

Abstract
The role of angiogenesis in tumor growth and metastasis is well established. Identification of small molecule that blocks tumor angiogenesis and is safe and affordable has been a challenge in drug development. In this study, we demonstrated that acetyl-11-keto-β-boswellic acid (AKBA), an active component from an Ayurvedic medicinal plant (Boswellia serrata), could strongly inhibit tumor angiogenesis. AKBA suppressed tumor growth in the human prostate tumor xenograft mice treated daily (10 mg/kg of AKBA) after solid tumors reached about 100 mm3 (n=5). The inhibitory effect of AKBA on tumor growth was well correlated with suppression of angiogenesis. When examined for the molecular mechanism, we found that AKBA significantly inhibited blood vessel formation in the Matrigel plug assay in mice and effectively and suppressed vascular endothelial growth factor (VEGF)-induced microvessel sprouting in rat aortic ring assay ex vivo. Furthermore, AKBA inhibited VEGF-induced cell proliferation, chemotactic motility, and the formation of capillary-like structures from primary cultured human umbilical vascular endothelial cells (HUVECs) in a dose-dependent manner. Western blot analysis and in vitro kinase assay revealed that AKBA suppressed VEGF-induced phosphorylation of VEGF receptor 2 kinase (KDR/Flk-1) with IC50 of 1.68 μmol/L. Specifically, AKBA suppressed the downstream protein kinases of VEGFR2, including Src family kinase, focal adhesion kinase, extracellular signal-related kinase, AKT, mTOR, and ribosomal protein S6 kinase. Our findings suggest that AKBA potently inhibits human prostate tumor growth through inhibition of angiogenesis induced by VEGFR2 signaling pathways.
Keywords: AKBA, KDR/Flk-1, tumor angiogenesis, mTOR, prostate tumor
Introduction
Angiogenesis plays an essential role in tumor growth, invasiveness, and metastasis(1–7). Vascular endothelial growth factor (VEGF), a glycoprotein that has mitogenic activity on vascular endothelial cells, is widely expressed in most cancers and is a critical component of tumor angiogenesis (2). VEGF exerts its biological actions by binding to its receptor tyrosine kinase. However, the VEGF signaling involved in angiogenesis is mainly mediated by VEGF receptor 2 (VEGFR2) (8). Specifically, VEGFR2 activation results in the activation of diverse intracellular signaling molecules, such as Src family kinase/focal adhesion kinase (FAK) (9), phosphoinositide 3-kinase/AKT kinase, mammalian target of rapamycin (mTOR)/ribosomal protein S6 kinase (p70S6K) (10), protein kinase C/protein kinase D (11), and mitogen extracellular kinase/extracellular signal-related kinase (ERK) (12), which promote the growth, migration, differentiation, and survival of endothelial cells in preexisting vasculature. The U.S. Food and Drug Administration has recently approved avastin, an antibody against the VEGF, for the treatment of cancer (3, 5, 6). Thus agents that are small, safe, affordable and efficacious in suppressing tumor angiogenesis will have great potential in treating tumor angigogeneis.
Acetyl-11-keto-β-boswellic acid (AKBA), is a derivative of boswellic acid, which is the main component of gum resin from Boswellia serrata and Boswellia carterii Birdw. Traditionally, boswellic acid has been used as an anti-inflammatory agent to treat arthritis, rheumatic disorders, and respiratory diseases. As an anti-inflammatory agent, boswellic acid directly interacts with IκB kinases (13) and suppresses nuclear factor-κB–regulated gene expression (14). Boswellic acid has been reported to noncompetitively inhibit 5-lipoxygenase (15–17), topoisomerase (18), and leukocyte elastase (19). In addition, boswellic acid been shown to potentiate apoptosis in several types of tumor cells, including colon cancer (20), prostate cancer (21), fibrosarcoma (22), hepatoma (23), and malignant glioma (24, 25) through caspase-8 activation (20) and death receptor 5–mediated signaling (21). However, whether AKBA can modulate prostate tumor growth and tumor angiogenesis in general is not understood. In this study, we investigated the functional roles of AKBA in suppressing human prostate cancer growth and angiogenesis. We demonstrate that AKBA potently inhibits angiogenesis and tumor growth by suppressing the VEGFR2 and the mTOR signaling pathways.
Materials and Methods
Reagents
Purified AKBA was supplied by Sabinsa Corporation (Piscataway, NJ). A 50-mmol/L stock solution of AKBA was prepared and then stored at −20°C as small aliquots until needed. Bacteria-derived recombinant human VEGF (VEGF165) was a gift from the Experimental Branch of the National Institutes of Health (NIH; Bethesda, MD). Growth factor–reduced Matrigel was purchased from BD Biosciences (San Jose, CA). Antibodies against FAK, Src, AKT, ERK1, β-actin, and poly (ADP-ribose) polymerase (PARP) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Caspase 3, mTOR and phospho-specific anti-Src (Tyr416), anti-FAK (Tyr567/577), anti-AKT (Ser473), anti-pERK1/2 (Thr202/Tyr204), anti-mTOR (Ser2448), anti-p70S6K (Thr389), and anti-VEGFR2 (Tyr1175) were purchased from Cell Signaling Technology (Danvers, MA).
Cell lines and cell culture
Primary human umbilical vascular endothelial cells (HUVECs) were kindly provided by Dr. Xinli Wang (Baylor College of Medicine, Houston, TX). HUVECs were cultured in endothelial cell growth medium (ECGM) as described previously (26). Human prostate cancer (PC-3) cells were purchased from American Type Culture Collection (Manassas, VA) and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). HUVECs and PC-3 cells were cultured at 37°C under a humidified 95%:5% (v/v) mixture of air and CO2.
Animal studies
Animals were maintained in a laminar airflow cabinet under specific pathogen-free conditions and a 12-h light-dark cycle. Mice were maintained according to the NIH standards established in the Guidelines for the Care and Use of Experimental Animals, and all of the experimental protocols were approved by the Animal Investigation Committee of the Institute of Biosciences and Technology, Texas A&M University Health Science Center.
Xenograft human prostate tumor mouse model
Xenograft mouse model assay was performed as previously described (27). Five-week-old male BALB/cA nude mice (National Rodent Laboratory Animal Resources, Shanghai, China) weighing about 25 g each were randomly divided into each group of 5. PC-3 cells were subcutaneously injected into the mice (2×106 cells per mouse). After tumors grew to about 100 mm3, mice were treated subcutaneously with or without AKBA (10 mg/kg/d) for a month. The body weight of each mouse was recorded every 5 d. At the same time, solid tumor volume was determined using Vernier caliper measurements and the formula of A×B2×0.52, where A is the longest diameter of the tumor and B is the shortest diameter of the tumor. After 30 d, mice with tumors no greater than 1.5 cm in diameter were sacrificed.
Histology and immunohistochemistry
Solid tumors were removed, fixed with 10% formaldehyde, and embedded in paraffin. A blood vessel staining kit (von Willebrand Factor; Millipore, Billerica, MA) was used to stain blood vessels in 5- μM tumor sections. Images of the stained blood vessels were taken using a Leica DM 4000B photo microscope (Solms, Germany). Blood vessels were counted (n=5).
In vivo Matrigel plug assay
Matrigel plug assay was performed and modified as described previously (28). Briefly, we subcutaneously injected 0.5 mL of Matrigel containing indicated amounts of AKBA, 80 ng of VEGF, and 20 units of heparin into the ventral area of 6-week-old C57BL/6 mice (NIH). Five mice were used for each group. After 6 d, the skin of each mouse was pulled back to expose an intact Matrigel plug. Hematoxylin and eosin staining was performed to identify the formation and infiltration of new, functional microvessels. Functional microvessels with intact red blood cells were quantified manually using a microscope (high-power field [HPF] ×200).
Rat aortic ring assay
Rat aortic ring assay was performed as described previously (28). In brief, 48-well plates were coated with 120 μL of Matrigel per well and polymerized in an incubator. Aortas isolated from 6-week-old male Sprague-Dawley rats were cleaned of periadventitial fat and connective tissues in cold phosphate-buffered saline and cut into rings of 1~1.5 mm in circumference. The aortic rings were randomized into wells and sealed with a 100- μL overlay of Matrigel. VEGF in 500 μL of serum-free ECGM with or without AKBA was added into the wells. As a control, ECGM alone was assayed, and the fresh medium was exchanged for every 2 d. After 6 d, microvessel sprouting was fixed and photographed using an inverted microscope (Olympus, Center Valley, PA; magnification ×100). The assay was scored from 0 (least positive) to 5 (most positive) in a double-blind manner. Each data point was assayed 6 times.
Cell viability assay
HUVECs or PC-3 cells (2×104 cells/well) were treated with or without VEGF (10 ng/mL) and various concentrations of AKBA for 24 h. To determine cell viability, we used a CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS; Promega; Madison, WI) and a VERSAmax microplate reader (Molecular Devices; Sunnyvale, CA).
Endothelial cell migration assay
HUVECs were allowed to grow to full confluence in 6-well plates precoated with 0.1% gelatin (Sigma, St. Louis, MO) and then starved with ECGM containing 0.5% FBS for 6 h to inactivate cell proliferation. The cells were then wounded with pipette tips and washed with phosphate-buffered saline. ECGM containing 0.5% FBS was added into the wells with or without 10 ng/mL VEGF and various concentration of AKBA. Images of the cells were taken after 8–10 h of incubation at 37°C in a 95%:5% (v/v) mixture of air and CO2. The migrated cells were counted manually, and the percentage of inhibition was expressed using untreated wells at 100%. Three independent experiments were performed.
Endothelial cell Transwell migration assay
The chemotactic motility of HUVECs was determined using a Transwell migration assay (BD Biosciences) with 6.5-mm diameter polycarbonate filters (8- μM pore size) as described previously (26). In brief, the filter of the Transwell plate was coated with 0.1% gelatin. The bottom chambers were filled with 500 μL of ECGM containing 0.5% FBS supplemented with 10 ng/mL VEGF. Inactivated HUVECs (4×104 cells) suspended in 100 μL of ECGM containing 0.5% FBS plus various concentrations of AKBA were seeded in the top chambers. Cells were allowed to migrate for 8–10 h. Non-migrated cells were removed with cotton swabs, and migrated cells were fixed with cold 4% paraformaldehyde and stained with 1% crystal violet. Images were taken using an inverted microscope (Olympus), and migrated cells were quantified by manual counting. The percentage of migrated cells inhibited by AKBA was expressed on the basis of untreated control wells.
Endothelial cell capillary-like tube formation assay
Tube formation was assessed as previously described (26). Growth factor–reduced Matrigel was pipetted into pre-chilled 24-well plates (100 μL Matrigel/well) and polymerized for 45 min at 37°C. HUVECs were first incubated in ECGM containing 0.5% FBS for 6 h and then treated with various concentrations of AKBA for 30 min before seeding. HUVECs were collected and placed onto the layer of Matrigel (1×105 cells/well) in 1 mL of ECGM containing 0.5% FBS, followed by the addition of 10 ng/mL of VEGF. After 6–8 h of incubating at 37°C in a 95%:5% (v/v) mixture of air and CO2, the endothelial cells were photographed using an inverted microscope (Olympus; magnification ×100). Three independent experiments were performed.
Western blot analysis
To determine the effects of AKBA on VEGFR2-dependent signaling cascade, HUVECs were first starved in serum-free ECGM for 6 h and then pretreated with or without various concentrations of AKBA for 30 min, followed by stimulation with 50 ng/mL of VEGF. Different stimulation time is responsible for different substrate phosphorylation by VEGF. The whole- cell extracts were prepared in RIPA buffer (20 mM Tris, 2.5 mM EDTA, 1% Triton X-100, 1% deoxycholate, 0.1% SDS, 40 mM NaF, 10 mM Na4P2O7, and 1 mM PMSF) supplemented with proteinase inhibitor cocktail (Calbiochem, San Diego, CA). Forty micrograms of cellular protein from each sample was applied to 8%–12% SDS-polyacrylamide gels and probed with specific antibodies, followed by exposure to a horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit antibody (Cell Signaling Technology). Protein concentrations were determined using bicinchoninic acid assay and equalized before loading.
In vitro VEGFR2 kinase inhibition assay
VEGFR2 kinase assay was performed using an HTScan VEGFR2 kinase kit (Cell Signaling Technology) combined with colorimetric ELISA detection. The final reaction system included 60 mM HEPES (pH 7.5), 5 mM MgCl2, 5 mM MnCl2, 3 μM Na3VO4, 1.25 mM DTT, 20 μM ATP, 1.5 μM substrate peptide, 100 ng of VEGF receptor kinase, and indicated concentrations of AKBA. The kinase assay was repeated 3 times independently.
Statistical analysis
Statistical comparisons between drug-treated group and untreated control group were performed using one-way analysis of variance followed by Dunnet’s test. Data were presented as means ± standard deviations. P values ≤ 0.05 were considered statistically significant.
Results
AKBA inhibits tumor growth and tumor angiogenesis in the xenograft mouse model
We used a xenograft prostate tumor model to investigate the effect of AKBA on tumor growth and angiogenesis and found that 10 mg/kg/d of AKBA significantly suppressed tumor volume (Fig. 1A) and tumor weight (Fig. 1C) but had no effect on the body weight of mice (Fig. 1B). In our present tumor inhibition model, the average tumor volume in the control mice increased from 95.83 ± 43.37 mm3 to 536.58 ± 241.801 mm3 after 30 d, whereas the average tumor volume in the AKBA-treated mice decreased from 92.43 ± 39.70 mm3 to 47.99 ± 35.49 mm3, indicating proliferation rate of tumor cells in treated mice was greatly inhibited by AKBA. Additionally, the average tumor weight in the control group was 0.194 ± 0.097 g, whereas the average tumor weight in the AKBA-treated group was only 0.049 ± 0.012 g (Fig. 1C), suggesting that AKBA strongly inhibited tumor growth in the xenograft prostate tumor mouse model.
Figure 1. AKBA inhibits tumor growth and tumor angiogenesis in a xenograft mouse model.
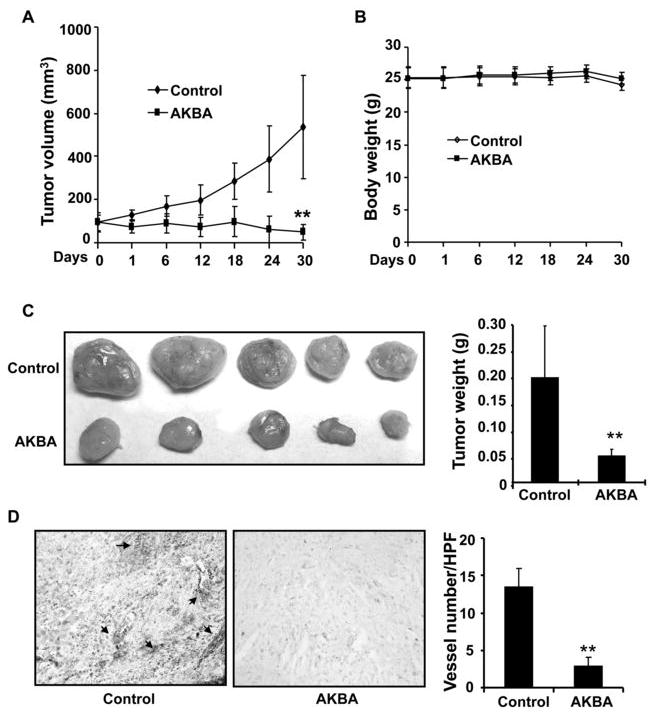
PC-3 cells were injected into 5-week-old BALB/cA nude mice (2×106 cells per mouse). After solid tumors grew to about 100 mm3, the mice were subcutaneously treated with or without AKBA (10 mg/kg/d). A, AKBA inhibited tumor growth as measured by tumor volume. B, As shown by the body weight change in mice, AKBA had little toxicity in the amount tested. There was no significant difference in body weight between the treated group and the control group. C, Solid tumors in the AKBA-treated mice were significantly smaller than those in the control mice. D, Blood vessel staining revealed that AKBA inhibited tumor angiogenesis. The 5- μM tumor sections from the control and AKBA-treated groups were stained using von Willebrand Factor blood vessel staining, and the number of blood vessels in an HPF was counted. The arrows indicate blood vessels. Columns, mean; bars, standard deviation; **, P < 0.01 vs control.
To further investigate whether AKBA inhibited tumor growth by suppressing tumor angiogenesis, we used a blood vessel staining kit to stain solid tumor sections. Our data show that the average number of blood vessels in AKBA-treated group is 2.83 ± 1.17 blood vessels/HPF (Fig. 1D) compared to 13.50 ± 2.43 blood vessels/HPF in the control group, indicating that AKBA significantly inhibited tumor angiogenesis.
AKBA inhibits VEGF-induced angiogenesis in vivo and vessel sprouting ex vivo
We further explored the anti-angiogenic activity of AKBA using in vivo and ex vivo angiogenesis models. First, we used Matrigel plug assay to determine the effects of AKBA on VEGF-induced angiogenesis in vivo. After 6 days, Matrigel plugs containing VEGF alone appeared dark (Fig. 2A) and were filled with intact red blood cells (RBCs), indicating that functional vasculatures had formed inside the Matrigel via angiogenesis triggered by VEGF. In contrast, addition of different concentrations of AKBA in the Matrigel plugs containing VEGF dramatically inhibited vascular formation (Fig. 2A). The color of the Matrigel plugs becomes pale due to the lack of RBCs. 60 μg AKBA totally blocked vasculature formation in the assays (Fig. 2A).
Figure 2. AKBA inhibits VEGF-induced angiogenesis in vivo.
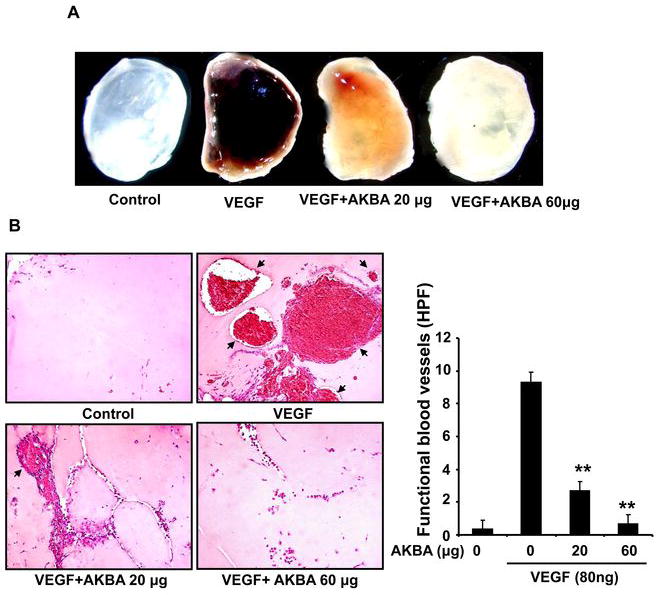
Six-week-old C57/BL/6 mice were injected with 0.5 mL of Matrigel containing indicated amounts of AKBA, 80 ng of VEGF, and 20 units of heparin into the ventral area (5 mice per group). After 6 d, the skin of mice was pulled back to expose the intact Matrigel plugs. A, Representative Matrigel plugs were photographed. B, AKBA inhibited blood vessel formation. The Matrigel plugs were fixed, sectioned, and stained with hematoxylin and eosin. Infiltrating microvessels with intact red blood cells were qualified by manual counting (magnification, ×200). Columns, mean; bars, standard deviation; **, P < 0.01 vs. VEGF alone.
We used hematoxylin and eosin (H&E) staining to quantify the amount of functional vasculature in Matrigel plugs (Fig. 2B) and found that 20 μg AKBA dramatically inhibited VEGF-induced vessel formation in vivo.
To further investigate whether AKBA inhibited VEGF-induced angiogenesis ex vivo, we examined the sprouting of vessels from aortic rings in the absence or presence of AKBA. VEGF (20 ng/mL) significantly stimulated microvessel sprouting, leading to the formation of a network of vessels around the aortic rings (Fig. 3). Treatment of AKBA antagonized the VEGF-induced sprouting in a dose-dependent manner (Fig. 3A). 10 μMol/L of AKBA can completely block the VEGF-induced microvessel sprouting of rat aortic rings (Fig. 3B).
Figure 3. AKBA inhibits VEGF-induced microvessel sprouting ex vivo.
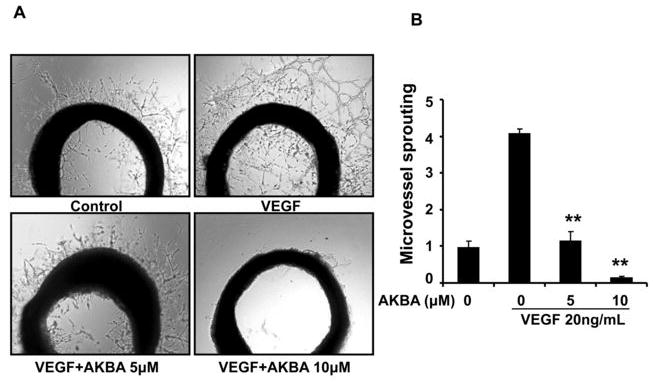
Aortic segments isolated from Sprague-Dawley rats were placed in growth factor reduced Matrigel-covered wells and treated with VEGF (20 ng/mL) in the presence or absence of AKBA for 6 d. A, Representative photographs of endothelial cell sprouts forming branching cords from the margins of aortic rings. B, Sprouts were scored from 0 (least positive) to 5 (most positive) in a double-blinded manner. Bars, standard deviation; **, P < 0.01 vs. VEGF alone.
AKBA inhibits VEGF-induced chemotactic motility and capillary structure formation of HUVECs
Endothelial cell migration is essential for blood vessels formation in angiogenesis, and also critical for tumor growth. To assess the antiangiogenic action of AKBA in vitro, we investigated AKBA’s inhibitory effects on the chemotactic motility of endothelial cells by using wound-healing migration assay (Fig. 4A) and Transwell assay (Fig. 4B), respectively. We found that AKBA inhibited VEGF-induced HUVEC migration (Fig. 4A) and cell invasion (Fig. 4B) in a dose-dependent manner, with significant inhibition at 1–5 μMol/L of AKBA.
Figure 4. AKBA inhibits VEGF-induced chemotactic motility and capillary-structure formation of endothelial cells.
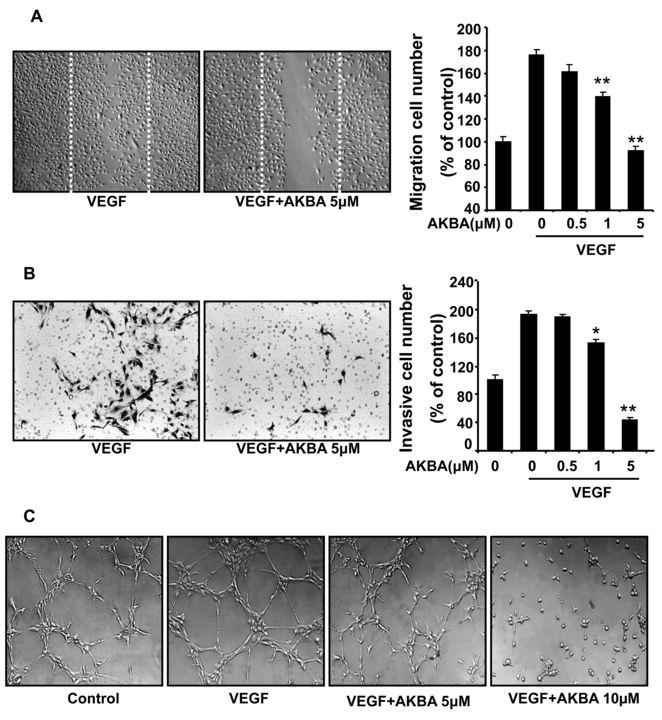
A, AKBA inhibited HUVEC migration. HUVECs were grown into full confluence in 6-well plates and then starved with 0.5% FBS ECGM for 6 h to inactivate cell proliferation. Cells were scratched by pipette and treated with or without 10 ng/mL VEGF and indicated concentrations of AKBA. The migrated cells were quantified by manual counting. B, AKBA inhibited HUVEC invasion. HUVECs were seeded in the upper chamber of a Transwell and treated with different concentrations of AKBA. The bottom chamber was filled with ECGM supplemented with VEGF. After 8–10 h, the HUVECs that migrated through the membrane were quantified. C, AKBA inhibited the VEGF-induced tube formation of HUVECs tubes. HUVECs were placed in 24-well plates coated with Matrigel (4×104 cells/well). After 6–8 h, cells were fixed, and tubular structures were photographed (magnification, ×100). Columns, mean from three different experiments with duplicates; bars, standard deviation; *, P < 0.05; **, P < 0.01 vs. VEGF alone.
Although angiogenesis is a very complex process involving several kinds of cells, tube formation of endothelial cells is one of the key steps of angiogenesis (29). We used two-dimensional Matrigel assays to examine the potential effects of AKBA on the tubular structure formation of endothelial cells. When HUVECs were seeded on the growth factor–reduced Matrigel, robust tubular-like structures were formed in the presence of VEGF. However, treatment with 5 μMol/L or 10 μMol/L AKBA significantly suppressed or terminated VEGF-induced tubular formation of endothelial cells (Fig. 4C). These results indicated that AKBA could block VEGF-induced angiogenesis in vitro by inhibiting cell motility and tubular structure formation of endothelial cells.
AKBA potentiates viability and apoptosis in endothelial cells and tumor cells
To further elucidate AKBA’s effects on endothelial cells, we used MTS assays to examine cell proliferation and survival. Because angiogenesis is primarily initiated by growth factors, we first tested whether AKBA decreased VEGF-mediated HUVEC proliferation and viability. We found that a concentration of 5 μMol/L AKBA significantly inhibited VEGF-mediated HUVEC survival (Fig. 5A). Under normal culture conditions (i.e., no VEGF), AKBA (20–50 μMol/L) also inhibited HUVEC proliferation (Fig. 5B). We also examined the inhibitory effects of AKBA on PC-3 cell proliferation and found that a much higher concentration of AKBA (about 50 μMol/L) was required to suppress PC-3 cell proliferation compared to the concentration of AKBA required to suppress endothelial cell proliferation (about 5 μMol/L), indicating that endothelial cells were more sensitive to AKBA than PC-3 cells (Fig. 5C).
Figure 5. AKBA potentiates viability and apoptosis in endothelial cells and tumor cells.
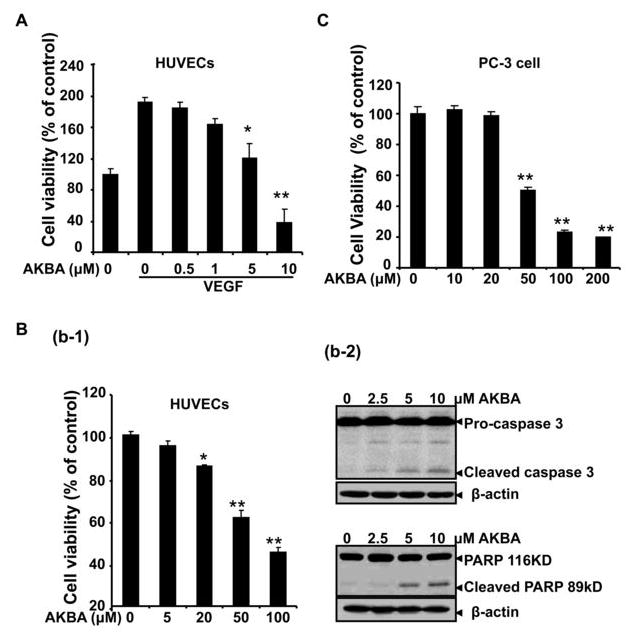
A, AKBA inhibited VEGF-induced cell viability in a dose-dependent manner. HUVECs (2×104 cells/well) were starved with serum-free ECGM and then treated with or without VEGF (10 ng/mL) and various concentrations of AKBA for 24 h. Cell viability was quantified by MTS assay *, P < 0.05; **, P < 0.01 vs. VEGF alone. B, AKBA inhibited HUVEC viability and induced cell apoptosis in normal culture condition. HUVECs (2×104 cells/well) were treated with different concentrations of AKBA in ECGM with 20% FBS for 24 h followed by MTS measurement. *, P < 0.05; **, P < 0.01 vs. media alone. C, AKBA inhibited PC-3 cell proliferation as measured by MTS. Columns, mean from three different experiments with six duplicates; bars, standard deviation. D. AKBA induced caspase-3 activation and cell apoptosis. Proteins from HUVECs treated with indicated concentrations of AKBA for 24 h were analyzed by Western blots analysis for cleaved caspase-3 and cleaved PARP determination.
To determine whether AKBA regulates endothelial cell death, we used Western blot analysis to examine caspase-3 activation and PARP cleavage in cells treated with different concentrations of AKBA. We found that addition of AKBA (2–5 μMol/L) led to the activation of caspase 3 and the cleavage of PARP from its intact form (116 kDa) to its cleaved form (89 kDa) in endothelial cells (Fig. 5D), suggesting that AKBA decreases cell viability and induces apoptosis through a caspase-dependent pathway.
AKBA is a potent VEGFR2 kinase inhibitor
To determine the molecular basis of AKBA-mediated antiangiogenesis, we examined whether AKBA could inhibit VEGFR2 activation. As shown in figure 6A, AKBA (5 μMol/L) strongly inhibited VEGF-activated VEGFR2 phosphorylation. To confirm our Western blot analysis data and determine whether AKBA directly inhibits VEGFR2 kinase activity, we performed in vitro kinase assays with different concentrations of AKBA using HTScan® VEGF receptor 2 kinase assay kit according to manufacturer suggested methods (Cell Signaling Technology and PerkinElmer Life Sciences, USA). Our data demonstrated that AKBA directly inhibited VEGFR2 kinase activity in a dose-dependent manner with a 50% inhibitory concentration of 1.68 μMol/L (Fig. 6B). These findings suggest that AKBA inhibits tumor angiogenesis by blocking VEGFR2 activation.
Figure 6. AKBA inhibits VEGFR2 kinase activity and its downstream signaling molecules.
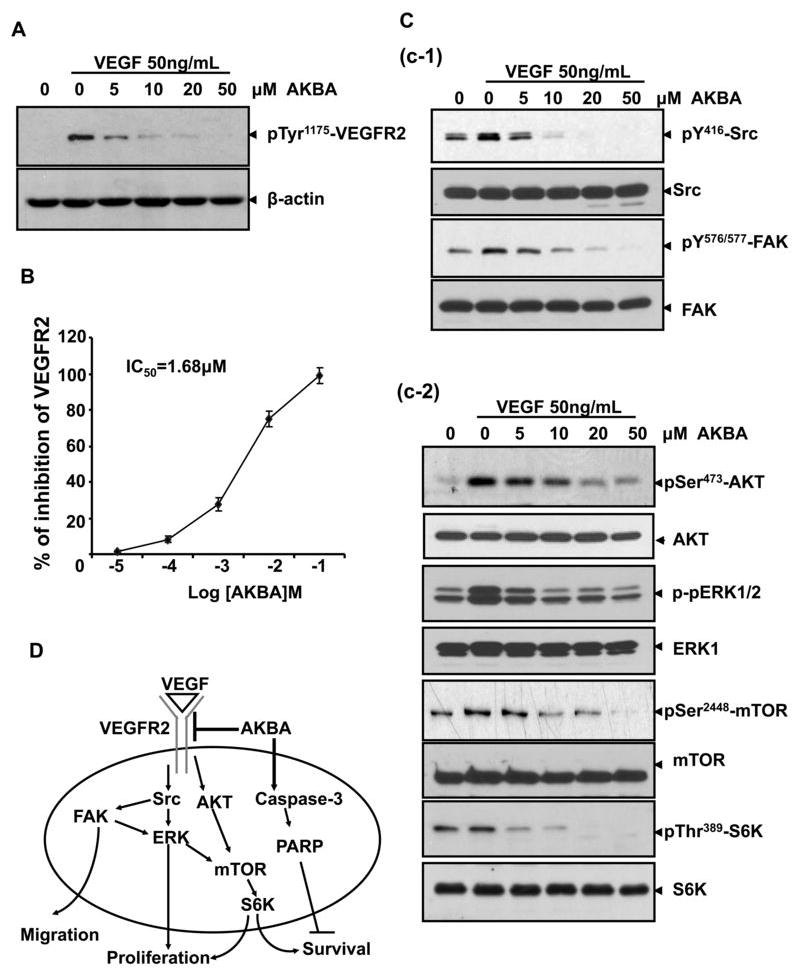
A, AKBA suppressed the activation of VEGFR2 triggered by VEGF in HUVECs. The activation of VEGFR2 from different treatments was analyzed by Western blotting and probed with anti-phospho-VEGFR2 antibody. B, AKBA inhibited VEGFR2 kinase activity in vitro. AKBA’s inhibition of VEGFR2 kinase activity was analyzed using an in vitro kinase assay kit according to the manufacturer’s instructions. Dots, from three different experiments with three duplicates; Bars, standard deviation. C, AKBA inhibited the activation of VEGFR2 downstream cascade, including mTOR pathway. Activation of Src and FAK complex (c-1), and AKT/mTOR/S6K signaling cascade were suppressed by AKBA (c-2). Proteins from different treatments were analyzed by Western blotting and probed with specific antibodies. D, The antiangiogenic signaling pathways regulated by AKBA in HUVECs.
AKBA inhibits the activation of VEGFR2-mediated signaling pathways
VEGFR2 activation leads to the activation of various downstream signaling substrates that are responsible for endothelial cell migration, proliferation, and survival. To determine whether AKBA inhibited downstream signaling of VEGFR2, we screened some key kinases involved in VEGFR2 signaling pathway. We found that 5 μMol/L of AKBA significantly suppressed the activation of c-Src/FAK (Fig 6C c-1) and AKT, ERK, mTOR and p70S6K (Fig 6C c-2), which suggested that AKBA exerted its antiangiogenic function through direct inhibiting VEGFR2 activation on the surface of endothelial cells and further suppressing VEGFR2-mediated downstream signaling cascade (Fig. 6D).
Discussion
In this study, we found that acetyl-11-keto-β-boswellic acid (AKBA) was a potent angiogenesis inhibitor and inhibited multiple steps of VEGF-mediated tumor angiogenesis, including cell proliferation, migration, invasion, and tube formation. As evidenced in our xenograft human prostate tumor mouse model experiments, tumor growth was significantly inhibited when AKBA antagonized tumor angiogenesis.
VEGF receptor inhibitors are a promising class of cancer treatment drugs (6, 8). VEGF is the primary and the most potent inducer of angiogenesis. VEGF activates cellular signaling pathways by binding to its receptor tyrosine kinase, which promotes several events required for angiogenesis, including endothelial cell survival, proliferation, and migration and vascular permeability (2). Currently, over 20 agents with antiangiogenic properties, including a VEGF-neutralizing antibody, soluble receptors, receptor antagonists, and tyrosine kinase inhibitors, are either already approved for cancer treatment or undergoing clinical (phase I–III) studies (2, 30). VEGF signaling events relevant to tumor growth and angiogenesis are mainly mediated by VEGFR2 (8, 31). In the present study, we found that a half-maximum inhibitory concentration of 1.68 μMol/L AKBA significantly blocked VEGFR2’s kinase activity, making AKBA a potent VEGFR2 inhibitor. Previous study also reported that AKBA inhibited basic fibroblast growth factor-induced signaling responses (32), suggesting that AKBA might be a broad receptor tyrosine kinase inhibitor.
In clinical trials, antiangiogenic therapy with VEGF antagonists has so far produced disappointing results (33). Successful antiangiogenic therapy may require the simultaneous blockade of signaling pathways downstream from multiple proangiogenic factor receptors (34). Previous studies have shown that the Src family kinase is substantially involved in VEGF-induced angiogenesis in vitro and in vivo (35, 36). By interacting with its downstream molecule, FAK, Src regulates cell motility (37) and vascular permeability (9). In the current study, we found that a low concentration of AKBA (5 μMol/L) effectively inhibited VEGF-triggered Src and FAK activation in endothelial cells.
Recently, AKT/mTOR/p70S6K signaling has been identified as a novel, functional mediator in angiogenesis (10, 38). In the current study, we found that AKBA significantly decreased the phosphorylation of mTOR and p70S6K, and its upstream kinase, AKT and ERK, indicating that AKBA suppresses tumor angiogenesis by inhibiting VEGFR2 and blocking its multiple downstream signaling components, thereby suppressing endothelial cell migration, proliferation, and survival (Fig. 6D). Interestingly, the mTOR/p70S6K pathway has been found to regulate the expression of proangiogenic factors such as interleukin-8 and VEGF in various human carcinomas by regulating hypoxia-inducible factor-1 α expression at the translational level (38–42), making the pathway a potential target for anticancer therapy. Therefore, AKBA may suppress tumor angiogenesis by inducing tumor cells to produce fewer proangiogenic molecules.
In the present studies, we demonstrate that 5 μMol/L AKBA was sufficient to inhibit VEGF-induced angiogenic responses in in vitro and ex vivo angiogenesis assays while 10 μMol/L AKBA completely blocked capillary-like structure formation and mircovessel sprouting. Our data indicate that AKBA’s anti-angiogenic activity inhibits tumor growth in vivo much earlier than its cytotoxic effects on tumor cells. Furthermore, we found that AKBA induced endothelial cell apoptosis through a caspase-dependent pathway, which suggests that AKBA inhibits endothelial cell survival not only by blocking VEGFR2 but also by blocking certain extrinsic death receptors on the cell surface or triggering the apoptotic cascade of the intrinsic mitochondrial pathway, as suggested by previous studies that higher concentrations of AKBA (30–50 μMol/L) directly interact with IκB kinase (13) to suppress nuclear factor-κB–regulated gene expression (14).
In conclusion, we found that AKBA potently inhibited tumor growth and angiogenesis by targeting VEGFR2 activation and mTOR signaling pathways, suggesting that AKBA has a potential role in cancer therapy.
Acknowledgments
Funding: This work is partially supported by the Science and Technology Commission of Shanghai Municipality, Research Platform for Cell Signaling Networks (06DZ22923), and National Institute of Health (NCI) (1R01CA106479).
We thank Joseph A. Munch for carefully proofreading the manuscript and providing valuable comments.
References
- 1.Compagni A, Christofori G. Recent advances in research on multistage tumorigenesis. Br J Cancer. 2000;83:1–5. doi: 10.1054/bjoc.2000.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–74. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 3.Quesada AR, Munoz-Chapuli R, Medina MA. Anti-angiogenic drugs: from bench to clinical trials. Med Res Rev. 2006;26:483–530. doi: 10.1002/med.20059. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 5.Zhong H, Bowen JP. Molecular design and clinical development of VEGFR kinase inhibitors. Curr Top Med Chem. 2007;7:1379–93. doi: 10.2174/156802607781696855. [DOI] [PubMed] [Google Scholar]
- 6.Takakura N. Basic science of angiogenesis and its progress in clinical application. Rinsho Ketsueki. 2008;49:1451–59. [PubMed] [Google Scholar]
- 7.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 8.Kowanetz M, Ferrara N. Vascular endothelial growth factor signaling pathways: therapeutic perspective. Clin Cancer Res. 2006;12:5018–22. doi: 10.1158/1078-0432.CCR-06-1520. [DOI] [PubMed] [Google Scholar]
- 9.Schlessinger J. New roles for Src kinases in control of cell survival and angiogenesis. Cell. 2000;100:293–6. doi: 10.1016/s0092-8674(00)80664-9. [DOI] [PubMed] [Google Scholar]
- 10.Liu LZ, Zheng JZ, Wang XR, Jiang BH. Endothelial p70 S6 kinase 1 in regulating tumor angiogenesis. Cancer Res. 2008;68:8183–8. doi: 10.1158/0008-5472.CAN-08-0819. [DOI] [PubMed] [Google Scholar]
- 11.Wong C, Jin ZG. Protein kinase C-dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. J Biol Chem. 2005;280:33262–9. doi: 10.1074/jbc.M503198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gollob JA, Wilhelm S, Carter C, Kelley SL. Role of Raf kinase in cancer: therapeutic potential of targeting the Raf/MEK/ERK signal transduction pathway. Semin Oncol. 2006;33:392–406. doi: 10.1053/j.seminoncol.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Syrovets T, Buchele B, Krauss C, Laumonnier Y, Simmet T. Acetyl-boswellic acids inhibit lipopolysaccharide-mediated TNF-alpha induction in monocytes by direct interaction with IkappaB kinases. J Immunol. 2005;174:498–506. doi: 10.4049/jimmunol.174.1.498. [DOI] [PubMed] [Google Scholar]
- 14.Takada Y, Ichikawa H, Badmaev V, Aggarwal BB. Acetyl-11-keto-beta-boswellic acid potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis by suppressing NF-kappa B and NF-kappa B-regulated gene expression. J Immunol. 2006;176:3127–40. doi: 10.4049/jimmunol.176.5.3127. [DOI] [PubMed] [Google Scholar]
- 15.Safayhi H, Mack T, Sabieraj J, Anazodo MI, Subramanian LR, Ammon HP. Boswellic acids: novel, specific, nonredox inhibitors of 5-lipoxygenase. J Pharmacol Exp Ther. 1992;261:1143–6. [PubMed] [Google Scholar]
- 16.Safayhi H, Sailer ER, Ammon HP. Mechanism of 5-lipoxygenase inhibition by acetyl-11-keto-beta-boswellic acid. Mol Pharmacol. 1995;47:1212–6. [PubMed] [Google Scholar]
- 17.Bishnoi M, Patil CS, Kumar A, Kulkarni SK. Potentiation of antinociceptive effect of NSAIDs by a specific lipooxygenase inhibitor, acetyl 11-keto-beta boswellic acid. Indian J Exp Biol. 2006;44:128–132. [PubMed] [Google Scholar]
- 18.Poeckel D, Werz O. Boswellic acids: biological actions and molecular targets. Curr Med Chem. 2006;13:3359–69. doi: 10.2174/092986706779010333. [DOI] [PubMed] [Google Scholar]
- 19.Safayhi H, Rall B, Sailer ER, Ammon HP. Inhibition by boswellic acids of human leukocyte elastase. J Pharmacol Exp Ther. 1997;281:460–3. [PubMed] [Google Scholar]
- 20.Liu JJ, Nilsson A, Oredsson S, Badmaev V, Zhao WZ, Duan RD. Boswellic acids trigger apoptosis via a pathway dependent on caspase-8 activation but independent on Fas/Fas ligand interaction in colon cancer HT-29 cells. Carcinogenesis. 2002;23:2087–93. doi: 10.1093/carcin/23.12.2087. [DOI] [PubMed] [Google Scholar]
- 21.Lu M, Xia L, Hua H, Jing Y. Acetyl-keto-beta-boswellic acid induces apoptosis through a death receptor 5-mediated pathway in prostate cancer cells. Cancer Res. 2008;68:1180–6. doi: 10.1158/0008-5472.CAN-07-2978. [DOI] [PubMed] [Google Scholar]
- 22.Zhao W, Entschladen F, Liu H, Niggemann B, Fang Q, Zaenker KS, et al. Boswellic acid acetate induces differentiation and apoptosis in highly metastatic melanoma and fibrosarcoma cells. Cancer Detect Prev. 2003;27:67–75. doi: 10.1016/s0361-090x(02)00170-8. [DOI] [PubMed] [Google Scholar]
- 23.Liu JJ, Nilsson A, Oredsson S, Badmaev V, Duan RD. Keto- and acetyl-keto-boswellic acids inhibit proliferation and induce apoptosis in Hep G2 cells via a caspase-8 dependent pathway. Int J Mol Med. 2002;10:501–5. [PubMed] [Google Scholar]
- 24.Glaser T, Winter S, Groscurth P, Safayhi H, Sailer ER, Ammon HP, et al. Boswellic acids and malignant glioma: induction of apoptosis but no modulation of drug sensitivity. Br J Cancer. 1999;80:756–65. doi: 10.1038/sj.bjc.6690419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hostanska K, Daum G, Saller R. Cytostatic and apoptosis-inducing activity of boswellic acids toward malignant cell lines in vitro. Anticancer Res. 2002;22:2853–62. [PubMed] [Google Scholar]
- 26.Pang X, Yi T, Yi Z, Cho SG, Qu W, Pinkaew D, et al. Morelloflavone, a biflavonoid, inhibits tumor angiogenesis by targeting rho GTPases and extracellular signal-regulated kinase signaling pathways. Cancer Res. 2009;69:518–25. doi: 10.1158/0008-5472.CAN-08-2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi T, Yi Z, Cho SG, Luo J, Pandey MK, Aggarwal BB, et al. Gambogic acid inhibits angiogenesis and prostate tumor growth by suppressing vascular endothelial growth factor receptor 2 signaling. Cancer Res. 2008;68:1843–50. doi: 10.1158/0008-5472.CAN-07-5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pyun BJ, Choi S, Lee Y, Kim TW, Min JK, Kim Y, et al. Capsiate, a nonpungent capsaicin-like compound, inhibits angiogenesis and vascular permeability via a direct inhibition of Src kinase activity. Cancer Res. 2008;68:227–35. doi: 10.1158/0008-5472.CAN-07-2799. [DOI] [PubMed] [Google Scholar]
- 29.Patan S. Vasculogenesis and angiogenesis. Cancer Treat Res. 2004;117:3–32. doi: 10.1007/978-1-4419-8871-3_1. [DOI] [PubMed] [Google Scholar]
- 30.Pourgholami MH, Morris DL. Inhibitors of vascular endothelial growth factor in cancer. Cardiovasc Hematol Agents Med Chem. 2008;6:343–7. doi: 10.2174/187152508785909528. [DOI] [PubMed] [Google Scholar]
- 31.Shibuya M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol. 2006;39:469–78. doi: 10.5483/bmbrep.2006.39.5.469. [DOI] [PubMed] [Google Scholar]
- 32.Singh SK, Bhusari S, Singh R, Saxena A, Mondhe D, Qazi GN. Effect of acetyl 11-keto beta-boswellic acid on metastatic growth factor responsible for angiogenesis. Vascul Pharmacol. 2007;46:333–7. doi: 10.1016/j.vph.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 33.Roodhart JM, Langenberg MH, Witteveen E, Voest EE. The molecular basis of class side effects due to treatment with inhibitors of the VEGF/VEGFR pathway. Curr Clin Pharmacol. 2008;3:132–43. doi: 10.2174/157488408784293705. [DOI] [PubMed] [Google Scholar]
- 34.Kanda S, Miyata Y, Kanetake H, Smithgall TE. Non-receptor protein-tyrosine kinases as molecular targets for antiangiogenic therapy. Int J Mol Med. 2007;20:113–21. [PubMed] [Google Scholar]
- 35.Chou MT, Wang J, Fujita DJ. Src kinase becomes preferentially associated with the VEGFR, KDR/Flk-1, following VEGF stimulation of vascular endothelial cells. BMC Biochem. 2002;3:32. doi: 10.1186/1471-2091-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ali N, Yoshizumi M, Fujita Y, Izawa Y, Kanematsu Y, Ishizawa K, et al. A novel Src kinase inhibitor, M475271, inhibits VEGF-induced human umbilical vein endothelial cell proliferation and migration. J Pharmacol Sci. 2005;98:130–41. doi: 10.1254/jphs.fp0040850. [DOI] [PubMed] [Google Scholar]
- 37.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–23. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 38.Matsuo M, Yamada S, Koizumi K, Sakurai H, Saiki I. Tumour-derived fibroblast growth factor-2 exerts lymphangiogenic effects through Akt/mTOR/p70S6kinase pathway in rat lymphatic endothelial cells. Eur J Cancer. 2007;43:1748–54. doi: 10.1016/j.ejca.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 39.Li W, Tan D, Zhang Z, Liang JJ, Brown RE. Activation of Akt-mTOR-p70S6K pathway in angiogenesis in hepatocellular carcinoma. Oncol Rep. 2008;20:713–9. [PubMed] [Google Scholar]
- 40.Verheul HM, Salumbides B, Van Erp K, Hammers H, Qian DZ, Sanni T, et al. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin Cancer Res. 2008;14:3589–97. doi: 10.1158/1078-0432.CCR-07-4306. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Maceira P, Mateo J. Silibinin inhibits hypoxia-inducible factor-1alpha and mTOR/p70S6K/4E-BP1 signalling pathway in human cervical and hepatoma cancer cells: implications for anticancer therapy. Oncogene. 2009;28:313–24. doi: 10.1038/onc.2008.398. [DOI] [PubMed] [Google Scholar]
- 42.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–64. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]