

Abstract
Side chain oxysterols are cholesterol derivatives thought to signal the abundance of cell cholesterol to homeostatic effector proteins. Here, we investigated how plasma membrane (PM) cholesterol might regulate 27-hydroxycholesterol (HC) biosynthesis in cultured fibroblasts. We showed that PM cholesterol was a major substrate for 27-HC production. Biosynthesis commenced within minutes of loading depleted cells with cholesterol, concurrent with the rapid inactivation of hydroxy-3-methylglutaryl CoA reductase (HMGR). 27-HC production rose ∼30-fold in normal and Niemann-Pick C1 fibroblasts when PM cholesterol was increased by ∼60%. 27-HC production was also stimulated by 1-octanol, which displaces PM cholesterol from its phospholipid complexes and thereby increases its activity (escape tendency) and elevates its intracellular abundance. Conversely, lysophosphatidylserine and U18666A inhibited 27-HC biosynthesis and the inactivation of HMGR, presumably by reducing the activity of PM cholesterol and, therefore, its circulation to mitochondria. We conclude that, in this in vitro system, excess (active) PM cholesterol rapidly reaches mitochondria where, as the rate-limiting substrate, it stimulates 27-HC biosynthesis. The oxysterol product then promotes the rapid degradation of HMGR, along with other homeostatic effects. The regulation of 27-HC production by the active excess of PM cholesterol can thus provide a feedback mechanism in the homeostasis of PM cholesterol.
Keywords: homeostasis, endoplasmic reticulum, oxysterol, feedback, Niemann-Pick
This study addresses the regulation of the biosynthesis of oxysterol signal molecules. Side chain oxysterols such as 24-, 25-, and 27-hydroxycholesterol (HC) are derived from cholesterol in the endoplasmic reticulum (ER) and mitochondria and trigger downregulation of the abundance of cell cholesterol through diverse pathways (1–4). In particular, the association of oxysterols with nuclear liver X receptors (LXR) promotes the expression of ATP-binding cassette (ABC) transport proteins that transfer excess cholesterol out of cells such as macrophages (5). Furthermore, because the oxysterol derivatives are more water-soluble than cholesterol, they readily exit cells and are transported via circulating lipoproteins to the liver for conversion to bile acids and excretion (6, 7). In addition, side-chain oxysterols promote the interaction of Insig proteins with sterol regulatory element-binding protein (SREBP) cleavage-activating protein-SREBP complexes; this action sequesters SREBP in the ER and, as a result, reduces the expression of genes for cholesterol accretion (8). By activating Insig, side-chain oxysterols also stimulate the proteolysis of hydroxy-3-methylglutaryl CoA reductase (HMGR) through a ubiquitination-proteasomal pathway (9, 10).
The mechanism by which oxysterol biosynthesis is set according to the level of cell cholesterol so as to execute these homeostatic functions is obscure. Indeed, despite the cogency of the oxysterol hypothesis, the physiological relevance of the underlying in vitro findings has long been debated (1, 11). To carry out its postulated role in vivo, oxysterol production should represent the level of cellular cholesterol, in particular, the major pool in the plasma membrane (PM). The present study therefore examined both the influence of the level of PM cholesterol on oxysterol biosynthesis and the feedback regulation of cholesterol biosynthesis by this oxysterol. Specifically, we tested the hypothesis that the rate of production of 27-HC is controlled by the flux of excess PM cholesterol to mitochondria. As taken up in the “Discussion,” excess PM cholesterol arises when the stoichiometric complexing capacity of its phospholipids is exceeded. In this way, the phospholipids set the physiologic level of cholesterol in the PM. The main criteria we employed to evaluate our hypothesis were the following: a) Is PM cholesterol a principal substrate for 27-HC biosynthesis? b) Does the rate of production of 27-HC respond to the abundance of excess PM cholesterol? That is, is 27-HC biosynthesis stimulated by small increments in PM cholesterol above its physiological set point and does its biosynthesis decline with small decrements in cholesterol below that point? c) Does the rate of 27-HC biosynthesis also vary in response to agents that increase and decrease free PM CH? d) Is the production of 27-HC evoked by cholesterol rapid and extensive enough to mediate adaptive responses? The experiments described below fulfill these criteria, adding support to the hypothesis that oxysterols can serve as homeostatic signals.
EXPERIMENTAL PROCEDURES
Materials
Cholesterol, 2-β-hydroxypropylcyclodextrin (HPCD), fetal calf lipoprotein-deficient serum, 1-palmitoyl-lysophosphatidylserine (LPS), and 1-octanol were purchased from Sigma; U18666A was from Biomol. We obtained hydroxymethylglutaryl-CoA bearing a dl-3-[glutaryl-3-14C] label from American Radiolabeled Chemicals, Inc.. Deuterated cholesterol (cholesterol-2,2,4,4,6-d5) was obtained from Medical Isotopes Inc.., Pelham, NH.
Preparation of cholesterol complexes
Aqueous solutions of ∼2.7 mg cholesterol/100 mg HPCD were prepared in water as described (12). Briefly, 1.5 ml of 30% HPCD in water was heated to 80°C. Then 120 µl of a 100 mg/ml solution of cholesterol in isopropanol:chloroform (2:1) was added dropwise, stirring continuously at 80°C. The mixture was allowed to clarify between additions of the cholesterol solution, and the final preparation was stored at room temperature.
Cell culture and treatments
Normal human foreskin fibroblasts were cultured in DMEM with 10% fetal bovine serum and antibiotics, as described (13). The mutant Niemann-Pick C type 1 (NPC1)-deficient human skin fibroblast line, GM 3123, was cultured as described (14). We used 25 cm2 flasks of confluent cells for HMGR experiments; 75 cm2 flasks of confluent cells were used for 27-HC experiments. A confluent 75 cm2 flask of wild-type fibroblasts typically yielded 850 µg protein and 30 μg cholesterol. Experiments were started by layering cells in flasks with medium A (DMEM containing 5% fetal calf lipoprotein-deficient serum). Time courses were initiated by adding agents or by altering cell cholesterol levels through incubation with HPCD ± cholesterol for 7 min at 37°C (12). Cells were dissociated from flasks for analysis by means of a 1 min incubation with 0.05% trypsin plus 0.02% EDTA at 37°C.
To remove ambient oxysterols, flasks were preincubated overnight in medium A. This treatment also lowered cell cholesterol by about 8%, on average, and caused a substantial increase in the level of cellular HMGR. Subsequent incubation of these starved cells for 7 min at 37°C with an extracellular cholesterol acceptor, 0.25–0.5% HPCD in PBS, further reduced the level of the oxysterol from 21.5 ± 4 (n = 3) to 4.6 ± 2 (n = 5) ng 27-HC/mg cell protein. The HPCD treatment also reduced cell sterol on average by an additional 4%. We therefore included HPCD in either the preincubation or the cholesterol-loading step in all experiments to minimize the 27-HC background.
Assays
Cholesterol mass was measured by HPLC (15). HMGR activity was assayed as described (16, 17). HMGR determinations were duplicates that agreed to within 5%; the means ± average deviations were expressed in pmol mevalonate/min/mg cell protein. Protein was determined in duplicate with a BCA kit (Pierce) using a BSA standard. We found that at least one-half of the 27-HC synthesized (50–89% of the total) was recovered in the medium at any given time. We therefore routinely harvested the incubation medium and extracted it with 3 vols of chloroform:methanol (2:1, v/v). The cells were dissociated, washed once with PBS, and resuspended; aliquots were then taken for cholesterol and protein determination. The remainder was extracted twice with 100 vols of hexane:isopropanol:water (3:2:9.1, v/v/v). The extracts of cells and media were pooled and stored under liquid nitrogen until the assay of oxysterols. This was performed in duplicate or triplicate by GC-MS, as described (18). 27-HC typically represented ∼90% of the side-chain oxysterols; hence, we did not report on the traces of 20(S)-, 24-, and 25-HC. The relatively high values for 24-HC reported previously (18) were not found here and are not characteristic of human fibroblasts. Furthermore, 27-HC can be metabolized to cholestenoic acid, so that the 27-HC values reported here may have underestimated its production. Deuterated 27-HC was determined by selective ion monitoring at m/z 456–461. (Medium A contained <1 ng 27-HC/mg cell protein.)
RESULTS
Is PM cholesterol a source of 27-HC production?
The rate of 27-HC synthesis was very low in fibroblasts depleted of PM cholesterol but was increased greatly by a brief pulse of exogenous cholesterol (see the following section). Oxysterol production in such enriched cells exhibited a variable lag of no more than 1 h, followed by more-or-less constant production over the following few h (Fig. 1; see also Fig. 3A). In all of our experiments, <0.1% of cell cholesterol was converted to 27-HC per h.
Fig. 1.

Time course of 27-HC biosynthesis in response to pulses of exogenous deuterated cholesterol. In each of these two replicate experiments, four 75 cm2 flasks of fibroblasts were preincubated overnight in medium A. The medium was replaced with 4 ml PBS and a pulse of 27 mg HPCD + 0.46 mg deuterated cholesterol was added to each flask for 7 min at 37°C. As a result, the mass of the deuterated cholesterol slightly exceeded the [1H]cholesterol in the loaded cells. The flasks were rinsed twice, and the cells layered with 4 ml medium A. Flasks were processed immediately or incubated at 37°C for 1, 2, or 4 h before the determination of 27-[1H]HC (O), deuterated 27-HC (▿), [1H]cholesterol, deuterated cholesterol, and protein. 27-HC values are the means of triplicate determinations ± SEM.
Fig. 3.

Time course of 27-HC biosynthesis and HMGR activity in response to PM cholesterol enrichment. A: 27-HC biosynthesis. Five 75 cm2 flasks of cells were incubated overnight in medium A. The medium was replaced with 4 ml PBS, a pulse of 18 mg HPCD + 0.43 mg cholesterol added to each, and the flasks incubated for 7 min at 37°C. This enriched the cells by ∼50%. The cells were rinsed, layered with 4 ml medium A, and incubated at 37°C. For the zero-time point, the cells were processed immediately after the cholesterol pulse. After the indicated incubation times, total 27-HC and cell protein were determined. Values are the means of duplicate assays that agreed to within 3%. B: HMGR activity. Six replicate 25 cm2 flasks were incubated overnight in medium A. The medium was replaced with 1.5 ml PBS, a pulse of 6 mg HPCD + 0.14 mg cholesterol added, and the flasks incubated for 7 min at 37°C. The buffer was replaced with 1.5 ml medium A and the flasks incubated at 37°C for the times indicated. HMGR activity and cell protein were then determined in duplicate. The enriched cells contained, on average, 43 μg cholesterol/mg cell protein, whereas unenriched cells normally contain ∼35 μg cholesterol/mg protein (23). The time course was fit to a first order expression with a half-time of 8.5 min. Data are from one of two similar experiments.
To test whether PM cholesterol was an important substrate for 27-HC biosynthesis, pulses of deuterated cholesterol were used as the stimulus in Fig. 1. Nearly one-half of the newly synthesized 27-HC was labeled with deuterium at all time points, indicating that the transfer of PM cholesterol to the mitochondria was rapid and extensive. On the other hand, the lag in the production of deuterated 27-HC compared with [1H]27-HC during the first h implies that several minutes were required for mitochondrial cholesterol to reach an exchange equilibrium with the PM pool. These data also suggest that the pool of cholesterol serving as a substrate for 27-HC production turns over with a halftime of <1 h. After 2 h of incubation, the isotope ratio in the 27-HC was about one-half that in the bulk cell cholesterol (not shown). Given that the specific content of deuterium in the 27-HC collected at the 2 h point was reduced by the presence of the less well-labeled oxysterol that had accumulated in the first h, it was estimated that ∼85% of the 27-HC synthesized in the second h was derived from PM cholesterol.
Dependence of 27-HC production on the level of PM cholesterol
Wild-type and NPC1-deficient fibroblasts were preincubated overnight in cholesterol-free medium. Their PM cholesterol was then variously adjusted with pulses of exogenous HPCD ± cholesterol and the rate of production of 27-HC determined. As shown in Fig. 2, reducing the PM cholesterol of the cells by ∼20% decreased their rate of 27-HC biosynthesis by approximately 5-fold, from 2.1 to about 0.4 ng/mg protein/h. Conversely, elevating cell cholesterol using HPCD-cholesterol complexes increased 27-HC production ∼6-fold, to as high as 13 ng/mg protein/h, with no sign of reaching a maximum. Thus, augmenting PM cholesterol by ∼60% from below to above its physiological set point stimulated 27-HC biosynthesis ∼30-fold overall.
Fig. 2.

Dependence of the biosynthesis of 27-HC on cholesterol load in wild-type and NPC1-deficient cells. Replicate flasks were preincubated overnight in medium A. The cells were layered with 4 ml PBS, pulses of HPCD ± cholesterol added, and the flasks incubated for 7 min at 37°C. For depletion, the pulses contained from 10 to 32 mg HPCD; for enrichment, the pulses contained mixtures ranging from 6.6 mg HPCD + 0.16 mg cholesterol to 19.5 mg HPCD + 0.47 mg cholesterol. This buffer was replaced with medium A, and the flasks incubated at 37°C for 4 h. Then, total 27-HC, cholesterol, and cell protein values were determined. (O) Wild-type fibroblasts (2 experiments); (Δ) NPC1-deficient fibroblasts (2 experiments). An average zero-time background value of 4.6 ng 27-HC/mg cell protein was subtracted from each data point. 27-HC and cholesterol values were normalized to those obtained for the cells treated with 10 mg HPCD (the 1.0/1.0 point). The average values for the 1.0/1.0 points for the two wild-type cells at 4 h were 35 ± 3 μg cholesterol/mg protein and 13 ± 1 ng 27-HC synthesized/mg protein. The corresponding average values for the two NPC1 flasks were 62 ± 1 μg cholesterol/mg protein and 18 ± 0.5 ng 27-HC synthesized/mg protein. The two lines are linear least square fits to the data below and above the 1.0/1.0 point, excluding the NPC1 outlier.
The dose-response curve for NPC1-deficient fibroblasts (triangles) in Fig. 2 was not significantly different from that observed for the wild-type (circles) in both the magnitude of the 27-HC biosynthetic response and in the position of the inflection point. It has been shown that ingested cholesterol accumulates to high levels in the endolysosomal compartments of these mutant cells both in vivo and in vitro but that the movement of PM cholesterol to the ER is intact under experimental conditions (19–22). This is presumably because, while the efflux of ingested LDL cholesterol is impeded in NPC1-deficient cells, the movement of PM cholesterol to the ER, as promoted by cholesterol loading via cyclodextrin or by 25-HC displacement, is functionally normal in the mutant cells (14, 23). Taken together, these findings suggest that the movement of PM cholesterol to ER and mitochondrial membranes is not limited by NPC1-mediated pathways; rather, the phenotype in NPC1 cells is related to impairment of the exit of endolysosomal cholesterol.
The dose-response curve in Fig. 2 inflected upward above the cholesterol level in the minimally depleted wild-type cells (set at 1.0/1.0). That is, the slope of the upper portion of this plot was 3.6-times higher than that of the lower portion (ignoring the high NPC1 outlier). Because the cell cholesterol in these experiments had been depleted by perhaps 10% in the preincubations, the inflection point (at a relative cholesterol value of ∼1.1 in Fig. 2) is actually close to the unmodified cholesterol content of these cells. This is noteworthy because the corresponding inflection points for the dependence of ER cholesterol on cholesterol loading in wild-type and NPC1-deficient fibroblasts are also near their physiological set point (12, 23).
Relationship of oxysterol biosynthesis to HMGR inactivation
Increasing PM cholesterol slightly above the physiological set point causes the acute inactivation of HMGR, an effect that requires sterol 27-hydroxylase activity (18, 24). To more closely examine the mediation of this relationship by endogenous oxysterol production, we compared the time course of 27-HC synthesis with that of HMGR inactivation following a pulse of cholesterol (Fig. 3). Prior to the addition of cholesterol, the basal level of 27-HC synthesis was very low, about 0.4–2.0 ng/mg protein/h (see Fig. 2), and HMGR activity was high. The production of 27-HC began immediately upon cholesterol enrichment in this experiment and proceeded more or less linearly for 2 h (Fig. 3A). The decline in HMGR activity also appeared to commence immediately when cell cholesterol was increased by ∼20% (Fig. 3B). The kinetics of inactivation were essentially first-order, with a half-time of ∼8.5 min. Thus, not only does the production of 27-HC commence within minutes of a jump in PM cholesterol, but the rate of inactivation of HMGR that it triggers becomes maximal after only a small amount of the oxysterol has been produced. Otherwise, the inactivation kinetics in Fig. 3B would not be first-order but, instead, would accelerate over time.
Effect of nonsterol intercalators on 27-HC production
The amphipath, 1-octanol, expands the ER cholesterol pool and stimulates the inactivation of HMGR, as if sending PM cholesterol to the intracellular membranes (17, 25). We reasoned that mitochondrial cholesterol and sterol 27-hydroxylase activity might be similarly affected by such agents. We therefore examined the effect of 1-octanol on the production of 27-HC. As shown in Fig. 4, treatment of fibroblasts with this bilayer intercalator led to a 2- to 3-fold increase in 27-HC; there was no corresponding change in cellular cholesterol (not shown).
Fig. 4.
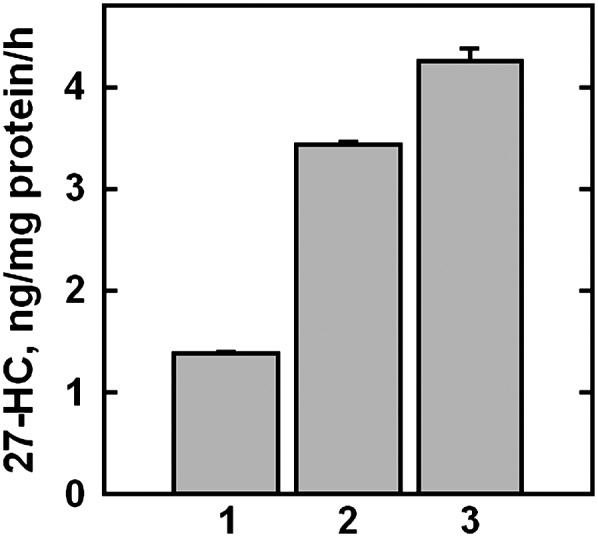
Effect of 1-octanol on 27-HC synthesis. Replicate flasks were incubated overnight in medium A. The cells were washed and the flasks incubated for 7 min at 37°C with 4 ml PBS containing 20 mg HPCD to reduce their background 27-HC. One flask was processed immediately (zero-time control), while three other flasks were incubated for 4 h at 37°C with 4 ml medium A plus these additions: bar 1, 0.6% ethanol (final); bars 2 and 3, 0.6% ethanol plus 0.6 mM or 0.8 mM 1-octanol, respectively. The values plotted were corrected for a zero-time value of 2.85 ng 27-HC/mg protein. Data are plotted as means ± average deviation for duplicate determinations. One of two similar experiments is shown.
The converse experiment was performed with lysophosphatides, intercalating agents known to rapidly reduce the level of cholesterol in the ER (17). We found that the pretreatment of fibroblasts with LPS reduced the production of 27-HC in response to loading cells with cholesterol (Fig. 5A). LPS also countered the rapid inactivation of HMGR accompanying the enrichment of cells with cholesterol (Fig. 5B).
Fig. 5.

Effect of lysophosphatidylserine (LPS) on 27-HC biosynthesis and HMGR activity. A: 27-HC biosynthesis. Four flasks of fibroblasts were incubated overnight in medium A. The cells were then preincubated for 10 min at 37°C in 4 ml PBS with or without 25 μM LPS. Then 10 mg HPCD alone (designated as minus CH) or 18 mg HPCD bearing 0.45 mg cholesterol (designated as plus CH) was added to the flasks for a 7 min incubation at 37°C. The buffer was replaced with 4 ml PBS ± 25 μM LPS and the flasks incubated at 37°C for an additional 1.5 h. 27-HC synthesis was determined in triplicate, a zero-time background of 4.6 ng 27-HC/mg protein subtracted, and the data expressed as means ± SEM relative to the control. A representative experiment. B: HMGR activity. The experiment was as in A except that the final incubation was for 2 h. HMGR activity was determined and data expressed as means ± average deviation relative to controls. One of two similar experiments is shown.
Effect of U18666A on 27-HC biosynthesis and HMGR activity
A diverse group of compounds referred to as class 2 amphipaths increase the cholesterol content of endolysosomes and reduce that in the ER (12, 21, 23, 26). Given the evidence that the cholesterol pool in the mitochondria seems to vary in parallel with that in the ER, we reasoned that treatment with class 2 amphipaths should counter the stimulation of 27-HC production in cells acutely loaded with cholesterol. We therefore incubated fibroblasts with U18666A, a prototypic class 2 amphipath, prior to pulsing the cells with exogenous cholesterol. We observed that U18666A greatly reduced 27-HC production (Fig. 6A). In parallel experiments, U18666A protected fibroblast HMGR against inactivation by cholesterol loading (Fig. 6B).
Fig. 6.

Effect of U18666A on the synthesis of 27-HC and HMGR activity. A: 27-HC biosynthesis. Four flasks were incubated overnight in medium A. Pairs of flasks were then incubated for 2 h at 37° C in medium A containing either 0.2% ethanol (black bar) or 0.2% ethanol + 4.5 μM U18666A (gray bar). To increase cell cholesterol, the medium in each flask was replaced with 4 ml PBS, a pulse of 18 mg HPCD bearing 0.43 mg cholesterol added, and the flasks incubated for 7 min at 37°C. The buffer was replaced with medium A containing 0.2% ethanol 4.5 μM U18666A and the flasks incubated at 37°C for an additional 3 h. 27-HC was then determined in triplicate on each of the duplicate flasks, a zero-time background of 4.6 ng 27-HC/mg protein subtracted and the data plotted as means ± SEM relative to the control. B: HMGR activity. The experiment was as in A except that single flasks were used, HMGR activity was determined in duplicate, and the data expressed as the mean ± average deviation relative to controls. Data are from one of two similar experiments.
DISCUSSION
Cell cholesterol regulates its own abundance through several feedback pathways involving different mechanisms that operate in diverse organelles on varied time scales. It is well understood that esterification in the ER serves to reversibly sequester excess cholesterol in lipid droplets (10, 12). This ER response is seen within a few minutes of pulse-labeling the cell surface with [3H]cholesterol (27). Cholesterol biosynthesis is inactivated just as rapidly following a small jump in PM cholesterol; this occurs in the ER through the proteolytic degradation of the rate limiting enzyme, HMGR [see Fig. 3B and (17)]. A surfeit of cholesterol also suppresses the expression of multiple proteins promoting sterol accretion. This inhibitory action is mediated by the sequestration of SREBP in ER complexes with SREBP cleavage-activating protein (SCAP) and Insig, stimulated by cholesterol and oxysterols, respectively (8–10). Conversely, a drop in cell cholesterol reduces the level of ER cholesterol (14), leading to the delivery of the active fragment of SREBP to the nuclei and the consequent expression of proteins mediating, among other functions, cholesterol biosynthesis and cholesterol endocytosis via LDL receptors (28).
Most of cell cholesterol is located in the PM and the endomembrane compartments with which it shares bilayer lipids (29, 30). On the other hand, the regulatory proteins that govern cholesterol homeostasis are mostly integral to the ER membrane (9, 10). Cholesterol briskly circulates between the PM and ER and, presumably, other cellular membranes, by a mechanism (or mechanisms) the molecular basis of which is not well established (31, 32). It may be the case that cholesterol partitions among available lipid compartments, including mitochondria, according to their relative sterol affinities; these, in turn, would be dependent on their phospholipid compositions (32–34). By these means, the homeostatic effectors in the ER and mitochondria can be informed of the sterol needs of the PM.
How the rate of oxysterol biosynthesis is regulated has not been systematically explored heretofore. Mitochondria do not synthesize their own cholesterol but rather partake of its intracellular circulation. While the cholesterol content of mitochondria has not been properly estimated because of PM contamination of those subcellular fractions, it appears that its level is quite low, thereby limiting the rate of synthesis of 27-HC. This supposition explains why bolstering the cholesterol in isolated mitochondria with cholesterol-cyclodextrin complexes promotes the production of 27-HC and why cyclodextrin itself has a similar effect in vitro: presumably it shuttles cholesterol present in contaminating PM fragments to the mitochondria (35–37). In keeping with these indirect findings, we find that, in intact cells, mitochondrial oxysterol production is greatly stimulated by augmenting PM cholesterol (Fig. 2) and that a principal source of this cholesterol substrate is derived from the PM (Fig. 1). These findings are consistent with earlier reports that the PM is a major source of cholesterol for the mitochondrial production of steroid hormones (38, 39).
The delivery of PM cholesterol to mitochondria depends upon the activity of intracellular sterol transport proteins (32). Nevertheless, Fig. 2 suggests that the rate of this process of delivery in cholesterol-depleted cultured fibroblasts varies acutely with the abundance of PM cholesterol relative to a threshold. That is, oxysterol biosynthesis increased about 30-fold with a ∼60% increase in cell (mostly, PM) cholesterol. Furthermore, this dose-response curve is inflected at the physiologic set point for PM cholesterol to which the fibroblasts home homeostatically. The shape of this “J-curve” parallels that seen earlier for the ER cholesterol pool and the inactivation of HMGR (12, 17).
The following mechanistic hypothesis can account for these data: cholesterol accumulates in complexes with PM phospholipids up to their stoichiometric capacity (34). The complexed sterol has a low escape tendency, fugacity, or chemical activity, simply referred to here as activity. Increments in PM cholesterol above this sterol/phospholipid equivalence point are not complexed and therefore have a greatly increased activity (31) [see also (40)]. The active cholesterol is thought to project from the bilayer surface more frequently and to escape to acceptors more readily. The predicted threshold in cholesterol activity at the equivalence point with phospholipids has been observed in the rate and extent of transfer of cholesterol to an exogenous cyclodextrin acceptor from both phospholipid-cholesterol monolayers (41) and biological membranes (17). Furthermore, small increments in the cholesterol of red cells or fibroblasts strongly enhance their susceptibility to attack by exogenous cholesterol oxidase (17, 42), presumably because the enzyme acts preferentially upon active sterol molecules projecting from the bilayer surface (43). That active cholesterol moves to cytoplasmic as well as extracellular (lipoprotein) acceptors explains why a slight elevation in PM cholesterol leads to a sharp rise in ER cholesterol, stimulation of cholesterol esterification, and the rapid inactivation of HMGR (12, 17). These diverse homeostatic mechanisms maintain PM cholesterol at the equivalence point of the cholesterol:phospholipid titration that defines the physiological set point.
The response of 27-HC biosynthesis to 1-octanol was tested because it is one of several amphipaths that appear to displace PM cholesterol from its association with phospholipids (25, 31). The displaced cholesterol is postulated to have high activity and, consequently, a propensity for transfer to other lipid compartments. Evidence for cholesterol activation by 1-octanol and other intercalating amphipaths includes their promotion of the interaction of red cell membrane cholesterol with cholesterol oxidase, saponins, and cyclodextrin as well as their ability to increase the pool of cholesterol in the ER (17, 25, 44). Consistent with these effects, 1-octanol and other amphipaths bring about the rapid inactivation of HMGR in fibroblasts (25); this is now known to depend upon the biosynthesis of 27-HC in mitochondria (18). The finding that 1-octanol stimulated 27-HC production is consistent with the premise that active PM cholesterol is the rate-limiting substrate for biosynthesis of the oxysterol.
LPS was found to block two effects of PM cholesterol loading; namely, its stimulation of 27-HC biosynthesis and the consequent inactivation of HMGR (Fig. 5). We employed LPS in this study because intercalated lysophosphatides apparently decrease the activity of PM cholesterol molecules, just as do the resident bilayer phospholipids (31). This hypothesis is supported by the fact that lysophosphatides reduce the susceptibility of PM cholesterol to cholesterol oxidase, reduce its transfer to cyclodextrin, reduce the size of the ER cholesterol pool, and inhibit the ability of intercalators such as 1-octanol to activate the sterol (17, 25). We therefore conclude from Fig. 5 that LPS countered the production of mitochondrial 27-HC by reducing the level of active PM cholesterol and that the proteolysis of HMGR is sensitive to oxysterols rather than to cholesterol itself (18).
We had originally reported evidence suggesting that lysophosphatidylcholine but not LPS reduces PM cholesterol activity (44). However, we have more recently shown in numerous tests that the potencies of these two lysophosphatides for reducing cholesterol activity are roughly comparable; for example, compare Fig. 5 to references 17 and 25. LPS is more convenient than lysophosphatidylcholine because of its greater aqueous solubility.
The stimulation of 27-HC biosynthesis by excess PM cholesterol was also inhibited by U18666A (Fig. 6). This agent was of relevance because it promotes the retention of endocytic PM and lipoprotein cholesterol in late endolysosomal compartments and reduces the level of cholesterol in the ER (12, 21, 45). The inhibition of the biosynthesis of 27-HC by U18666A can therefore be ascribed to a block in the requisite expansion of the mitochondrial cholesterol pool, similar to its effect on the ER.
U18666A treatment has been shown to mimic the defect in NPC1-deficient cells in many respects; furthermore, it may act upon the same pathway (21, 23, 46). In both systems, there is massive accumulation of cholesterol and other membrane lipids in the endolysosomal pathway as well as diverse and incompletely understood alterations in cellular cholesterol management. However, our experiments revealed characteristic differences between these two systems. In NPC1-deficient fibroblasts, the dependence on PM cholesterol of both 27-HC biosynthesis (Fig. 2) and ER cholesterol pool size is normal (12, 23). In contrast, U18666A treatment significantly reduces 27-HC biosynthesis (Fig. 6) and the size of the ER cholesterol pool (12). It could be that the two systems report on different aspects of cholesterol management. In particular, the compartments bearing cholesterol in unperturbed NPC1-deficient cells are presumably in a long-term steady-state relationship; indeed, the longevity of subjects with this disease suggests that PM, ER, and mitochondrial cholesterol pools function relatively normally despite the chronic expansion of the endolysosomal lipid compartment. In contrast, perturbations by U18666A are acute, so that the membrane lipids do not have time to adjust through homeostatic mechanisms. In particular, by causing the short-term accumulation of cholesterol in the endolysosomes, U18666A could reduce the active fraction of PM cholesterol and, consequently, diminish the size of the ER and mitochondrial cholesterol pools. Alternatively, compounds such as U18666A might have direct effects on membrane sites other than endolysosomes or the NPC1 protein (46).
Oxysterols signal an excess of PM cholesterol to homeostatic proteins by representing the magnitude of its superabundance above the physiological set point. It is 27-HC, rather than cholesterol, that stimulates the rapid inactivation of HMGR (18). This effect is mediated by the activation of Insig through its binding of oxysterol molecules (8) and/or through oxysterol interactions with the ER bilayer (47). From Fig. 3, we infer that it takes only a small rise in 27-HC to rapidly downregulate HMGR, here, that which accumulated in the cells during the few minutes that followed a cholesterol pulse. Similarly, it has been shown that incrementing resting fibroblast PM cholesterol by a few percent triggers a marked inactivation of HMGR (17). To get a rough idea of how much endogenous oxysterol is required to inhibit cholesterol biosynthesis, we calculated from the values in Fig. 3 that the production of 20 ng of 27-HC/mg cell protein over 30 min can inactivate most of the HMGR. Because the cells retain about one-half of this oxysterol and contain ∼4 mg water/mg protein, the overall cytoplasmic concentration of 27-HC would be roughly 6 nM. This value is far less than the concentration of exogenous oxysterol needed to elicit the same degree of HMGR inhibition (18, 25), suggesting an efficient delivery system.
Converting trace amounts of excess cell cholesterol into the more water-soluble oxysterol form creates potent homeostatic signals (11). This is most clearly seen in the case of the activation of nuclear LXR receptors by oxysterols. The high intermembrane mobility of oxysterols and their rapid egress from cells also can serve in homeostatic regulation by curtailing the duration of their intracellular action. However, conversion to oxysterols probably does not constitute a high-capacity cholesterol disposal system, at least in these fibroblasts, because even at the highest rate of 27-HC biosynthesis we observed, ∼50 ng 27-HC/mg cell protein/h, the elimination of just 1% of cell cholesterol, which amounts to ∼35 μg/mg cell protein, would take perhaps 10 h. The situation could be different in cholesterol-loaded scavenger cells like macrophages. In any case, 27-HC would not seem to be essential to acute cholesterol management, given the relatively benign phenotype of individuals with cerebrotendinous xanthomatosis who lack the sterol 27-hydroxylase and the fibroblasts cultured therefrom (18). Presumably, parallel pathways and/or redundant oxysterol messengers maintain cholesterol homeostasis in that case.
Our findings provide this general hypothesis for the regulation of 27-HC biosynthesis: the physiological set point for PM cholesterol is its stoichiometric equivalence with its phospholipid partners, above which active cholesterol accumulates (31). 27-HC production in the mitochondria derives from the active excess of PM cholesterol, parallels its magnitude, and enhances its feedback signal to limit further cholesterol accretion. The time frame for these effects is a matter of minutes. Although obtained in cultured human fibroblasts under nonphysiologic conditions, the evidence that active PM cholesterol regulates 27-HC biosynthesis deepens our appreciation of the role side-chain oxysterols might play in cholesterol homeostasis (1, 11).
Acknowledgments
We thank David Scherrer for assistance with MS analysis.
Footnotes
Abbreviations:
- ER
- endoplasmic reticulum
- HC
- hydroxycholesterol
- HMGR
- hydroxy-3-methylglutaryl CoA reductase
- HPCD
- 2-β-hydroxypropylcyclodextrin
- LPS
- 1-palmitoyl-lysophosphatidylserine
- medium A
- DMEM plus 5% lipoprotein-deficient serum
- NPC1
- Niemann-Pick C type 1
- PBS
- 0.15 M NaCl + 5 mM NaPi, pH 7.4
- PM
- plasma membrane
- SREBP
- sterol regulatory element-binding protein
This work was supported by National Institutes of Health Grants HL-28448 (Y.L.) and HL-67773 (D.S.O.).
REFERENCES
- 1.Gill S., Chow R., Brown A. J. 2008. Sterol regulators of cholesterol homeostasis and beyond: the oxysterol hypothesis revisited and revised. Prog. Lipid Res. 47: 391–404. [DOI] [PubMed] [Google Scholar]
- 2.Ory D. S. 2004. Nuclear receptor signaling in the control of cholesterol homeostasis: have the orphans found a home? Circ. Res. 95: 660–670. [DOI] [PubMed] [Google Scholar]
- 3.Schroepfer G. J., Jr. 2000. Oxysterols: modulators of cholesterol metabolism and other processes. Physiol. Rev. 80: 361–554. [DOI] [PubMed] [Google Scholar]
- 4.Wong J., Quinn C. M., Gelissen I. C., Brown A. J. 2008. Endogenous 24(S),25-epoxycholesterol fine-tunes acute control of cellular cholesterol homeostasis. J. Biol. Chem. 283: 700–707. [DOI] [PubMed] [Google Scholar]
- 5.Chawla A., Boisvert W. A., Lee C. H., Laffitte B. A., Barak Y., Joseph S. B., Liao D., Nagy L., Edwards P. A., Curtiss L. K., et al. 2001. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell. 7: 161–171. [DOI] [PubMed] [Google Scholar]
- 6.Babiker A., Andersson O., Lund E., Xiu R. J., Deeb S., Reshef A., Leitersdorf E., Diczfalusy U., Bjorkhem I. 1997. Elimination of cholesterol in macrophages and endothelial cells by the sterol 27-hydroxylase mechanism. Comparison with high density lipoprotein-mediated reverse cholesterol transport. J. Biol. Chem. 272: 26253–26261. [DOI] [PubMed] [Google Scholar]
- 7.Meaney S., Bodin K., Diczfalusy U., Bjorkhem I. 2002. On the rate of translocation in vitro and kinetics in vivo of the major oxysterols in human circulation: critical importance of the position of the oxygen function. J. Lipid Res. 43: 2130–2135. [DOI] [PubMed] [Google Scholar]
- 8.Radhakrishnan A., Ikeda Y., Kwon H. J., Brown M. S., Goldstein J. L. 2007. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. USA. 104: 6511–6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein J. L., DeBose-Boyd R. A., Brown M. S. 2006. Protein sensors for membrane sterols. Cell. 124: 35–46. [DOI] [PubMed] [Google Scholar]
- 10.Chang T. Y., Chang C. C., Ohgami N., Yamauchi Y. 2006. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 22: 129–157. [DOI] [PubMed] [Google Scholar]
- 11.Bjorkhem I. 2009. Are side-chain oxidized oxysterols regulators also in vivo? J. Lipid Res. 50 (Suppl.): S213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lange Y., Ye J., Rigney M., Steck T. L. 1999. Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. J. Lipid Res. 40: 2264–2270. [PubMed] [Google Scholar]
- 13.Lange Y., Steck T. L. 1997. Quantitation of the pool of cholesterol associated with acyl-CoA:cholesterol acyltransferase in human fibroblasts. J. Biol. Chem. 272: 13103–13108. [DOI] [PubMed] [Google Scholar]
- 14.Frolov A., Zielinski S. E., Crowley J. R., Dudley-Rucker N., Schaffer J. E., Ory D. S. 2003. NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. J. Biol. Chem. 278: 25517–25525. [DOI] [PubMed] [Google Scholar]
- 15.Lange Y., Echevarria F., Steck T. L. 1991. Movement of zymosterol, a precursor of cholesterol, among three membranes in human fibroblasts. J. Biol. Chem. 266: 21439–21443. [PubMed] [Google Scholar]
- 16.Brown M. S., Dana S. E., Goldstein J. L. 1973. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts by lipoproteins. Proc. Natl. Acad. Sci. USA. 70: 2162–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lange Y., Ye J., Steck T. L. 2004. How cholesterol homeostasis is regulated by plasma membrane cholesterol in excess of phospholipids. Proc. Natl. Acad. Sci. USA. 101: 11664–11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lange Y., Ory D. S., Ye J., Lanier M. H., Hsu F-F., Steck T. L. 2008. Effectors of rapid homeostatic responses of endoplasmic reticulum cholesterol and 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem. 283: 1445–1455. [DOI] [PubMed] [Google Scholar]
- 19.Vance J. E. 2006. Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett. 580: 5518–5524. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J., Dudley-Rucker N., Crowley J. R., Lopez-Perez E., Issandou M., Schaffer J. E., Ory D. S. 2004. The steroidal analog GW707 activates the SREBP pathway through disruption of intracellular cholesterol trafficking. J. Lipid Res. 45: 223–231. [DOI] [PubMed] [Google Scholar]
- 21.Koh C. H. V., Cheung N. S. 2006. Cellular mechanism of U18666A-mediated apoptosis in cultured murine cortical neurons: bridging Niemann-Pick disease type C and Alzheimer’s disease. Cell. Signal. 18: 1844–1853. [DOI] [PubMed] [Google Scholar]
- 22.Lange Y., Ye J., Rigney M., Steck T. L. 2002. Dynamics of lysosomal cholesterol in Niemann-Pick type C and normal human fibroblasts. J. Lipid Res. 43: 198–204. [PubMed] [Google Scholar]
- 23.Lange Y., Ye J., Rigney M., Steck T. 2000. Cholesterol movement in Niemann-Pick type C cells and in cells treated with amphiphiles. J. Biol. Chem. 275: 17468–17475. [DOI] [PubMed] [Google Scholar]
- 24.Axelson M., Larsson O. 1995. Low density lipoprotein (LDL) cholesterol is converted to 27-hydroxycholesterol in human fibroblasts. Evidence that 27-hydroxycholesterol can be an important intracellular mediator between LDL and the suppression of cholesterol production. J. Biol. Chem. 270: 15102–15110. [DOI] [PubMed] [Google Scholar]
- 25.Lange Y., Ye J., Steck T. L. 2005. Activation of membrane cholesterol by displacement from phospholipids. J. Biol. Chem. 280: 36126–36131. [DOI] [PubMed] [Google Scholar]
- 26.Lange Y., Steck T. L. 1994. Cholesterol homeostasis. Modulation by amphiphiles. J. Biol. Chem. 269: 29371–29374. [PubMed] [Google Scholar]
- 27.Lange Y., Ye J., Strebel F. 1995. Movement of 25-hydroxycholesterol from the plasma membrane to the rough endoplasmic reticulum in cultured hepatoma cells. J. Lipid Res. 36: 1092–1097. [PubMed] [Google Scholar]
- 28.Radhakrishnan A., Goldstein J. L., McDonald J. G., Brown M. S. 2008. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 8: 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maxfield F. R., Menon A. K. 2006. Intracellular sterol transport and distribution. Curr. Opin. Cell Biol. 18: 379–385. [DOI] [PubMed] [Google Scholar]
- 30.van Meer G., Voelker D. R., Feigenson G. W. 2008. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9: 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lange Y., Steck T. L. 2008. Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Prog. Lipid Res. 47: 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prinz W. A. 2007. Non-vesicular sterol transport in cells. Prog. Lipid Res. 46: 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wattenberg B. W., Silbert D. F. 1983. Sterol partitioning among intracellular membranes. Testing a model for cellular sterol distribution. J. Biol. Chem. 258: 2284–2289. [PubMed] [Google Scholar]
- 34.Radhakrishnan A., McConnell H. M. 2000. Chemical activity of cholesterol in membranes. Biochemistry. 39: 8119–8124. [DOI] [PubMed] [Google Scholar]
- 35.Petrack B., Latario B. J. 1993. Synthesis of 27-hydroxycholesterol in rat liver mitochondria: HPLC assay and marked activation by exogenous cholesterol. J. Lipid Res. 34: 643–649. [PubMed] [Google Scholar]
- 36.Li X., Hylemon P., Pandak W. M., Ren S. 2006. Enzyme activity assay for cholesterol 27-hydroxylase in mitochondria. J. Lipid Res. 47: 1507–1512. [DOI] [PubMed] [Google Scholar]
- 37.Stravitz R. T., Vlahcevic Z. R., Russell T. L., Heizer M. L., Avadhani N. G., Hylemon P. B. 1996. Regulation of sterol 27-hydroxylase and an alternative pathway of bile acid biosynthesis in primary cultures of rat hepatocytes. J. Steroid Biochem. Mol. Biol. 57: 337–347. [DOI] [PubMed] [Google Scholar]
- 38.Gocze P. M., Freeman D. A. 1993. Plasma membrane cholesterol is utilized as steroidogenic substrate in Y-1 mouse adrenal tumor cells and normal sheep adrenal cells. Exp. Cell Res. 209: 21–25. [DOI] [PubMed] [Google Scholar]
- 39.Freeman D. A., Romero A., Choi Y. S. 1998. Plasma membrane steroidogenic cholesterol: the relative importance of membrane internalization rate and cholesterol extraction rate of internalized membrane. Endocr. Res. 24: 619–622. [DOI] [PubMed] [Google Scholar]
- 40.Huang J., Feigenson G. W. 1999. A microscopic interaction model of maximum solubility of cholesterol in lipid bilayers. Biophys. J. 76: 2142–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radhakrishnan A., McConnell H. M. 2002. Thermal dissociation of condensed complexes of cholesterol and phospholipid. J. Phys. Chem. B. 106: 4755–4762. [Google Scholar]
- 42.Lange Y., Cutler H. B., Steck T. L. 1980. The effect of cholesterol and other intercalated amphipaths on the contour and stability of the isolated red cell membrane. J. Biol. Chem. 255: 9331–9337. [PubMed] [Google Scholar]
- 43.Ahn K. W., Sampson N. S. 2004. Cholesterol oxidase senses subtle changes in lipid bilayer structure. Biochemistry. 43: 827–836. [DOI] [PubMed] [Google Scholar]
- 44.Lange Y., Matthies H., Steck T. L. 1984. Cholesterol oxidase susceptibility of the red cell membrane. Biochim. Biophys. Acta. 769: 551–562. [DOI] [PubMed] [Google Scholar]
- 45.Liscum L., Sturley S. L. 2004. Intracellular trafficking of Niemann-Pick C proteins 1 and 2: obligate components of subcellular lipid transport. Biochim. Biophys. Acta. 1685: 22–27. [DOI] [PubMed] [Google Scholar]
- 46.Ko D. C., Gordon M. D., Jin J. Y., Scott M. P. 2001. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol. Biol. Cell. 12: 601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gale S. E., Westover E. J., Dudley N., Krishnan K., Merlin S., Scherrer D. E., Han X., Zhai X., Brockman H. L., Brown R. E., et al. 2009. Side chain oxygenated cholesterol regulates cellular cholesterol homeostasis through direct sterol-membrane interactions. J. Biol. Chem. 284: 1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]