

Abstract
We have investigated the production of reactive oxygen species (ROS) by Complex I in isolated open bovine heart submitochondrial membrane fragments during forward electron transfer in presence of NADH, by means of the probe 2′,7′-Dichlorodihydrofluorescein diacetate. ROS production by Complex I is strictly related to its inhibited state. Our results indicate that different Complex I inhibitors can be grouped into two classes: Class A inhibitors (Rotenone, Piericidin A and Rolliniastatin 1 and 2) increase ROS production; Class B inhibitors (Stigmatellin, Mucidin, Capsaicin and Coenzyme Q2) prevent ROS production also in the presence of Class A inhibitors. Addition of the hydrophilic Coenzyme Q1 as an electron acceptor potentiates the effect of Rotenone-like inhibitors in increasing ROS production, but has no effect in the presence of Stigmatellin-like inhibitors; the effect is not shared by more hydrophobic quinones such as decylubiquinone. This behaviour relates the prooxidant CoQ1 activity to a hydrophilic electron escape site. Moreover the two classes of Complex I inhibitors have an opposite effect on the increase of NADH–DCIP reduction induced by short chain quinones: only Class B inhibitors allow this increase, indicating the presence of a Rotenone-sensitive but Stigmatellin-insensitive semiquinone species in the active site of the enzyme. The presence of this semiquinone was also suggested by preliminary EPR data. The results suggest that electron transfer from the iron–sulphur clusters (N2) to Coenzyme Q occurs in two steps gated by two different conformations, the former being sensitive to Rotenone and the latter to Stigmatellin.
Keywords: Complex I inhibitor; Reactive oxygen species; Iron-sulphur cluster; 2′, 7′-Dichlorodihydrofluorescein diacetate
1. Introduction
Complex I is a very large enzyme catalyzing at the entry point of the mitochondrial electron transport chain [1–3]. The total number of subunits in the bovine heart enzyme is 45 [4] for a molecular mass of about 1000 KDa. Seven subunits are products of the mitochondrial genome [5,6] that correspond to hydrophobic subunits named ND1–ND6 and ND4 L. The molecular mechanism of catalysis of this enzyme is not completely understood. The main reason is the lack of detailed structural information of the membrane part of Complex I, although X-ray structure of the extramembrane part was determined recently by Sazanov and Hinchliffe [7] utilizing Thermus thermophilus HB-8 enzyme. The minimal active form of Complex I is that found in bacteria, composed of 14 subunits, all of which are homologous to their mitochondrial counterparts. Based on this comparison, all other subunits are called “accessory subunits” and their functional role in the mitochondrial enzyme is not yet clear. The Complex I enzyme oxidizes NADH transferring electrons to a lipid soluble electron carrier, namely Ubiquinone or Coenzyme Q (CoQ). Based on the thermodynamic profiles of redox active groups, the FMN is considered to be the direct electron acceptor of NADH and subsequently electrons are transferred to the iron–sulphur clusters. Bovine heart Complex I contains 8 distinct iron–sulphur clusters (cluster N1a, N3, N1b, N4, N5, N6a, N6b, N2). Clusters N3–N6 are considered to share the same midpoint redox potential (Em) values (− 250 mV), and are called the isopotential group. Two clusters have different characteristics: N1a, that is the [2Fe–2S] type cluster, and has the lowest midpoint potential (Em = −370 mV) and cluster N2, that is the [4Fe–4S] type cluster which has the highest Em value (between −150 mV and −50 mV), and is located close to the interface between the peripheral and the membrane arms [7]. Thus cluster N2 is considered to be the direct electron donor to ubiquinone. In the tightly coupled bovine heart SMP, initially three distinct EPR semiquinone (SQ) signals were proposed as Complex I components [8], but subsequently revised to two species of SQ signals [9]; one is uncoupler sensitive the other is insensitive. In the presence of reduced cluster N2, the former SQ species shows extremely fast spin relaxation (thus designated as SQNf) while the latter shows much slower spin relaxation (designated as SQNs). Direct spin–spin interaction between cluster N2 and SQNf was demonstrated and their mutual distance was estimated to be 12 Å [10,11]. In uncoupled SMP only the slowly relaxing SQ species is observed [8,9].
Complex I is inhibited by more than 60 different families of compounds [12] starting from Rotenone, the prototype of this series, to a number of synthetic insecticides/acaricides. These inhibitors were grouped into three classes based on their effects on the kinetic behaviour of the enzyme: Class I/A (the prototype of which is Piericidin A), Class II/B (the prototype of which is Rotenone) and Class C (the prototype of which is Capsaicin). Nevertheless, from kinetic studies it has not been possible to assign different binding sites for these three classes of inhibitors. Thus it is commonly accepted that they share the same large hydrophobic pocket in the enzyme [13].
Complex I is also involved in the formation of the trans-membrane proton gradient with a stoichiometry of 4H+/2e−. The limited knowledge about the 3D-structure and the function of the whole Complex I makes it difficult to predict the proton pumping mechanism of Complex I across the inner mitochondrial membrane [9,14].
Besides its well known redox role in the electron transport chain, Complex I is also considered to be one of the main sites of reactive oxygen species (ROS) production; electrons leaked at Complex I can reduce oxygen and give rise to superoxide anion [15]. The mechanism of superoxide production by Complex I is not yet clear probably because of the lack of knowledge on the exact sequence of the electron carriers and how electron transfer is coupled to proton translocation. The sites of ROS production in the mitochondrial electron transport chain have been localized in Complex I and Complex III [16]. Whereas the site of electron escape in Complex III has been identified in the so called center “o”, the direct oxygen reductant site in Complex I has not been established yet.
Recently, using different Complex I inhibitors to functionally dissect the enzyme, it was suggested that iron–sulphur cluster N2 could be the site of the electron leak [17], but N2–SQNf region [18], ubisemiquinone (SQNf) [19], FMN [15,20,21] and iron–sulphur cluster N1a [22] have also been proposed as electron donors to oxygen. In addition, it was found that defective Complex I produces more reactive oxygen species (ROS) [16], suggesting that structural modifications of the enzyme may play a crucial role in the ROS production process.
The superoxide production by Complex I is much higher during the reverse electron transport from succinate to NAD+ [19,23], than during the forward electron transport. The reasons of this discrepancy are still not understood.
An understanding of the detailed mechanism of reaction of Complex I is required not only for advancement in basic knowledge but also in biomedical research. In fact, a number of devastating neurodegenerative disorders are associated with Complex I deficiency, resulting in a decline of energy production by the respiratory chain and in increased production of reactive oxygen species (ROS) (for reviews see [24–28]).
Based upon the latter observations we have studied the effect of different Complex I inhibitors on the ROS production to elucidate the mechanism by which Complex I transfers electrons to molecular oxygen, with the additional aim to exploit superoxide generation to shed light on the mechanism of electron transfer to the natural acceptor, Coenzyme Q10.
2. Materials and methods
2.1. Materials
2′,7′-Dichlorodihydrofluorescein diacetate (DCFDA) was purchased from Molecular probes, Invitrogen, Milano Italy. Mucidin (Strobilurin A) was a kind gift from Dr. F. Nerud of the Academy of Sciences in Prague, Czech Republic. Rolliniastatin-1 and -2 were gifts from Dr E. Estornell of the University of Valencia, Spain. Piericidin A and Stigmatellin were purchased from Fluka, Sigma-Aldrich, Milano, Italy. All other chemicals were purchased from Sigma-Aldrich, Milano, Italy.
2.2. Preparations
Submitochondrial particles (SMP) were prepared from bovine heart mitochondria (BHM) by sonic irradiation of the frozen and thawed BHM [29]; the particles were essentially broken membrane fragments [30]. Protein was evaluated by the Biuret method of Gornall et al. [31] with addition of 10% sodium deoxycholate and using bovine serum albumin (BSA) as the standard.
2.2.1. Measure of hydrogen peroxide production
The method used to measure H2O2 production in submitochondrial particles (SMP) is based on the fluorogenic probe 2′,7′-Dichlorodihydrofluorescein diacetate (DCFDA or H2DCFDA) which emits an intense green fluorescence only after deacylation and subsequent oxidation [32,33]. The advantage to use this probe is that it does not inhibit the activity of Complex I [34]. Alternatively H2O2 production was measured using Amplex Red. ROS production by SMP was measured in a fluorescence plate reader using a 96-well microtiter plate. In each well were present 0.5 mg/ml SMP (pretreated with 1.8 μM Mucidin) and 5 μM DCFDA or 10 μM Amplex Red to a final volume of 0.2 ml with KCl, 10 mM TRIS, 1 mM EDTA buffer, pH 7.5, 25 °C. The reaction was started by the addition of 150 μM NADH, in presence and in absence of different respiratory inhibitors and/or quinone acceptors.
2.2.2. Enzyme assays
NADH–CoQ reductase was assayed essentially as described by Yagi [35] and modified by Degli Esposti et al. [36] in the presence of 2 mM KCN and 2 μM Antimycin A to block Complexes IV and III, respectively. Determination of the kinetic constants was accomplished at saturating concentration of NADH (150 μM) and 150 μM of CoQ1 following the decrease in absorbance at 340 minus 380 nm, in a Jasco V550 spectrophotometer equipped with dual wavelength device, using an extinction coefficient of 3.5 mM−1 cm−1.
NADH–O2 reductase activity was assayed essentially in the same conditions avoiding only KCN and Antimycin A in the assay mixture. To compare the inhibition effect of different Complex I inhibitors with ROS production, we performed the NADH–CoQ1 reductase assays with high protein concentration (0.25 mg/ml of SMP).
NADH–DCIP reductase activity was assayed as above, following the reduction of DCIP absorbance at 748 nm using an extinction coefficient of 0.8 mM−1 cm−1 with 40 μg/ml of SMP. Activity was recorded in the presence and absence of Complex I inhibitors and CoQ1 (25 μM) or DB (25 μM).
2.2.3. EPR sample preparation
EPR samples were prepared as follows. Submitochondrial particles were suspended in the reaction buffer (Sucrose 0.25 M, TRIS 10 mM, EDTA 1 mM) to be 30 mg/mL. The suspension in a glass test tube was kept on ice. Antimycin A 5 μM, Carboxin 100 μM and Mucidin 1.8 μM were added and the mixtures were incubated on ice for at least 5 min.
SMP samples were treated with different Complex I inhibitors to completely block the enzyme: 10 μM Rotenone and 80 μM Stigmatellin. The reaction was initiated by adding 150 μM NADH.
These treated SMP were rapidly transferred into EPR tubes and immediately frozen in dry ice/ethanol mixture, typically within 10 s after adding the substrate and were stored in liquid nitrogen until analysis.
2.2.4. EPR measurements
EPR experiments were performed at the Department of Chemical Sciences, University of Padova, Italy, with a Bruker ER 200D spectrometer operating at X-band (9.4 GHz), equipped with a rectangular cavity ER4102ST, and a variable-temperature controller Bruker ER 4111 VT; the microwave frequency was measured by a frequency counter (model HP 5342A).
All spectra were obtained using the following parameters: microwave power 0.66 mW; modulation amplitude 0.5 mT; modulation frequency 100 kHz; time constant 41 ms; conversion time 82 ms; scan width 10 mT; 1024 points; temperature 180 K; sample volume 300 μl. All spectra have an average of 9 scans and have been corrected by subtraction of the oxidized SMP background.
3. Results
3.1. Suitability of fluorescent probes to investigate ROS production in SMP
Fluorescent probes are widely used for ROS detection in biological systems; at present several such probes are available: dihydro-compounds such as 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH–DA), dihydroethidium (HE) and 10-acetyl-3,7-dihydroxyphenoxazine (AmplexRed) are mostly used. DCFH and Amplex Red are suggested as specific probes for H2O2, while dihydroethidium seems to be more suitable for detection. Anyway all fluorescent probes for ROS detection suffer a lack of selectivity and it is generally thought that they react with various types of ROS [22,23,27], although they are generally used for detecting total oxidative activity in living cells or tissues.
DCFDA is routinely used in intact cells, being taken up and deacetylated by endogenous hydrolases to a form (DCFH) that is then oxidized by peroxides (including H2O2) to fluorescent 2′,7′-Dichlorofluorescein (DCF). It has been shown [34] that mitochondria and sub-mitochondrial particles can deacetylate the probe and oxidize it by ROS.
Using DCFDA or Amplex Red for reliable superoxide detection in SMP, it is required that deacetylation of DCFDA probe and conversion of superoxide to hydrogen peroxide proceed at a rate that is not rate-limiting with respect to superoxide production.
Fig. 1 shows that addition of hydrogen peroxide enhances the probe fluorescence to an extent largely exceeding that one obtained with respiratory substrates, suggesting that the non-reactive acetyl ester is cleaved at a rate higher than that of natural H2O2 production.
Fig. 1.
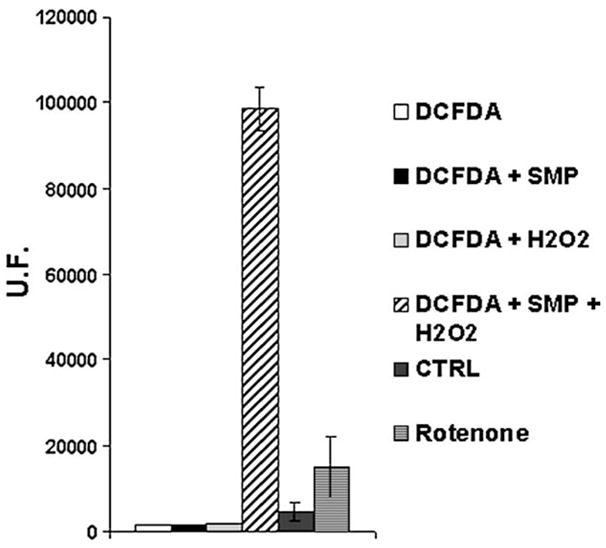
Suitability of DCFDA probe (5 μM) for H2O2 determination in presence of SMP (0.5 mg/ml) supplemented with 150 μM NADH (CTRL) and treated with 1 μM Rotenone (Rotenone). The amount of the deacetylated probe by SMP is largely exceeding that one oxidized by respiratory substrates as indicated by high fluorescence achieved with 5 μM of H2O2. Fluorescence intensity was detected after 2400 s from NADH addition. No fluorescence was detected by addition of 5 μM hydrogen peroxide in absence of SMP. Data are the mean of at least five different determinations±standard deviation.
Considering DCFH more specific for peroxide than for superoxide we have evaluated the effect of SOD on the fluorescence levels detected. The conversion of superoxide anion to hydrogen peroxide catalyzed by SOD induces a modest fluorescence increase both in control and in Complex I inhibited particles (i.e. +30% in presence of Rotenone plus SOD vs Rotenone alone, data not shown) without substantial alterations of their relative ratio. This feature suggests that our system is suitable for ROS detection even in the absence of SOD.
Amplex Red is a non-fluorescent molecule that originates resorufin, a highly fluorescent product when oxidized by H2O2 [37,38]. As an advantage over DCFH, Amplex Red presents low background fluorescence as well as stability and high fluorescence power on oxidation. Furthermore, its excitation and emission maximum wavelengths subsist in a spectral zone that has little susceptibility to interference from autofluorescence in assays where biological samples are used [37,38].
Amplex Red is normally used in association with HRP (horse radish peroxidase). In our system, oxidation of the probe is achieved even without HRP addition.
Addition of KCN completely prevents Amplex Red oxidation suggesting that in mitochondrial membranes are present enzymes with KCN sensitive peroxidase activity. In fact cyanide has been widely used as an inhibitor of many peroxidases and oxidases [39].
Nevertheless, it must be taken into account that in biological systems NADH may interfere with Amplex Red/peroxidase assay system resulting in a decreased fluorescence [40].
Dihydroethidium has been used as a fluorescent probe for detecting [41–44]. Indeed, when HE is oxidized by superoxide, it originates ethidium (E+), a fluorescent compound [45].
Even this probe shows some limitations: cytochrome c is able to oxidize HE and the superoxide detection might not be quantitative by this method because HE increases the superoxide/hydrogen peroxide dismutation rate [42,46].
In addition E+ fluorescence is enhanced in presence of DNA. For this reason HE is not suitable for our DNA-free system.
Because of the different limitations showed by these probes we have carried out experiments using all three probes, with superimposable results. Nevertheless, the systematic studies reported were performed mainly using DCFDA.
3.2. ROS production by Complex I in presence of reducing and oxidizing substrates
To study ROS production by Complex I in situ, we have used mitochondrial membrane fragments (SMP) derived by ultrasonic irradiation of Bovine Heart Mitochondria (BHM). SMP used in this study have been shown to be broken membrane fragments devoid of permeability barriers and of membrane potential [30]. For this reason they are not coupled and are incapable of reverse electron transfer from succinate to NAD+. Since the latter reaction is considered to be a major source of ROS, these particles represent an ideal system to investigate direct electron transfer in Complex I from NADH to CoQ without interference by the reverse reaction.
Mucidin is an inhibitor of Complex III at center “o” (or P) that completely prevents ROS formation by this complex [17] even in presence of Antimycin A.
Addition of NADH to SMP inhibited with 1.8 μM Mucidin, that completely inhibits electron transfer in Complex III, does not induce electron escape from Complex I to molecular oxygen, suggesting that a fully reduced state of Complex I is necessary but not sufficient for ROS production.
For this reason Mucidin can functionally isolate Complex I from further segments of the respiratory chain and we used 1.8 μM Mucidin-treated SMP as control in all experiments.
Addition of oxidized Coenzyme Q1, or decylubiquinone (DB) to such Mucidin-inhibited particles is not able to stimulate ROS production.
On the other hand Complex I inhibition with Rotenone induces a strong increase in ROS generation. Moreover addition of CoQ1 to the Rotenone-inhibited enzyme stimulates ROS production, while addition of decylubiquinone has no effect or even induces a slight decrease of ROS production (Fig. 2). Because both CoQ1 and DB are good oxidizing substrates for Complex I activity, this difference is very puzzling and needs an explanation. The reason for this different behaviour may be due to the higher water solubility of CoQ1 with respect to DB [47], suggesting that the prooxidant activity of oxidized quinones is due to their interaction with an hydrophilic site for electron escape.
Fig. 2.

ROS production in SMP (0.5 mg/ml) treated with 1.8 μM Mucidin, 10 μM DPI, 75μM CoQ1, 1 μM Rotenone, 1 μM Rotenone plus 75 μM CoQ1 and 1 μM Rotenone plus 50 μM DB. ROS production was detected following the fluorescence variations in presence of 5 μM DCFDA. Each value was detected after 2400 s from 150 μM NADH addition and is expressed as percent of fluorescence change with respect to the control. Data are the mean of at least four different determinations.
3.2.1. Effect of Complex I inhibitors on ROS production
Complex I activity is very sensitive to a large spectra of compounds (cf. [12] for a review), among which we can find short chain quinones (i.e. CoQ2) and Complex III center “o” inhibitors (i.e. Stigmatellin). For this reason we have undertaken a deep analysis on the effect of different Complex I inhibitors on ROS production. The results depicted in Table 1 and Fig. 3 allow a distinction of inhibitors into two classes: Piericidin A and Rolliniastatin-1 and -2 behave like Rotenone, whereas Stigmatellin, Capsaicin, Mucidin and Coenzyme Q2 (CoQ2) prevent the oxidation of the probe and rather decrease it below control levels. The effect of an inhibitor of the former class to stimulate ROS production is abolished by the combined presence of an inhibitor of the latter class.
Table 1.
Effect of different Complex I inhibitors on ROS production in SMP supplemented with 150 μM NADH
| Inhibitor | DCFDA | Amplex Red |
|---|---|---|
| Rotenone (2 μM) | +53%±23 | +62%±3 |
| Piericidin A (1 μM) | +101%±30 | +53%±5 |
| Rolliniastatin 1 (30 μM) | +17%±4 | |
| Rolliniastatin 2 (30 μM) | +17%±9 | |
| Stigmatellin (60 μM) | −11%±5 | −34%±3 |
| Mucidin (80 μM) | −7%±6 | −8%±3 |
| Capsaicin (50 μM) | −49%±3 | |
| CoQ2 (20 μM) | −39%±23 |
The results are expressed as percent of fluorescence variation with respect to the control treated with 1.8 μM Mucidin. Fluorescence intensity was collected after 2400 s from NADH addition. Each value is the mean of at least 10 different determinations.
Fig. 3.

Panel A: Representative experiment of ROS detection in SMP (0.5 mg/ml) inhibited with Class A (i.e. 2 μM Rotenone) and Class B (i.e. 60 μM Stigmatellin) inhibitors. All samples were treated with 1.8 μM Mucidin (to block electron transfer and to avoid ROS production by Complex III) and supplemented with 150 μM NADH. Panel B: As panel A except for SMP concentration (1.5 mg/ml) and Mucidin concentration: 5.4 μM. Inserts show the full time course of the experiments.
Analyzing the compounds listed in these two classes we can observe that the distinction between ROS-inducing and ROS-preventing inhibitors resembles the classic Complex I inhibitors distinction based on their antagonistic effect with respect to quinone or quinol.
The response of the probe to the ROS detection is not linear and is weak in the first 15 min using 0.5 mg/ml protein as described in Materials and Methods (Fig. 3A). For this reason we have chosen a time of 40 min as standard time for our measurements. In parallel experiments (Fig. 3B), using higher amount of protein (1.5 mg/ml), we show that the ratio between inhibited and control samples is retained both at 10 and at 40 min, suggesting that the fluorescence increase is representative of the amount of ROS produced in our system.
ROS production is a typical chain reaction, described by a non linear fluorescence increase of the probes. This behaviour does not allow a quantitative measure of ROS production rate, however the fluorescence value depends on the total amount of ROS produced and remains proportional throughout the time course of the experiment.
Complex I activity can be also affected by compounds acting on the FMN site such as DPI and pHMB (para-hydroxymercuribenzoate). Both these inhibitors block the electron input to the redox centers inside Complex I, also preventing electron delivery to molecular oxygen. Moreover, addition of CoQ1 to a SMP sample inhibited by DPI in presence of NADH has no effect on ROS production, suggesting that the site involved in the CoQ1 prooxidant effect is located downstream the DPI inhibition site (data not shown).
The ability of Complex I to produce ROS is strictly related to the percent of inhibition exerted by Rotenone-like inhibitors as well as the decrease of ROS generation by Stigmatellin is strictly related to the extent of Stigmatellin inhibition of NADH–CoQ1 reductase activity (Fig. 4A, B, C, D).
Fig. 4.

Correlation between percentage of DCFDA fluorescence variation and percentage of NADH–CoQ1 activity inhibition in presence of different Complex I inhibitors: Panel A) Correlation between ROS production and Complex I inhibition by Rotenone. Panel B) Correlation between ROS production and Complex I inhibition by Piericidin A. Samples were prepared as follows: SMP were incubated with increasing amounts of Rotenone or Piericidin A. For each sample we measured NADH:CoQ1 activity and ROS production. Panel C) Effect of increasing amounts of Stigmatellin on ROS produced by 100% Rotenone inhibited Complex I. Panel D) Effect of increasing amount of Stigmatellin on ROS produced by 100% Piericidin A inhibited Complex I. Samples were prepared as follows: SMP completely inhibited with 2 μM of Rotenone or Piericidin A were treated with increasing amounts of Stigmatellin and ROS production was detected. The percentage of inhibition of NADH–CoQ1 activity exerted by the same amounts of Stigmatellin was determined in a parallel experiment. Each value is the mean of at least ten different determinations.
Inspection of panels A and B shows that Class A inhibitors Rotenone and Piericidin A at concentrations inducing low extents of inhibition fail to induce ROS generation; moreover, Rotenone and Piericidin A exhibit a different behaviour, with a much more pronounced lag in presence of the latter inhibitor.
Some experimental factors like low probe sensitivity or NADH interference with the fluorescent probe may partially explain the lack of ROS production in presence of low inhibition levels.
In fact in a control experiment using AAPH as radical source we have detected a lower DCF fluorescence intensity in presence of NADH 150 μM.
Nevertheless Piericidin A requires a higher extent of Complex I inhibition to start ROS production in comparison with Rotenone (60–70% for Piericidin A vs 20–30% for Rotenone).
This behaviour may be due to the presence of two different partially overlapping binding sites for Piericidin A, one with high affinity and the other with low affinity and shared by Rotenone [48]. Piericidin A at low concentration is able to block electron transfer without inducing ROS formation, whereas at high concentration it behaves like Rotenone, both blocking electron transfer and inducing ROS production.
In this scenario Piericidin A needs to occupy the Rotenone binding site to trigger ROS production. Moreover titration of ROS production induced by Rotenone in SMP partially pre-inhibited with Piericidin A shows loss of the lag phase, suggesting an additive effect between these two inhibitors (Fig. 5).
Fig. 5.
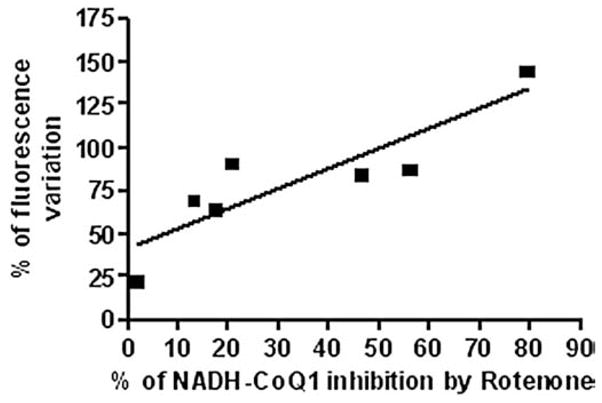
Correlation between ROS production (indicated as percentage of DCFDA fluorescence variation) and percentage of NADH–CoQ1 inhibition by Rotenone in SMP pretreated with 20 pmol/mg of Piericidin A. Addition of 20 pmol/mg of Piericidin A to SMP results in 30% of inhibition of NADH–CoQ1 activity without triggering massive ROS production.
The overlapping between different inhibition sites inside Complex I is also consistent with the inhibition of ROS production observed when Stigmatellin is added in presence of either Rotenone or Piericidin A. (Fig. 4C, D). In the first case we observe a linear correlation (Fig. 4C) between Complex I inhibition and decrease of ROS production, in the second case this correlation follows a sigmoidal behaviour (Fig. 4D) suggesting that Piericidin A binding sites may partially overlap with both Rotenone and Stigmatellin binding sites. Evidences about the different effects on ROS production and electron transport in Complex I related to different sites of inhibition for acetogenin derivatives were recently described by Miyoshi and coworkers [61].
3.3. Kinetic analysis of Complex I inhibitors on electron transfer to DCIP
DCIP is a hydrophilic electron acceptor widely used to test Complex I reductase activity; only 20–30% of this activity is sensitive to Rotenone, while the remaining is both Rotenone and DPI insensitive. This suggests the presence of at least two sites for DCIP reduction: one independent of the DPI inhibition site, the second corresponding to the physiological ubiquinone reducing site.
Short chain ubiquinone analogues like CoQ1 and DB increase NADH–DCIP reductase activity in a Rotenone sensitive way, whereas they do not increase activity in presence of DPI.
The stimulation of NADH–DCIP reductase activity by CoQ1 or DB offers a new tool to investigate the electron transfer mechanism inside the active site.
The two classes of Complex I inhibitors affect the stimulation of DCIP reduction by short chain quinones in a different way. The results listed in Fig. 6 show that Rotenone-like inhibitors prevent this stimulation, while Stigmatellin-like inhibitors allow it.
Fig. 6.

NADH–DCIP reductase activity related to the physiological quinone reducing site. Data were obtained subtracting the DPI insensitive activity from the total DCIP reductase activity. The stimulation effect of 20 μM DB on NADH–DCIP reductase activity is maintained in presence of Stigmatellin but is abolished by Rotenone. Each value is the mean of at least 5 different determinations. *P<0.005.
Since both classes of inhibitors prevent the reduction of quinones to quinols, the extra DCIP reduction in presence of CoQ1 or DB might be due to the presence of an intermediate species between the fully oxidized and the fully reduced one: thus it must be the semiquinone form of CoQ1 or DB generated inside the active site of Complex I. The new finding resulting from this observation allows us to hypothesize the presence of a semiquinone in the enzyme active site insensitive to the Stigmatellin-like inhibitors. To test this hypothesis we started to study semiquinone EPR spectra in SMP treated with Rotenone-like or Stigmatellin-like inhibitors.
3.4. Preliminary semiquinone EPR data
It is known from the literature that Rotenone and Piericidin A strongly reduce the semiquinone EPR signal intensity from Complex I [8], while there are no data on the effect of Stigmatellin and Stigmatellin-like inhibitors. We recorded EPR spectra of uncoupled SMP treated with Rotenone, Stigmatellin, or both. Results are reported in Fig. 7: our spectra show a Rotenone-sensitive signal centered at g=2.005, identifying it as a semiquinone radical [11]. The spectra confirm a strong signal reduction in the presence of Rotenone while the signal intensity is only slightly reduced in samples treated with Stigmatellin (Fig. 7, left); in the presence of both inhibitors, the signal intensity is equal to that detected when only Stigmatellin is present (Fig. 7, right).
Fig. 7.

EPR signals of the semiquinone radical in Complex I at 180 K. The systems contained 30 mg/ml of SMP in 300 μl, 150 μM CoQ1. The ubisemiquinone formation was initiated by the addition of 150 μM NADH. Spectra were obtained at a microwave frequency of 9.4121 GHz, obtaining a value of g=2.005 for the semiquinone radical.
4. Discussion
Complex I is the most debated enzyme of the mitochondrial respiratory chain, due to its high structural complexity and many redox centers involved in the electron transfer from NADH to ubiquinone. From the analysis of the midpoint redox potentials of the enzyme prosthetic groups, FMN is the entry point of electrons from NADH, while N2 iron–sulphur center is considered to be the direct electron donor to endogenous ubiquinone.
A large number of compounds inhibit Complex I: Rotenone, as well as other classic Complex I inhibitors (Piericidin A, Rolliniastatin-1 and -2, Capsaicin, etc.), block electron transfer from iron–sulphur clusters to the ubiquinone pool. Despite the different chemical structure of Complex I inhibitors it has not been possible to identify different binding sites in the enzyme, and it is commonly accepted that these inhibitors share a rather large hydrophobic pocket with partially overlapping binding sites [13].
We have exploited the ability of Complex I to transfer electrons directly to molecular oxygen with the aim of elucidating not only the site of electron escape but also the electron transfer pathway. The results depicted in this work allow us to divide Complex I inhibitors into two distinct classes depending on their effect on ROS production:
Class A inhibitors: inducing strong increase in ROS production.
Class B inhibitors: preventing ROS production.
Class A inhibitors include Rotenone, Piericidin A, Rolliniastatin-1 and -2, while Class B includes Stigmatellin, Capsaicin, Mucidin and Coenzyme Q2.
Most of Class B compounds are also classical Complex III inhibitors, acting at the so called center “o”, where they block electron transfer from ubiquinol to the Rieske iron–sulphur protein, while CoQ2 is known to be a poor electron acceptor from Complex I, on which it exerts an inhibitory effect ascribed to the quinol form [47].
Class A inhibitors are thought to prevent access of physiological CoQ10 to its reduction site [49], allowing the release of one electron to molecular oxygen. On the other hand, Class B inhibitors appear to directly prevent oxygen reduction presumably acting on the electron escape site [12].
This behaviour raises the question of the identification of the direct reductant of molecular oxygen.
One of the possible candidates is the ubisemiquinone species [19]; EPR data reported by the Ohnishi group [8] showed that Complex I inhibitors such as Rotenone and Piericidin A turn off the EPR signals from semiquinone species.
From our results on the ROS production it appears that inhibitors shutting down the semiquinone signals are also most efficient in the direct transfer of electrons to molecular oxygen. These results would suggest that the endogenous semiquinone formed during the redox cycle of the enzyme is not involved in ROS production. This conclusion is in line with a previous report showing that in CoQ-depleted mitochondria, Complex I is able to produce oxygen radicals at a rate comparable with the enzyme in non-extracted mitochondria [17].
A second major candidate as the electron donor to oxygen has been proposed to be FMN [15,20,21]; recently Brandt and his coworkers showed that ROS production was still present in a mutant Complex I from Yarrowia lipolytica lacking iron–sulphur cluster N2, concluding a direct involvement of FMN in this activity [50]. On the other hand Ohnishi and coworkers showed that DPI inhibits ROS production in the forward electron transfer, while enhanced it in the reverse electron transfer [18]. The loss of ROS detection in the presence of DPI seems to exclude any involvement of FMN in favor of a direct involvement of iron–sulphur clusters. In fact DPI inhibits the reduction of iron–sulphur clusters while the reduced state of protein-bound FMN is stabilized [51]. The FMN involvement in ROS production remains an open question and the discrepancy found in literature should be ascribed to the difficulty encountered in achieving a complete inhibition in the NADH–O2 activity, moreover the NAD+/NADH ratio seems to be crucial for electron escape from FMN [52].
Nevertheless the results reported in this work allow us to distinguish Complex I inhibitors, both acting downstream the FMN moiety, in two classes with opposite effect on ROS production. For this reason it is reasonable to conclude that FMN is not directly involved in electron escape to oxygen in our experimental conditions of forward electron transfer in intact membranes. Previous investigations demonstrating FMN as the electron donor to oxygen in forward electron transfer were mainly performed in isolated Complex I or subfragments thereof; a recent study by Ohnishi et al. [53] showed that ROS were generated at the FMN site in isolated Complex I only in absence of CoQ acceptors.
Another major candidate as direct oxygen reductant is the iron–sulphur cluster N2 because of its highest midpoint potential. The electron transfer from NADH to the ubiquinone in Complex I requires the presence of at least eight iron–sulphur clusters, seven of which are well protected from reacting with oxygen with the exception of the N2 center. From structural and functional studies the iron–sulphur cluster N2 seems to be localized in a region that should be accessible to protein bound ubisemiquinone, to H+ ions and to water, hence this region should be also accessible to molecular oxygen [54,55]. Moreover the mid point potential of cluster N2 is around − 0.15 to − 0.05 V [56] and therefore it is compatible with the reduction of oxygen to superoxide anion (mid point potential for the couple superoxide/oxygen is − 0.14 V) [57,58].
We favor the hypothesis indicating the cluster N2 as the direct reductant of the molecular oxygen. To allow electron escape from Complex I to oxygen the reduced state of the enzyme is not sufficient, as indicated by the lack of ROS production in the presence of 1.8 μM Mucidin. In this condition and in the presence of saturating concentration of NADH, Complex III is completely inhibited while Complex I is fully reduced [59]. ROS production by Complex I requires the presence of a Class A inhibitor besides the reduced state of the enzyme. On the other hand, using Mucidin at high concentration (80 μM) we achieve full inhibition of both NADH–CoQ1 activity and ROS production even in presence of Class A inhibitors.
It might be guessed that Class A inhibitors induce an enzyme conformational change, making the electron escape site more accessible to molecular oxygen, whereas Class B inhibitors would either directly block this site, or make it less accessible by way of a conformational change.
In this scenario the different behaviour of Coenzyme Q1 can be explained referring to its higher water solubility with respect to the physiological CoQ10 or the more hydrophobic analogue Decylubiquinone. It may be postulated that CoQ1, when added to a non-inhibited enzyme, is transformed to the antioxidant quinol form in the physiological quinone reducing site, thus explaining its lack of promotion of ROS generation. However, in the presence of a Class A inhibitor, CoQ1 cannot reach the physiological Q-binding site. In this condition the enzyme is completely reduced and CoQ1 can react with a hydrophilic site located upstream the physiological one. This reaction results in a thermodynamically unstable CoQ1 semiquinone radical, able to readily react with molecular oxygen in the presence of protons [60]. This behaviour would explain the prooxidant role of CoQ1, not shared by other more hydrophobic analogues such as DB.
CoQ1 and DB increase NADH–DCIP reductase activity in a way sensitive to Class A inhibitors, suggesting that this effect is mediated by the physiological site. On the other hand this increased activity is insensitive to Class B inhibitors. Since both classes of inhibitors completely prevent quinol formation, the increase of NADH–DCIP activity observed even in the presence of Class B inhibitors must be ascribed to the presence of a semiquinone form in the active site.
Preliminary results obtained by EPR analysis of submitochondrial particles treated with Class A and Class B inhibitors confirm the decrease of the semiquinone signal in presence of Class A inhibitors but clearly show the presence of semiquinone in samples treated with Class B inhibitors either alone or in presence of Class A inhibitors. This behaviour is completely in line with the results obtained with DCIP.
5. Conclusions
The results of this investigation allow us to draw conclusions on the mechanism of electron transfer from the iron–sulphur clusters to ubiquinone. It is generally believed that center N2 is the direct electron donor to CoQ10 in a two steps mechanism by which two electrons are consecutively delivered to quinone to achieve its fully reduced form. This hypothesis is supported by recent findings published by Sazanov and coworkers, describing a linear disposition for Fe–S clusters inside Complex I [7]. In a simple linear scheme considering N2 center as the only direct electron donor to quinone, our results suggest that Class B inhibitors would act upstream, while Class A inhibitors would block electron flow downstream N2 center. However, this scheme is incompatible with a series of observations.
Class B inhibitors, normally considered quinol antagonists, cannot act upstream the quinone reducing site [12].
Class B inhibitors, while blocking quinol formation, do not prevent semiquinone formation (our observations).
To explain our results we would need a mechanism of bifurcated electron transfer, in which an iron–sulphur cluster located upstream N2 center would act as a “switch” for electron delivery in such a way that one-electron quinone reduction to semiquinone and semiquinone reduction to quinol would be accomplished by two different electron donors. Since it is highly unlikely that quinone can reach iron–sulphur clusters other than N2, that is the only center not deeply buried in the protein, the delivery of both electrons by N2 requires that the switch between the two gated states is represented by a suitable conformational change.
The presence of oxidized CoQ10 in the Q-pocket induces an enzyme conformation, allowing electron delivery to reduce CoQ10 to semiquinone. The semiquinone formation induces a conformational change now allowing the delivery of the second electron to the semiquinone to produce the fully reduced form. This mechanism is schematically represented in Fig. 8A.
Fig. 8.
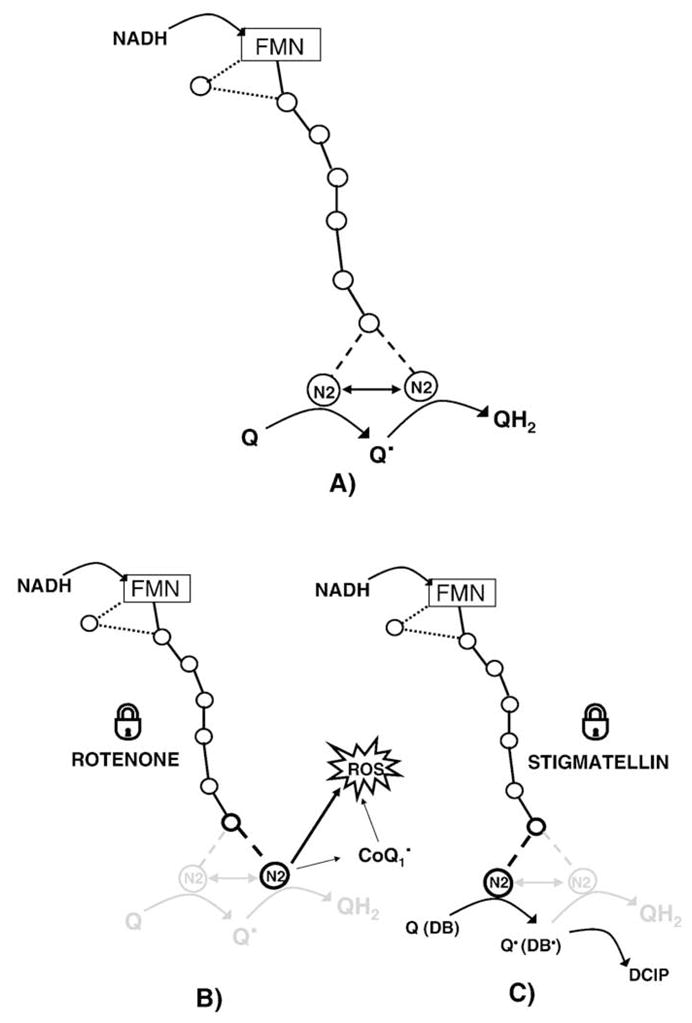
Proposed two step mechanism for electron transfer from NADH to quinone in Complex I (A), in presence of Class A inhibitors (B) and in presence of Class B inhibitors (C). The role of hydrophilic (CoQ1) and hydrophobic (DB) quinones is highlighted. CoQ1 can react with the physiological ubiquinone reducing site and, because of its higher water solubility, it can also react with the electron escape site, increasing superoxide production.
Class A inhibitors (Fig. 8B), not allowing access of the quinone to the active site, would block the enzyme in a conformation that does not allow quinone reduction but only permits electron delivery from N2 to oxygen; on the other hand, Class B inhibitors (Fig. 8C) would block the enzyme in a conformation allowing the first electron delivery to form the semiquinone, but the incapability to further reduction to quinol; such conformation would not allow reaction of N2 with oxygen.
This working hypothesis requires further experiments and EPR characterization of the redox state of all prosthetic groups in the enzyme to clarify the mechanism of quinone reduction by Complex I.
Acknowledgments
This work was supported by MIUR-Rome (Italy).
Dr. Christian Bergamini was supported by Marco Polo Fellowship for a stage in Tomoko Ohnishi’s laboratory from June to September 2007.
Abbreviations
- DCFDA
2,7 ′,′-Dichlorodihydrofluorescein diacetate
- NADH
β-Nicotinamide adenine dinucleotide
- ROS
Reactive oxygen species
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide
- FMN
Flavin mononucleotide
- BHM
Bovine heart mitochondria
- BSA
Bovine serum albumine
- DOC
Deoxycholate
- SMP
Submitochondrial particles from bovine heart
- DPI
Diphenylene iodonium
- DB
Decylubiquinone
- DCIP
2,6-dichloroindo-phenol
- CoQ1
2,3-Dimethoxy-5-methyl-6-(3-methyl-2-butenyl)-1,4-benzoquinone
- FCCP
Carbonylcyanide-p-trifluoromethoxy-phenylhydrazone
- AAPH
α,α′-Azodiisobutyramidine dihydrochloride
References
- 1.Matsuno-Yagi A, Yagi T. Introduction: Complex I—an L-shaped black box. J Bioenerg Biomembr. 2001;33:155–157. doi: 10.1023/a:1010702116348. [DOI] [PubMed] [Google Scholar]
- 2.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 3.Schultz BE, Chan SI. Structures and proton-pumping strategies of mitochondrial respiratory enzymes. Annu Rev Biophys Biomol Struct. 2001;30:23–65. doi: 10.1146/annurev.biophys.30.1.23. [DOI] [PubMed] [Google Scholar]
- 4.Carroll J, Fearnley IM, Shannon RJ, Hirst J, Walker JE. Analysis of the subunit composition of Complex I from bovine heart mitochondria. Mol Cell Proteomics. 2003;2:117–126. doi: 10.1074/mcp.M300014-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Chomyn A, Cleeter MW, Ragan CI, Riley M, Doolittle RF, Attardi G. URF6, last unidentified reading frame of human mtDNA, codes for an NADH dehydrogenase subunit. Science. 1986;234:614–618. doi: 10.1126/science.3764430. [DOI] [PubMed] [Google Scholar]
- 6.Chomyn A, Mariottini P, Cleeter MW, Ragan CI, Matsuno-Yagi A, Hatefi Y, Doolittle RF, Attardi G. Six unidentified reading frames of human mitochondrial DNA encode components of the respiratory-chain NADH dehydrogenase. Nature. 1985;314:592–597. doi: 10.1038/314592a0. [DOI] [PubMed] [Google Scholar]
- 7.Sazanov LA, Hinchliffe P. Structure of the hydrophilic domain of respiratory Complex I from Thermus thermophilus. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 8.Magnitsky S, Toulokhonova L, Yano T, Sled VD, Hagerhall C, Grivennikova VG, Burbaev DS, Vinogradov AD, Ohnishi T. EPR characterization of ubisemiquinones and iron–sulfur cluster N2, central components of the energy coupling in the NADH–ubiquinone oxidoreductase (Complex I) in situ. J Bioenerg Biomembr. 2002;34:193–208. doi: 10.1023/a:1016083419979. [DOI] [PubMed] [Google Scholar]
- 9.Ohnishi T, Johnson JE, Jr, Yano T, Lobrutto R, Widger WR. Thermodynamic and EPR studies of slowly relaxing ubisemiquinone species in the isolated bovine heart Complex I. FEBS Lett. 2005;579:500–506. doi: 10.1016/j.febslet.2004.11.107. [DOI] [PubMed] [Google Scholar]
- 10.Yano T, Dunham WR, Ohnishi T. Characterization of the delta muH+-sensitive ubisemiquinone species (SQ(Nf)) and the interaction with cluster N2: new insight into the energy-coupled electron transfer in Complex I. Biochemistry. 2005;44:1744–1754. doi: 10.1021/bi048132i. [DOI] [PubMed] [Google Scholar]
- 11.Ohnishi T, Salerno JC. Conformation-driven and semiquinone-gated proton-pump mechanism in the NADH–ubiquinone oxidoreductase (Complex I) FEBS Lett. 2005;579:4555–4561. doi: 10.1016/j.febslet.2005.06.086. [DOI] [PubMed] [Google Scholar]
- 12.Degli Esposti M. Inhibitors of NADH–ubiquinone reductase: an overview. Biochim Biophys Acta. 1998;1364:222–235. doi: 10.1016/s0005-2728(98)00029-2. [DOI] [PubMed] [Google Scholar]
- 13.Okun JG, Lummen P, Brandt U. Three classes of inhibitors share a common binding domain in mitochondrial Complex I (NADH:ubiquinone oxidoreductase) J Biol Chem. 1999;274:2625–2630. doi: 10.1074/jbc.274.5.2625. [DOI] [PubMed] [Google Scholar]
- 14.Brandt U. Energy converting NADH:quinone oxidoreductase (Complex I) Annu Rev Biochem. 2006;75:69–92. doi: 10.1146/annurev.biochem.75.103004.142539. [DOI] [PubMed] [Google Scholar]
- 15.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH: ubiquinone oxidoreductase (Complex I) from bovine heart mitochondria. Proc Natl Acad Sci U S A. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. 2000;25:502–508. doi: 10.1016/s0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 17.Genova ML, Ventura B, Giuliano G, Bovina C, Formiggini G, Parenti Castelli G, Lenaz G. The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron–sulfur cluster N2. FEBS Lett. 2001;505:364–368. doi: 10.1016/s0014-5793(01)02850-2. [DOI] [PubMed] [Google Scholar]
- 18.Ohnishi ST, Ohnishi T, Muranaka S, Fujita H, Kimura H, Uemura K, Yoshida K, Utsumi K. A possible site of superoxide generation in the Complex I segment of rat heart mitochondria. J Bioenerg Biomembr. 2005;37:1–15. doi: 10.1007/s10863-005-4117-y. [DOI] [PubMed] [Google Scholar]
- 19.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (Complex I) J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 21.Vinogradov AD. Catalytic properties of the mitochondrial NADH–ubiquinone oxidoreductase (Complex I) and the pseudo-reversible active/inactive enzyme transition. Biochim Biophys Acta. 1998;1364:169–185. doi: 10.1016/s0005-2728(98)00026-7. [DOI] [PubMed] [Google Scholar]
- 22.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation–reduction state. Biochem J. 2002;368:545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (Complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bailey SM, Landar A, Darley-Usmar V. Mitochondrial proteomics in free radical research. Free Radic Biol Med. 2005;38:175–188. doi: 10.1016/j.freeradbiomed.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 25.DiMauro S, Hirano M. Mitochondrial encephalomyopathies: an update. Neuromuscul Disord. 2005;15:276–286. doi: 10.1016/j.nmd.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Kerscher S, Kashani-Poor N, Zwicker K, Zickermann V, Brandt U. Exploring the catalytic core of Complex I by Yarrowia lipolytica yeast genetics. J Bioenerg Biomembr. 2001;33:187–196. doi: 10.1023/a:1010726818165. [DOI] [PubMed] [Google Scholar]
- 27.Yagi T, Seo BB, Di Bernardo S, Nakamaru-Ogiso E, Kao MC, Matsuno-Yagi A. NADH dehydrogenases: from basic science to biomedicine. J Bioenerg Biomembr. 2001;33:233–242. doi: 10.1023/a:1010787004053. [DOI] [PubMed] [Google Scholar]
- 28.Zeviani M, Di Donato S. Mitochondrial disorders. Brain. 2004;127:2153–2172. doi: 10.1093/brain/awh259. [DOI] [PubMed] [Google Scholar]
- 29.Beyer RE. The isolation, Propertier, and Assay of ATP Synthetase II. Methods in Enzymol. 1967;10:519–522. [Google Scholar]
- 30.Fato R, Cavazzoni M, Castelluccio C, Parenti Castelli G, Palmer G, Degli Esposti M, Lenaz G. Steady-state kinetics of ubiquinol–cytochrome c reductase in bovine heart submitochondrial particles: diffusional effects. Biochem J. 1993;290(Pt 1):225–236. doi: 10.1042/bj2900225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gornall AG, Bardawill CJ, David MM. Determination of serum proteins by means of the Biuret reaction. J Biol Chem. 1949;177:751–766. [PubMed] [Google Scholar]
- 32.Black MJ, Brandt RB. Spectrofluorometric analysis of hydrogen peroxide. Anal Biochem. 1974;58:246–254. doi: 10.1016/0003-2697(74)90464-3. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. 1997;272:11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- 34.Degli Esposti M. Measuring mitochondrial reactive oxygen species. Methods. 2002;26:335–340. doi: 10.1016/S1046-2023(02)00039-7. [DOI] [PubMed] [Google Scholar]
- 35.Yagi T. Inhibition by capsaicin of NADH–quinone oxidoreductases is correlated with the presence of energy-coupling site 1 in various organisms. Arch Biochem Biophys. 1990;281:305–311. doi: 10.1016/0003-9861(90)90448-8. [DOI] [PubMed] [Google Scholar]
- 36.Degli Esposti M, Ghelli A, Crimi M, Estornell E, Fato R, Lenaz G. Complex I and Complex III of mitochondria have common inhibitors acting as ubiquinone antagonists. Biochem Biophys Res Commun. 1993;190:1090–1096. doi: 10.1006/bbrc.1993.1161. [DOI] [PubMed] [Google Scholar]
- 37.Mohanty JG, Jaffe JS, Schulman ES, Raible DG. A highly sensitive fluorescent micro-assay of H2O2 release from activated human leukocytes using a dihydroxyphenoxazine derivative. J Immunol Methods. 1997;202:133–141. doi: 10.1016/s0022-1759(96)00244-x. [DOI] [PubMed] [Google Scholar]
- 38.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 39.Solomonson LP. In: Cyanide in Biology. Vennesland B, Conn EE, Knowles CJ, Westley J, Wissing I, editors. Academic Press; New York: 1981. [Google Scholar]
- 40.Gomes A, Fernandes E, Lima JL. Fluorescence probes used for detection of reactive oxygen species. J Biochem Biophys Methods. 2005;65:45–80. doi: 10.1016/j.jbbm.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 41.Barbacanne MA, Souchard JP, Darblade B, Iliou JP, Nepveu F, Pipy B, Bayard F, Arnal JF. Detection of superoxide anion released extracellularly by endothelial cells using cytochrome c reduction, ESR, fluorescence and lucigenin-enhanced chemiluminescence techniques. Free Radic Biol Med. 2000;29:388–396. doi: 10.1016/s0891-5849(00)00336-1. [DOI] [PubMed] [Google Scholar]
- 42.Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med. 1998;25:826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- 43.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walrand S, Valeix S, Rodriguez C, Ligot P, Chassagne J, Vasson MP. Flow cytometry study of polymorphonuclear neutrophil oxidative burst: a comparison of three fluorescent probes. Clin Chim Acta. 2003;331:103–110. doi: 10.1016/s0009-8981(03)00086-x. [DOI] [PubMed] [Google Scholar]
- 45.Munzel T, Afanasev IB, Kleschyov AL, Harrison DG. Detection of superoxide in vascular tissue. Arterioscler Thromb Vasc Biol. 2002;22:1761–1768. doi: 10.1161/01.atv.0000034022.11764.ec. [DOI] [PubMed] [Google Scholar]
- 46.Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol. 2004;286:R431–R444. doi: 10.1152/ajpregu.00361.2003. [DOI] [PubMed] [Google Scholar]
- 47.Fato R, Estornell E, Di Bernardo S, Pallotti F, Parenti Castelli G, Lenaz G. Steady-state kinetics of the reduction of coenzyme Q analogs by Complex I (NADH: ubiquinone oxidoreductase) in bovine heart mitochondria and submitochondrial particles. Biochemistry. 1996;35:2705–2716. doi: 10.1021/bi9516034. [DOI] [PubMed] [Google Scholar]
- 48.Gutman M, Singer TP, Casida JE. Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase. XVII. Reaction sites of piericidin A and Rotenone. J Biol Chem. 1970;245:1992–1997. [PubMed] [Google Scholar]
- 49.Friedrich T, Ohnishi T, Forche E, Kunze B, Jansen R, Trowitzsch W, Hofle G, Reichenbach H, Weiss H. Two binding sites for naturally occurring inhibitors in mitochondrial and bacterial NADH:ubiquinone oxidoreductase (Complex I) Biochem Soc Trans. 1994;22:226–230. doi: 10.1042/bst0220226. [DOI] [PubMed] [Google Scholar]
- 50.Galkin A, Brandt U. Superoxide radical formation by pure Complex I (NADH: ubiquinone oxidoreductase) from Yarrowia lipolytica. J Biol Chem. 2005;280:30129–30135. doi: 10.1074/jbc.M504709200. [DOI] [PubMed] [Google Scholar]
- 51.Majander A, Finel M, Wikstrom M. Diphenyleneiodonium inhibits reduction of iron–sulfur clusters in the mitochondrial NADH–ubiquinone oxidoreductase (Complex I) J Biol Chem. 1994;269:21037–21042. [PubMed] [Google Scholar]
- 52.Barker CD, Reda T, Hirst J. The flavoprotein subcomplex of Complex I (NADH: ubiquinone oxidoreductase) from bovine heart mitochondria: insights into the mechanisms of NADH oxidation and NAD+ reduction from protein film voltammetry. Biochemistry. 2007;46:3454–3464. doi: 10.1021/bi061988y. [DOI] [PubMed] [Google Scholar]
- 53.Ohnishi ST, Shinzawa-Itoh K, Yoshikawa S, Ohnishi T. In: Porter RK, Davey G, editors. 15th European Bioenergetics Conference; Dublin. 2008. p. S36. [Google Scholar]
- 54.Garofano A, Zwicker K, Kerscher S, Okun P, Brandt U. Two aspartic acid residues in the PSST-homologous NUKM subunit of Complex I from Yarrowia lipolytica are essential for catalytic activity. J Biol Chem. 2003;278:42435–42440. doi: 10.1074/jbc.M305819200. [DOI] [PubMed] [Google Scholar]
- 55.Grgic L, Zwicker K, Kashani-Poor N, Kerscher S, Brandt U. Functional significance of conserved histidines and arginines in the 49-kDa subunit of mitochondrial Complex I. J Biol Chem. 2004;279:21193–21199. doi: 10.1074/jbc.M313180200. [DOI] [PubMed] [Google Scholar]
- 56.Ohnishi T. Iron–sulfur clusters/semiquinones in Complex I. Biochim Biophys Acta. 1998;1364:186–206. doi: 10.1016/s0005-2728(98)00027-9. [DOI] [PubMed] [Google Scholar]
- 57.Muller F. The nature and mechanism of superoxide production by the electron transport chain: its relevance to aging. J Am Aging Assoc. 2000;23:227–253. doi: 10.1007/s11357-000-0022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petlicki J, van de Ven TGM. J Chem Soc Faraday Trans. 1998;94:2763–2767. [Google Scholar]
- 59.Kroger A, Klingenberg M. Further evidence for the pool function of ubiquinone as derived from the inhibition of the electron transport by Antimycin. Eur J Biochem. 1973;39:313–323. doi: 10.1111/j.1432-1033.1973.tb03129.x. [DOI] [PubMed] [Google Scholar]
- 60.Nohl H, Gille L, Schonheit K, Liu Y. Conditions allowing redox-cycling ubisemiquinone in mitochondria to establish a direct redox couple with molecular oxygen. Free Radic Biol Med. 1996;20:207–213. doi: 10.1016/0891-5849(95)02038-1. [DOI] [PubMed] [Google Scholar]
- 61.Murai M, Ichimaru N, Abe MA, Nishioka T, Miyoshi H. Mode of inhibitory action of Δlac-Acetogenins, a new class of inhibitors of bovine heart mitochondrial Complex I. Biochemistry. 2006;45:9778–9787. doi: 10.1021/bi060713f. [DOI] [PubMed] [Google Scholar]