

Abstract
CCAAT/enhancer binding protein (C/EBP) transcription factors play essential roles in regulating an array of cellular processes, including differentiation, energy metabolism, and inflammation. In this report we demonstrate that both C/EBPα and C/EBPβ activate the promoter driving transcription of the tumor necrosis factor receptor 1 (TNFR1). TNFR1 is the major receptor for tumor necrosis factor (TNF), a critical cytokine mediator of the inflammatory response. Although the TNFR1 protein has been shown to be regulated through post-translational modifications, very little is known about the transcriptional regulation of the TNFR1 gene. Here we have identified a specific C/EBP binding site within the TNFR1 promoter, and shown that this site is required for both C/EBPα and C/EBPβ activation of the promoter in reporter gene assays. Furthermore, we show that both C/EBPα and C/EBPβ are bound to the TNFR1 promoter in cells using chromatin immunoprecipitation assays. Finally, we demonstrate that reducing the level of C/EBPα and C/EBPβ expression in cells using siRNA technology leads to decreased expression of the TNFR1 protein. These results suggest that the C/EBPα and C/EBPβ transcription factors enhance expression of the TNFR1 protein in cells. Given that TNF and C/EBPβ are known to activate each other's expression, C/EBPβ may greatly amplify the initial TNF signal through a positive auto-regulatory mechanism.
Keywords: Transcription factor, cytokine, gene regulation, inflammation, siRNA
1. Introduction
The tumor necrosis factor (TNF, formerly TNFα) cytokine is a key member of the TNF superfamily that activates expression of a wide range of genes crucial to the immune and inflammatory response such as chemokines, cytokines, and major histocompatibility complex (MHC) molecules (MacEwan, 2002). TNF, which is secreted mainly by macrophages (but also by lymphocytes, natural killer cells, and epithelial cells), can induce apoptosis in cells infected with viruses and other pathogens (Wallach et al., 1999). The importance of TNF anti-microbial activities has led a number of pathogens to evolve ways to evade the effects of this cytokine (Benedict and Ware, 2001). However, in addition to being a guardian of health, TNF can also be an agent of destruction. Since it is a key immune system modulator that has far-reaching effects, abnormal expression can cause significant damage. A delicate balance exists between beneficial immune stimulation versus pathogenesis. Several autoimmune and inflammatory diseases, including rheumatoid arthritis, have been associated with the effects of excessive TNF (G. Chen and Goeddel, 2002). Therefore, understanding the regulation of TNF and its signaling is essential for dissecting how TNF can either prevent, or induce, various diseases.
TNF mediates its effects by binding to its major receptor, TNFR1 (also known as p55 TNFR, TR60, and CD120a), located on the surface of most cells (Wallach et al., 1999). Binding of TNFR1 by TNF initiates a complex signaling cascade inside the cell that can lead to several different outcomes, depending on which proteins associate with the intracellular portion of the receptor. Recruitment of TRADD (TNFR-associated death domain protein) can activate NF-κB, leading to cell survival and release of inflammatory mediators; however, recruitment of TRADD can also induce caspase-dependent apoptosis (Bertazza and Mocellin, 2008). Alternatively, the death domain (DD) of TNFR1 can associate with RIP1 (receptor-interacting protein kinase 1) to signal cell necrosis (Zheng et al., 2006). What determines the major outcome of the TNFR1 signaling pathway (death versus survival) in various situations is still poorly understood. The results of TNFR1 signaling can be amplified by impacting nearby cells in a paracrine fashion.
Despite the critical role that TNFR1 plays in TNF mediated signaling, very little is known about the regulation of its promoter. It has been suggested by some investigators that the TNFR1 promoter is constitutively active, similar to the promoters of “housekeeping” genes. Although the TNFR1 is expressed in a variety of cell types, the cellular transcription factors and promoter sequences required for this activity have not been well defined. Our laboratory previously showed that activity of the TNFR1 promoter is dramatically decreased by an Epstein-Barr virus encoded protein, BZLF1, thereby helping the virus to evade the anti-viral effects of TNF (Morrison et al., 2004). Similarly, both murine and human cytomegalovirus decrease cell surface expression of TNFR1 (Baillie et al., 2003; Popkin and Virgin, 2003). In addition, many tumors cells do not express TNFR1. Therefore, mechanisms for increasing, or reducing, TNFR1 expression in some cell types must exist.
In this report, we have examined the potential role of C/EBPα and C/EBPβ in regulation of the TNFR1 promoter. The C/EBP family of transcription factors is expressed in a wide range of cell types and plays roles in many cellular processes. All C/EBP proteins have a conserved basic leucine zipper (bZIP) domain that allows them to form homo- or hetero-dimers and to bind a set of related DNA recognition sequences (Osada et al., 1996; Poli, 1998). Members of this family (including C/EBPα, C/EBPβ, and C/EBPδ) have important roles in regulation of lineage commitment, growth, differentiation, and immune response induction in specific cell types (Nerlov, 2007; Poli, 1998; Ramji and Foka, 2002). C/EBPα is highly expressed in adipose tissue, liver, intestine, lung, adrenal gland, peripheral-blood mononuclear cells, and placenta, and is important for adipocyte, monocyte, and granulocyte differentiation (Friedman, 2007; Poli, 1998) as well as being a key inhibitor of mitotic growth in many cell types (Schuster and Porse, 2006). C/EBPβ is also expressed in many tissues (including lung, liver, intestine, adipose tissue, spleen, kidney, and myelomonocytic cells) and is important for macrophage differentiation and inflammatory responses (Pham et al., 2007; Poli, 1998).
Here we report that both C/EBPα and C/EBPβ regulate the TNFR1 promoter. We show that the TNFR1 promoter contains a C/EBP binding motif located at position -88 to -80 (relative to the transcriptional start site) that efficiently binds to C/EBPα and C/EBPβ in vitro. Furthermore, we find that both C/EBPα and C/EBPβ activate the TNFR1 promoter in reporter gene assays in vivo, and this effect is greatly diminished following site-directed mutation of the C/EBP binding motif. C/EBPα and C/EBPβ can be co-immunoprecipitated with the TNFR1 promoter in ChIP assays, confirming that these transcription factors bind to the promoter when expressed at endogenous levels within the cell. Finally, we show that inhibition of C/EBP expression using siRNA technology reduces constitutive expression of TNFR1 protein. These results suggest that C/EBP proteins are important activators of TNFR1 expression.
2. Materials and methods
2.1 Cell Culture
HeLa cervical carcinoma cells and HEK-293 embryonic kidney cells were maintained in DMEM (Gibco, Carlsbad, CA, USA). HONE-1 clone 39, a nasopharyngeal carcinoma line (a gift from Dr. Ronald Glaser at Ohio State University), was maintained in RPMI 1640 (Gibco). The gastric carcinoma cell lines, AGS and AGS-B95.8, were both cultured in F12 (Gibco). The AGS-B95.8 line was constructed as previously described (Jones et al., 2007) and supplemented with 100μg/mL hygromycin B. A latently infected subline was used in these studies. All media was supplemented with 10% fetal bovine serum (Gemini Bio-Products, West Sacramento, CA, USA), 100 units/mL penicillin, and 100μg/mL streptomycin.
2.2 Plasmids
The TNFR1p-CAT plasmids have the TNFR1 promoter region from -619, -338 or -154 through +35 (relative to the transcriptional start site) inserted upstream of the chloramphenicol acetyltransferase (CAT) reporter gene (Morrison et al., 2004). Site-directed mutagenesis (using QuikChange XL Site-Directed Mutagenesis Kit, #200517 Stratagene, La Jolla, CA, USA) was performed to create the -154/+35 TNFR1p-CAT ΔC/EBP plasmid. C/EBPα and C/EBPβ expression plasmids were gifts from Dr. Ormond MacDougald at the University of Michigan. Each plasmid has the full-length mouse C/EBPα or C/EBPβ gene sequence inserted into a pcDNA3.1+ expression vector.
2.3 Chloramphenicol acetyltransferase (CAT) Assays
Cells were transiently transfected with the TNFR1p-CAT plasmid, along with combinations of C/EBPα, C/EBPβ, and empty vector (control) plasmids. After 48 or 72 hours, cells were harvested in Reporter Lysis Buffer (#E397A Promega, Madison, WI, USA) and subjected to freeze/thaw and centrifugation. The cell lysates were incubated at 37°C with acetyl coenzyme A and 14C-labeled chloramphenicol (Amersham Biosciences, Piscataway, NJ, USA), as described previously (Gorman et al., 1982). Activity of the TNFR1 promoter was measured by acetylation of chloramphenicol, and the percent acetylation was quantitated by thin layer chromatography followed by phosphorimager screening.
2.4 Electrophoretic Mobility Shift Assay (EMSA)
The Klenow fragment DNA polymerase I (Roche, Indianapolis, IN, USA) and α-32P dATP/dCTP (Amersham Biosciences) were used to label double-stranded, annealed DNA oligonucleotides for use in DNA/protein binding experiments. Oligonucleotide probes contained 100 base-pair (bp) and 50 bp sequences spanning the TNFR1 promoter (-529 to -419, -438 to -329, -348 to -239, -258 to -115, -164 to -114, -124 to -74, -84 to -34, -44 to +6, -4 to +46). Additional oligonucleotides consisted of a 20bp sequence containing a potential C/EBP site (underlined) in the TNFR1 promoter (TNFR1 -94 to -75: forward 5′-GAT CTC CCG CTG TTG CAA CAC TGC, reverse 5′-GAT CGC AGT GTT GCA ACA GCG GGA), or the same oligonucleotide with the C/EBP motif mutated (TNFR1-94 to -75 ΔC/EBP: forward 5′-GAT CTC CCG CTG CTG CGC CAC TGC, reverse 5′-GAT CGC AGT GGC GCA GCA GCG GGA). An oligonucleotide containing the consensus C/EBP binding sequence was also synthesized (C/EBP consensus: forward 5′-GAT CCT AGC TGC AGA TTG CGC AAT CTG CAG, reverse 5′-GAT CCT GCA GAT TGC GCA ATC TGC AGC TAG). The protein samples used in EMSAs were either nuclear protein extracts harvested from transfected cells, or in vitro translated protein (IVP). Protein samples (2μg nuclear extract or 2μL IVP protein) were incubated with binding buffer (10mM Hepes pH 7.9, 50mM KCl, 2.5mM MgCl2, 10% glycerol, 1μg BSA, 1mM dithiothreitol, 2μg poly(dI-dC)) for 5 minutes at room temperature before addition of radiolabeled probes (20,000 cpm/20μL reaction). After incubating for 20 minutes at room temperature the samples were loaded onto a 4% polyacrylamide gel and run in 0.5X Tris-borate-EDTA buffer at room temperature. Supershift experiments (using 2μg goat polyclonal C/EBPα [sc-9314 Santa Cruz Biotechnology, Santa Cruz, CA, USA] or mouse monoclonal C/EBPβ [Santa Cruz sc-7962] antibodies added to the protein/DNA complexes) were performed to confirm the identity of the binding protein.
2.5 Preparation of proteins for EMSA
To make in vitro translated proteins, C/EBPα and C/EBPβ plasmids were transcribed/translated with TNT T7 Quick Coupled Transcription/Translation System (Promega) according to the manufacturer's instructions. Reticulocyte protein with no DNA added was used as control for EMSA. To make nuclear extracts, cells were transfected with various combinations of C/EBPα, C/EBPβ, and empty vector (control) using Fugene 6 (Roche) for HeLa cells and Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for 293 cells. After 48 or 72 hours, 107 cells were washed twice with 1X PBS and harvested in 350μL buffer CE (10mM HEPES pH7.6, 60mM KCl, 1mM EDTA, 1mM dithiothreitol, 0.075% NP40, protease inhibitors). The samples were incubated on ice 10 minutes, vortexed, and then spun at 1500RPM for 10 minutes. The supernatant was discarded and the pellet resuspended in 50μL NE buffer (20mM Tris pH8.0, 420mM NaCl, 1.5mM MgCl2, 0.2mM EDTA, 25% glycerol, protease inhibitors). Before samples were mixed, salt concentration was adjusted to 400mM with 5M NaCl and an additional 50μL NE buffer was added. Samples were incubated on ice 10 minutes with occasional vortexing, and then spun at 13,000RPM at 4°C for 10 minutes. Supernatant was saved as nuclear extract and assayed for protein concentration.
2.6 siRNA Experiments
Cells were transiently transfected with siRNA against C/EBPα (Santa Cruz sc-37047), C/EBPβ (Santa Cruz sc-44251), or control siRNA (#AM4613 Ambion, Austin, TX, USA) using X-tremeGENE (Roche). Cells were transfected twice (on days 1 and 3) and harvested on day 5. Cells were harvested in buffers I and II (see below) for immunoblot assay.
2.7 Immunoblot Analysis
Cells were washed twice with phosphate buffered saline and resuspended in a 1:3 mixture of buffer I (5% sodium dodecyl sulfate [SDS], 0.15M Tris-HCl pH 6.8, 30% glycerol) and buffer II (25mM Tris-Cl pH8.3, 50mM NaCl, 0.5% NP-40, 0.5% deoxycholate, 0.1% SDS, and protease inhibitors). The cells were briefly sonicated then centrifuged, and the resulting supernatant was assayed for protein concentration. 5-20μg of protein was loaded in each lane of a 10% SDS polyacrylamide gel. After electrophoresis, protein was transferred to nitrocellulose. The membrane was rocked in blocking buffer (1X PBS, 5% milk, 0.1% Tween-20) for thirty minutes then incubated in primary antibody for 1 hour at room temperature or overnight at 4°C (monoclonal mouse anti-TNFR1 [Santa Cruz sc-8436], monoclonal rabbit anti-C/EBPα [#1704-1 Epitomics, Burlingame, CA, USA], polyclonal goat anti-C/EBPα [Santa Cruz sc-9314], monoclonal mouse anti-C/EBPβ [Santa Cruz sc-7962], or monoclonal mouse anti-β-actin [#A5441 Sigma, Saint Louis, MO, USA]). The membrane was washed in 1X PBS-0.1% Tween 20 for 20 minutes then incubated in secondary antibody for 1 hour at room temperature (goat anti-mouse Ig horseradish peroxidase conjugate [#31430 Thermo Scientific, Waltham, MA, USA], goat anti-rabbit Ig horseradish peroxidase conjugate [Promega W401B], donkey anti-goat horseradish peroxidase conjugate [Santa Cruz sc-2020]). The membrane was then washed and the results visualized using the Thermo Scientific ECL chemiluminescence kit (#32106).
2.8 Chromatin immunoprecipitation (ChIP) assay
ChIP was performed using the ChIP-IT Enzymatic Kit (#53006 Active Motif, Carlsbad, CA, USA) according to the manufacturer's instructions, with a few alterations. Fixation, harvest, and lysis of cells, followed by digestion of DNA (10 minutes at 37° with provided enzyme cocktail), were performed according to the Active Motif protocol. The digested DNA was pre-cleared with a 50% slurry of Protein A/G PLUS Agarose beads (Santa Cruz sc-2003) in Triton lysis buffer (50mM Tris-HCl pH 7.4, 150mM NaCl, 1% Triton X-100, 1% bovine serum albumin, 10μg/mL salmon sperm DNA), then incubated overnight at 4°C with 2μg of antibody (Epitomics rabbit anti-C/EBPα, Santa Cruz mouse anti-C/EBPβ, Santa Cruz normal rabbit IgG sc-2027, and Santa Cruz normal mouse IgG sc-2025). Beads were added to the samples for an additional 2 hour incubation at 4°C. The beads were then washed sequentially with low salt immune complex wash buffer, high salt immune complex wash buffer, LiCl immune complex wash buffer (recipes from Upstate ChIP Assay Kit #17-295, Billerica, MA, USA), and TE (two washes). The DNA/protein complexes were eluted from the beads, crosslinks reversed, protein degraded, and DNA purified according to Active Motif protocol. DNA was resuspended in 50μL (500μL for unprecipitated DNA control) and 4μL were used for each PCR reaction. PCR was performed using two sets of primers specific for the region of the TNFR1 promoter with the potential C/EBP binding site (TNFR1 -223 to -29: forward 5′-GAT TGG TGG GTT GGG GGC ACA, reverse 5′-ATT AAA GCA GAG AGG AGG GGA GAG A; TNFR1 -154 to -35: forward 5′-AGT TAA AGA ACG TTG GGC CTC CT, reverse 5′-GCA GAG AGG AGG GGA GAG AAG G), another region of the TNFR1 promoter further upstream as a negative control (TNFR1 -608 to -466: forward 5′-TTC CCA AGA AAG AGG GAG ACT AGG A, reverse 5′-CTG GGG TTC CTG TAA GGA TTT GTT C), and the cyclooxygenase-2 (COX-2) promoter as a positive control for C/EBPα (COX-2: forward 5′-GGT GGA ACT CGG GGA GGA GA, reverse 5′-GAC TGA AAA CCA AGC CCA TGT GAC). PCR products were run on a 2-2.5% agarose gel. After pre-clearing (before addition of antibody) a small amount of DNA was set aside to serve as an input control. This input DNA was kept at -80°C until reentering the protocol at the crosslink reversal step.
3. Results
3.1 Both C/EBPα and C/EBPβ activate the TNFR1 promoter
To determine if either C/EBPα and/or C/EBPβ enhance TNFR1 promoter activity, the human TNFR1 promoter sequences from -619 to +35, -338 to +35, or -154 to +35 (relative to the transcriptional start site) were inserted upstream of the chloramphenicol acetyltransferase (CAT) gene to create a group of reporter gene constructs (TNFR1p-CAT). HeLa cells were transfected with the various TNFR1p-CAT constructs and either vector control DNA or C/EBP expression vector DNA, and CAT activity was measured 48 to 72 hours later. As shown in Fig. 1A, co-transfection with a C/EBPα expression vector increased the activity of each of the three different TNFR1 promoter constructs. As shown in Fig. 1B, both the C/EBPβ and C/EBPα expression vector significantly enhanced the activity of the TNFR1p-CAT (-154 to +35) promoter construct, although the C/EBPβ vector activated the TNFR1p-CAT construct to a lesser extent (6-fold) than the C/EBPα vector (33-fold). The lesser effect of the C/EBPβ expression vector may reflect the fact that HeLa cells already have a high level of endogenous C/EBPβ expression. The combination of both C/EBPα and C/EBPβ produced only slightly more promoter activity than that produced by C/EBPα alone (data not shown). These results suggest that both C/EBPα and C/EBPβ positively regulate TNFR1 transcription, and that TNFR1 promoter sequences located between -154 and +35 are sufficient for this activation.
Fig. 1.

The TNFR1 promoter is responsive to C/EBPα and C/EBPβ. (A) TNFR1 promoter constructs (containing the regions of the TNFR1 promoter indicated linked to the CAT gene) were co-transfected into HeLa cells with control vector DNA or a C/EBPα expression vector. CAT activity was measured 2-3 days later. The fold increase in CAT activity induced by C/EBPα (relative to the vector control) for each promoter construct is shown. (B) The -154 to +35 TNFR1p-CAT construct was co-transfected with C/EBPα, C/EBPβ, or empty vector control into HeLa cells. The relative CAT activity produced by each condition is shown (activity of promoter plus vector control DNA is set at 1). All values in (A) and (B) are given as mean ± standard deviation of duplicate conditions. Three independent experiments showed similar results.
3.2 The TNFR1 promoter contains a C/EBP binding motif
C/EBP binding motifs can vary, but two accepted consensus sequences are T(T/G)NNG(C/T)AA(T/G) and (A/G)TTGCG(C/T)AA(C/T) (Osada et al., 1996). Inspection of the TNFR1 promoter sequence suggested the presence of a potential C/EBP binding motif, TGTTGCAAC, located from -88 to -80 (relative to the transcriptional start site). To determine if either C/EBPα or C/EBPβ binds to this motif in vitro, we performed electrophoretic mobility shift assays (EMSAs). C/EBPα and C/EBPβ were in vitro translated using reticulocyte lysate and incubated with a 32P –labeled DNA oligonucleotide probe containing the TNFR1 promoter sequence between -94 and -75. As shown in Fig. 2A, both C/EBPα and C/EBPβ bound to this probe. No other C/EBP binding sites were identified in the TNFR1 promoter using a series of oligonucleotide probes spanning -554 to +29 (data not shown). EMSA studies performed using nuclear extracts harvested from 293 cells transfected with either vector control or a C/EBPα expression plasmid confirmed that C/EBP alpha binds to the TNFR1 -94 to -75 sequence, in addition to a positive control probe containing a consensus C/EBP binding motif (Fig. 2B). These results indicate that the TNFR1 promoter contains one C/EBP binding site located between -94 and -75.
Fig. 2.
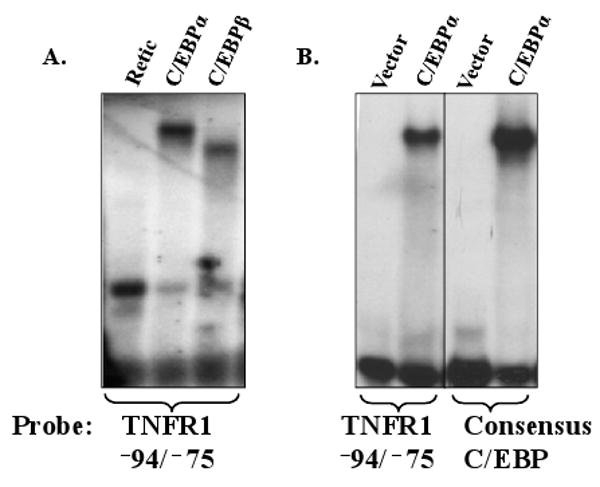
C/EBPα and C/EBPβ bind to the TNFR1 promoter. (A) In vitro translated C/EBPα or C/EBPβ, or reticulocyte lysate control, were incubated with a radiolabeled oligonucleotide probe containing TNFR1 promoter sequences between -94 and -75 in an EMSA. The positions of the bound C/EBPα or C/EBPβ proteins are shown. (B) Nuclear extracts from 293 cells transfected with a C/EBPα expression vector or empty vector control were incubated with the TNFR1 -94/-75 probe or a positive control C/EBP consensus binding sequence. All lanes were run on the same gel, with a vertical black line representing a lane deletion.
3.3 C/EBPα and C/EBPβ bind to the TGTTGCAAC sequence in the TNFR1 promoter
To further define the location of the C/EBP binding site in the TNFR1 promoter, we synthesized an oligonucleotide probe in which several base pairs of the most likely C/EBP binding motif were altered (sequences switched from TGTTGCAAC to TGCTGCGCC), and determined how this change affected C/EBPα and C/EBPβ binding to the promoter in EMSA assays. As shown in Fig. 3A, C/EBPα bound to the wildtype probe but not the mutant probe. Likewise, endogenous and transfected C/EBPβ bound the wildtype TNFR1 probe, but not the probe in which the most likely C/EBP motif was mutated (Fig. 3B). To further confirm that the complexes binding to the wildtype TNFR1 promoter probe contain C/EBPα and C/EBPβ, we performed binding assays in the presence of antibodies directed against C/EBPα and C/EBPβ (or control antibodies). The protein/DNA complexes binding to the promoter probe were supershifted using the appropriate antibody to C/EBPα or C/EBPβ, confirming the identity of the binding proteins. These results indicate that C/EBPα and C/EBPβ bind to the sequence TGTTGCAAC in the TNFR1 promoter.
Fig. 3.
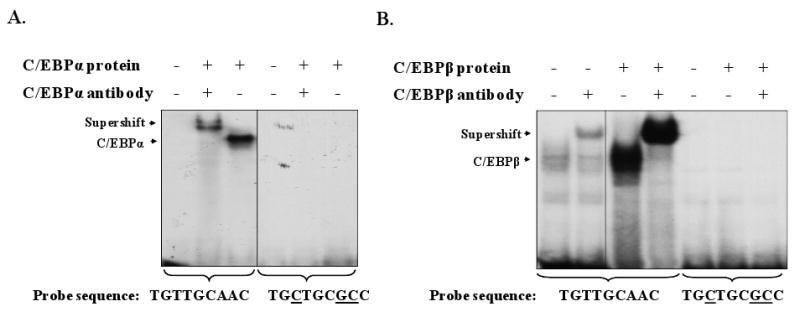
C/EBPα and C/EBPβ bind to the TGTTGCAAC sequence in the TNFR1 promoter. EMSA was performed using the wild-type −94 to −75 TNFR1 promoter probe, or the same probe with the probable C/EBP site mutated, and nuclear extracts from 293 cells transfected with vector control, C/EBPα (A), or C/EBPβ (B) expression vectors. In some conditions C/EBPα-specific or C/EBPβ-specific antibodies were added as indicated to confirm the identity of each protein in the binding complex. The low level C/EBPβ binding in lanes 1 and 2 of B represents binding of endogenous C/EBPβ present in the vector control nuclear extract. Parts A and B each represent one gel with lanes removed for clarity (marked by vertical black lines).
3.4 The C/EBP binding motif in the TNFR1 promoter is required for C/EBPα and C/EBPβ activation
C/EBPα and C/EBPβ could potentially activate the TNFR1 promoter through a direct binding mechanism, or indirectly through effects on other transcription factors. To determine if the C/EBP binding site in the TNFR1 promoter is required for C/EBPα or C/EBPβ activation, we performed site-directed mutagenesis of the -154/+35 TNFR1p-CAT construct to alter the C/EBP site from TGTTGCAAC to TGCTGCGCC (identical to the mutant oligonucleotide sequence used for the EMSA in Fig. 3). As shown in Fig. 4, mutation of the C/EBP motif dramatically reduced the ability of both C/EBPα (4A) and C/EBPβ (4B) to activate the TNFR1 promoter. These results indicate that the C/EBP binding site is required for C/EBPα and C/EBPβ to activate the TNFR1 promoter.
Fig. 4.

The C/EBP site in the TNFR1 promoter is required for activation by C/EBPα and C/EBPβ. HeLa cells were transfected with the wildtype TNFR1p-CAT construct (-154 to +35) or a mutant construct (TNFR1p-CAT ΔC/EBP) in which the C/EBP site was specifically altered, in the presence or absence of co-transfected C/EBPα (A), or C/EBPβ (B) expression vectors. The relative CAT activity in each condition is shown (activity of promoter plus vector control is set as 1). All values in (A) and (B) are given as mean ± standard deviation of duplicate conditions. Two independent experiments showed similar results.
3.5 Reduction of endogenous C/EBPα and C/EBPβ expression decreases TNFR1 expression
To determine if endogenous C/EBPα and/or C/EBPβ activity contributes to TNFR1 expression, AGS cells were transfected twice with siRNA directed against C/EBPα and/or C/EBPβ, or a control siRNA. Four days later, the level of TNFR1 expression was examined by immunoblot analysis (Fig. 5). AGS gastric carcinoma cells were chosen for this assay as they have been reported to express both C/EBPα and C/EBPβ (Huang et al., 2006) and have low level constitutive TNFR1 expression (Morrison et al., 2004). When C/EBPβ protein expression was reduced using the C/EBPβ siRNA, there was a concomitant decrease in TNFR1 protein, but not actin expression. Similar results were observed using siRNA against C/EBPα. Additional siRNA experiments using two other control siRNAs and different C/EBPα and C/EBPβ siRNAs had similar outcomes (data not shown), confirming that these results were not due to “off-target” effects of the siRNAs. Unexpectedly, however, the siRNA against C/EBPα also decreased the level of C/EBPβ expression (although it did not affect actin expression), allowing for the possibility that the decrease in TNFR1 protein in cells treated with the C/EBPα siRNA was primarily due to decreased C/EBPβ expression rather than decreased C/EBPα expression. This result also suggests that C/EBPα activates C/EBPβ expression in AGS cells. Members of the C/EBP family can affect each other's expression through direct and indirect mechanisms (C. Chen et al., 2005; Ramji and Foka, 2002); for example, during adipogenesis C/EBPβ activates expression from the C/EBPα promoter (Zuo et al., 2006). Our results suggest that cellular levels of C/EBPβ (and possibly C/EBPα) contribute to constitutive TNFR1 promoter activity.
Fig. 5.
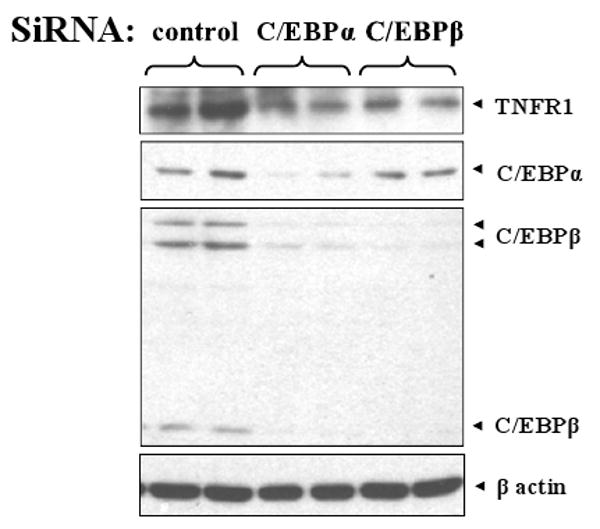
siRNAs directed against C/EBPα and C/EBPβ reduce TNFR1 protein expression. AGS cells were transfected with C/EBPα, C/EBPβ, or control siRNA as indicated. Four days later, immunoblots were performed to examine C/EBPβ, C/EBPα, TNFR1, and β-actin expression.
3.6 Endogenous C/EBPα and C/EBPβ are complexed to the TNFR1 promoter
To determine if C/EBPα and/or C/EBPβ are complexed with the TNFR1 promoter in vivo, we performed chromatin immunoprecipitation assays in AGS-B95.8 gastric carcinoma cells (Fig. 6C-D) as well as another cell line with substantial levels of both C/EBP proteins, HONE-1 nasopharyngeal carcinoma cells (6A-B). Formaldehyde-fixed cells were harvested and their chromatin digested to 200-1000 bp fragments (most fewer than 500bp). The resulting DNA/protein extract was then immunoprecipitated with antibodies directed against the C/EBPα or C/EBPβ proteins, or appropriate control antibodies. A small amount of digested chromatin (no antibody) was also saved as an input control. PCR was performed using primers sets corresponding to distinct regions of the TNFR1 promoter to determine if C/EBP protein was bound to the region of the promoter containing the C/EBP binding site. As shown in Figure 6, C/EBPα and C/EBPβ both bound to the promoter sequences spanning -223 to -29 and -154 to -35, which contain the site shown to bind to C/EBPα and C/EBPβ in vitro. In contrast, C/EBPα and C/EBPβ binding to TNFR1 promoter sequences located further upstream, at least 400 bp away from the C/EBP site (-608 to -466), was not observed. Similar results were observed in both HONE-1 cells and AGS-B95.8 cells. Binding of C/EBPα to the COX-2 promoter was used as a positive control in the HONE-1 cells (Jeong et al., 2007). These results indicate that both C/EBPα and C/EBPβ are complexed with the TNFR1 promoter in vivo.
Fig. 6.
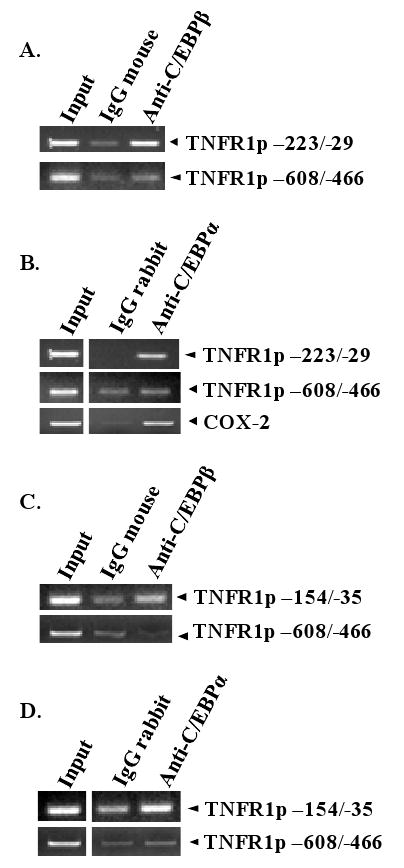
C/EBPα and C/EBPβ bind to the TNFR1 promoter in vivo. ChIP assays were performed in HONE-1 (A, B) and AGS-B95.8 (C, D) cells using control IgG mouse antibody, mouse anti-C/EBPβ antibody, control rabbit IgG antibody, or rabbit anti-C/EBPα antibody. PCR was performed using two sets of primers for the TNFR1 promoter encompassing the region containing the C/EBP binding motif (-223/-29 and -154/-35), a region of the TNFR1 promoter which does not bind to C/EBP (-608/-466), or a region of the COX-2 promoter known to bind to C/EBPα. Input DNA for each condition is shown in the first lane.
4. Discussion
Although TNF acquired its name from its demonstrated antitumor capabilities, it also is thought to contribute, in some cases, to cancer development and progression via its involvement in inflammation (Mocellin and Nitti, 2008). Despite its role in fighting infection, TNF can also contribute to the pathogenesis of septic shock, diabetes, arthritis, hepatitis, and other inflammatory and autoimmune diseases (G. Chen and Goeddel, 2002). In the last decade, a revolutionary advance in the treatment for rheumatoid arthritis has been the development of agents which block the actions of TNF (Lipsky et al., 2000). In the same vein, by better understanding how expression of the TNF receptor is regulated, additional therapies which block TNFR1 expression could potentially be developed. Several viruses already use this approach for their own survival: cytomegalovirus relocalizes the receptor from the cell surface (Baillie et al., 2003; Popkin and Virgin, 2003); herpes simplex virus 1 degrades TNFR1 mRNA (Liang and Roizman, 2006); and Epstein-Barr virus decreases expression from the TNFR1 promoter (Morrison et al., 2004). In this paper, we demonstrate that both C/EBPα and C/EBPβ directly bind to, and activate, the promoter driving expression of the TNFR1 gene. Furthermore, we show that C/EBPα and C/EBPβ contribute to constitutive expression of the TNFR1 protein in cells. Thus, therapies which inhibit the activity of either C/EBPα and/or C/EBPβ might be useful for inhibiting TNF signaling.
Despite a wealth of knowledge regarding post-translational modifications of the TNFR1 protein which affect its localization and function, much less is known about the regulation of the TNFR1 gene promoter. While it has been generally assumed that TNFR1 promoter expression is constitutive and not inducible, there is an increasing number of reports that suggest otherwise. For example, interferon-γ can induce TNFR1 mRNA and increase receptor expression (Kost et al., 1999). TNF and ultraviolet radiation have been shown to have a biphasic effect on TNFR1 mRNA and protein expression (Trefzer et al., 1993). Treatment with a demethylating agent enhanced expression of TNFR1 in a human melanoma cell line (Kaminski et al., 2004), suggesting that promoter methylation may be a mechanism for inhibiting its activity in some cell lines. In addition, a genetic polymorphism in which the guanine at position -329 (upstream of the transcription start site) is switched to a thymine results in decreased TNFR1 expression (Kim et al., 2008), suggesting that this base change may alter the binding of an undetermined cellular transcription factor.
In this report, we have demonstrated that the TNFR1 promoter contains a functionally important C/EBP binding site which contributes to the constitutive activity of the promoter, and may also increase expression of the TNFR1 protein during inflammation. Although it was previously suggested that the human TNFR1 promoter may contain binding sites for NFκB, Ap-2, and interferon-responsive transcription factors, to date these factors have not been shown to bind to, or regulate, the TNFR1 promoter, and these sites are not present in the murine promoter (Kemper and Wallach, 1993). Using EMSA and ChIP assays, we found that both C/EBPα and C/EBPβ bind the TNFR1 promoter, and that a sequence located between -88 and -80 (TGTTGCAAC) is necessary for this binding. In reporter gene assays, we showed that both C/EBPα and C/EBPβ can activate the TNFR1 promoter, and this activation requires the C/EBP binding site. Furthermore, by knocking-down either C/EBPα or C/EBPβ protein expression using siRNA, we confirmed that C/EBPβ (and possibly C/EBPα) expression contributes to constitutive TNFR1 protein expression in AGS gastric carcinoma cells. Furthermore, the C/EBP motif is conserved in the mouse TNFR1 promoter save for one base change (human: TGTTGCAAC, mouse: TCTTGCAAC) (Kemper and Wallach, 1993).
Our finding that C/EBPβ activates expression of the TNFR1 promoter, combined with a previous report indicating that C/EBPβ also binds to and activates the TNF promoter (Pope et al., 1994), suggests that C/EBPβ might be a particularly potent factor for activating TNF signaling during the inflammatory response. Interestingly, interferon-γ has been shown to increase the expression and activity of C/EBPβ (Salmenpera et al., 2003), suggesting a mechanism for the previously observed finding that interferon-γ activates TNFR1 expression (Kost et al., 1999). In addition, since TNF has been reported to induce C/EBPβ expression (Cardinaux et al., 2000) and increase its nuclear localization (Jain et al., 1999; Yin et al., 1996), it is likely that C/EBPβ may greatly amplify the initial TNF signal through a positive auto-regulatory mechanism.
Interestingly, it was recently reported that differentiation of human peripheral blood monocytes into macrophage or dendritic cells increases TNFR1 mRNA levels (Schling et al., 2006). In contrast, differentiation of colon epithelial cells decreases TNFR1 mRNA levels (Schling et al., 2006). Given that the abundance and activity of the C/EBP proteins is known to be involved in myeloid differentiation (Friedman, 2007), as well as the differentiation of other cell types (Nerlov, 2007), it is tempting to speculate that alterations in C/EBPα and/or C/EBPβ activity may regulate the expression of the TNF receptor during certain forms of differentiation.
Numerous post-translational modifications of C/EBP proteins, including phosphorylation, sumoylation, and ubiquitination, have been shown to regulate their activities. Many of these functional effects depend on the specific C/EBP protein, function, and cell type (Nerlov, 2008). For example, phosphorylation can repress or activate C/EBPα depending on the sites phosphorylated and the activity, be it lipogenic gene induction or granulopoiesis regulation (Khanna-Gupta, 2008). Phosphorylation and sumoylation can also affect subcellular or even subnuclear localization in the case of C/EBPβ (Nerlov, 2008). SUMO modification may also selectively change protein-protein interactions (Nerlov, 2008). Acetylation of C/EBP proteins can decrease DNA binding affinity, but an acetyl group at certain sites can also increase the transactivation potential of the protein (Nerlov, 2008). Ubiquitination in most cases targets C/EBP proteins for proteasomal degradation, though there are exceptions (Hasselgren, 2007). Any one of these alterations to the C/EBP proteins has the potential to modulate their effect on TNFR1 expression.
Intriguingly, the histone deacetylase (HDAC) inhibitors, suberoylanilide hydroxamic acid (SAHA) and trichostatin A (TSA), were recently shown to decrease TNFR1 mRNA and protein levels in two non-small cell lung carcinoma lines, thereby inhibiting TNFR1-mediated activation of the cell-survival protein NF-κB following TNF treatment (Imre et al., 2006). Although the mechanism for this effect is so far unknown, it is noteworthy that C/EBPα and C/EBPβ both interact with HDACs and their functions can be impacted by HDAC inhibitors. C/EBPα and C/EBPβ together with HDAC1 bind to the peroxisome proliferator-activated receptor beta (PPARβ) promoter in keratinocytes and induce histone deacetylation, decreasing promoter activity (Di-Poi et al., 2005). In pre-adipocytes, C/EBPβ associates with HDAC1 in a transcriptional repressor complex that promotes deacetylation of histone H4 on the C/EBPα promoter (Wiper-Bergeron et al., 2003). Both TSA and another HDAC inhibitor, valproic acid, targeted HDAC1 within the complex for degradation, stimulating C/EBPα expression (Wiper-Bergeron et al., 2003). HDACs also modify C/EBP proteins directly. For example, HDAC1 can deacetylate C/EBPβ in a pro-B cell line, increasing C/EBPβ's binding affinity for the Id-1 promoter (Xu et al., 2003). These observations together suggest that HDAC inhibitors could be working through one or both C/EBP proteins to affect TNFR1 expression. It is possible that HDAC inhibitors, by either inducing the degradation of HDACs complexed with C/EBP or preventing the deacetylation of C/EBP proteins (or both), could be used to modify C/EBP transcriptional activation of TNFR1.
Acknowledgments
This work was supported by National Institutes of Health grants R01-CA66519, P01-CA022443, and P01-CA019014.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baillie J, Sahlender DA, Sinclair JH. Human cytomegalovirus infection inhibits tumor necrosis factor alpha (TNF-alpha) signaling by targeting the 55-kilodalton TNF-alpha receptor. J Virol. 2003;77:7007–7016. doi: 10.1128/JVI.77.12.7007-7016.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict CA, Ware CF. Virus targeting of the tumor necrosis factor superfamily. Virology. 2001;289:1–5. doi: 10.1006/viro.2001.1109. [DOI] [PubMed] [Google Scholar]
- Bertazza L, Mocellin S. Tumor necrosis factor (TNF) biology and cell death. Front Biosci. 2008;13:2736–2743. doi: 10.2741/2881. [DOI] [PubMed] [Google Scholar]
- Cardinaux JR, Allaman I, Magistretti PJ. Pro-inflammatory cytokines induce the transcription factors C/EBPbeta and C/EBPdelta in astrocytes. Glia. 2000;29:91–97. [PubMed] [Google Scholar]
- Chen C, Dudenhausen E, Chen H, Pan YX, Gjymishka A, Kilberg MS. Amino-acid limitation induces transcription from the human C/EBPbeta gene via an enhancer activity located downstream of the protein coding sequence. Biochem J. 2005;391:649–658. doi: 10.1042/BJ20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Goeddel DV. TNF-R1 signaling: A beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- Di-Poi N, Desvergne B, Michalik L, Wahli W. Transcriptional repression of peroxisome proliferator-activated receptor beta/delta in murine keratinocytes by CCAAT/enhancer-binding proteins. J Biol Chem. 2005;280:38700–38710. doi: 10.1074/jbc.M507782200. [DOI] [PubMed] [Google Scholar]
- Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene. 2007;26:6816–6828. doi: 10.1038/sj.onc.1210764. [DOI] [PubMed] [Google Scholar]
- Gorman CM, Moffat LF, Howard BH. Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol Cell Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselgren PO. Ubiquitination, phosphorylation, and acetylation--triple threat in muscle wasting. J Cell Physiol. 2007;213:679–689. doi: 10.1002/jcp.21190. [DOI] [PubMed] [Google Scholar]
- Huang J, Liao G, Chen H, Wu FY, Hutt-Fletcher L, Hayward GS, et al. Contribution of C/EBP proteins to epstein-barr virus lytic gene expression and replication in epithelial cells. J Virol. 2006;80:1098–1109. doi: 10.1128/JVI.80.3.1098-1109.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imre G, Gekeler V, Leja A, Beckers T, Boehm M. Histone deacetylase inhibitors suppress the inducibility of nuclear factor-kappaB by tumor necrosis factor-alpha receptor-1 down-regulation. Cancer Res. 2006;66:5409–5418. doi: 10.1158/0008-5472.CAN-05-4225. [DOI] [PubMed] [Google Scholar]
- Jain R, Police S, Phelps K, Pekala PH. Tumour necrosis factor-alpha regulates expression of the CCAAT-enhancer-binding proteins (C/EBPs) alpha and beta and determines the occupation of the C/EBP site in the promoter of the insulin-responsive glucose-transporter gene in 3T3-L1 adipocytes. Biochem J. 1999;338(Pt 3):737–743. [PMC free article] [PubMed] [Google Scholar]
- Jeong HG, Pokharel YR, Han EH, Kang KW. Induction of cyclooxygenase-2 by ginsenoside rd via activation of CCAAT-enhancer binding proteins and cyclic AMP response binding protein. Biochem Biophys Res Commun. 2007;359:51–56. doi: 10.1016/j.bbrc.2007.05.034. [DOI] [PubMed] [Google Scholar]
- Jones RJ, Dickerson S, Bhende PM, Delecluse HJ, Kenney SC. Epstein-barr virus lytic infection induces retinoic acid-responsive genes through induction of a retinol-metabolizing enzyme, DHRS9. J Biol Chem. 2007;282:8317–8324. doi: 10.1074/jbc.M608667200. [DOI] [PubMed] [Google Scholar]
- Kaminski R, Kozar K, Niderla J, Grzela T, Wilczynski G, Skierski JS, et al. Demethylating agent 5-aza-2′-deoxycytidine enhances expression of TNFRI and promotes TNF-mediated apoptosis in vitro and in vivo. Oncol Rep. 2004;12:509–516. [PubMed] [Google Scholar]
- Kemper O, Wallach D. Cloning and partial characterization of the promoter for the human p55 tumor necrosis factor (TNF) receptor. Gene. 1993;134:209–216. doi: 10.1016/0378-1119(93)90095-k. [DOI] [PubMed] [Google Scholar]
- Khanna-Gupta A. Sumoylation and the function of CCAAT enhancer binding protein alpha (C/EBP alpha) Blood Cells Mol Dis. 2008;41:77–81. doi: 10.1016/j.bcmd.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Moon SM, Kim YS, Kim JJ, Ryu HJ, Kim YJ, et al. TNFR1 promoter - 329G/T polymorphism results in allele-specific repression of TNFR1 expression. Biochem Biophys Res Commun. 2008;368:395–401. doi: 10.1016/j.bbrc.2008.01.098. [DOI] [PubMed] [Google Scholar]
- Kost ER, Mutch DG, Herzog TJ. Interferon-gamma and tumor necrosis factor-alpha induce synergistic cytolytic effects in ovarian cancer cell lines-roles of the TR60 and TR80 tumor necrosis factor receptors. Gynecol Oncol. 1999;72:392–401. doi: 10.1006/gyno.1998.5257. [DOI] [PubMed] [Google Scholar]
- Liang L, Roizman B. Herpes simplex virus 1 precludes replenishment of the short-lived receptor of tumor necrosis factor alpha by virion host shutoff-dependent degradation of its mRNA. J Virol. 2006;80:7756–7759. doi: 10.1128/JVI.00587-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky PE, van der Heijde DM, Clair EW, Furst DE, Breedveld FC, Kalden JR, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. anti-tumor necrosis factor trial in rheumatoid arthritis with concomitant therapy study group. N Engl J Med. 2000;343:1594–1602. doi: 10.1056/NEJM200011303432202. [DOI] [PubMed] [Google Scholar]
- MacEwan DJ. TNF receptor subtype signalling: Differences and cellular consequences. Cell Signal. 2002;14:477–492. doi: 10.1016/s0898-6568(01)00262-5. [DOI] [PubMed] [Google Scholar]
- Mocellin S, Nitti D. TNF and cancer: The two sides of the coin. Front Biosci. 2008;13:2774–2783. doi: 10.2741/2884. [DOI] [PubMed] [Google Scholar]
- Morrison TE, Mauser A, Klingelhutz A, Kenney SC. Epstein-barr virus immediate-early protein BZLF1 inhibits tumor necrosis factor alpha-induced signaling and apoptosis by downregulating tumor necrosis factor receptor 1. J Virol. 2004;78:544–549. doi: 10.1128/JVI.78.1.544-549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlov C. The C/EBP family of transcription factors: A paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007;17:318–324. doi: 10.1016/j.tcb.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Nerlov C. C/EBPs: Recipients of extracellular signals through proteome modulation. Curr Opin Cell Biol. 2008;20:180–185. doi: 10.1016/j.ceb.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Osada S, Yamamoto H, Nishihara T, Imagawa M. DNA binding specificity of the CCAAT/enhancer-binding protein transcription factor family. J Biol Chem. 1996;271:3891–3896. doi: 10.1074/jbc.271.7.3891. [DOI] [PubMed] [Google Scholar]
- Pham TH, Langmann S, Schwarzfischer L, El Chartouni C, Lichtinger M, Klug M, et al. CCAAT enhancer-binding protein beta regulates constitutive gene expression during late stages of monocyte to macrophage differentiation. J Biol Chem. 2007;282:21924–21933. doi: 10.1074/jbc.M611618200. [DOI] [PubMed] [Google Scholar]
- Poli V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem. 1998;273:29279–29282. doi: 10.1074/jbc.273.45.29279. [DOI] [PubMed] [Google Scholar]
- Pope RM, Leutz A, Ness SA. C/EBP beta regulation of the tumor necrosis factor alpha gene. J Clin Invest. 1994;94:1449–1455. doi: 10.1172/JCI117482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popkin DL, Virgin HW., 4th Murine cytomegalovirus infection inhibits tumor necrosis factor alpha responses in primary macrophages. J Virol. 2003;77:10125–10130. doi: 10.1128/JVI.77.18.10125-10130.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramji DP, Foka P. CCAAT/enhancer-binding proteins: Structure, function and regulation. Biochem J. 2002;365:561–575. doi: 10.1042/BJ20020508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmenpera P, Hamalainen S, Hukkanen M, Kankuri E. Interferon-gamma induces C/EBP beta expression and activity through MEK/ERK and p38 in T84 colon epithelial cells. Am J Physiol Cell Physiol. 2003;284:C1133–9. doi: 10.1152/ajpcell.00293.2002. [DOI] [PubMed] [Google Scholar]
- Schling P, Rudolph C, Heimerl S, Fruth S, Schmitz G. Expression of tumor necrosis factor alpha and its receptors during cellular differentiation. Cytokine. 2006;33:239–245. doi: 10.1016/j.cyto.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Schuster MB, Porse BT. C/EBPalpha: A tumour suppressor in multiple tissues? Biochim Biophys Acta. 2006;1766:88–103. doi: 10.1016/j.bbcan.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Trefzer U, Brockhaus M, Lotscher H, Parlow F, Budnik A, Grewe M, et al. The 55-kD tumor necrosis factor receptor on human keratinocytes is regulated by tumor necrosis factor-alpha and by ultraviolet B radiation. J Clin Invest. 1993;92:462–470. doi: 10.1172/JCI116589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and fas signaling mechanisms. Annu Rev Immunol. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- Wiper-Bergeron N, Wu D, Pope L, Schild-Poulter C, Hache RJ. Stimulation of preadipocyte differentiation by steroid through targeting of an HDAC1 complex. EMBO J. 2003;22:2135–2145. doi: 10.1093/emboj/cdg218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Nie L, Kim SH, Sun XH. STAT5-induced id-1 transcription involves recruitment of HDAC1 and deacetylation of C/EBPbeta. EMBO J. 2003;22:893–904. doi: 10.1093/emboj/cdg094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M, Yang SQ, Lin HZ, Lane MD, Chatterjee S, Diehl AM. Tumor necrosis factor alpha promotes nuclear localization of cytokine-inducible CCAAT/enhancer binding protein isoforms in hepatocytes. J Biol Chem. 1996;271:17974–17978. doi: 10.1074/jbc.271.30.17974. [DOI] [PubMed] [Google Scholar]
- Zheng L, Bidere N, Staudt D, Cubre A, Orenstein J, Chan FK, et al. Competitive control of independent programs of tumor necrosis factor receptor-induced cell death by TRADD and RIP1. Mol Cell Biol. 2006;26:3505–3513. doi: 10.1128/MCB.26.9.3505-3513.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Qiang L, Farmer SR. Activation of CCAAT/enhancer-binding protein (C/EBP) alpha expression by C/EBP beta during adipogenesis requires a peroxisome proliferator-activated receptor-gamma-associated repression of HDAC1 at the C/ebp alpha gene promoter. J Biol Chem. 2006;281:7960–7967. doi: 10.1074/jbc.M510682200. [DOI] [PubMed] [Google Scholar]