

Abstract
A rhodium(I)-xylyl-BINAP catalyzed asymmetric [2 + 2 + 2] cycloaddition of achiral conjugated aryl ynamides with various diynes is described here. This asymmetric cycloaddition provides a series of structurally interesting chiral N,O-biaryls with excellent enantioselectivity along with a modest diastereoselectivity with respect to both C-C and C-N axial chirality.
Keywords: Aryl ynamides, asymmetric [2 + 2 + 2] cycloaddition, chiral N, O-biaryls, rhodium(I) catalyst and (S)-xylyl-BINAP, C-N axial chirality, and double asymmetric induction of non-point stereocenters
1. Introduction
In the last 15 years, ynamides have captured much interest from the synthetic community. An immense amount of efforts has led to a number of elegant synthetic methodologies to be developed adopting ynamides as a de novo functional group,1–4 and culminated in several total syntheses employing ynamides as a versatile building block.5,6 Among these efforts [Figure 1], both Witulski’s work on [2 + 2 + 2] cycloadditions of ynamides in the synthesis of indoles and carbazoles,7,8 and Rainier’s formal [2 + 2 + 2] cycloaddition9 inspired us10,11 to develop a new approach toward chiral N,O-biaryls through a [2 + 2 + 2] cycloaddition of conjugated aryl ynamides with diynes.12–14 This concept was envisioned at the heel of our success in the synthesis of conjugated aryl ynamides via Sonogashira cross-coupling.15 During our pursuit, Tanaka16,17 beautifully demonstrated the feasibility of an asymmetric [2 + 2 + 2] cycloaddition of ynamides in the synthesis of anilides with an axially chiral C-N bond.18–20
Figure 1.

Ynamide-[2 + 2 + 2] Cycloadditions.
Because our diastereoselective ynamide-[2 + 2 + 2] cycloaddition only resulted in modest selectivity through the use of chiral aryl ynamide 1 [Scheme 1],10 we began contemplating the possibility of pursuing an asymmetric cycloaddition employing achiral aryl ynamides 3 and a suitable chiral rhodium(I) complex.11 If successful, asymmetry could be induced at both axially chiral C-C biaryl bond [in blue] and C-N anilide bond [in red] in cycloadducts 5, thereby constituting a rare example of double asymmetric induction of non-point stereocenters by a single catalyst.12,20–22 Moreover, the coordinating ability of the achiral amido-carbonyl group could play a role in the asymmetric induction, thereby rendering this design an achiral-template directed asymmetric catalysis.23 We report here our development of an asymmetric ynamide-[2 + 2 + 2] cycloaddition.
Scheme 1.

An Asymmetric Ynamide-[2 + 2 + 2] Cycloaddition.
2. Results and Discussions
2.1. Establishing the Feasibility
In our earlier work, we were intrigued by the observation that while we could not improve the diastereoselectivity in our RhCl(Ph3P)3-catalyzed [2 + 2 + 2] cycloadditions of chiral ynamide 1, we could readily diminish the ratio from 4:1 by using an external phosphine ligand such as BINAP [Scheme 2]. To account for the modest diastereoselectivity, we had proposed pro-M- and pro-P-TS models, which consist of the key bidentate coordination [in green] of Rh(III) from both the oxazolidinone carbonyl oxygen and the anisol oxygen atom. We concluded that neither really possesses a distinct advantage over the other except with pro-P-TS suffering from a remote steric interaction [red arrows] between the cyclopentyl and the phenyl group on the oxazolidinone ring. Based on these same models, the above finding suggests that an external chiral ligand [in blue] exerts a stronger influence on this cycloaddition than the stereocenter on the chiral ynamide, thereby implying the possibility of an asymmetric cycloaddition manifold employing achiral ynamides.
Scheme 2.

Recognition of An External Ligand Controlled Process.
Toward this goal, we synthesized achiral conjugated aryl ynamides 6 and 724–26 containing a sulfonamide and an acyclic amide, respectively, as shown in Scheme 3. However, to our disappointment, after screening several catalytic systems, we were unable to even find the desired cycloaddition products 8 and 9 let alone analyzing possible enantioselectivity. Recognizing that ynamides with the amido group cyclic in nature may be better suited for the cycloaddition, we proceeded to prepare aryl ynamide 10a containing the 2-oxazolidinone ring [Scheme 4].
Scheme 3.

Cycloadditions of Ynamides with an Acyclic Amido-Motif.
Scheme 4.

Cycloadditions of Ynamides with a Cyclic Amido-Motif.
After screening again conditions such as Rh(PPh3)3Cl/AgSbF6, [Rh(CO)2Cl]2, [Rh(cod)Cl]2/AgSbF6, and [Rh(cod)2]BF4 activated with H2, we found that the usage of 10 mol% [Rh(cod)2]BF4 and 10 mol% (S)-xylyl-BINAP at 85 °C in ClCH2CH2Cl with 4Å MS provided the best outcome when reacting 10a with diyne 11. While the resulting diastereomeric N,O-biaryls 12-M,p and 12-P,p27 were separable by TLC, they rapidly inter-converted to the enantiomer of each other: 12-M,p led to 12-M,m with 12-P,p yielding 12-P,m via C-N bond rotation [in red]. Even after a clean and facile separation on a silica gel column, NMR analysis revealed that the two diastereomers of 12 have already scrambled likely during the removal of the solvent under reduced pressure even without heating. Thus, this complication prevented us from meaningfully determining dr and ee.
Ultimately, cycloadditions of achiral conjugated aryl ynamides 10b and 10c allowed us to analyze possible enantioselectivity. As shown in Scheme 5, N,O-biaryl atropisomers 14-M,p and 14-P,p were attained in 95% yield from 10b when reacting with diyne 13a. Although potential diastereomers of 14 due to restricted C-N bond rotation were detectable in proton NMR, they were not separable physically. Consequently, we were able to cleanly assess the enantiomeric excess using Chiral HPLC.
Scheme 5.

An Asymmetric Ynamide-[2 + 2 + 2] Cycloaddition.<br>[a] The ee was determined using chiral HPLC [CHIRALCEL OD-H; Size: 250 × 4.6mm (L × I.D.); eluent: i-PrOH in hexanes].
Furthermore, aryl ynamide 10c containing the 6-membered 2-oxazinone ring also led to N,O-biaryls 15 and 16 when reacting with diynes 13a and 13b, respectively. There was even less complication in these two reactions, as only a single diastereomer was detectable in 1H NMR presumably due to rapid free rotation at the C-N bond. Thus, we were able to concisely calibrate ee values for both 15 and 16, which are lower than that of 14. It is noteworthy that since BINAP had given lower ee and yield than xylyl-BINAP, and with Tanaka’s success using xylyl-BINAP, we did not screen beyond these two chiral bidentate phosphines.
2.2. Syntheses of Chiral N,O-Biaryls
Having established an asymmetric protocol, a range of chiral N,O-biaryls could be prepared as shown in Figure 1. Reactions of aryl ynamide 10a with diyne 11 led to the respective diastereomeric N,O-biaryl isomers 17 and 18 in good yields. Notably, we observed a diastereoselectivity as high as 8:1 in favor of the isomer 17-M,p, which also possesses an enantiomeric excess of 95%, while the minor isomer 18-P,p has a 54% ee. The usage of (R)-xylyl-BINAP led to chiral N,O-biaryls ent-17 and ent-18 but in slightly lower ee and dr. Moreover, reactions of ynamide 10b containing the 5-membered 2-oxazolidinone ring led to N,O-biaryls generally with higher ee for both [P,p] and [M,p] diastereomers. For example, while diastereomeric isomers 23-M,p and 24-P,p were attained with a dr of 6:1, both diastereomers possess 99% ee. Finally, while the ratio dropped for diastereomeric N,O-biaryls 27 and 28, as well as for 29 and 30, which all contain a naphthyl ring, their respective enantioselectivity remained high.
2.3. Assignment of Absolute Configurations
The minor diastereomer 18-P,p is crystalline, and its absolute configuration could be readily resolved through X-ray structure of a single crystal [Figure 3].28 However, assignments of the major isomer were not as trivial. As shown in Scheme 6, absolute configurations of major isomers 17-M,p containing the 6-membered 2-oxazinone ring, and 23-M,p containing the 5-membered 2-oxazolidinone ring, had to be assigned via X-ray structures of their respective camphor-sulfonyl derivatives 32-M,p and 34-M,p prepared in two steps. X-Ray structures of 32-M,p and 34-M,p are shown in Figure 4. Correlations of aromatic protons on the anisyl ring allow for the assignment of all other isomeric N,O-biaryls.
Figure 3.
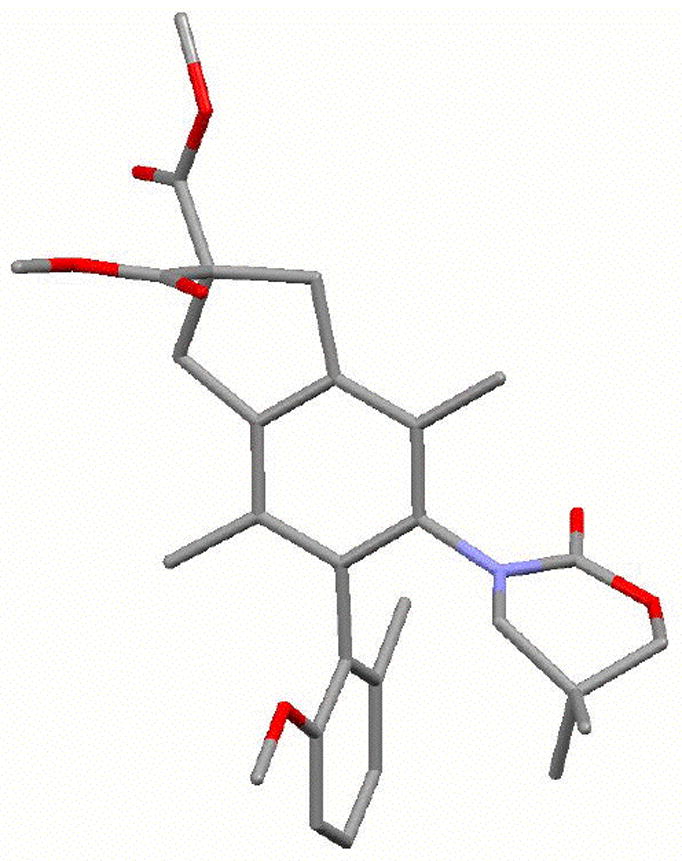
X-Ray Structures Minor Atropisomer 18-P,p.
Scheme 6.

Syntheses of 32-M,p and 34-M,p.
Figure 4.
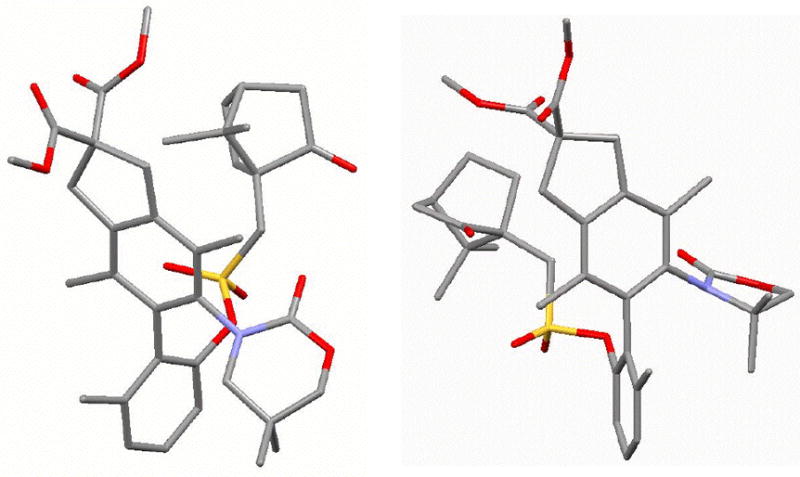
X-Ray Structures: 18-P,p 32-M,p [left] and 34-M,p [right].
2.4. Synthesis of a Chiral Amino-Biaryl
To demonstrate that these chiral N,O-biaryls can be useful in the venue of designing new chiral N,O-biaryl ligands, we pursue the follow asymmetric cycloaddition, As shown in Scheme 7, cycloaddition of ynamide 35 containing the 2-oxazolidinone ring with gem-diphenyl substitutions11,25 with diyne 36 gave cycloadducts 37-M,p and 38-P,p in 1:1 ratio with 82% ee for each diastereomer. We took 37-M,p on and removed the achiral 2-oxazolidinone ring via hydrogenations to afford aniline 39-M. Although we were unable to clearly discern the final enantiomeric access of 39-M through chiral HPLC, with the C-C bond rotational barrier being 37.7 kcal mol−1 [PM3 calculations], we believe there should be very little racemization during the hydrogenation at rt.
Scheme 7.

Synthesis of Chiral Amino-Biaryl 38-M.
2.5. Equilibration Studies
We were fascinated with the unique structural feature of these de novo N,O-biaryls. Spartan™ B3LYP/6-31G* calculations revealed a ΔE of 1.11 Kcal mol−1 in favor of the minor isomer 18, thereby implying that the observed selectivity for 17 is kinetic. In addition, B3LYP/6-31G* calculations provided Eact of 34.0 Kcal mol−1 and 94.4 Kcal mol−1, respectively, for the axially chiral C-N and C-C bonds. These calculations would suggest that any thermal equilibrations would first lead to epimerization through the C-N rotation. That is the equilibration between 17-M,p and ent-18-M,m, or between 18-P,p and ent-17-P,m [see Scheme 8], should occur faster at lower temperature than that between 17-M,p and 18-P,p, or between ent-18-M,m and ent-17-P,m. To confirm these assertions, we carried out following equilibration studies.
Scheme 8.

Thermal Equilibration and Rotational Barriers.
As shown in Table 1, heating pure biaryl 17-M,p with a 95% ee at 85 °C in toluene-d8 for 24 h [the original reaction temperature] did not result in any epimerization or loss of optical integrity. At 120 °C, epimerization occurred and led to a significant loss of 17-M,p and an increase in the amount of ent-18-M,m. However, there was no loss of enantiomeric access [94% ee] for both 17-M,p and ent-18-M,m, thereby suggesting that at 120 °C, the epimerization was occurring solely through rotation of the axially chiral C-N bond [assuming enantiomers in general possess the same rate of epimerization in first order]. At 165 °C and 200 °C, while 17-M,p continued to epimerize to ent-18-M,m with the ratio gravitating toward 23:77, there was a noticeable loss in the ee for both 17-M,p and ent-18-M,m. Thus, at these high temperatures, epimerization was taking place through free rotations at both C-N and C-C bonds, which should then lead to racemization.
Table 1.
Heating of Pure 17-M,p Diastereomer
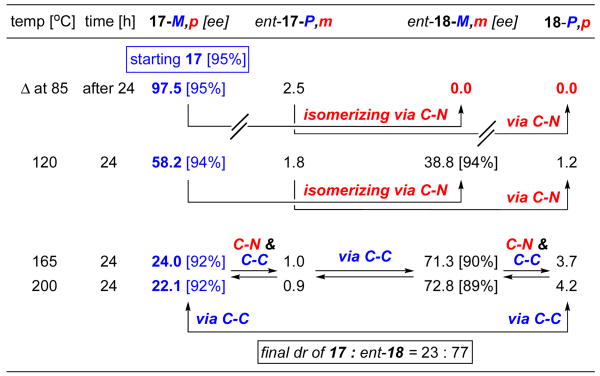
|
Equilibrations of pure ent-17-P,m with a 86% ee led to a very similar outcome and likewise with equilibrations of 18-P,p even starting at a 52% ee [Table 2]. It is noteworthy that all the equilibration studies led to a final resting thermodynamic ratio of 22:78 for 17:18. This appears to be the thermodynamic ratio, which is consistent with the calculation in which isomer 18 is favored by 1.11 kcal mol−1.
Table 2.
Heating of Pure ent-17-P,m and 18-P,p Diastereomers.
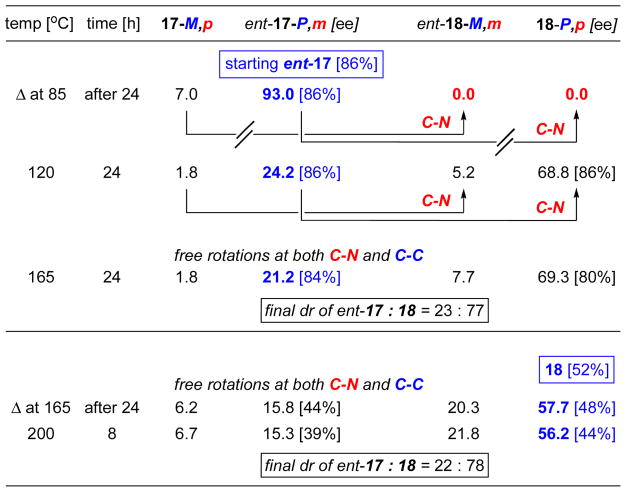
|
2.6. Mechanistic Considerations
Given the stereochemical outcome, we proposed a mechanistic model that could provide a unified rationale for the observed asymmetric inductions. As shown in Scheme 9, with the rhodio-cyclopentadiene intermediate complexed to (S)-xylyl-BINAP, a respective ynamide could approach the metal more favorably as shown in complex-A1 with the anisole ring sliding into the less hindered space near Ar2 with a smaller amido group assuming the relatively more crowded space next to Ar1. This binding orientation would be the opposite in complex-B1 and would lead to a less favorable fit in the two respective “binding pockets.”
Scheme 9.

A Proposed Asymmetric Model.
With complex-A1 in hand, a key bidentate chelation of the Rh metal via both the anisyl OMe and the carbonyl group could occur to assist the complexation of the ynamide unto the metal center. The ensuing cycloaddition, in a stepwise or Diels-Alder manner, would lead to the major diastereomer [M,p]. Because the enantiomer of the major isomer would come from complex-B1, which is not favored in these reactions, enantiomeric access is high in almost all cases for atropisomer [M,p].
If the bidentate chelation fails due to the slipping of the weaker coordination from the anisole oxygen atom through a C-C rotation, one would obtain complex-A2, which would give the observed minor diastereomer [P,p] after the cycloaddition. On the other hand, if the C-N bond in A1 rotates and one loses the stronger coordination from the carbonyl oxygen, complex-A3 would be present to give corresponding the [M,m] isomer, which is the enantiomer of the minor diastereomer. Since the carbonyl oxygen in general possesses better coordinating ability than a phenolic ethereal oxygen, it is reasonable to see much less formation of [M,m], thereby rendering [P,p] as the major enantiomer for the minor diastereomer.
Finally, the importance of the carbonyl oxygen in this asymmetric cycloaddition can also further underscored from the fact that 2-oxazolidinone is better chelator than 2-oxazinone. Consequently, ynamides substituted with 2-oxazolidinone gave higher ee. Overall, the current mechanistic model provides another excellent example for showcasing the versatility of enantioselective catalysis through an asymmetric template that employs an achiral auxiliary.23
3. Conclusion
We have described here a rhodium(I)-xylyl-BINAP catalyzed asymmetric [2 + 2 + 2] cycloaddition of achiral conjugated aryl ynamides with various diynes. This asymmetric cycloaddition represents a rare example of double asymmetric induction of non-point stereocenters by a single catalyst, and provides a series of structurally interesting chiral N,O-biaryls with excellent enantioselectivity in most cases along with a modest diastereomeric selectivity with respect to both C-C and C-N axial chirality.
4. Experimental Section
All reactions were performed in flame-dried glassware under a nitrogen atmosphere. Solvents were distilled prior to use. Reagents were used as purchased (Aldrich, Acros), except where noted. Chromatographic separations were performed using Bodman 60 Å SiO2. 1H and 13C NMR spectra were obtained on Varian VI-300, VI-400, and VI-500 spectrometers using CDCl3 (except where noted) with TMS or residual CHCl3 in the solvent as standard. Melting points were determined using a Laboratory Devices MEL-TEMP and are uncorrected/calibrated. Infrared spectra were obtained using NaCl plates on a Bruker Equinox 55/S FT–IR Spectrophotometer, and relative intensities are expressed qualitatively as s (strong), m (medium), and w (weak). TLC analysis was performed using Aldrich 254 nm polyester-backed plates (60 Å, 250 μm) and visualized using UV and a suitable chemical stain. Low-resolution mass spectra were obtained using an Agilent-1100-HPLC/MSD and can be either APCI or ESI, or an IonSpec HiRes-MALDI FT-Mass Spectrometer. High-resolution mass spectral analyses were performed at University of Wisconsin Mass Spectrometry Laboratories. All spectral data obtained for new compounds are reported.
4.1. A General Procedure for the Asymmetric Ynamide-[2 + 2+ 2] Cycloaddition
To a solution of [Rh(cod)2]BF4 (10 mol%) and (S)-xylyl-BINAP (10 mol%) in anhyd ClCH2CH2Cl (5.0 mM) was added 4Å Molecular Sieves in a sealed tube. The mixture was stirred at rt for 10 min before a respective ynamide (1.00 mmol) and diyne (2.00 mmol) were added. The solution was heated to 85°C and followed by LCMS. After the reaction was complete, the solution was cooled to RT and filtered through a short pad of silica gel. Elution with EtOAc/hexanes (1:1) followed by concentration in vacuo afforded a crude mixture of diastereomers. Separation and purification of the resulting crude residue via silica gel flash column chromatography (gradient eluent: EtOAc in hexanes) afforded the desired N,O-biaryl diastereomers. Diastereomeric ratios were found in crude 1H NMR, and enantiomeric excess of each diastereomer was determined via chiral HPLC [CHIRALCEL OD-H; Size: 250 × 4.6mm (L × I.D.); eluent: iso-propyl alcohol in Hexanes].
4.1.1. Establishing the Feasibility
Chiral Biaryl 12-Major
Rf = 0.52 [20% EtOAc in chloroform]; thick oil; 1H NMR (500 MHz, CDCl3) δ 1.81 (s, 3H), 1.96 (s, 3H), 2.16 (s, 3H), 3.35 (td, J = 8.9, 4.6 Hz, 1H), 3.54–3.59 (m, 2H), 3.61–3.74 (m, 4H), 3.68 (s, 3H), 3.792 (s, 3H), 3.797 (s, 3H), 4.22 (td, J = 8.6, 4.6 Hz, 1H), 6.77 (d, J = 8.5 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 15.0, 16.4, 20.0, 40.66, 40.68, 46.9, 53.36, 53.43, 55.6, 59.4, 62.9, 107.7, 123.4, 126.8, 128.6, 129.9, 131.1, 133.2, 136.2, 139.3, 139.4, 139.7, 156.6, 156.8, 172.4, 172.8; IR (film) cm−1 2946w, 2354w, 1730s, 1688s, 1461m, 1439m, 1235s, 1183m; mass spectrum (APCI): m/e (% relative intensity) 468 (100) (M + H)+; 12-Minor. Rf = 0.52 [20% EtOAc in chloroform]; thick oil; 1H NMR (500 MHz, CDCl3) δ 1.88 (s, 3H), 1.93 (s, 3H), 2.17 (s, 3H), 3.01 (td, J = 8.6, 4.7 Hz, 1H), 3.48 (q, J = 8.8 Hz, 1H), 3.54–3.75 (m, 5H), 3.67 (m, 3H), 3.78 (s, 3H), 3.81 (s, 3H), 4.18 (td, J = 8.7, 4.7 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 6.85 (d, J = 7.2 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 15.0, 16.4, 19.9, 30.0, 40.7, 46.3, 53.4, 55.9, 59.4, 62.3, 108.5, 122.1, 126.3, 126.6, 128.8, 130.06, 130.08, 131.1, 135.6, 139.5, 139.8, 156.4, 157.8, 172.2, 173.0 [missing one carbon due to overlap]; IR (film) cm−1 2896w, 2344w, 1724s, 1679s, 1443m, 1425m, 1245s, 1190m; mass spectrum (APCI): m/e (% relative intensity) 468 (100) (M + H)+; HRMS of the isomeric mixture (ESI, m/e) calcd for C26H30NO7 468.2017, found 468.2013.
Chiral Biaryl 14-M
Rotamers observed on the NMR timescale; Rf = 0.34 [60% EtOAc in hexanes]; pale solid; mp 185–188 °C; [α]D20 = − 16.7 (c 2.14, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.53 (s, 3H), 0.73 (s, 3H), 1.25 (s, 3H), 1.26 (s, 3H), 2.07 (s, 3H), 2.17 (s, 3H), 3.66 (d, J = 8.4 Hz, 1H), 3.68 (s, 6H), 3.75 (d, J = 8.4 Hz, 1H), 3.97 (d, J = 8.0 Hz, 1H), 3.99 (d, J = 8.0 Hz, 1H), 5.09–5.20 (m, 8H), 6.73 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 8.4 Hz, 1H), 6.85 (d, J = 6.8 Hz, 1H), 6.87 (d, J = 7.6 Hz, 1H), 7.13 (s, 2H), 7.15 (s, 1H), 7.19 (s, 1H), 7.22 (t, J = 8.0 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 20.2, 21.1, 23.5, 23.8, 24.9, 55.3, 55.6, 62.2, 62.5, 73.5, 73.6, 75.0, 75.3, 107.7, 107.8, 122.69, 122.72, 124.5, 125.0, 125.2, 125.5, 127.9, 128.2, 128.7, 128.9, 133.8, 134.1, 137.4, 137.5, 138.0, 139.76, 139.83, 139.91, 139.95, 157.3, 157.6, 158.4 [missing four carbons due to overlap]; IR (film) cm−1 2935w, 1746s, 1466m, 1389s, 1259s, 1068s, 1029s, 734s; mass spectrum (APCI): m/e (% relative intensity) 354 (100) (M + H)+; HRMS (ESI, m/e) calcd for C21H23NO4 353.1622, found 353.1626.; HPLC (80:20 hexane/2-propanol): tr = major 15.6 min, and minor 12.0 min.
Chiral Biaryl 15-M
Rf = 0.10 [50% EtOAc in hexanes]; pale solid; mp 96–100 °C; [α]D20 = − 27.5 (c 2.0, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 0.58 (broad s, 3H), 0.92–0.96 (broad m, 3H), 2.09 (s, 3H), 3.02–3.09 (broad m, 2H), 3.62–3.70 (broad m, 1H), 3.70 (s, 3H), 3.84–3.87 (broad m, 1H), 5.12 (s, 2H), 5.16 (s, 2H), 6.78 (d, J = 8.5 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 7.04 (s, 1H), 7.23 (t, J = 8.0 Hz, 1H), 7.27 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 20.8, 22.0, 22.8, 29.2, 31.1, 55.9, 59.9, 73.9, 76.3, 108.0, 122.0, 123.2, 124.7, 127.4, 129.5, 135.6, 139.4, 140.7, 140.9, 141.5, 152.3, 156.8; IR (film) cm−1 2959w, 2927w, 2361w, 2249w, 1735s, 1698s, 1476m, 1356m; mass spectrum (APCI): m/e (% relative intensity) 368 (100) (M + H)+.
Chiral Biaryl 16-M
Rf = 0.38 [60% EtOAc in hexanes]; pale solid; mp 74–80 °C; [α]D20 = − 25.3 (c 2.16, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.56 (broad s, 3H), 0.98 (broad m, 3H), 2.09 (s, 3H), 2.12 (quintet, J = 7.6 Hz, 2H), 2.89–3.08 (broad m, 6H), 3.65 (broad m, 1H), 3.69 (s, 3H), 3.86–3.87 (broad m, 1H), 6.76 (d, J = 8.4 Hz, 1H), 6.87 (d, J = 7.6 Hz, 1H), 7.01 (s, 1H), 7.20 (t, J = 8.0 Hz, 1H), 7.22 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 20.4, 21.8, 22.9, 25.8, 29.0, 32.8, 33.0, 55.6, 59.9, 76.4, 107.9, 123.1, 124.6, 127.4, 128.0, 128.5, 133.6, 139.3, 140.5, 144.2, 145.0, 152.7, 156.9; IR (film) cm−1 2959w, 2839w, 1791s, 1473s, 1371m, 1260s, 1200s, 1148m, 1057m; mass spectrum (APCI): m/e (% relative intensity) 366 (100) (M + H)+.
4.1.2. Synthesis Chiral N,O-Biaryls
Chiral Biaryl 17-M,p
Rf = 0.28 [60% EtOAc in hexanes]; pale solid; mp 111–113 °C; [α]D20 = − 46.7 (c 1.0, CH2Cl2); ent-17-P,m: [α]D20 = + 39.5 (c 0.72, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 0.53 (s, 3H), 1.01 (s, 3H), 1.85 (s, 3H), 1.91 (s, 3H), 2.15 (s, 3H), 2.57 (d, J = 11.0 Hz, 1H), 2.82 (dd, J = 11.2, 1.2 Hz, 1H), 3.34 (d, J = 10.5 Hz, 1H), 3.59 (ABq, Δν = 19.7 Hz, J = 17.0 Hz, 2H), 3.65 (ABq, Δν = 18.2 Hz, J = 17.0 Hz, 2H), 3.67 (s, 3H), 3.70 (dd, J = 10.5, 1.0 Hz, 1H), 3.78 (s, 3H), 3.80 (s, 3H), 6.79 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 7.5 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 14.9, 16.5, 20.0, 22.4, 23.2, 29.2, 40.7, 40.8, 53.3, 53.4, 55.9, 58.7, 59.4, 75.4, 108.3, 122.0, 126.7, 128.7, 129.6, 131.2, 134.7, 137.7, 137.9, 138.9, 139.2, 151.6, 158.3, 172.2, 173.1; IR (Neat) cm−1 2958w, 2360m, 2342m, 1734m, 1696m, 1433w, 1265s, 1109m, 734s; mass spectrum (APCI): m/e (% relative intensity) 510 (100) (M + H)+; HRMS (ESI, m/e) calcd for C29H35NO7 510.2487, found 510.2482; HPLC (95:5 hexane/2-propanol): tr = major 11.4 min, and minor 14.7 min.
Chiral Biaryl 18-P,p
Rf = 0.35 [60% EtOAc in hexanes]; pale solid; mp 187–190 °C; [α]D20 = + 5.9 (c 2.40, CH2Cl2); ent-18-M,m: [α]D20 = − 3.2 (c 0.12, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.51 (s, 3H), 0.95 (s, 3H), 1.78 (s, 3H), 2.01 (s, 3H), 2.15 (s, 3H), 2.92 (d, J = 11.6 Hz, 1H), 2.99 (dd, J = 11.8, 1.6 Hz, 1H), 3.47 (dd, J = 10.4, 1.6 Hz, 1H), 3.65 (ABq, Δν = 71.6 Hz, J = 16.8 Hz, 2H), 3.66 (ABq, Δν = 24.1 Hz, J = 17.2 Hz, 2H), 3.67 (s, 3H), 3.789 (s, 3H), 3.795 (s, 3H), 3.80 (d, J = 10.5 Hz, 1H), 6.75 (d, J = 8.0 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 7.21 (t, J = 8.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 14.9, 16.4, 20.2, 21.9, 23.2, 29.1, 40.7, 40.8, 53.3, 53.4, 55.3, 58.8, 59.4, 76.0, 107.7, 123.5, 126.8, 128.5, 129.2, 131.4, 135.1, 138.0, 138.7, 139.2, 140.5, 152.1, 156.7, 172.4, 172.9; IR (Neat) cm−1 2957w, 2360w, 1734s, 1697s, 1467m, 1427m, 1251s, 1172s, 1061s; mass spectrum (APCI): m/e (% relative intensity) 510 (100) (M + H)+; HRMS (ESI, m/e) calcd for C29H35NO7 510.2487, found 510.2494.; HPLC (95:5 hexane/2-propanol): tr = major 40.9 min, and minor 40.0 min.
19-M,p
Rf = 0.34 [60% EtOAc in hexanes]; pale solid; mp 80–83 °C; [α]D20 = − 65.1 (c 0.723, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.54 (s, 3H), 1.02 (s, 3H), 1.86 (s, 3H), 1.89–1.92 (m, 1H), 1.94 (s, 3H), 2.00–2.15 (m, 2H), 2.16 (s, 3H), 2.60 (d, J = 10.4 Hz, 1H) 2.83–2.94 (m, 4H), 3.35 (d, J = 10.0 Hz, 1H), 3.69 (s, 3H), 3.71 (dd, J = 6.8, 1.2 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 6.84 (d, J = 7.6 Hz, 1H), 7.22 (t, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 15.0, 16.6, 20.0, 22.3, 23.2, 24.4, 29.2, 32.67, 32.70, 55.9, 58.7, 75.4, 108.3, 121.9, 127.2, 128.4, 129.3, 131.0, 133.5, 136.6, 137.9, 143.0, 143.3, 151.7, 158.3; IR (film) cm−1 2949s, 2923s, 2852s, 2362m, 2344m, 1685s, 1469s, 1373m, 1256m; mass spectrum (APCI): m/e (% relative intensity) 394 (100) (M + H)+; HRMS of mixture of 19-M,p and 20-P,p (ESI, m/e) calcd for C25H31NO3 393.2299, found 393.2303; HPLC (95:5 hexane/2-propanol): tr = major 26.7 min, and minor 25.5 min.
Chiral Biaryl 20-P,p
Rf = 0.48 [60% EtOAc in hexanes]; thick oil; [α]D20 = + 7.5 (c 0.185, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.44 (s, 3H), 0.88 (s, 3H), 1.70–1.76 (m, 1H), 1.72 (s, 3H), 1.97 (s, 3H), 2.05 (quintet, J = 7.4 Hz, 2H), 2.10 (s, 3H), 2.80–2.95 (m, 4H), 3.50 (dd, J = 11.6, 1.8 Hz, 1H), 3.41 (dd, J = 10.6, 1.8 Hz, 1H), 3.62 (s, 3H), 3.74 (d, J = 10.8 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 7.6 Hz, 1H), 7.13 (t, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 15.0, 16.5, 20.2, 21.9, 23.2, 24.3, 29.0, 29.9, 32.67, 32.75, 55.4, 58.9, 76.0, 107.7, 123.4, 127.3, 128.3, 131.2, 133.8, 137.0, 140.6, 142.9, 143.5, 152.2, 156.7; IR (film) cm−1 2969s, 2918s, 2850s, 2360m, 2340m, 1686s, 1467s, 1378m, 1256m; mass spectrum (APCI): m/e (% relative intensity) 394 (100) (M + H)+; HRMS of mixture of 19-M,p and 20-P,p (ESI, m/e) calcd for C25H31NO3 393.2299, found 393.2303; HPLC (95:5 hexane/2-propanol): tr = major 26.7 min, and minor 25.5 min.
Chiral Biaryl 21-M,p
Rf = 0.17 [60% EtOAc in hexanes]; pale solid; mp 174–176 °C; [α]D20 = − 71.3 (c 1.42, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.55 (s, 3H), 1.03 (s, 3H), 1.83 (s, 3H), 1.95 (s, 3H), 2.14 (s, 3H), 2.62 (d, J = 10.4 Hz, 1H), 2.85 (dd, J = 11.2, 1.6 Hz, 1H), 3.37 (d, J = 10.4, Hz, 1H), 3.69 (s, 3H), 3.72 (dd, J = 10.6, 1.4 Hz, 1H) 5.07–5.16 (m, 4H), 6.82 (d, J = 8.0 Hz, 1H), 6.86 (d, J = 7.6 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.9, 16.3, 19.8, 22.3, 23.0, 29.1, 55.8, 58.6, 74.3, 75.4, 108.3, 122.0, 126.0, 127.3, 128.8, 128.9, 135.2, 137.7, 137.99, 138.02, 138.5, 151.5, 158.1 [missing one carbon due to overlap]; IR (film) cm−1 3418w, 2967w, 2360w, 1692s, 1578w, 1468s, 1373m, 1259m, 1175m; mass spectrum (APCI): m/e (% relative intensity) 396 (100) (M + H)+; HRMS (ESI, m/e) calcd for C24H29NO4 395.2092, found 395.2093; HPLC (95:5 hexane/2-propanol): tr = major 27.0 min, and minor 26.5 min.
Chiral Biaryl 22-P,p
Rf = 0.26 [60% EtOAc in hexanes]; thick oil; [α]D20 = + 21.4 (c 0.075, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.52 (s, 3H), 0.96 (s, 3H), 1.76 (s, 3H), 2.04 (s, 3H), 2.14 (s, 3H), 2.95 (d, J = 11.6 Hz, 1H), 3.01 (dd, J = 11.6, 1.6 Hz, 1H), 3.50 (dd, J = 10.8, 2.0 Hz, 1H), 3.70 (s, 3H), 3.83 (d, J = 10.4 Hz, 1H) 5.14–5.19 (m, 4H), 6.77 (d, J = 8.4 Hz, 1H), 6.95 (d, J = 7.6 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.9, 16.3, 20.1, 21.8, 23.2, 29.0, 55.3, 58.7, 74.3, 76.0, 77.4, 107.8, 123.5, 126.2, 127.0, 128.7, 129.1, 135.6, 137.9, 138.4, 138.6, 140.4, 152.0, 156.6; IR (film) cm−1 2969w, 2884w, 2839w, 2360m, 2342m, 1754s, 1699s, 1468s, 1258s; mass spectrum (APCI): m/e (% relative intensity) 396 (100) (M + H)+; HRMS (ESI, m/e) calcd for C24H29NO4 395.2092, found 395.2081; HPLC (95:5 hexane/2-propanol): tr = major 24.3 min, and minor 25.8 min.
Chiral Biaryl 23-M,p
Rf = 0.32 [60% EtOAc in hexanes]; pale solid; mp 96–100 °C; [α]D20 = − 6.8 (c 1.18, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 0.45 (s, 3H), 1.14 (s, 3H), 1.88 (s, 3H), 2.00 (s, 3H), 2.14 (s, 3H), 3.61 (s, 2H), 3.65 (ABq, Δν = 17.1 Hz, J = 16.8 Hz, 2H), 3.66 (s, 3H), 3.75 (d, J = 8.0 Hz, 1H), 3.79 (s, 3H), 3.80 (s, 3H), 4.00 (d, J = 8.0 Hz, 1H), 6.77 (d, J = 8.0 Hz, 1H), 6.81 (d, J = 7.5 Hz, 1H), 7.21 (d, J = 8.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 16.9, 17.5, 20.2, 24.7, 25.8, 40.9, 41.2, 53.38, 53.40, 55.8, 59.2, 62.0, 77.0, 108.2, 122.1, 127.7, 128.8, 131.79, 131.85, 133.1, 137.0, 139.3, 139.7, 139.8, 157.2, 158.0, 172.2, 173.1; IR (film) cm−1 2972w, 2360w, 1735s, 1434m, 1374m, 1264s, 1061m, 733s; mass spectrum (APCI): m/e (% relative intensity) 496 (100) (M + H)+; HRMS (ESI, m/e) calcd for C28H33NO7 496.2330, found 496.2339; HPLC (80:20 hexane/2-propanol): tr = major 28.2 min, and minor 25.8 min.
Chiral Biaryl 24-P,p
Rf = 0.44 [60% EtOAc in hexanes]; thick oil; [α]D20 = + 29.8 (c 0.33, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.61 (s, 3H), 1.10 (s, 3H), 1.77 (s, 3H), 2.01 (s, 3H), 2.12 (s, 3H), 3.65 (ABq, Δν = 61.3, J = 16.8 Hz, 2H), 3.67 (ABq, Δν = 31.7, J = 17.2 Hz, 2H),3.69 (s, 3H), 3.796 (s, 3H), 3.804 (s, 3H), 3.89 (d, J = 8.0 Hz, 1H), 4.05 (d, J = 8.0 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 6.87 (d, J = 7.6 Hz, 1H), 7.20 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 16.8, 17.0, 20.1, 23.1, 26.1, 40.9, 41.1, 53.3, 53.4, 55.1, 59.1, 61.9, 107.3, 123.2, 127.9, 128.6, 131.1, 131.7, 133.2, 137.0, 139.2, 139.5, 139.8, 157.8, 157.9, 172.4, 172.9, [missing one carbon due to overlap]; IR (film) cm−1 2956w, 2360w, 1735s, 1698s, 1468m, 1433m, 1252s, 1173m; mass spectrum (APCI): m/e (% relative intensity) 496 (100) (M + H)+; HRMS (ESI, m/e) calcd for C28H33NO7 496.2330, found 496.2320; HPLC (95:5 hexane/2-propanol): tr = major 25.5 min, and minor 19.3 min.
Chiral Biaryl 25-M,p
Rf = 0.35 [60% EtOAc in hexanes]; pale solid; mp 110–113 °C; [α]D20 = − 4.3 (c 3.15, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.48 (s, 3H), 1.17 (s, 3H), 1.86 (s, 3H), 2.04 (s, 3H), 2.13 (s, 3H), 3.68 (s, 3H), 3.79 (d, J = 8.4 Hz, 1H), 4.03 (d, J = 8.0 Hz, 1H), 5.07–5.19 (m, 4H), 6.79 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 7.6 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 16.7, 17.2, 20.1, 24.6, 25.8, 55.8, 61.9, 74.5, 74.6, 108.3, 122.2, 127.0, 129.0, 129.5, 129.6, 133.6, 137.5, 139.0, 139.06, 139.1, 157.1, 157.9 [missing one carbon due to overlap]; IR (film) cm−1 2968w, 2858w, 2369w, 2335w, 1745s, 1582w, 1397m, 1210m; mass spectrum (APCI): m/e (% relative intensity) 382 (100) (M + H)+; HRMS (ESI, m/e) calcd for C23H27NO4 381.1935, found 381.1938; HPLC (95:5 hexane/2-propanol): tr = major 27.0 min, and minor 26.0 min.
Chiral Biaryl 26-P,p
Rf = 0.41 [60% EtOAc in hexanes]; thick oil; [α]D20 = + 65.0 (c 0.85, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.63 (s, 3H), 1.13 (s, 3H), 1.76 (s, 3H), 2.05 (s, 3H), 2.11 (s, 3H), 3.72 (s, 3H), 3.92 (d, J = 8.4 Hz, 1H), 4.07 (d, J = 8.0 Hz, 1H), 5.12–5.21 (m, 4H), 6.72 (d, J = 8.4 Hz, 1H), 6.90 (d, J = 7.6 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 16.6, 16.9, 20.1, 23.1, 26.1, 55.2, 62.0, 74.5, 74.6, 77.7, 107.4, 123.3, 127.3, 128.8, 128.9, 129.6, 133.7, 137.6, 138.5, 139.0, 139.7, 157.80, 157.85; IR (film) cm−1 2956w, 2360w, 1735s, 1698s, 1468m, 1433m, 1252s, 1173m; mass spectrum (APCI): m/e (% relative intensity) 496 (100) (M + H)+; HRMS (ESI, m/e) calcd for C23H27NO4 382.2013, found 382.2013; HPLC (95:5 hexane/2-propanol): tr = major 24.3 min, and minor 25.8 min.
Chiral Biaryl 27-M,p
Rf = 0.17 [10% EtOAc in CH2Cl2]; yellow solid; mp 110–114 °C; [α]D20 = − 24.4 (c 1.53, CHCl3); 1H NMR (400 MHz, CDCl3) δ −0.096 (s, 3H), 0.99 (s, 3H), 1.73 (s, 3H), 2.18 (s, 3H), 3.65 (d, J = 7.8 Hz, 1H), 3.66 (s, 2H), 3.71 (ABq, Δν = 19.9 Hz, J = 17.0 Hz, 2H), 3.81 (s, 3H), 3.82 (s, 6H), 3.94 (d, J = 8.0 Hz, 1H), 7.23 (dd, J = 8.4, 0.8 Hz, 1H), 7.27–7.35 (m, 2H), 7.35 (d, J = 9.2 Hz, 1H), 7.79 (dd, J = 7.9, 1.4 Hz, 1H), 7.87 (d, J = 9.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 16.9, 18.0, 24.1, 26.4, 40.9, 41.3, 53.4, 56.4, 59.1, 61.9, 77.0, 77.5, 113.3, 121.3, 123.3, 125.1, 127.0, 128.6, 128.7, 129.8, 131.9, 132.6, 133.9, 134.9, 135.8, 139.8, 140.1, 154.9, 157.3, 172.2, 173.1; IR (film) cm−1: 2930w, 1735s, 1623w, 1595w, 1436m, 1397w, 1371m, 1351m, 1337m, 1272s, 1249s, 1167s, 1060s; mass spectrum (APCI): m/e (% relative intensity) 532 (100) (M + H)+, 474 (7); HRMS (ESI, m/e) calcd for C31H33NO7 532.2330, found 532.2320; HPLC (95:5 hexane/2-propanol): tr = major 26.4 min, and minor 25.2 min.
Chiral Biaryl 28-P,p
Rf = 0.39 [10% EtOAc in CH2Cl2]; yellow solid; mp 245–250 °C; [α]D20 = − 19.6 (c 1.13, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.38 (s, 3H), 1.12 (s, 3H), 1.80 (s, 3H), 2.17 (s, 3H), 3.50 (d, J = 8.2 Hz, 1H), 3.60–3.80 (m [two ABq], 4H), 3.81 (s, 3H), 3.82 (s, 3H), 3.87 (s, 3H), 3.89 (d, J = 8.0 Hz, 1H), 7.26 (d, J = 9.2 Hz, 1H), 7.28–7.35 (m, 2H), 7.50–7.52 (m, 1H), 7.73–7.75 (m, 1H), 7.88 (d, J = 9.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 16.9, 17.2, 23.9, 25.7, 41.0, 41.2, 53.3, 53.4, 55.7, 59.1, 62.0, 77.7, 111.8, 122.2, 124.3, 126.6, 126.9, 127.2, 129.0, 129.9, 131.6, 132.7, 133.3, 133.6, 135.8, 139.5, 139.9, 154.5, 157.2, 172.4, 172.9; IR (film) cm−1: 3003w, 1730s, 1592w, 1512w, 1462w, 1433w, 1397w, 1368w, 1268s, 1246s, 1199m, 1168m, 1062s; mass spectrum (APCI): m/e (% relative intensity) 532 (100) (M + H)+; HRMS (ESI, m/e) calcd for C31H33NO7 532.2330, found 532.2327; HPLC (95:5 hexane/2-propanol): tr = major 20.7 min, and minor 15.7 min.
Chiral Biaryl 29-M,p
Rf = 0.25 [60% EtOAc in hexanes]; yellow solid; mp 105–110 °C; [α]D20 = − 40.8 (c 2.055, CHCl3); 1H NMR (400 MHz, CDCl3) δ −0.052 (s, 3H), 1.02 (s, 3H), 1.71 (s, 3H), 2.17 (s, 3H), 3.70 (d, J = 8.0 Hz, 1H), 3.85 (s, 3H), 3.97 (d, J = 8.0 Hz, 1H), 5.12–5.25 (m, 4H), 7.26–7.38 (m, 3H), 7.37 (d, J = 9.2 Hz, 1H), 7.82 (dd, J = 7.8, 1.2 Hz, 1H), 7.90 (d, J = 9.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 16.7, 17.9, 24.1, 26.4, 56.5, 61.9, 74.5, 74.6, 77.1, 113.3, 120.7, 123.4, 124.9, 127.2, 128.69, 128.73, 129.6, 130.1, 130.5, 134.4, 134.8, 136.4, 139.1, 139.5, 155.0, 157.3; IR (film) cm−1: 2933w, 2839w, 1747s, 1620w, 1593w, 1507w, 1457w, 1416w, 1367m, 1345m, 1296w, 1274m, 1261m; mass spectrum (APCI): m/e (% relative intensity) 418 (100) (M + H)+; HRMS of mixture of 29-M,p and 30-P,p (ESI, m/e) calcd for C26H27NO4 417.1935, found 417.1928; HPLC (80:20 hexane/2-propanol): tr = major 17.1 min, and minor 13.2 min.
Chiral Biaryl 30-P,p
Rf = 0.42 [60% EtOAc in hexanes]; yellow solid; mp 115–120 °C; [α]D20 = − 57.8 (c 0.79, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.38 (s, 3H), 1.15 (s, 3H), 1.78 (s, 3H), 2.16 (s, 3H), 3.53 (d, J = 8.2 Hz, 1H), 3.89 (s, 3H), 3.91 (d, J = 8.2 Hz, 1H), 5.15–5.25 (m, 4H), 7.28 (d, J = 9.2 Hz, 1H), 7.30–7.38 (m, 2H), 7.52–7.55 (m, 1H), 7.75–7.78 (m, 1H), 7.91 (d, J = 9.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 16.8, 17.1, 23.9, 25.7, 55.8, 62.0, 74.57, 74.64, 77.0, 111.8, 121.5, 124.4, 126.3, 127.1, 127.4, 129.0, 129.4, 130.2, 130.6, 133.2, 134.1, 136.4, 138.7, 139.4, 154.5, 157.2; IR (film) cm−1: 2840w, 1743m, 1592w, 1510w, 1462w 1362w, 1345w, 1300w, 1258m, 1244m; mass spectrum (APCI): m/e (% relative intensity) 418 (100) (M + H)+; HRMS of mixture of 29-M,p and 30-P,p (ESI, m/e) calcd for C26H27NO4 417.1935, found 417.1928; HPLC (80:20 hexane/2-propanol): tr = major 18.7 min, and minor 15.2 min.
4.1.3. Assignment of Absolute Configurations
Demethylation Procedure
A solution of 17-M,p (30.0 mg, 0.059 mmol) in anhyd CH2Cl2 was cooled to −78 °C under an argon atmosphere. To tthis reaction mixture was added BBr3 (1.0 M soln in CH2Cl2, 5.0 equiv) carefully dropwise. The reaction mixture was warmed to −25 °C and stirred for 24 h. After the reaction was complete, the solution was quenched with H2O and the reaction mixture was warmed to RT. The layers were separated and the organic layer extracted with CH2Cl2 (2 × equal volume). The combined organic layers were dried with Na2SO4, filtered, and concentrated under reduced pressure.
The crude product was re-dissolved in anhyd acetone and 2.5 equiv of Me2SO4, and anhyd solid NaHCO3 (2.5 equiv) was added under an argon atmosphere. The reaction mixture was stirred at RT for 48 h. After the reaction was complete, the mixture filtered through a short pad of Celite.™ Acetone was removed under reduced pressure and the crude mixture was re-dissolved in EtOAc. The reaction mixture was washed with water followed by sat aq NaCl (equal volume). The organic layer was dried with MgSO4, filtered, and concentrated under reduced pressure. Purification of the crude residue by silica gel flash column chromatography [isocratic eluent: 50% EtOAc in hexanes] yielded the desired phenol 31-M,p in 73% yield. 31-M,p: Rf = 0.52 [50% EtOAc in hexanes]; white solid; mp 165–168 °C; [α]D20 = + 7.8 (c 2.05, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.50 (s, 3H), 0.96 (s, 3H), 1.84 (s, 3H), 1.94 (s, 3H), 2.16 (s, 3H), 2.69 (dd, J = 11.2, 2.0 Hz, 1H), 3.08 (d, J = 11.2 Hz, 1H), 3.57–3.71 (m, 5H), 3.79 (s, 3H), 3.80 (s, 3H), 3.94 (d, J = 10.8 Hz, 1H), 6.28 (s, 1H), 6.82 (d, J = 7.2 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 7.14 (t, J = 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.5, 16.4, 20.2, 21.3, 23.0, 29.1, 40.6, 53.3, 53.4, 58.4, 59.3, 60.6, 76.1, 116.2, 122.5, 125.8, 129.0, 129.2, 132.5, 133.5, 136.7, 137.8, 139.9, 140.3, 154.1, 155.0, 172.0, 172.7 [missing one carbon due to overlap]; IR (film) cm−1 3332s, 2959w, 2889w, 2360w, 2254w, 1734s, 1697s, 1467m, 1402m; mass spectrum (APCI): m/e (% relative intensity) 496 (100) (M + H)+; HRMS (ESI, m/e) calcd for C28H33NO7 496.2330, found 496.2318.
Preparation of Camphor Sulfonyl Derivative
A solution of 31-M, p (55.0 mg, 0.11 mmol) in anhyd CH2Cl2 was cooled to 0 °C under an argon atmosphere. To the reaction mixture was added (1S)-(+)-10-camphorsulfonyl chloride (31.0 mg, 0.122 mmol) followed by catalytic DMAP and Et3N (0.122 mmol). The reaction was warmed to RT and stirred for 12 h. After the reaction was complete, the solution was quenched with pH 7.0 aqueous buffer and the layers were separated. The organic layer was extracted with CH2Cl2 (2 × equal volume), dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification of the crude residue by silica gel flash column chromatography [isocratic eluent: 40% EtOAc in hexanes] afforded the product 32-M,p in 85% yield. 32-M,p: Rf = 0.46 [50% EtOAc in hexanes]; clear crystals; mp 230–233 °C; [α]D20 = − 39.2 (c 1.66, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 0.49 (s, 3H), 0.73 (s, 3H), 0.79 (s, 3H), 0.97 (s, 3H), 1.20–1.28 (m, 1H), 1.36–1.43 (m, 1H), 1.52–1.59 (m, 1H), 1.71–1.80 (m, 1H), 1.80 (d, J = 18.4 Hz, 1H), 1.89 (s, 3H), 1.97 (t, J = 4.4 Hz, 1H), 2.01 (s, 3H), 2.15 (s, 3H), 2.27 (ddd, J = 17.2, 4.4, 3.2 Hz, 1H), 2.47 (d, J = 10.8 Hz, 1H), 2.84 (d, J = 10.8 Hz, 1H), 3.40 (ABq, Δν = 85.5 Hz, J = 15.2 Hz, 2H), 3.49 (d, J = 10.0 Hz, 1H), 3.58 (ABq, Δν = 33.3 Hz, J = 17.0 Hz, 2H), 3.66 (ABq, Δν = 54.1 Hz, J = 16.6 Hz, 2H), 3.78 (d, J = 9.2 Hz, 1H), 3.79 (s, 3H), 3.80 (s, 3H), 7.17 (d, J = 7.6 Hz, 1H), 7.29 (t, J = 8.0 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H); 13C NMR (400 MHz, CDCl3) δ 15.1, 16.7, 19.4, 19.6, 20.0, 22.1, 23.1, 24.0, 27.1, 29.1, 40.7, 42.6, 43.1, 48.3, 49.7, 53.3, 53.4, 58.3, 58.8, 59.0, 75.6, 120.1, 127.9, 129.0, 130.1, 130.8, 132.0, 132.8, 138.3, 138.6, 139.1, 139.8, 147.4, 151.4, 172.2, 172.7, 214.1; IR (film) cm−1 2959w, 2889w, 2360w, 2254w, 1734s, 1697s, 1467m, 1402m; mass spectrum (APCI): m/e (% relative intensity) 709 (100) (M + H)+; HRMS (ESI, m/e) calcd for C38H47NO10S 710.2994, found 710.3004.
Free Phenol 33-M,p
1H NMR (400 MHz, CDCl3) δ 0.66 (s, 3H), 1.17 (s, 3H), 1.87 (s, 3H), 1.99 (s, 3H), 2.14 (s, 3H), 3.59–3.72 (m, 3H), 3.81 (s, 3H), 3.81 (s, 3H), 6.05 (s, 1H), 6.79 (d, 1H, J = 7.6 Hz), 6.89 (d, J = 8.0 Hz), 7.14 (t, J = 8.0 Hz, 1H); mass spectrum (APCI): m/e (% relative intensity) 496 (10) (M+H)+, 483 (30), 482 (100); 34-M,p. 1H NMR (500 MHz, CDCl3) δ 0.47 (s, 3H), 0.75 (s, 3H), 0.84 (s, 3H), 1.11 (s, 3H), 1.40 (ddd, J = 4.4, 9.2, 13.6 Hz, 1H), 1.65–1.83 (m, 4H), 1.92 (s, 3H), 1.97 (t, J = 4.4 Hz, 1H), 2.11 (s, 3H), 2.15 (s, 3H), 2.30 (ddd, J = 2.8, 4.4, 18.4 Hz, 1H), 2.93 (d, J =15.2 Hz, 1H), 3.61 (t, J =6.4 Hz, 2H), 3.66 (d, J = 6.8 Hz, 2H), 3.80 (s, 6H), 4.03 (d, J = 8.0 Hz, 1H), 7.15 (d, J = 7.2 Hz, 1H), 7.28 (t, J = 9.2 Hz, 1H), 7.36 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 17.0,17.5, 19.5, 19.6, 20.3, 24.4, 25.9, 27.1, 40.9, 41.1, 42.6, 43.3, 48.0, 49.6, 53.3, 53.4, 58.4, 58.8, 62.1, 120.8, 128.1, 129.0, 132.2, 132.3, 132.5, 133.5, 135.3, 137.2, 139.5, 140.3, 140.6, 147.3, 153.9, 157.2, 169.4, 172.2, 172.8; IR (neat) cm−1 3474w, 2967m, 2893m, 23841w, 2340w, 1737s, 1686m, 1578w, 1481m, 1433s, 1398m, 1374m, 1355m; mass spectrum (APCI): m/e (% relative intensity) 695 (40) (M+H)+, 694 (100), 481 (20), 480 (90), 422 (20), 231(20); HRMS (ESI, m/e) calcd for C37H45NNaO10S, 718.2657, found 718.2643.
4.1.4. Synthesis of a Chiral Amino Biaryl Ligand
Ynamide 35
Rf = 0.58 [50% EtOAc in hexanes]; 1H NMR (400 MHz, CDCl3) δ 2.12 (s, 3H), 3.72 (s, 3H), 4.88 (s, 2H), 6.62 (d, J = 8.0 Hz, 1H), 6.70 (d, J = 7.6 Hz, 1H), 7.07 (t, J = 8.0 Hz, 1H), 7.36–7.41 (m, 6H), 7.41–7.46 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 20.9, 53.7, 56.0, 70.4, 71.8, 76.6, 86.0, 108.0, 111.9, 121.8, 127.7, 128.7, 128.9, 139.7, 142.1, 155.3, 160.5 [missing eight carbons due to symmetry and overlap]; IR (film) cm−1 2969w, 2900w, 2839w, 2360w, 2248m, 1777w, 1716s, 1472s; mass spectrum (APCI): m/e (% relative intensity) 384 (100) (M + H)+; HRMS (ESI, m/e) calcd for C38H47NO10S 383.1516, found 383.1505.
Chiral Biaryl 37-M,p
Rf = 0.39 [60% EtOAc in hexanes]; pale solid; mp 103–106 °C; [α]D20 = − 21.6 (c 0.98, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 3H), 1.42 (s, 3H), 1.64 (s, 3H), 3.78 (s, 3H), 4.40 (d, J = 8.4 Hz, 1H), 4.95 (ABqd, Δν = 29.9, J = 12.4, 2.2 Hz, 2H), 5.10 (ABqd, Δν = 37.2, J = 12.8, 1.6 Hz, 2H), 5.20 (d, J = 8.4 Hz, 1H), 6.19 (d, J = 7.6 Hz, 1H), 6.51 (d, J = 7.6 Hz, 2H), 6.78 (d, J = 8.0 Hz, 1H), 6.84 (t, J = 8.0 Hz, 2H), 7.01 (d, J = 8.0 Hz, 3H), 7.05 (t, J = 8.0 Hz, 1H), 7.23–7.27 (m, 2H), 7.35 (t, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 15.8, 17.03, 20.0, 55.9, 71.2, 74.4, 74.6, 77.0, 107.8, 123.1, 126.5, 126.7, 127.1, 128.07, 128.15, 128.4, 128.6, 128.7, 129.8, 130.9, 135.9, 137.4, 137.9, 138.6, 138.7, 139.2, 144.0, 157.7, 158.0 [missing four carbons due to symmetry]; IR (film) cm−1 3058w, 2840w, 2360w, 1760s, 1579w, 1469m, 1262m, 1069m, 908w; mass spectrum (APCI): m/e (% relative intensity) 506 (100) (M + H)+; HRMS (ESI, m/e) calcd for C33H31NO4 505.2253, found 505.2250; HPLC (95:5 hexane/2-propanol): tr = major 27.5 min, and minor 26.4 min.
Chiral Biaryl 38-P,p
Rf = 0.62 [60% EtOAc in hexanes]; thick oil; [α]D20 = + 74.5 (c 0.98, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 1.43 (s, 3H), 1.62 (s, 3H), 2.19 (s, 3H), 2.92 (s, 3H), 4.31 (d, J = 8.0 Hz, 1H), 4.92 (ABq, Δν = 49.9, J = 12.2 Hz, 2H), 5.12 (ABq, Δν = 28.7, J = 12.5 Hz, 2H), 5.29 (d, J = 3.2 Hz, 1H), 5.31 (d, J = 4.0 Hz, 1H), 5.89 (d, J = 8.4 Hz, 1H), 6.43 (broad d, J = 7.2 Hz, 2H), 6.86 (t, J = 7.6 Hz, 1H), 6.88 (d, J = 6.8 Hz, 1H), 7.01 (dt, J = 6.8, 0.8 Hz, 1H), 7.04 (d, J = 8.0 Hz, 1H), 7.34–7.35 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 15.5, 16.8, 20.2, 53.9, 71.0, 74.2, 74.5, 77.1, 107.7, 122.7, 125.4, 126.8, 127.0, 127.9, 128.2, 128.5, 128.7, 129.5, 129.9, 135.3, 136.4, 138.2, 138.9, 139.0, 139.2, 145.2, 156.7, 158.9 [missing four carbons due to symmetry and one carbon due to overlap]; IR (film) cm−1 3058w, 2839w, 2360w, 2341w, 1753s, 1598w, 1492m, 1468m, 1301m; mass spectrum (APCI): m/e (% relative intensity) 506 (100) (M + H)+; HRMS (ESI, m/z) calcd for C33H31NO4 505.2253, found 505.2257; HPLC (95:5 hexane/2-propanol): tr = major 11.5 min, and minor 16.3 min.
Preparation of Chiral Amino-Biaryl 39-M
To a solution of chiral biaryl 37-M, p [13.0 mg, 82% ee) in MeOH (3 mL) flushed with argon was added 10 mol% Pd/C (3.0 mg). The reaction mixture was flushed with H2 gas and then stirred under a balloon of H2 for 3 d. The resulting mixture was filtered though a short pad of Celite™ and solvent was removed under reduced pressure. The crude product was purified by preparative TLC (1:1 Hex/EtOAc) and 3.0 mg of the desired amino biaryl 39-M was isolated (42% yield). 39-M: Rf = 0.55 [10% CH2Cl2/EtOAc]; [α] D 23 = +25.8° [c 0.0039 CH2Cl2];1H NMR (500 MHz, CDCl3) δ 1.75 (s, 3H), 1.98 (s, 3H), 2.05 (s, 3H), 3.21–3.56 (bs, 1H), 3.66 (d, 1H, J = 3.0 Hz), 3.73 (s, 3H), 5.15 (d, 4H, J = 21.5 Hz), 6.87 (d, 1H, J = 10.5 Hz), 6.96 (d, J = 9.5 Hz), 7.25–7.29 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 14.2, 16.5, 19.7, 29.9, 56.1, 74.4, 74.4, 108.8, 112.7, 114.1, 122.5, 123.1, 125.9, 127.5, 127.8, 128.8, 137.6, 139.3, 141.5, 141.7, 157.7; IR (neat) cm−1 2918s, 2849s, 2361m, 2340w, 1732w, 1618w, 1578w, 1467s, 1367m; mass spectrum (APCI): m/e (% relative intensity) 352 (100) (M+H)+, 285 (20), 284 (100).
Figure 2. Scope of Asymmetric Ynamide-[2 + 2 + 2] Cycloadditions.

[a] The dr was determined using 1H/13C NMR. [b] Isolated yields. [c] The ee was determined using chiral HPLC [CHIRALCEL OD-H; Size: 250 × 4.6 mm]; eluent: i-PrOH in hexanes. [d](R)-Xylyl-BINAP was used.
Acknowledgments
Authors thank NIH-NIGMS [GM066055]. Authors also thank Dr. Victor Young and Mr. Ben Kucera [University of Minnesota] for providing X-ray structural analysis. Finally, we thank Professor Marisa Kozlowski for invaluable discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For reviews on ynamides, see: Zificsak CA, Mulder JA, Hsung RP, Rameshkumar C, Wei LL. Tetrahedron. 2001;57:7575.Mulder JA, Kurtz KCM, Hsung RP. Synlett. 2003:1379.Katritzky AR, Jiang R, Singh SK. Heterocycles. 2004;63:1455.
- 2.For a special issue dedicated to the chemistry of ynamides, see: Tetrahedron-Symposium-In-Print. Chemistry of Electron-Deficient Ynamines and Ynamides. Tetrahedron. 2006;62(16)
- 3.For chemistry of ynamides in the last two years, see: Couty S, Liegault B, Meyer C, Cossy J. Tetrahedron. 2009;65:3882.Deweerdt K, Birkedal H, Ruhland T, Skrydstrup T. Org Lett. 2009;11:221. doi: 10.1021/ol802477d.Dooleweerdt K, Birkedal H, Ruhland T, Skrydstrup T. J Org Chem. 2008;73:9447. doi: 10.1021/jo801935b.Saito N, Katayama T, Sato Y. Org Lett. 2008;10:3829. doi: 10.1021/ol801534e.Yasui H, Yorimitsu H, Oshima K. Bull Chem Soc Jpn. 2008;81:373.Yasui H, Yorimitsu H, Oshima K. Chem Lett. 2008;37:40.Istrate FM, Buzas AK, Jurberg ID, Odabachian Y, Gagosz F. Org Lett. 2008;10:925. doi: 10.1021/ol703077g.Martínez-Esperón MF, Rodríguez D, Castedo L, Saá C. Tetrahedron. 2008;64:3674.Kim JY, Kim SH, Chang S. Tetrahedron Lett. 2008;49:1745.Yavari I, Sabbaghan M, Hosseini N, Hossaini Z. Synlett. 2007;20:3172.Hashimi ASK, Salathe R, Frey W. Synlett. 2007:1763.Rodríguez D, Martínez-Esperón MF, Castedo L, Saá C. Synlett. 2007:1963.Couty S, Meyer C, Cossy J. Synlett. 2007:2819.Movassaghi M, Hill MD, Ahmad OK. J Am Chem Soc. 2007;129:10096. doi: 10.1021/ja073912a.
- 4.For our own efforts, see: Zhang Y, DeKorver KA, Lohse AG, Zhang YS, Hsung RP. Org Lett. 2009;11:899. doi: 10.1021/ol802844z.Al-Rashid ZF, Johnson WL, Hsung RP, Wei Y, Yao PY, Liu R, Zhao K. J Org Chem. 2008;73:8780. doi: 10.1021/jo8015067.Yao PY, Zhang Y, Hsung RP, Zhao K. Org Lett. 2008;10:4275. doi: 10.1021/ol801711p.Zhang X, Hsung RP, Li H, Zhang Y, Johnson WL, Figueroa R. Org Lett. 2008;10:3477. doi: 10.1021/ol801257j.Al-Rashid ZF, Hsung RP. Org Lett. 2008;10:661. doi: 10.1021/ol703083k.You L, Al-Rashid ZF, Figueroa R, Ghosh SK, Li G, Lu T, Hsung RP. Synlett. 2007:1656.Li H, You L, Zhang X, Johnson WL, Figueroa R, Hsung RP. Heterocycles. 2007;74:553.Zhang X, Hsung RP, Li H. Chem Commun. 2007:2420. doi: 10.1039/b701040k.Oppenheimer J, Johnson WL, Tracey MR, Hsung RP, Yao PY, Liu R, Zhao K. Org Lett. 2007;9:2361. doi: 10.1021/ol0707362.
- 5.Couty S, Liegault B, Meyer C, Cossy J. Tetrahedron. 2006;62:3882. [Google Scholar]
- 6.Zhang Y, Hsung RP, Zhang X, Huang J, Slafer BW, Davis A. Org Lett. 2005;7:1047. doi: 10.1021/ol0473391. [DOI] [PubMed] [Google Scholar]
- 7.(a) Witulski B, Stengel T. Angew Chem, Int Ed. 1999;38:2426. [PubMed] [Google Scholar]; (b) Witulski B, Stengel T, Fernàndez-Hernàndez JM. Chem Commun. 2000:1965. [Google Scholar]; (c) Witulski B, Alayrac C. Angew Chem, Int Ed. 2002;41:3281. doi: 10.1002/1521-3773(20020902)41:17<3281::AID-ANIE3281>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 8.For a review, see: Zhang Y, Hsung RP. ChemTracts. 2004;17:442.
- 9.(a) Rainier JD, Imbriglio JE. J Org Chem. 2000;65:7275. doi: 10.1021/jo001044t. [DOI] [PubMed] [Google Scholar]; (b) Rainier JD, Imbriglio JE. Org Lett. 1999;1:2037. [Google Scholar]
- 10.Tracey MR, Oppenheimer J, Hsung RP. J Org Chem. 2006;71:8629. doi: 10.1021/jo061683p. [DOI] [PubMed] [Google Scholar]
- 11.For a preliminary communication for this work, see: Oppenheimer J, Hsung RP, Figueroa R, Johnson WL. Org Lett. 2007;9:3969. doi: 10.1021/ol701692m.
- 12.For recent reviews, see: Wallace TW. Org Biomol Chem. 2006;4:3197. doi: 10.1039/b608470m.Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Graner J, Breuning M. Angew Chem Int Ed. 2005;44:5384. doi: 10.1002/anie.200462661.Varela JA, Saá C. Chem Rev. 2003;103:3787. doi: 10.1021/cr030677f.Rubin M, Sromek AW, Gervorgyan V. Synlett. 2003:2265.Aubert C, Buisine O, Malacria M. Chem Rev. 2002;102:813. doi: 10.1021/cr980054f.Saito S, Yamamoto Y. Chem Rev. 2000;100:2901. doi: 10.1021/cr990281x.Frühauf HW. Chem Rev. 1997;97:523. doi: 10.1021/cr941164z.Ojima I, Tzamarioudaki M, Li Z, Donovan R. J Chem Rev. 1996;96:635. doi: 10.1021/cr950065y.Lautens M, Klute W, Tam W. Chem Rev. 1996;96:49. doi: 10.1021/cr950016l.
- 13.(a) Nishida G, Noguchi K, Hirano M, Tanaka K. Angew Chem Int Ed. 2007;46:3951. doi: 10.1002/anie.200700064. [DOI] [PubMed] [Google Scholar]; (b) Yamamoto Y, Ishii JI, Nishiyama H, Itoh K. J Am Chem Soc. 2005;127:9625. doi: 10.1021/ja051377d. [DOI] [PubMed] [Google Scholar]; (c) Sato Y, Tamura T, Mori M. Angew Chem Int Ed. 2004;43:2436. doi: 10.1002/anie.200453809. [DOI] [PubMed] [Google Scholar]; (d) Gandon V, Leca D, Aechtner T, Vollhardt KPC, Malacria M, Aubert C. Org Lett. 2004;6:3405. doi: 10.1021/ol0485823. [DOI] [PubMed] [Google Scholar]; (e) Kinoshita H, Shinokubo H, Oshima K. J Am Chem Soc. 2003;125:7784. doi: 10.1021/ja035438o. [DOI] [PubMed] [Google Scholar]; (f) Tanaka K, Shirasaka K. Org Lett. 2003;5:4697. doi: 10.1021/ol035963s. [DOI] [PubMed] [Google Scholar]; (g) Bonaga LVR, Zhang HC, Gauthier DA, Reddy I, Maryanoff BE. Org Lett. 2003;5:4537. doi: 10.1021/ol030096c. [DOI] [PubMed] [Google Scholar]; (h) Deaton KR, Gin MS. Org Lett. 2003;5:2477. doi: 10.1021/ol034720x. [DOI] [PubMed] [Google Scholar]; (i) Sung MJ, Pang JH, Park SB, Cha JK. Org Lett. 2003;5:2137. doi: 10.1021/ol034592c. [DOI] [PubMed] [Google Scholar]
- 14.Also see: Boñaga LVR, Zhang HC, Moretto AF, Ye H, Gauthier DA, Li J, Leo GC, Maryanoff BE. J Am Chem Soc. 2005;127:3473. doi: 10.1021/ja045001w. and references therein.Tanaka K, Nishida G, Ogino M, Hirano M, Noguchi K. Org Lett. 2005;7:3119. doi: 10.1021/ol0511880.Stará IG, Alexandrováá Z, Teply F, Sehnal P, Stary I, Saman D, Budesinsky M, Cvacka J. Org Lett. 2005;7:2547. doi: 10.1021/ol047311p.Shibata T, Fujimoto T, Yokota K, Takagi K. J Am Chem Soc. 2004;126:8382. doi: 10.1021/ja048131d.Gutnov A, Heller B, Fischer C, Drexler HJ, Spannenberg A, Sundermann B, Sundermann C. Angew Chem, Int Ed. 2004;43:3795. doi: 10.1002/anie.200454164.Tanaka K, Nishida G, Wada A, Noguchi K. Angew Chem, Int Ed. 2004;43:6510. doi: 10.1002/anie.200461533.Nishii Y, Wakasugi K, Koga K, Tanabe Y. J Am Chem Soc. 2004;126:5358. doi: 10.1021/ja0319442.
- 15.Tracey MR, Zhang Y, Frederick MO, Mulder JA, Hsung RP. Org Lett. 2004;6:2209. doi: 10.1021/ol0493251. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka K, Takeishi K, Noguchi K. J Am Chem Soc. 2006;128:4586. doi: 10.1021/ja060348f. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka K, Takeishi K. Synthesis. 2007:2920. [Google Scholar]
- 18.For some elegant examples of axially chiral amido-aryl bonds: Kitagawa O, Takahashi M, Yoshikawa M, Taguchi T. J Am Chem Soc. 2005;127:3676. doi: 10.1021/ja042216x.Bennett DJ, Pickering PL, Simpkins NS. Chem Commun. 2004:1392. doi: 10.1039/b403193h.Terauchi J, Curran DP. Tetrahedron Asy. 2003;14:587.Kitagawa O, Kohriyama M, Taguchi T. J Org Chem. 2002;67:8682. doi: 10.1021/jo0264271.Ates A, Curran DP. J Am Chem Soc. 2001;123:5130. doi: 10.1021/ja010467p.Hata T, Koide H, Taniguchi N, Uemura M. Org Lett. 2000;2:1907. doi: 10.1021/ol0001057.Shimizu KD, Freyer HO, Adams RD. Tetrahedron Lett. 2000;41:5431.Kondo K, Fujita H, Suzuki T, Murakami Y. Tetrahedron Lett. 1999;40:5577.Ahmed A, Bragg RA, Clayden J, Lai LW, McCarthy C, Pink JH, Westlund N, Yasin SA. Tetrahedron. 1998;54:13277.
- 19.For pioneering work, see: Kitagawa O, Izawa H, Sato K, Dobashi A, Taguchi T, Shiro M. J Org Chem. 1998;63:2634. doi: 10.1021/jo9721711.Curran DP, Qi H, Geib SJ, DeMello NC. J Am Chem Soc. 1994;116:3131.
- 20.For leading reviews, see: Clayden J. Chem Soc Rev. 2009;38:817. doi: 10.1039/b801639a.Clayden J. Org Biomol Chem. 2006;4:2667. doi: 10.1039/b604548k.Jorgensen KA. Angew Chem Int Ed. 2000;39:3558.Clayden J. Angew Chem Int Ed. 1997;36:949.
- 21.For an excellent book on concepts of asymmetric catalysis, see: Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. University Science Books: Sausalito, CA; 2008.
- 22.For leading examples, see: Xie X, Phuan PW, Kozlowski MC. Angew Chem Int Ed. 2003;42:2168. doi: 10.1002/anie.200250325.Shibata T, Fujimoto T, Yokota K, Takagi K. J Am Chem Soc. 2004;126:8382. doi: 10.1021/ja048131d.For a diastereoselective example, see: Fogel L, Hsung RP, Wulff WD, Sommer RD, Rheingold AL. J Am Chem Soc. 2001;123:5580. doi: 10.1021/ja010091f.
- 23.For elegant examples of achiral template based asymmetric catalysis, see: Sibi MP, Soeta T. J Am Chem Soc. 2007;129:4522. doi: 10.1021/ja069312d.Sibi MP, Stanley LM, Nie X, Venkatraman L, Liu M, Jasperse CP. J Am Chem Soc. 2007;129:395. doi: 10.1021/ja066425o.Sibi MP, Manyem S, Palencia H. J Am Chem Soc. 2006;128:13660. doi: 10.1021/ja064472a.Sibi MP, Stanley LM, Jasperse CP. J Am Chem Soc. 2005;127:8277. doi: 10.1021/ja051650b.Sibi MP, Zhang R, Manyem S. J Am Chem Soc. 2003;125:9306. doi: 10.1021/ja035979d.
- 24.For a review on synthesis of ynamides, see: Tracey MR, Hsung RP, Antoline JA, Kurtz KCM, Shen L, Slafer BW, Zhang Y. In: Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. Steve M Weinreb., editor. Stuttgart, Germany: Georg Thieme Verlag KG; 2005. Chapter 21.4.
- 25.For synthesis of ynamides through Ullmann-type coupling, see: Zhang Y, Hsung RP, Tracey MR, Kurtz KCM, Vera EL. Org Lett. 2004;6:1151. doi: 10.1021/ol049827e.Dunetz JR, Danheiser RL. Org Lett. 2003;5:4011. doi: 10.1021/ol035647d.Frederick MO, Mulder JA, Tracey MR, Hsung RP, Huang J, Kurtz KCM, Shen L, Douglas CJ. J Am Chem Soc. 2003;125:2368. doi: 10.1021/ja021304j.Zhang X, Zhang Y, Huang J, Hsung RP, Kurtz KCM, Oppenheimer J, Petersen ME, Sagamanova IK, Tracey MR. J Org Chem. 2006;71:4170. doi: 10.1021/jo060230h.Sagamanova IK, Kurtz KCM, Hsung RP. Organic Syn. 2007;84:359.Kohnen AL, Dunetz JR, Danheiser RL. Organic Syn. 2007;84:88.Riddell N, Villeneuve K, Tam W. Org Lett. 2005;7:3681. doi: 10.1021/ol0512841.
- 26.For a recent oxidative cross-coupling, see: Hamada T, Ye X, Stahl SS. J Am Chem Soc. 2008;130:833. doi: 10.1021/ja077406x.
- 27.Throughout the paper, the capital M and P denote C-C axial chirality while small m and p denote C-N-axial chirality and are listed second.
- 28.CCDC for compounds 32-M,p, 18-P,p and 34-M,p are 665008, 665009 & 722530, respectively. CCDC 665008, 665009 and 722530 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallopgraphic Data Centre viawww.ccdc.cam.ac.uk/data_request.cif.