3-NP across all 5 sampling sites (repeated-measures ANOVA; p < 0.03). Post hoc analysis showed reduced release at sites 1, 3, 4 (*p < 0.01) and 2 (**p < 0.02).
Balance beam.
Rats were tested for their ability to walk across a rod before injection of 3-NP and then again before being killed at the 24 or 48 h post-3-NP-injection time point as a measure of motor coordination and balance (Friedemann and Gerhardt, 1992). Each rat received an injection of saline 24 h before its pre-3-NP, control test on the balance beam. The walking apparatus was a 60-cm-long wooden dowel, 5 cm in diameter, that was grooved and suspended between two platforms 60 cm above a padded surface. Rats were placed in the center of the rod facing one platform, and each rat was tested three times to reach the escape platform with a maximum time limit of 120 s. A rank score was determined using the following scale: 0, fall; 1, clasp; 2, all paws on top; 3, takes steps; 4, reach the platform (Friedemann and Gerhardt, 1992). The rat was then given a score that reflected the maximum of the three trials.
Accelerating rotarod test.
Rats were tested before injection with 3-NP and before they were killed at the 24 or 48 h post-3-NP-injection time point. Rats were first individually habituated to a stationary rod for 180 s and then tested on an accelerating rod (rotarod) (Rotamex; Columbus Instruments). The rod accelerated from 2 to 20 rpm with the rotation increasing by 2 rpm every 10 s. The final speed and the latency to fall were recorded. Three trials were performed with a 10 min interval between trials. The rat was given the mean score of the three trials.
Brain slice preparation.
Rats were anesthetized with halothane and decapitated immediately at either 24 or 48 h after intraperitoneal injection of 3-NP. Their brains were removed and placed in cooled (1–4°C), modified-oxygenated artificial CSF (aCSF). In the modified aCSF, some of the sodium was replaced with sucrose to reduce tissue excitability during brain slice cutting (124 mm sucrose and 62 mm NaCl). This solution maintained the osmotic balance found in normal aCSF. Normal aCSF contained (in mm) 124 NaCl, 1.3 MgSO4, 3.0 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2.4 CaCl2, and 10.0 glucose, equilibrated with a 95% O2–5% CO2 mixture to obtain a pH value of 7.3–7.4.
Hemicoronal striatal slices were cut at a thickness of 400 μm with a Vibratome 1000 (Vibratome). The slices were immediately placed in an oxygenated aCSF solution and were slowly brought to room temperature (23°C). Bicuculline methiodide (BIC; 30 μm; Sigma) was always present during recordings and used to block GABAA receptor-mediated inhibition as a method to isolate excitatory synaptic events. Slices remained in this solution for 2 h before and throughout all recording sessions. Single slices were transferred to the recording chamber (Haas ramp style gas interface chamber) and bathed continuously with the oxygenated BIC–aCSF solution maintained at a temperature of 32°C. In some experiments, drugs were added directly to the bathing media to block D1 dopamine receptors [R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine (SCH 23390); Sigma], D2 dopamine receptors (l-sulpiride; Sigma), and NMDA receptors [5-amino phosphonovaleric acid (APV); Sigma].
Intracellular recording and extracellular stimulation.
Bipolar insulated tungsten wire (50 μm diameter) stimulating electrodes were used for delivering paired and tetanizing extracellular stimuli to the border between the striatum and the overlying corpus callosum. Intracellular records were obtained at a 1 mm distance from the extracellular electrodes. Test synaptic responses were delivered as pairs separated by an interstimulus interval of 50 ms, with constant current stimulus intensity (10–500 μA) and duration (0.1 ms). The paired-pulse ratio (PPR) was then determined for each recording as EPSP2/EPSP1. Responses to paired stimuli were sampled every 20 s for a control period of 10 min before tetanization and during a posttetanus sampling period of at least 20 min. Tetanic stimulation consisted of four trains of stimuli separated by 10 s. Each train lasted 1 s and was delivered at a frequency of 100 Hz. The tetanus stimulation intensity was set to equal the threshold for orthodromic induction of action potentials. The intensity used to sample EPSPs was then set to half the intensity of the orthodromic threshold. The orthodromic threshold and the threshold for first detection of corticostriatal EPSPs were determined by performing an input–output relationship between stimulation intensity and EPSP amplitude.
Intracellular records were obtained with glass microelectrodes pulled by a Flaming-Brown P-87 pipette puller. Electrodes filled with 2 m potassium acetate had resistance values ranging from 100 to 160 MΩ. Intracellular electrodes contained 2% biocytin (Sigma) in some experiments to verify the cell type based on morphology. Intracellular signals were amplified with an Axoclamp 2A amplifier, digitized with a Digidata 1200, and stored on disk using pCLAMP software (Molecular Devices). Established electrophysiological criteria for striatal medium spiny neurons were used for including cells in this study, which included resting membrane potentials greater than −80 mV, a stable input resistance >20 MΩ, A-current delayed firing in response to suprathreshold depolarizing injections of current, and nonadapting firing of action potentials in response to stronger injections of depolarizing current injections (Calabresi et al., 1992; Akopian et al., 2000). These characteristics were determined for each cell at the beginning of each experiment using 500-ms-long injections of current.
To obtain the ratio of NMDA receptor-mediated (EPSCNMDA) to AMPA receptor-mediated (EPSCAMPA) synaptic currents in striatal medium spiny neurons, some recordings were performed in discontinuous single-electrode voltage clamp (dSEVC) mode using an Axoclamp 2A amplifier with sharp intracellular electrodes filled with 2 m CsCl and 100 mm QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide]. After impaling a cell in current-clamp mode, the amplifier was switched to discontinuous current clamp to adjust optimal capacitance compensation and sampling rate. The gain was increased carefully after switching to dSEVC mode followed by further sampling rate correction. This process was observed on the separate oscilloscope to avoid instability and overshooting. It was usually possible to use a sampling rate to 5–7 kHz. Currents were recorded with a low-pass filter set at 3 kHz and sampled at 10 kHz. After establishing a holding voltage (Vh) at 0 mV (no or minimal responses on extracellular synaptic stimulation), Vh was stepped to either −80 mV or +40 mV 5 s before synaptic stimulation. Synaptic responses were sampled three times at each holding potential using 20 s interpulse intervals for each holding potential. Synaptic responses from these three trials were averaged during the analysis of synaptic ratios. The EPSCNMDA/EPSCAMPA ratios were obtained by dividing the currents recorded at a Vh of +40 mV by the synaptic currents measured at Vh of −80 mV. AMPA synaptic currents were measured as the peak current recorded at Vh of −80 mV, and NMDA synaptic currents were measured as the synaptic current measured 60 ms after the stimulus artifact for the synaptic response generated at a holding potential of +40 mV (Kretizer and Malenka, 2007).
Electrophysiological analysis of EPSPs and EPSCs.
The peak amplitude of EPSPs were measured with respect to the potential measured just before the stimulus artifact off-line using Clampfit analysis software (Molecular Devices). This procedure reduced, but did not eliminate, the possibility of error in calculating the amplitude of the second response of the pair created by summation between the second response and the falling phase of the first response when studying paired-pulse synaptic plasticity. Another way of minimizing this problem is to measure the ascending slope of the response, but previous work from our laboratory and others has shown identical outcomes for measurement of response amplitude and response ascending slope (Dos Santos Villar and Walsh, 1999; Kerr and Wickens, 2001).
Synaptic responses were sampled at 20 s intervals, and the average of three samples (1 min) was plotted for each minute of the experiment. The baseline paired-pulse plasticity, determined for each recording, was the average of paired-pulse plasticities generated over the entire 10 min baseline recording period. Each control pretetanus three-sample time point (see above) was then normalized to the average value obtained over the entire 10 min baseline recording period. Cells were included in the study if they (1) maintained stability in EPSP amplitude over the entire 10 min baseline recording period (±5% of the original EPSP amplitude) and (2) maintained stable responses to current injection, including their pattern of action potential discharge generated by depolarizing injections of current. Posttetanic changes in response amplitude were calculated by expressing the amplitude of each 1 min average as a percentage of the average response amplitude generated during the 10 min baseline sampling period in each cell. Paired-pulse synaptic plasticity was calculated by expressing the amplitude of the second response of each pair as a percentage of the amplitude of the first response of the pair (PPR). Individual cells were categorized for the plasticity they expressed 15–20 min after the tetanus by comparing the average amplitude of the EPSP expressed by the cell at 15–20 min after the tetanus to the average control amplitude of the EPSP for that cell calculated during from the 10 min baseline pretetanus sampling period. Cells showing a 5% increase in EPSP amplitude 15–20 min after the tetanus were categorized as expressing LTP.
Amplitude descriptive statistics (i.e., mean ± SEM) were calculated for paired-pulse, 3–4 min posttetanic and long-term posttetanic samples. Differences in tetanus-induced plasticity were determined through repeated-measures ANOVA performed across the entire posttetanus sampling period. Each cell was sampled every 60 s for 20 min after the tetanus, and thus the repeated-measures ANOVA had 20 sampling time points. Degrees of freedom (df), F-score (F), and probability (p) are reported for repeated-measures ANOVAs. Post hoc comparisons were also performed between each group at 0–3 min after tetanus (posttetanic plasticity) and at 16–20 min after tetanus (long-term plasticity) using post hoc Student's t tests assuming unequal variance. The 0–3 and 16–20 min values were calculated by averaging the 1 min samples occurring in the posttetanic and long-term plasticity ranges (0–3 and 15–20 min). Thus, post hoc tests consisted of single values from each cell for posttetanic and long-term plasticity. To examine distribution of response patterns, posttetanic responses were categorized as potentiation (>100% of pretetanus average) or depression (<100% of pretetanus average) for short-term plasticity (STP) (3–4 min after tetanus) and long-term plasticity (15–20 min after tetanus) time points as done previously in our laboratory (Akopian and Walsh, 2006). Distributions were plotted as bar graphs, and analysis of distributions was performed using Fisher's exact test. The small sample size dictated the use of the Fisher's exact test over χ2 analysis to study distributions.
Fast-scan cyclic voltammetry quantification of striatal dopamine release.
Disc carbon fiber electrodes (CFEs) were made from 7 mm unsized carbon fibers (Goodfellow) by electrophoretic anodic deposition of paint (ALA Scientific Instruments) (Schulte and Chow, 1996). Extracellular dopamine was monitored at the carbon fiber microelectrode every 100 ms by applying a triangular waveform (−0.4 to +1.0 V vs Ag/AgCl, 300 V/s). Currents were recorded with a modified VA-10X Voltammetric and Amperometric Amplifier (NPI Electronic). Data acquisitions were controlled by Clampex 7.0 software (Molecular Devices). Electrical stimulation of the brain slice surface across a twisted, bipolar, nichrome electrode was used to evoke dopamine release. Single constant current pulses of 250 μA and 0.1 ms duration were obtained by using an A360R Constant Current Stimulus Isolator (World Precision Instruments) and a Master-8 pulse generator (A.M.P.I.). Stimulus intervals between pulses were not less than 5 min. The CFEs were inserted 75–100 μm into the slice at a position 100–200 μm from the stimulating electrode pair (Miles et al., 2002). Each slice was sampled for dopamine at five sites, which represented medial-to-lateral and dorsal-to-ventral dimensions (see Fig. 6). Changes in extracellular dopamine were determined by monitoring the current over a 200 mV window at the peak oxidation potential for dopamine (for review, see Patel and Rice, 2006). Subtracting the current obtained before stimulation from the current obtained in the presence of dopamine created background-subtracted cyclic voltammograms. Electrodes were calibrated with 5 μm dopamine solutions in aCSF after each experiment to convert the oxidation current to dopamine concentration.
Figure 6.

Systemic injection of 3-NP creates regionally dependent decreases in dopamine release in the striatum. The amount of dopamine release was determined at 5 regions within the striatum (at approximately bregma level 1.00–0.80) including (1) midstriatum, (2) dorsomedial, (3) dorsal, (4) dorsolateral, and (5) ventrolateral in representative mice from all four groups (see brain slice inset). The bar graph illustrates peak dopamine released by a single intrastriatal stimulus (Calibration: 200 μA, 0.1 ms) applied to striatal brain slices at each of the above indicated regions for saline-injected rats, rats injected 24 h earlier with 3-NP, and rats injected 48 h earlier with 3-NP. Inset, Voltammogram response for dopamine release. Fifty traces are shown at a sampling rate of every 100 ms (10 Hz) after delivery of a single 0.1 ms, 200 μA intrastriatal stimulus (*p < 0.05 for comparison of saline control vs 24 after 3-NP injection).
HPLC analysis of brain slice dopamine and DA metabolites.
Brain slices (400 μm) were removed from incubation chambers containing oxygenated aCSF (see above), and the dorsomedial region of the striatum was selectively removed for analysis, because this was the site for all intracellular recordings performed in this study. Care was taken in each experiment to select the same slice in the rostral–caudal dimension (slices are cut in the coronal plane) at the same postcutting time point (average time of intracellular recording). Slices were placed in prelabeled squares of aluminum foil and rapidly transferred to dry ice. The folded-over pieces of foil containing brain slices were stored at −80°C until the dopamine content assay was performed. The pieces of foil (containing brain slices) were coded to ensure that the dopamine analysis was performed blind. Following the procedure of Crawford et al. (2000), frozen slices were sonicated in 10 volumes of 0.1N HClO4 and then centrifuged at 20,000 × g for 20 min at 4°C. The supernatant was then filtered through a 0.22 mm centrifugation unit for 5 min at 2000 × g at 4°C. The remaining pellet from each tissue sample was then resuspended and used for protein determination using the Bio-Rad Protein Assay (Bio-Rad Laboratories) based on the methods of Bradford (1976) and Lowry et al. (1951). Twenty microliters of the resulting extracts were assayed for dopamine content using HPLC (ESA; 582 pump with a MD-150 column) with electrochemical detection (ESA; Coulochem II EC detector). The mobile phase consisted of 75 mm NaH2PO4, 1.4 mm 1-octane sulfonic acid, 10 mm EDTA, and 10% acetonitrile at a pH of 3.1 (MD-TM Mobile Phase; ESA) pumped at a rate of 0.6 ml/min. Equipment for HPLC was as follows: Waters Binary Pump (model 1525), Autosampler (Waters 717plus), and Electrochemical Detector (ESA Coulochem II). The 24 and 48 h post-3-NP groups were compared with saline-injected controls using separate one-way ANOVAs.
Homogenate [3H]MK-801 binding assay.
On the day of assay, frozen striatal tissue was thawed on ice and homogenized in 15 volumes of 5 mm Tris-HCl buffer, pH 7.4, for ∼10 s using a Brinkmann Polytron. Homogenates were then centrifuged at 40,000 × g for 20 min. The supernatant was discarded and the pellet washed twice under the conditions described above. The final pellet was suspended in ∼10 volumes of buffer, pH 7.4. Protein concentrations for the final pellet were determined using the Bio-Rad Protein Assay with BSA as the standard.
Striatal homogenates were assayed in duplicate in test tubes containing 5 nm [3H] (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine (MK-801), 0.3 mm glycine, 2 mm glutamate, and 5 mm Tris-HCl buffer, pH 7.4. Nonspecific binding was determined in the presence of 100 μm unlabled (+) MK-801. Incubation time was 90 min at 23°C. Incubation was terminated by vacuum filtration over glass fiber filters (Whatman GF/B, pretreated with 0.1% polyethylenimine). Filters were washed twice with ice-cold Tris-HCl buffer, and radioactivity was measured by liquid scintillation spectrometry.
Immunohistochemical staining for tyrosine hydroxylase and dopamine transporter proteins and Nissl staining.
Rats (3 per group) were anesthetized (pentobarbital, 30 mg/kg) and perfused transcardially with 100 ml of ice-cold 0.9% saline followed by 250 ml of 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.2 (PFA/PBS). Brains were removed, postfixed in PFA/PBS at 4°C for 24 h, immersed in 20% sucrose/PBS, and frozen in isopentane cooled on dry ice and stored at −80°C until sectioning. Each brain was sectioned using a sliding microtome (Leica CM-1900) cryostat at 25 μm thickness encompassing the entire striatum. Sections were collected free-floating in PBS containing thimerosal and stored at 4°C until used for Nissl or immunohistochemical staining. Tissue sections at the level of the midstriatum (bregma 0.60) were selected for immunohistochemical staining for tyrosine hydroxylase (TH) (polyclonal antibody made in rabbit; Millipore Bioscience Research Reagents) and dopamine transporter (monoclonal made in rat; Millipore Bioscience Research Reagents) or stained for Nissl substance. Briefly, sections were rinsed in TBS (50 mm Tris, pH 7.2, with 0.9% NaCl), quenched in 10% methanol/3% H2O2/50 mm Tris, pH 7.2, blocked in 4% normal serum, exposed to antibody (concentration of 1:1000) for 48 h at 4°C, rinsed in TBS, and then exposed to secondary antibody using the ABC Elite Kit (Vectastain; Vector Laboratories). Antibody staining was visualized by development in 0.1% diaminobenzoic acid/3% H2O2. For Nissl staining, tissue sections were placed on gelatin-coated acid-washed microscope slides and allowed to dry overnight. Sections were stained with thionin, dried in alcohol, cleared in xylenes, and coverslipped. For quantification of immunostaining, a region of interest of 0.8 mm × 1.4 mm within the dorsomedial striatum (where electrophysiological recordings were made) was captured at 10× using an Olympus BX-51 microscope equipped with the computer-assisted image analysis program Bioquant (Bioquant Imaging). The relative optical density resulting from immunostaining within the dorsomedial striatum was determined by densitometry after subtraction of background determined from staining within the adjacent corpus callosum. The number of Nissl-stained cell bodies was determined within the same region. At 40× objective magnification, the image was digitized in the Bioquant program, and the stained neurons (cells >10 μm in diameter) displaying a large Nissl-stained cytoplasm and nucleus were counted manually. For each analysis, at least six sections from each of the three rats per treatment group were used.
Results
Behavioral effects of systemic 3-NP
Young rats (2 months old) given a single injection of 3-NP (15–16.5 mg/kg) showed variable sensitivity based on changes in their behavior noted during the first 48 h after the injection. We found that higher doses of 3-NP injections (18 mg/kg) caused death, whereas doses <15 mg/kg did not reliably result in visible motor abnormalities. Injection of 15.0–16.5 mg/kg 3-NP into 64 young rats (2 months old) resulted in death in 15 rats (23.4%) within 24 h. All surviving rats were examined at 6–8 h after 3-NP injection for signs of motor system failure according to the rating scale developed by Ouary et al. (2000) (see Materials and Methods). The distribution of scores for animals receiving a single 3-NP injection was 26.5% rated as 1, 26.5% rated as 2, 30.6% rated as 3, 12.2% rated as 5, and 4.2% rated as 6 (n = 49), with a rating of 1 being the least affected and a rating of 6 being the most affected. The most obvious and consistent problem observed was hindlimb paralysis. All animals showed complete recovery from the motor failure, as demonstrated by hindlimb paralysis, by 24 h after the injection.
Rats were next examined 24–48 h after 3-NP injection for motor behavior. The hindlimb paralysis seen at 6–8 h after 3-NP subsided in all surviving rats. Rats were tested before (saline-injected control) and then again 24 or 48 h after 3-NP injection for their ability to walk across a rod. The 24 h post-3-NP-injection rats fell often, showed decreased ability to navigate the rod, and were never able to reach the safe platform. Their control rating was 3.70 ± 0.11, whereas their behavior at 24 h after 3-NP was 1.60 ± 0.29 (p < 0.0001, paired t test; n = 49) (Fig. 1A). The 48 h post-3-NP-injection rats also showed motor deficits with a behavioral rating of 2.43 ± 0.24 (n = 7; p = 0.012, paired t test). Comparison of behavioral ratings between the 24 and 48 h post-3-NP-injection rats did not reveal a significant difference (p = 0.137, unpaired t test).
Figure 1.
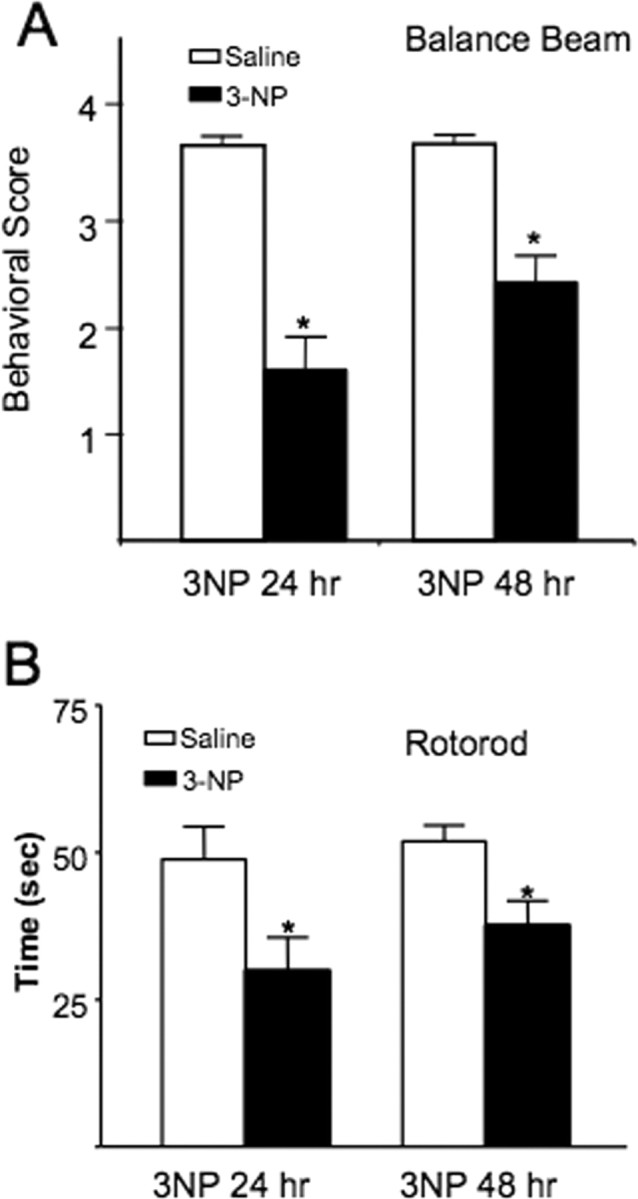
Systemic 3-NP causes impairment in balance beam and rotarod performance during the first 48 h in rats. A, Rats were ranked according to a five-point scale on their ability to navigate a balance beam according to the methods outlined by Friedemann and Gerhardt (1992). Rats showed impairment at both 24 and 48 h after systemic injection with 3-NP (*p < 0.01). B, Rats were tested on a slowly accelerating rotarod test and both the 24 and 48 h after 3-NP rats showed impairment relative to their performance before being exposed to 3-NP (*p < 0.01).
A subset of rats was also tested on a separate rotarod apparatus to see whether the behavioral problems created by 3-NP generalized to another motor task (Fig. 1B). Rats were tested before being injected with 3-NP, and the same rats were tested again at 24 or 48 h post-3-NP-injection times. Saline-injected rats stayed on the rotarod for 48.77 ± 5.57 s (n = 17). In contrast, the same rats at 24 h after 3-NP injection stayed on the rotarod for 29.85 ± 5.68 s. The 48 h post-3-NP-injection rats stayed on the rotarod for 37.57 ± 4.2 s (n = 16). A main effect for 3-NP exposure on rotarod performance was found for both the 24 and 48 h groups (F(1,62) = 5.56; p < 0.01).
Systemic exposure to 3-NP changes the expression of corticostriatal long-term synaptic plasticity during the first 24 h after injection
Rats injected 24 h earlier with 3-NP were compared with age-matched control rats for tetanus-induced long-term plasticity at dorsomedial corticostriatal synapses. Comparison between control and 24 h post-3-NP-injection animals revealed a significant difference across the entire posttetanus sampling period (p < 0.032; df = 1, 34; F = 5.054; repeated-measures ANOVA) (Fig. 2). Post hoc analysis of posttetanic plasticity (average 0–3 min posttetanus plasticity) also revealed a difference (p < 0.05, unpaired t test), as did long-term plasticity (average 15–20 min posttetanus) (p < 0.025, unpaired t test). Each cell contributed a single value for the posttetanic and long-term plasticity post hoc tests. This same method of repeated-measures ANOVA followed by post hoc tests for posttetanic and long-term plasticity averages was performed for all corticostriatal tetanus experiments.
Figure 2.

Systemic exposure to 3-NP increases LTP expression at dorsomedial corticostriatal synapses. A, Example traces of EPSPs selected from indicated time points for each experimental group. B, Plots of tetanus-induced long-term synaptic plasticity at dorsomedial corticostriatal synapses are shown. 3-NP injected intraperitoneally 24 h earlier increased the expression of LTP induced in vitro. Control rats were injected with saline and examined 24 h later. Long-term plasticity switched to LTD by 48 h after the 3-NP injection. EPSPs were measured using sharp electrode intracellular recordings. C, Bar graphs showing distribution of cells expressing tetanus-induced potentiation (>100%) and depression (<100%) for STP and LTP under each experimental condition (control, 24 and 48 h after 3-NP).
Individual cells were categorized for the posttetanic or short-term plasticity (0–3 min average) and long-term plasticity they expressed (15–20 min average) by comparing these averages to the average control amplitude of the EPSP for that cell calculated from the 10 min baseline pretetanus sampling period. Cells were then categorized for expressing an EPSP amplitude of 100% or greater than their own pretetanus EPSP amplitude, and we found that 50% (10 of 20) of control cells expressed LTP. In contrast, 14 of 16 (87.5%) cells recorded from animals injected 24 h earlier with 3-NP expressed LTP. Analysis of potentiation versus depression distributions between control and 24 h post-3-NP did not show a difference for short-term plasticity (0–3 min posttetanus; p = 0.1, Fisher's exact test), but did approach a significant difference for long-term plasticity (15–20 min posttetanus; p = 0.06, Fisher's exact test) (Fig. 2C). With a two-sided Fisher's exact test, our existing sample size (control = 20; 3-NP = 16) achieved 68% power to detect a difference of 36% in proportion of long-term plasticity between groups at a 0.05 significant level. To achieve a desired power of 80%, we need to at least have 25 in the control and 20 in 3-NP group. Similarly, our existing sample size has 65% power to detect a difference of 28% in proportion of short-term plasticity between the two groups at a 0.05 significant level. An additional 9 in the control and 11 in the 3-NP group would be needed to gain a sufficient power of 80%.
No difference was found between saline-injected controls and rats injected 24 h earlier with 3-NP in the PPR at corticostriatal synapses. The PPR for saline-injected controls was 108.7 ± 5 (SEM; n = 20) and for 24 h post-3-NP-injection rats was 104.8 ± 4 (SEM; n = 16).
The same experiment was repeated in a separate set of rats injected 48 h earlier with 3-NP, and no difference was found in the tetanus-induced short- or long-term plasticity when the 48 h group was compared with age-matched, saline-injected controls (Fig. 2). Comparison between 24 h (n = 16) and 48 h (n = 7) post-3-NP-injection animals revealed a significant difference across the entire posttetanus sampling period (p < 0.004; df = 1, 21; F = 10.6; repeated-measures ANOVA). Post hoc analysis of posttetanic plasticity (average 0–3 min posttetanus plasticity) also revealed a difference (p < 0.04, unpaired t test), as did long-term plasticity (average 15–20 min posttetanus; p < 0.03, unpaired t test). No difference in distributions was found between control and 48 h post-3-NP groups. Comparison between 24 and 48 h post-3-NP groups did not reveal a difference in short-term plasticity, but there was a trend for difference in distributions for long-term plasticity (p = 0.09, Fisher exact test) (Fig. 2C). The PPR for corticostriatal synapses in rats injected 48 h earlier with 3-NP was 94 ± 5 (SEM; n = 7), which was reduced compared with saline-injected controls (p < 0.05, unpaired t test).
Enhanced LTP seen 24 h after systemic 3-NP is NMDA receptor mediated
Brain slices taken from rats injected 24 h earlier with 3-NP were bathed in the selective NMDA receptor antagonist AP-5, and long-term plasticity was examined to test whether the enhanced LTP seen in these rats was NMDA receptor dependent. AP-5 completely blocked the enhancement of short-term potentiation and LTP created by systemic 3-NP (Fig. 3A). Comparison between aCSF (n = 16)-bathed and AP-5 (n = 5)-bathed slices taken from rats injected 24 h earlier with 3-NP revealed a significant difference across the entire posttetanus sampling period (p < 0.002; df = 1, 19; F = 12.74; repeated-measures ANOVA). Post hoc analysis of short-term plasticity (average 0–3 min posttetanus plasticity) also revealed a difference (p < 0.001, unpaired t test), as did long-term plasticity (average 15–20 min posttetanus; p < 0.001, unpaired t test). The PPR for corticostriatal synapses from slices bathed in AP-5 was similar to that seen in slices from 3-NP-injected rats bathed in aCSF only (107.8 ± 2.7; SEM; n = 5).
Figure 3.
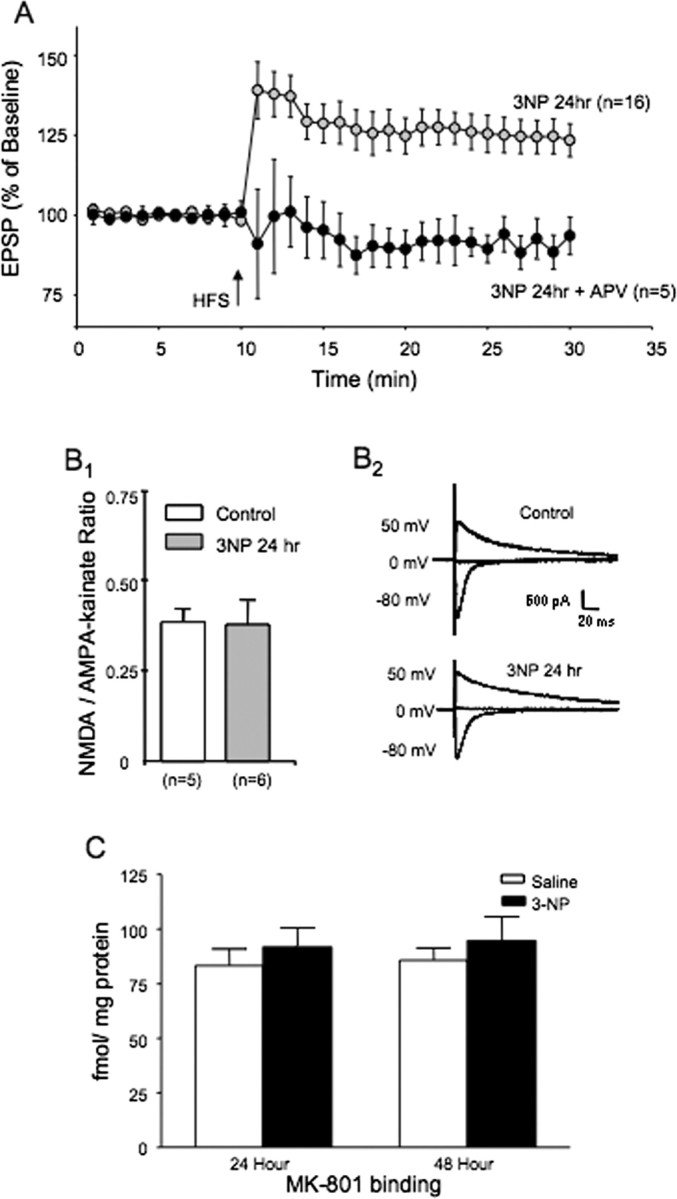
LTP produced by systemic 3-NP is NMDA receptor dependent. A, Plots of tetanus-induced long-term synaptic plasticity in rats injected 24 h earlier with 3-NP. The 3-NP group is the same as that shown in Figure 1, where brain slices were perfused with normal aCSF. Perfusion of brain slices taken from rats injected 24 h earlier with 3-NP with the NMDA receptor antagonist APV blocked the induction of LTP seen in normal aCSF. B, The NMDA/AMPA-kainate synaptic current ratio does not change at corticostriatal synapses from rats injected 24 h earlier with 3-NP. B1, Bar graph of NMDA/AMPA-kainate current synaptic ratio from saline and 3-NP-injected rats. B2, Examples of corticostriatal synaptic currents evoked at the indicated holding potentials using single electrode switch clamp methods. C, MK-801 binding to NMDA receptors from the dorsomedial striatum does not change 24 h after systemic injection with 3-NP. Bar graphs illustrate MK-801 binding in saline-injected and 3-NP-injected rats (24 and 48 h after 3-NP).
The increased expression of NMDA receptor-dependent LTP is not attributable to a change in properties of NMDA receptors
We examined NMDA receptor binding and NMDA receptor-mediated synaptic currents in animals injected 24 h earlier with 3-NP to determine whether the enhancement of NMDA receptor-dependent LTP created by 3-NP was attributable to an intrinsic change in NMDA receptor expression or physiological function. EPSCNMDA/EPSCAMPA-K ratios were calculated in dorsomedial striatal neurons from control slices and in cells from slices cut from rats injected with 3-NP 24 h earlier. The synaptic ratios were determined by sampling corticostriatal synapse, while holding cells at a membrane potential of either −80 mV (AMPA/kainate receptor response) or +40 mV (NMDA receptor response). The NMDA response was measured 60 ms after the stimulus artifact (Kreitzer and Malenka, 2007). No difference was found between cells from control slices and those sampled from 3-NP-treated rats in the EPSCNMDA/EPSCAMPA-K ratio. The synaptic ratio was 0.38 ± 0.03 (n = 5) in striatal neurons from control rats and 0.37 ± 0.07 (n = 6) in striatal neurons from 3-NP-treated rats (Fig. 3B).
Homogenates were made from the dorsomedial striatum from saline- and 3-NP-injected rats and examined for [3H]MK-801 binding as a measure of plasma membrane NMDA receptor expression. No differences were found between saline-injected and 3-NP-injected rats for [3H]MK-801 binding at either 24 or 48 h after injection. [3H]MK-801 binding was 83.25 ± 7.56 fmol/mg protein 24 h after saline control injections and 91.57 ± 8.95 fmol/mg protein 24 h after a single 16.5 mg/kg intraperitoneal injection of 3-NP. At 48 h after injection, [3H]MK-801 binding was 85.65 ± 5.68 fmol/mg protein for saline controls and 94.42 ± 11.2 for 3-NP-injected rats (Fig. 3C). All values are mean ± SEM, and each group sample size was n = 6.
Enhanced LTP seen 24 h after systemic 3-NP requires activation of D1 receptors
The LTP seen at corticostriatal synapses bathed in Mg2+-free aCSF has been shown to be D1 receptor dependent (Centonze et al., 2001a; Kerr and Wickens, 2001). To determine whether the enhanced LTP seen at corticostriatal synapses from rats injected 24 h earlier with 3-NP was also D1 receptor dependent, we bathed slices in the selective D1 receptor antagonist SCH 23390 (10 μm). It should be noted, however, that this experiment differed from those described by Centonze et al. (1999, 2001a) and Kerr and Wickens (2001) in our use of normal levels of extracellular Mg2+. Comparison between neurons from slices bathed in aCSF alone (n = 16) or aCSF + SCH 23390 (n = 8) in rats injected 24 h earlier with 3-NP revealed a significant difference across the entire posttetanus sampling period (p < 0.05; df = 1, 18; F = 4.64; repeated-measures ANOVA) (Fig. 4). Post hoc analysis of posttetanic plasticity (average 0–3 min posttetanus plasticity) also revealed a difference (p < 0.02, unpaired t test), and a similar trend was seen for long-term plasticity (average 15–20 min posttetanus; p < 0.09, unpaired t test). The PPR for corticostriatal synapses from slices bathed in SCH 23390, which were cut from rats injected 24 h earlier with 3-NP, was not different from that seen in slices from 3-NP-injected rats bathed in aCSF only (95.2 ± 2; SEM; n = 5).
Figure 4.

LTP produced by systemic injection of 3-NP 24 h earlier requires dopamine activation of D1 receptors. A, LTP produced by systemic injection of 3-NP is eliminated by addition of the D1 receptor antagonist SCH 23990 or dopamine. Plots of tetanus-induced plasticity in brain slices taken from rats injected 24 h earlier with 3-NP. Brain slices were bathed in either aCSF, aCSF + SCH 23390 (10 μm), or aCSF + 30 μm dopamine. B, LTP produced by systemic injection of 3-NP 24 h earlier is slightly enhanced by block of D2 receptors with l-sulpiride (Sulp) (1 μm) and completely eliminated by activation of D2 receptors with the D2 receptor agonist quinpirole (Quin) (10 μm).
Enhanced LTP seen 24 h after systemic 3-NP is reversed by adding dopamine or the D2 receptor agonist quinpirole to brain slices
Dopamine was added to brain slices taken from rats injected 24 h earlier to test the hypothesis that, although the enhanced LTP was D1 receptor dependent, it occurred because dopamine levels were reduced by 3-NP exposure. Brain slices taken from rats injected 24 h earlier with 3-NP were bathed in either aCSF alone or with dopamine (30 μm) added, and long-term plasticity was examined. The antioxidant sodium metabisulfite (30 μm) was added to dopamine-containing solution to keep dopamine from oxidizing. Dopamine completely blocked the enhancement of short-term plasticity and LTP created by systemic 3-NP and actually facilitated the expression of LTD (Fig. 4A). Comparison between neurons from slices bathed in aCSF (n = 16) or dopamine (n = 8) from rats injected 24 h earlier with 3-NP revealed a significant difference across the entire posttetanus sampling period (p < 0.0001; df = 1, 22; F = 18.01; repeated-measures ANOVA). Post hoc analysis of posttetanic plasticity (average 0–3 min posttetanus plasticity) also revealed a difference (p < 0.05, unpaired t test), as did long-term plasticity (average 15–20 min posttetanus; p < 0.025, unpaired t test). The PPR for corticostriatal synapses in rats injected 24 h earlier with 3-NP and bathed in dopamine was 89.1 ± 2.4 (SEM; n = 8), which was reduced compared with the PPR seen in slices from 3-NP-injected rats bathed in aCSF only (p < 0.05, unpaired t test).
The ability of exogenous dopamine to reverse the D1 receptor-mediated enhancement of corticostriatal LTP suggested that systemic 3-NP reduced endogenous dopamine, which altered the normal balance between D1 and D2 receptor activation during tetanic conditioning. To test this hypothesis, we manipulated D2 receptor activation and examined the impact of this manipulation on long-term synaptic plasticity in 3-NP-treated rats. Slices taken from rats injected 24 h earlier with 3-NP were bathed in the D2 receptor antagonist l-sulpiride (1 μm), which resulted in a slight but insignificant enhancement in the potentiation produced by tetanizing conditioning of corticostriatal synapses (Fig. 4B) (l-sulpiride, n = 3; aCSF, n = 16; df = 1, 17; F = 0.64; p = 0.43; repeated-measures ANOVA). Post hoc analysis of posttetanic plasticity (average 0–3 min posttetanus plasticity) showed a trend toward increased LTP in sulpiride-treated slices (p = 0.09, unpaired t test), with no difference in long-term plasticity (average 15–20 min posttetanus; p = 0.88, unpaired t test). We next took brain slices from rats injected 24 h earlier with 3-NP and bathed them in the D2 receptor agonist quinpirole (10 μm; n = 3). Quinpirole treatment caused a significant reduction in the 3-NP-induced expression of LTP at corticostriatal synapses (quinpirole, n = 3; aCSF, n = 16; df = 1, 17; F = 4.75; p < 0.05; repeated-measures ANOVA) (Fig. 4B). Post hoc comparison between aCSF- and quinpirole-treated slices from animals injected earlier with 3-NP revealed differences in posttetanic (average 0–3 min posttetanus plasticity; p < 0.009, unpaired t test), and long-term plasticity (average 15–20 min posttetanus; p < 0.03, unpaired t test).
Dopamine content is reduced at 24 h but not 48 h after 3-NP–HPLC
The ability of dopamine applied to brain slices to reverse the enhanced LTP expression at dorsomedial corticostriatal synapses in rats injected 24 h earlier with 3-NP suggested that the LTP may be facilitated by a reduction in striatal dopamine. To test this hypothesis, we specifically measured striatal dopamine content using HPLC from the dorsomedial striatum and found that DA was decreased at 24 h after a single intraperitoneal injection of 3-NP (16.5 mg/kg; 90.50%; n = 5, control and 3-NP; 3-NP main effect, F(1,8) = 6.349; p < 0.05; one-way ANOVA) (Fig. 5A). No differences were seen for 3,4-dihydroxy-phenylacetic acid (DOPAC) or homovanillic acid (HVA) levels between saline-injected controls and rats injected 24 h earlier with 3-NP. HPLC measurements for saline-injected rats were as follows: dopamine, 158.95 ± 5.02; DOPAC, 22.611 ± 1.41; HVA, 10.072 ± 0.899 (n = 5). HPLC measures for rats injected with 3-NP 24 h earlier were as follows: dopamine, 143.83 ± 4.76; DOPAC, 23.071 ± 1.73; HVA, 9.342 ± 0.647 (n = 5). We next examined dopamine, DOPAC, and HVA levels 48 h after a single injection of 3-NP (16.5 mg/kg) and found that they returned to the normal. HPLC measures for the 48 h saline controls were as follows: dopamine, 148.52 ± 5.45; DOPAC, 23.172 ± 1.59; HVA, 9.231 ± 0.546 (n = 6). HPLC measures for the 48 h post-3-NP group were as follows: dopamine, 149.26 ± 8.10; DOPAC, 24.192 ± 1.63; HVA, 9.749 ± 0.637 (n = 6). Values obtained for the 24 and 48 h post-3-NP groups are normalized to saline-injected controls in Figure 3. All data were obtained from the dorsomedial striatum, because this was the site used for all electrophysiological recordings.
Figure 5.

Systemic 3-NP causes a reduction in dopamine content in the dorsomedial striatum 24 h after the injection. A, Bar graphs of dopamine content, DOPAC, and HVA obtained from the dorsomedial striatum of rats injected with 3-NP (16.5 mg/kg) 24 and 48 h earlier. The single injection of 3-NP caused a significant reduction in dopamine content 24 h after the injection, but this change reversed by 48 h after the injection. B, Injection of 3-NP for 2 consecutive days causes significant changes in dorsomedial striatal dopamine, DOPAC, and HVA. The double injection protocol was 15 mg/kg on day 1 and 10 mg/kg on day 2, and the rat was killed 3 h later for analysis of dopamine and its metabolites in total striatal tissue. Dopamine content was dramatically reduced, and this change was associated with large changes in DOPAC and HVA. Note the change in scale for the DOPAC measurement for the double injection protocol. The single injection data from the dorsomedial striatum shown in A are illustrated for comparison. All values are normalized to saline-injected control rats. *p < 0.05; **p < 0.03.
Previous 3-NP-mediated lesion studies have used a paradigm of injecting Sprague Dawley rats with 20 mg/kg 3-NP for 5 consecutive days, and HPLC measures were reported for the entire striatum (Beal et al., 1993b). We attempted to replicate this strategy for our dorsomedial striatum study, but we found we had to limit 3-NP injections to 2 d because of the extreme sensitivity of the Fischer 344 rat to 3-NP. We also needed to use a smaller first-day dose of 10 mg/kg combined with a second day's dose of 15 mg/kg. The rats were near death after the second dose and were therefore killed 4 h later for dopamine, DOPAC, and HVA detection using HPLC. Significant differences were found in the dorsomedial striatum for dopamine, DOPAC, and HVA between saline-injected controls and Fischer 344 rats receiving two 3-NP injections over 2 d as indicated above (dopamine, F(1,12) = 6.057, p < 0.030; DOPAC, F(1,12) = 9.542, p < 0.009; HVA, F(1,12) = 11.623, p < 0.005) (Fig. 5B). HPLC measures for the double-injection saline controls were as follows: dopamine, 147.72 ± 3.23; DOPAC, 15.627 ± 0.768; HVA, 7.150 ± 0.557 (n = 6). HPLC measures for values obtained after 2 d of 3-NP injection were as follows: dopamine, 86.927 ± 21.04; DOPAC, 111.455 ± 26.58; HVA, 10.651 ± 0.778 (n = 8). All HPLC data were reported in nanograms per milligram protein. Values obtained for the 2 d 3-NP group are normalized to saline-injected controls in Figure 5, and the data were obtained from the dorsomedial striatum.
Dopamine release is reduced at 24 h but not 48 h after 3-NP
Dopamine release was measured via fast-scan cyclic voltammetry (FSCV) in brain slices taken from rats injected either 24 or 48 h earlier with 3-NP. Dopamine release was measured in each slice at five different sites spanning medial-to-lateral and dorsal-to-ventral dimensions of coronal brain slices. Comparison of dopamine release between slices from saline-injected rats and slices from rats injected 24 h earlier with 3-NP revealed a difference across all five sampling sites (p < 0.015; df = 1, 13; F = 8.207; repeated-measures ANOVA) (Fig. 6). Post hoc analyses revealed that dopamine release was significantly reduced at central as well as dorsal and dorsolateral sites of striatum relative to that seen in saline controls. The strongest effect of 3-NP in rats injected 24 h earlier on evoked dopamine release was found at central striatum [position (pos) 1; 0.07 ± 0.03 μm dopamine (n = 9) vs control, 3.14 ± 0.39 μm DA (n = 6); p = 9.2 × 10−6, unpaired t test], whereas less, but significant, reduction of dopamine release was also found at dorsal striatum (position 3; 1.29 ± 0.61 μm dopamine vs 2.93 ± 0.38 μm dopamine in control; p = 0.042, unpaired t test) and at dorsolateral striatum (position 4; 1.04 ± 0.71 μm dopamine vs 2.93 ± 0.41 μm dopamine in control; p = 0.040, unpaired t test). The ventrolateral (position 5; 1.05 ± 0.71 μm dopamine vs 2.32 ± 0.56 μm dopamine in control; p = 0.182, unpaired t test) and medial (position 2; 2.36 ± 0.68 μm dopamine vs 3.82 ± 0.34 μm dopamine in control; p = 0.080, unpaired t test) sites were less affected.
Interestingly, levels of evoked dopamine release recovered rapidly and by 48 h after injection were slightly elevated compared with saline-injected controls (Fig. 6). The 48 h post-3-NP dopamine release values were as follows: central striatum, 4.11 ± 0.80 μm; dorsomedial striatum, 4.33 ± 0.45 μm; dorsal striatum, 4.27 ± 0.56 μm; dorsolateral striatum, 3.51 ± 0.48 μm; and ventrolateral striatum, 3.82 ± 0.59 μm (n = 7). Comparison of dopamine release between slices from rats injected 24 h earlier with 3-NP and slices from rats injected with 3-NP 48 h earlier revealed a difference across all five sampling sites (p < 0.003; df = 1, 14; F = 14.611; repeated-measures ANOVA) (Fig. 6). Post hoc analyses revealed differences at sites 1–5. The outcomes of unpaired t tests performed at each position are as follows: central striatum (pos 1; p < 0.002), medial striatum (pos 2; p < 0.04), dorsal striatum (pos 3; p < 0.003), dorsolateral striatum (pos 4; p < 0.02), and lateral striatum (pos 5; p < 0.05).
Comparison of dopamine release between slices from control saline-injected rats and slices from rats injected with 3-NP 48 h earlier revealed a trend toward increased dopamine release in rats injected 48 h earlier with 3-NP across all five sampling sites (p = 0.159; df = 1, 11; F = 2.283; repeated-measures ANOVA). Post hoc analysis demonstrated that position 3 (the dorsal striatum) showed the greatest difference (p = 0.075).
Single injection of 3-NP does not alter striatal TH levels or show evidence of striatal cell loss in Nissl-stained sections
Immunostaining for tyrosine hydroxylase protein in sections through the midstriatum showed no evidence of loss of nigrostriatal fibers based on the degree of TH immunostaining (Fig. 7), nor DA transporter immunostaining (data not shown). The relative optical density of TH immunostaining at either 1 or 2 d post-3-NP-treatment rats was not statistically different from the saline-treated group (F = 0.28; p = 0.76). We also examined for evidence of striatal cell loss by counting the number of Nissl-stained neurons (large cell bodies) within the dorsomedial striatum, the analogous region used for analysis of TH immunostaining and electrophysiological recording. Comparison between the saline-treated rats and the 3-NP-treated rats at 1 and 2 d after lesioning showed no statistically significant differences between the groups (F = 2.28; p = 0.76). Similar outcomes were found in the dorsolateral striatum.
Figure 7.

Staining of striatal tissue for tyrosine hydroxylase protein or Nissl substance. A–L, Tissue sections from the midstriatum of rats treated with either saline or 3-NP (collected at 1 or 2 d after lesioning) were immunostained for either TH protein (A–F) or Nissl substance (G–L). The top panels show sections at low magnification of the entire striatum in coronal sections, whereas the bottom panels show a higher magnification (20×) of the dorsal region of the striatum. Scale bars: (top) 1.7 mm; (bottom) 100 μm. Relative optical density of the dorsomedial striatum (graph below TH panels) showed no evidence of loss of TH immunostaining, and cell counting (graph below Nissl panels) showed no reduced number of Nissl-stained striatal neurons.
Sprague Dawley rats show reduced sensitivity to 3-NP
Our data illustrate the sensitivity of Fischer 344 rats to 3-NP; however, a majority of studies investigating the action of 3-NP have been performed on the Sprague Dawley rat strain. We therefore studied corticostriatal synaptic plasticity and dopamine release 24 h after systemic injection of 3-NP in Sprague Dawley rats age-matched to the Fischer 344 rats used in this study. We began with a much higher dose of 3-NP in Sprague Dawley rats (25 mg/kg) than what was used for Fischer 344 rats (15 mg/kg), because previous studies have shown Sprague Dawley rats to be less sensitive to 3-NP (Ouary et al., 2000). We found that doses as high as 35 mg/kg did not produce a significant change in long-term plasticity at corticostriatal synapses (saline, n = 9; single 3-NP injection, n = 6; df = 1, 15; F = 1.87; p = 0.192; repeated-measures ANOVA) (Fig. 8A). Brain slices from the same animals studied for corticostriatal synaptic plasticity indicated above were also studied for changes in evoked dopamine release using FSCV, and no change was found in dopamine release in Sprague Dawley rats injected with 25–35 mg/kg 3-NP (Fig. 8B). We therefore applied a repeated 3-NP injection dosing strategy in Sprague Dawley rats based on previous lesioning studies, which used large daily injections of 20 mg/kg 3-NP over a 5 d period (Beal et al., 1993a,b). However, our goal was to find a dose for inducing changes in dopaminergic and glutamatergic synaptic function. We found that injecting 3-NP once a day for 3 d using a dosing regimen of 15, 10, and 10 mg/kg caused significant changes in dopamine release across all five sampling sites between saline- (n = 6) and 3-NP- (n = 3) injected rats (df = 1, 7; F = 7.5; p < 0.03; repeated-measures ANOVA). Post hoc analysis demonstrated significant reductions in dopamine release at sites 1, 3, 4 (p < 0.01), and 2 (p < 0.02) (unpaired t test) (Fig. 8B). We also found a significant difference in long-term plasticity at corticostriatal synapses, with the 3× 3-NP-injected rats showing reduced expression of LTD (3× 3-NP, n = 8; saline, n = 9; df = 1, 15; F = 7.785; p < 0.02; repeated-measures ANOVA). Post hoc analysis of posttetanic plasticity (average 0–3 min posttetanus plasticity) showed the same trend (p = 0.06, unpaired t test), as did long-term plasticity (average 15–20 min posttetanus; p < 0.02, unpaired t test). Comparison of distributions of short- (p = 0.66) and long- (p = 0.34, Fisher exact test) term plasticity did not reveal a difference between recordings from control and 3× 3-NP Sprague Dawley rats. We further examined strain differences in synaptic function by comparing long-term plasticity and dopamine release between saline-injected Sprague Dawley (n = 9) and Fischer 344 (n = 20) rats. Saline-injected Sprague Dawley rats showed increased expression of LTD across the 20 min posttetanus sampling period (df = 1, 27; F = 7.88; p < 0.01; repeated-measures ANOVA) and increased dopamine release across the five sampling sites (df = 1, 10; F = 11.24; p < 0.008; repeated-measures ANOVA; Sprague Dawley, n = 6; Fischer 344, n = 6).
Discussion
3-NP is a plant mycotoxin known epidemiologically for outbreaks of dystonia and cardiac arrest after ingestion of moldy sugarcane in China (Liu et al., 1992; Ming, 1995). Pathology studies revealed that 3-NP inhibition of mitochondria complex II caused necrosis in the putamen (Ludolph et al., 1991). 3-NP animal models corroborated this finding, revealing a pattern of cell loss similar to that seen in Huntington's disease and hypoxia (Hamilton and Gould, 1987; Beal et al., 1993b; Beal, 1994; Borlongan et al., 1995; Nishino et al., 2000). Our analysis focused on changes in striatal circuitry created by exposure to low doses of 3-NP, which limited the chance of striatal lesions. We discovered a 3-NP-induced change in the relationship between striatal glutamatergic and dopaminergic synapses during the first 24 h after 3-NP exposure.
Behaviorally, adult Fischer 344 rats responded to a single injection of 3-NP with hindlimb dystonia 4 h after injection (ranked at 6–8 h after injection), and they performed poorly on the balance beam and rotarod 24 h after injection (Fig. 1). These findings are in agreement with other behavioral studies performed after multiple systemic injections of 3-NP, which created striatal lesions (Koutouzis et al., 1994; Ouary et al., 2000). Unlike lesion studies, the hindlimb dystonia subsided by 24 h after a single 3-NP injection.
A single 3-NP injection increased the expression of corticostriatal LTP 24 h after injection in Fischer 344 rats. Dalbem et al. (2005) found that multiple, high-dose systemic injections of 3-NP increased LTP at corticostriatal synapses for 48–60 h in Wistar rats. In contrast, Picconi et al. (2006) reported that a 4 d cycle of multiple 3-NP injections did not change Mg2+-free saline-dependent LTP. However, their use of Mg2+-free saline may have masked the 3-NP-mediated increase in corticostriatal LTP observed under normal Mg2+ and Ca2+ conditions (Fig. 1) (Dalbem et al., 2005). Our study also differed in using a single injection of 3-NP, which did not create striatal lesions, and we recorded from the less vulnerable dorsomedial striatum.
The increase in LTP that we observed 24 h after systemic 3-NP exposure was NMDA receptor dependent (Fig. 3). In vitro ischemia also produces NMDA receptor-dependent potentiation of corticostriatal synapses, and acute application of 3-NP to striatal brain slices produces a direct increase in NMDA, but not AMPA receptor-mediated EPSPs (Calabresi et al., 2001, 2003; Centonze et al., 2001a). Furthermore, the NMDA receptor antagonist memantine blocks changes in synaptic function created by in vitro ischemia and bath application of 3-NP (Tozzi et al., 2007). We therefore reasoned that increased NMDA receptor function contributed to enhanced LTP observed 24 h after systemic 3-NP. However, we did not find a change in NMDA/AMPA-kainate receptor ratio 24 h after systemic 3-NP exposure (Thomas et al., 2001; Borgland et al., 2004). We also did not find a change in MK-801 binding to NMDA receptors 24 h after 3-NP injection. This finding differs from the increase in MK-801 binding seen 3 h after 3-NP exposure (Wüllner et al., 1994), which may reflect a process temporally distinct from that examined in this study.
We next examined 3-NP-induced changes in dopamine modulation of corticostriatal synaptic function as a mechanism to explain changes in synaptic plasticity created by systemic 3-NP because (1) dopamine influences corticostriatal long-term plasticity and (2) nigrostriatal dopamine release underlies 3-NP-induced striatal degeneration (Calabresi et al., 1992; Reynolds et al., 1998; Fernegut et al., 2002; Wang et al., 2006; Kreitzer and Malenka, 2007). Dopamine content was reduced 24 h after a single 3-NP injection in the dorsomedial striatum (HPLC), and corroborating earlier multiple injection studies in Sprague Dawley rats, two injections over 2 d in Fischer 344 rats produced profound changes in total striatal dopamine content (Beal et al., 1993b) (Fig. 5).
FSCV revealed that systemic 3-NP created regionally specific decreases in evoked dopamine release 24 h after 3-NP. The largest effect was in the center of the striatum, which corresponds to the site most vulnerable for 3-NP-induced lesions (Beal et al., 1993b). A smaller decrease in dopamine release was found in the dorsomedial recording site used to study corticostriatal synaptic plasticity. Previous work from our laboratory and others has shown that dopamine release created by intrastriatal stimulation is Ca2+ and TTX sensitive (Cragg and Greenfield, 1997; Rice et al., 1997; Petzinger et al., 2007). HPLC demonstrated a return in dopamine content, and FSCV showed a slight but insignificant increase in dopamine release compared with saline-injected rats 48 h after 3-NP. The 48 h time point thus provides an estimate of the time to restore dopamine levels in nigrostriatal terminals (Reynolds et al., 1998; Marti et al., 2003). TH content did not change, indicating that a low dose of 3-NP did not cause permanent damage to nigrostriatal terminals as reported for larger doses (Nakao et al., 1999; Blum et al., 2004).
We found that LTP induced 24 h after systemic 3-NP exposure was D1 receptor dependent. Corticostriatal LTP enabled by Mg2+-free aCSF is also D1 receptor dependent (Kerr and Wickens, 2001). D1 receptors modulate NMDA receptors via the adenylate cyclase–protein kinase A–DARPP-32 pathway in the striatum, cerebral cortex, and hippocampus (Huang and Kandel, 1995; Otmakhova and Lisman, 1996; Gurden et al., 2000; Flores-Hernández et al., 2002; Otani et al., 2003). However, bath application of 3-NP is reported to enhance NMDA receptor function via D2 receptors when slices are bathed in Mg2+-free aCSF (Calabresi et al., 2001).
The coincidence of increased LTP expression and reduced dopamine release in our study suggests that these observations may be related. Trantham-Davidson et al. (2004) theorized that D1 receptors play a stronger role in the prefrontal cortex when dopamine levels are low, and D2 receptors take over when dopamine is high. Reynolds et al. (2001) and Reynolds and Wickens (2002) proposed a similar theory to explain D2 receptor-dependent LTD and D1 receptor-dependent LTP at corticostriatal synapses. Our data support these theories, because (1) LTP is enhanced when 3-NP reduces dopamine release in the striatum and (2) exogenous addition of dopamine or the D2 receptor agonist quinpirole eliminates the 3-NP enhanced LTP and increases LTD. More LTP is seen in the dorsomedial striatum, as are more D1 receptors, and thus, when dopamine is reduced, it has a greater probability of binding to D1 receptors (Maeno, 1982; Ariano et al., 1989; Partridge et al., 2000; Smith et al., 2001). In contrast, normal dopamine release in control slices or addition of dopamine to dopamine-depleted brain slices increases the probability of dopamine binding to D2 receptors, which increases LTD expression at corticostriatal synapses (Calabresi et al., 1992; Akopian and Walsh, 2006; Wang et al., 2006; Kreitzer and Malenka, 2007).
Our study cannot address differences that may exist in D2- (indirect pathway) versus D1- (direct pathway) expressing striatal neurons (Wang et al., 2006; Kreitzer and Malenka, 2007). However, our selection of the dorsomedial striatum increased the likelihood of D1 receptor participation over D2 receptors (Maeno, 1982; Ariano et al., 1989). Wang et al. (2006) found that dopamine triggers D2 receptor-dependent LTD by acting presynaptically to reduce acetylcholine (ACh) release from cholinergic interneurons. Our voltammetry data indicate that dopamine release is dramatically reduced 24 h after 3-NP, which would (1) reduce D2 receptor-mediated presynaptic inhibition of ACh release and consequently (2) enhance M1 receptor-dependent LTP (Calabresi et al., 1999). Our ability to reverse 3-NP-induced LTP to LTD by adding dopamine adds further support to this hypothesis. Last, widespread D2 receptor activation created by bath application of dopamine may cause a diffuse “spill-over” of messengers, like endocannabinoids, which act presynaptically to reduce glutamate release (Wang et al., 2006; Kreitzer and Malenka, 2007).
This study was performed in the Fischer 344 rat strain, identified as a model for aging studies as well as stroke or hypoxia (Coleman et al., 1977; Aspey et al., 1998, 2000). Oaury et al. (2000) found the Fischer 344 strain to be far more sensitive to chemical hypoxia via 3-NP poisoning than Sprague Dawley or Lewis rat strains. However, much of the previous work examining 3-NP-induced lesions of the striatum used Sprague Dawley rats. We thus wanted to know whether our findings on synaptic change generalized to the Sprague Dawley strain and found, much like Ouary et al. (2000), that Sprague Dawley rats were far less sensitive to 3-NP. Single injections of 3-NP had little effect on corticostriatal synapses or dopamine release at a dose that was larger than that used in Fischer 344 rats (25–35 mg/kg vs 15 mg/kg). Simpson and Isacson (1993) also found a single systemic dose of 30 mg/kg to be below the threshold creating lesions in Sprague Dawley rats. However, multiple injections of 3-NP in Sprague Dawley rats over 3 d created changes in corticostriatal synaptic plasticity and dopamine release (Fig. 8). Our findings thus add to the literature demonstrating strain-dependent differences in sensitivity to metabolic challenge in mice and rats (Aspey et al., 1998, 2000; Ouary et al., 2000; Schauwecker, 2005; Mclin et al., 2006). We also found saline-injected Sprague Dawley rats had greater dopamine release and increased LTD relative to saline-injected Fischer 344 rats, further supporting the role for enhanced dopamine release and LTD expression.
In conclusion, mild exposure to 3-NP alters both dopamine and glutamate physiology in the striatum of the Fischer 344 rat. Systemic 3-NP increased LTP at corticostriatal synapses, which may be linked, at least in part, to changes in the nigrostriatal pathway. These changes occur without a pathological loss of striatal neurons or nigrostriatal terminals, suggesting alterations in striatal circuitry may be sufficient to alter behavior (Newcomb et al., 1995). The link between reduced dopamine release and increased D1-dependent NMDA receptor function could also explain the cell death created by exposure to low doses of 3-NP combined with NMDA (Simpson and Isacson, 1993), and it is a mechanism likely operating in excitotoxicity and delayed cell death (apoptosis) seen with more severe exposure to 3-NP (Beal et al., 1993b; Beal, 1994; Wüllner et al., 1994; Kim et al., 2000).
Footnotes
This work was supported by National Institutes of Health Grant AG 21937 and a gift from Anita Wilson.
References
- Akopian G, Walsh JP. Reduced expression of short- and long-term facilitation at aged corticostriatal synapses. Synapse. 2006;60:223–238. [Google Scholar]
- Akopian G, Musleh W, Smith R, Walsh JP. Functional state of presynaptic terminals influences the expression of short- and long-term plasticity at corticostriatal synapses. Synapse. 2000;38:271–280. doi: 10.1002/1098-2396(20001201)38:3<271::AID-SYN6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Ariano MA, Monsma FJ, Jr, Barton AC, Kang HC, Haugland RP, Sibley DR. Direct visualization and cellular localization of D1 and D2 dopamine receptors in rat forebrain by use of fluorescent ligands. Proc Natl Acad Sci U S A. 1989;86:8570–8574. doi: 10.1073/pnas.86.21.8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspey BS, Cohen S, Patel Y, Terruli M, Harrison MJ. Middle cerebral artery occlusion in the rat: consistent protocol for a model of stroke. Neuropathol Appl Neurobiol. 1998;24:487–497. doi: 10.1046/j.1365-2990.1998.00146.x. [DOI] [PubMed] [Google Scholar]
- Aspey BS, Taylor FL, Terruli M, Harrison MJ. Temporary middle cerebral artery occlusion in the rat: consistent protocol for a model of stroke and reperfusion. Neuropathol Appl Neurobiol. 2000;26:232–242. doi: 10.1046/j.1365-2990.2000.00221.x. [DOI] [PubMed] [Google Scholar]
- Beal MF. Neurochemistry and toxin models in Huntington's disease. Curr Opin Neurol. 1994;7:542–547. doi: 10.1097/00019052-199412000-00012. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins B, Henshaw R, Rosen B, Hyman BT. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993a;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993b;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum D, Galas MC, Gall D, Cuvelier L, Schiffmann SN. Striatal and cortical neurochemical changes induced by chronic metabolic compromise in the 3-nitropropionic model of Huntington's disease. Neurobiol Dis. 2002;10:410–426. doi: 10.1006/nbdi.2002.0512. [DOI] [PubMed] [Google Scholar]
- Blum D, Galas MC, Cuvelier L, Schiffmann SN. Chronic intoxication with 3-nitropropionic acid in rats induces the loss of striatal dopamine terminals without affecting nigral cell viability. Neurosci Lett. 2004;354:234–238. doi: 10.1016/j.neulet.2003.10.034. [DOI] [PubMed] [Google Scholar]
- Bogdanov MB, Ferrante RJ, Kuemmerle S, Klivenyi P, Beal MF. Increased vulnerability to 3-nitropropionic acid in an animal model of Huntington's disease. J Neurochem. 1998;71:2642–2644. doi: 10.1046/j.1471-4159.1998.71062642.x. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlongan CV, Koutouzis TK, Randall TS, Freeman TB, Cahill DW, Sanberg PR. Systemic 3-nitropropionic acid: behavioral deficits and striatal damage in adult rats. Brain Res Bull. 1995;36:549–556. doi: 10.1016/0361-9230(94)00242-s. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Burke RE, Franklin SO, Inturrisi CE. Acute and persistent suppression of preproenkephalin mRNA expression in the striatum following developmental hypoxic-ischemic injury. J Neurochem. 1994;62:1878–1886. doi: 10.1046/j.1471-4159.1994.62051878.x. 1994. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Bernardi G. Activation of M1-like muscarinic receptors is required for the induction of corticostriatal LTP. Neuropharmacology. 1999;38:323–326. doi: 10.1016/s0028-3908(98)00199-3. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Gubellini P, Picconi B, Centonze D, Pisani A, Bonsi P, Greengard P, Hipskind RA, Borrelli E, Bernardi G. Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine. J Neurosci. 2001;21:5110–5120. doi: 10.1523/JNEUROSCI.21-14-05110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Cupini L, Bernardi G. Synaptic plasticity in the ischaemic brain. Lancet Neurol. 2003;2:622–629. doi: 10.1016/s1474-4422(03)00532-5. [DOI] [PubMed] [Google Scholar]
- Callaway JK, Lawrence AJ, Jarrott B. AM-36, a novel neuroprotective agent, profoundly reduces reactive oxygen species formation and dopamine release in the striatum of conscious rats after endothelin-1-induced middle cerebral artery occlusion. Neuropharmacol. 2003;44:787–800. doi: 10.1016/s0028-3908(03)00068-6. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2:396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Gubellini P, Picconi B, Calabresi P, Giacomini P, Bernardi G. Unilateral dopamine denervation blocks corticostriatal LTP. J Neurophysiol. 1999;82:3575–3579. doi: 10.1152/jn.1999.82.6.3575. [DOI] [PubMed] [Google Scholar]
- Centonze D, Picconi B, Gubellini P, Bernardi G, Calabresi P. Dopaminergic control of synaptic plasticity in the dorsal striatum. Eur J Neurosci. 2001a;13:1071–1077. doi: 10.1046/j.0953-816x.2001.01485.x. [DOI] [PubMed] [Google Scholar]
- Centonze D, Saulle E, Bernardi G, Calabresi P. Receptor and post-receptor mechanisms of ischemic long-term potentiation in the striatum. Functional Neurology. 2001b;16:149–152. [PubMed] [Google Scholar]
- Coleman GL, Barthold W, Osbaldiston GW, Foster SJ, Jonas AM. Pathological changes during aging in barrier-reared Fischer 344 male rats. J Gerontol. 1977;32:258–278. doi: 10.1093/geronj/32.3.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ, Greenfield SA. Differential autoreceptor control of somatodendritic and axon terminal dopamine release in substantia nigra, ventral tegmental area, and striatum. J Neurosci. 1997;17:5738–5746. doi: 10.1523/JNEUROSCI.17-15-05738.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford CA, Zavala AR, Karper PE, McDougall SA. Long-term effects of postnatal amphetamine treatment on striatal protein kinase A activity, dopamine D(1)-like and D(2)-like binding sites, and dopamine content. Neurotoxicol Teratol. 2000;22:799–804. doi: 10.1016/s0892-0362(00)00109-4. [DOI] [PubMed] [Google Scholar]
- Dalbem A, Silveira CV, Pedroso MF, Breda RV, Werne Baes CV, Bartmann AP, da Costa JC. Altered distribution of striatal activity-dependent synaptic plasticity in the 3-nitropropionic acid model of Huntington's disease. Brain Res. 2005;1047:148–158. doi: 10.1016/j.brainres.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Dos Santos Villar F, Walsh JP. Modulation of long-term synaptic plasticity at corticostriatal synapses. Neuroscience. 1999;90:1031–1041. doi: 10.1016/s0306-4522(98)00504-1. [DOI] [PubMed] [Google Scholar]
- Fancellu R, Armentero MT, Nappi G, Blandini F. Neuroprotective effects mediated by dopamine receptor agonists against malonate-induced lesion in the rat striatum. Neurol Sci. 2003;24:180–181. doi: 10.1007/s10072-003-0119-x. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Diguet E, Jaber M, Bioulac B, Tison F. Dopamine transporter knock-out mice are hypersensitive to 3-nitropropionic acid-induced striatal damage. Eur J Neurosci. 2002;15:2053–2056. doi: 10.1046/j.1460-9568.2002.02047.x. [DOI] [PubMed] [Google Scholar]
- Ferriero DM, Arcavi LJ, Sagar SM, McIntosh TK, Simon RP. Selective sparing of NADPH-diaphorase neurons in neonatal hypoxia-ischemia. Ann Neurol. 1988;24:670–676. doi: 10.1002/ana.410240512. [DOI] [PubMed] [Google Scholar]
- Flores-Hernández J, Cepeda C, Hernández-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS. Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: role of D1 receptors and DARPP-32. J Neurophysiol. 2002;88:3010–3020. doi: 10.1152/jn.00361.2002. [DOI] [PubMed] [Google Scholar]
- Friedemann MN, Gerhardt GA. Regional effects of aging on dopaminergic function in the Fisher-344 rat. Neurobiol Aging. 1992;13:325–332. doi: 10.1016/0197-4580(92)90046-z. [DOI] [PubMed] [Google Scholar]
- Golden WC, Brambrink AM, Traystman RJ, Shaffner DH, Martin LJ. Nitration of the striatal Na,K-ATPase alpha3 isoform occurs in normal brain development but is not increased during hypoxia-ischemia in newborn piglets. Neurochem Res. 2003;28:1883–1889. doi: 10.1023/a:1026132110850. [DOI] [PubMed] [Google Scholar]
- Gu W, Zhao H, Yenari MA, Sapolsky RM, Steinberg GK. Catalase over-expression protects striatal neurons from transient focal cerebral ischemia. Neuroreport. 2004;15:413–416. doi: 10.1097/00001756-200403010-00006. [DOI] [PubMed] [Google Scholar]
- Gubellini P, Centonze D, Tropepi D, Bernardi G, Calabresi P. Induction of corticostriatal LTP by 3-nitropropionic acid requires the activation of mGluR1/PKC pathway. Neuropharmacol. 2004;46:761–769. doi: 10.1016/j.neuropharm.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Gurden H, Takita M, Jay TM. Essential role of D1 but not D2 receptors in the NMDA receptor-dependent long-term potentiation at hippocampal-prefrontal cortex synapses in vivo. J Neurosci. 2000;20:RC106. doi: 10.1523/JNEUROSCI.20-22-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton BF, Gould DH. Nature and distribution of brain lesions in rats intoxicated with 3-nitropropionic acid: a type of hypoxic (energy deficient) brain damage. Acta Neuropathol. 1987;72:286–297. doi: 10.1007/BF00691103. [DOI] [PubMed] [Google Scholar]
- Hashimoto N, Matsumoto T, Mabe H, Hashitani T, Nishino H. Dopamine has inhibitory and accelerating effects on ischemia-induced neuronal cell damage in the rat striatum. Brain Res Bull. 1994;33:281–288. doi: 10.1016/0361-9230(94)90195-3. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. D1/D5 receptor agonists induce a protein synthesis-dependent late potentiation in the CA1 region of the hippocampus. Proc Natl Acad Sci U S A. 1995;92:2446–2450. doi: 10.1073/pnas.92.7.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JN, Wickens JR. Dopamine D-1/D-5 receptor activation is required for long-term potentiation in the rat neostriatum in vitro. J Neurophysiol. 2001;85:117–124. doi: 10.1152/jn.2001.85.1.117. [DOI] [PubMed] [Google Scholar]
- Kim GW, Chan PH. Involvement of superoxide in excitotoxicity and DNA fragmentation in striatal vulnerability in mice after treatment with the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab. 2002;22:798–809. doi: 10.1097/00004647-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Kim GW, Copin JC, Kawase M, Chen SF, Sato S, Gobbel GT, Chan PH. Excitotoxicity is required for induction of oxidative stress and apoptosis in mouse striatum by the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab. 2000;20:119–129. doi: 10.1097/00004647-200001000-00016. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, Andreassen OA, Ferrante RJ, Dedeoglu A, Mueller G, Lancelot E, Bogdanov M, Andersen JK, Jiang D, Beal MF. Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. J Neurosci. 2000;20:1–7. doi: 10.1523/JNEUROSCI.20-01-00001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutouzis TK, Borlongan CV, Scorcia T, Creese I, Cahill DW, Freeman TB, Sanberg PR. Systemic 3-nitropropionic acid: long-term effects on locomotor behavior. Brain Res. 1994;646:242–246. doi: 10.1016/0006-8993(94)90085-x. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lancelot E, Callebert J, Revaud ML, Boulu RG, Plotkine M. Detection of hydroxyl radicals in rat striatum during transient focal cerebral ischemia: possible implication in tissue damage. Neurosci Lett. 1995;197:85–88. doi: 10.1016/0304-3940(95)11901-8. [DOI] [PubMed] [Google Scholar]
- Liu X, Luo X, Hu W. Studies on the epidemiology and etiology of moldy sugarcane poisoning in China. Biomed Environ Sci. 1992;5:161–177. [PubMed] [Google Scholar]
- Lowry OH, Rosenbrough NG, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;93:265–275. [PubMed] [Google Scholar]
- Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M. 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J Neurolog Sci. 1991;18:492–498. doi: 10.1017/s0317167100032212. [DOI] [PubMed] [Google Scholar]
- Maeno H. Dopamine receptors in canine caudate nucleus. Mol Cell Biochem. 1982;43:65–80. doi: 10.1007/BF00423094. [DOI] [PubMed] [Google Scholar]
- Marti M, Mela F, Ulazzi L, Hanau S, Stocchi S, Paganini F, Beani L, Bianchi C, Morari M. Differential responsiveness of rat striatal nerve endings to the mitochondrial toxin 3-nitropropionic acid: implications for Huntington's disease. Eur J Neurosci. 2003;18:759–767. doi: 10.1046/j.1460-9568.2003.02806.x. [DOI] [PubMed] [Google Scholar]
- McLin JP, Thompson LM, Steward O. Differential susceptibility to striatal neurodegeneration induced by quinolinic acid and kainate in inbred, outbred and hybrid mouse strains. Eur J Neurosci. 2006;24:3134–3140. doi: 10.1111/j.1460-9568.2006.05198.x. [DOI] [PubMed] [Google Scholar]
- Miles PR, Mundorf ML, Wightman RM. Release and uptake of catecholamines in the bed nucleus of the stria terminalis measured in the mouse brain slice. Synapse. 2002;44:188–197. doi: 10.1002/syn.10069. [DOI] [PubMed] [Google Scholar]
- Ming L. Moldy sugarcane poisoning—a case report with a brief review. J Toxicol Clin Toxicol. 1995;33:363–367. doi: 10.3109/15563659509028924. [DOI] [PubMed] [Google Scholar]
- Nakao N, Nakai E, Nakai K, Itakura T. Ablation of the subthalamic nucleus supports the survival of nigral dopaminergic neurons after nigrostriatal lesions induced by the mitochondrial toxin 3-nitropropionic acid. Ann Neurol. 1999;45:640–651. [PubMed] [Google Scholar]
- Newcomb JD, Brown WD, Rodriguez AI, Garbuzova-Davis S, Saporta S, Sanberg PR, Willing AE. Behavioral alterations in Lewis rats following two-day continuous 3-nitropropionic acid administration. Neurotox Res. 2005;8:259–266. doi: 10.1007/BF03033979. [DOI] [PubMed] [Google Scholar]
- Nishino H, Hida H, Kumazaki M, Shimano Y, Nakajima K, Shimizu H, Ooiwa T, Baba H. The striatum is the most vulnerable region in the brain to mitochondrial energy compromise: a hypothesis to explain its specific vulnerability. J Neurotrauma. 2000;17:251–260. doi: 10.1089/neu.2000.17.251. [DOI] [PubMed] [Google Scholar]
- Otani S, Daniel H, Roisin MP, Crepel F. Dopaminergic modulation of long-term synaptic plasticity in rat prefrontal neurons. Cereb Cortex. 2003;13:1251–1256. doi: 10.1093/cercor/bhg092. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 1996;16:7478–7486. doi: 10.1523/JNEUROSCI.16-23-07478.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouary S, Bizat N, Altairac S, Ménétrat H, Mittoux V, Condé F, Hantraye P, Brouillet E. Major strain differences in response to chronic systemic administration of the mitochondrial toxin 3-nitropropionic acid in rats: implications for neuroprotection studies. Neuroscience. 2000;97:521–530. doi: 10.1016/s0306-4522(00)00020-8. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Ling GY, Gajendiran M, Xu ZC. Asymmetrical changes of excitatory synaptic transmission in dopamine-denervated striatum after transient forebrain ischemia. Neuroscience. 2002;114:317–326. doi: 10.1016/s0306-4522(02)00309-3. [DOI] [PubMed] [Google Scholar]
- Partridge JG, Tang KC, Lovinger DM. Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum. J Neurophysiol. 2000;84:1422–1429. doi: 10.1152/jn.2000.84.3.1422. [DOI] [PubMed] [Google Scholar]
- Patel J, Rice ME. Monitoring dopamine release in brain slices. In: Grimes CA, Dickey EC, Pishko MV, editors. Encyclopedia of sensors. Stevenson Ranch, CA: American Scientific; 2006. pp. 313–334. [Google Scholar]
- Petzinger GM, Walsh JP, Akopian G, Hogg E, Abernathy A, Arevalo P, Turnquist P, Vucković M, Fisher BE, Togasaki DM, Jakowec MW. Effects of treadmill exercise on dopaminergic transmission in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-(MPTP)-lesioned mouse model of basal ganglia injury. J Neurosci. 2007;27:5291–5300. doi: 10.1523/JNEUROSCI.1069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi B, Passino E, Sgobio C, Bonsi P, Barone I, Ghiglieri V, Pisani A, Bernardi G, Ammassari-Teule M, Calabresi P. Plastic and behavioral abnormalities in experimental Huntington's disease: a crucial role for cholinergic interneurons. Neurobiol Dis. 2006;22:143–152. doi: 10.1016/j.nbd.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Reynolds DS, Carter RJ, Morton AJ. Dopamine modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of Huntington's disease. J Neurosci. 1998;18:10116–10127. doi: 10.1523/JNEUROSCI.18-23-10116.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JN, Wickens JR. Dopamine-dependent plasticity of corticostriatal synapses. Neural Netw. 2002;15:507–521. doi: 10.1016/s0893-6080(02)00045-x. [DOI] [PubMed] [Google Scholar]
- Reynolds JN, Hyland BI, Wickens JR. A cellular mechanism of reward-related learning. Nature. 2001;413:67–70. doi: 10.1038/35092560. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ, Greenfield SA. Characteristics of electrically evoked somatodendritic release in substanitia nigra and ventral tegmental area in vitro. J Neurophysiol. 1997;77:853–862. doi: 10.1152/jn.1997.77.2.853. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. Susceptibility to excitotoxic and metabolic striatal neurodegeneration in the mouse is genotype dependent. Brain Res. 2005;1040:112–120. doi: 10.1016/j.brainres.2005.01.067. [DOI] [PubMed] [Google Scholar]
- Schulte A, Chow RH. A simple method for insulating carbon-fiber microelectrodes using anodic electrophoretic deposition of paint. Anal Chem. 1996;68:3054–3058. doi: 10.1021/ac960210n. [DOI] [PubMed] [Google Scholar]
- Simpson JR, Isacson O. Mitochondrial impairment reduces the threshold for in vivo NMDA-mediated neuronal death in the striatum. Exp Neurol. 1993;121:57–64. doi: 10.1006/exnr.1993.1071. [DOI] [PubMed] [Google Scholar]
- Smith R, Musleh W, Akopian W, Buckwalter G, Walsh JP. Regional differences in the expression of corticostriatal synaptic plasticity. Neuroscience. 2001;106:95–101. doi: 10.1016/s0306-4522(01)00260-3. [DOI] [PubMed] [Google Scholar]
- Ste-Marie L, Vachon P, Vachon L, Bémeur C, Guertin MC, Montgomery J. Hydroxyl radical production in the cortex and striatum in a rat model of focal cerebral ischemia. Can J Neurol Sci. 2000;27:152–159. [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nature Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Toner CC, Stamford JA. Effects of metabolic alterations on dopamine release in an in vitro model of neostriatal ischaemia. Brain Res Bull. 1999;48:395–399. doi: 10.1016/s0361-9230(99)00016-7. [DOI] [PubMed] [Google Scholar]
- Tozzi A, Costa C, Di Filippo M, Tantucci M, Siliquini S, Belcastro V, Parnetti L, Picconi B, Calabresi P. Memantine reduces neuronal dysfunctions triggered by in vitro ischemia and 3-nitropropionic acid. Exp Neurol. 2007;207:218–226. doi: 10.1016/j.expneurol.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Trantham-Davidson H, Neely LC, Lavin A, Seamans JK. Mechanisms underlying differential D1 versus D2 dopamine receptor regulation of inhibition in prefrontal cortex. J Neurosci. 2004;24:10652–10659. doi: 10.1523/JNEUROSCI.3179-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Wüllner U, Young AB, Penney JB, Beal MF. 3-Nitropropionic acid toxicity in the striatum. J Neurochem. 1994;63:1772–1781. doi: 10.1046/j.1471-4159.1994.63051772.x. [DOI] [PubMed] [Google Scholar]