

Abstract
Background
Benzyl isothiocyanate (BITC), a compound found in cruciferous vegetables, has been reported to have anticancer properties, but the mechanism whereby it inhibits growth of human pancreatic cancer cells is incompletely understood.
Methods
Human pancreatic cancer cells (BxPC-3, AsPC-1, Capan-2, MiaPaCa-2, and Panc-1) and immortalized human pancreatic cells (HPDE-6) were treated with vehicle or with BITC at 5–40 μM, cell survival was evaluated by sulforhodamine B assay, and apoptosis by caspase-3 and poly-ADP ribose polymerase cleavage or by a commercial assay for cell death. Total and activated signal transducer and activator of transcription-3 (STAT-3) protein expression in the cells were examined by western blotting, STAT-3 mRNA levels by reverse transcription–polymerase chain reaction, and STAT-3 DNA-binding and transcriptional activity by commercially available binding and reporter assays. The effects of BITC treatment on tumor growth, apoptosis, and STAT-3 protein expression in vivo were studied in xenografts of BxPC-3 pancreatic tumor cells in athymic nude mice. All statistical tests were two-sided.
Results
BITC treatment reduced cell survival and induced apoptosis in BxPC-3, AsPC-1, Capan-2, and MiaPaCa-2 cells, and to a much lesser extent in Panc-1 cells, but not in HPDE-6 cells. It also reduced levels of activated and total STAT-3 protein, and as a result, STAT-3 DNA-binding and transcriptional activities. Overexpression of STAT-3 in BxPC-3 cells inhibited BITC-induced apoptosis and restored STAT-3 activity. In mice that were fed BITC (60 μmol/wk, five mice, 10 tumors per group), growth of BxPC-3 pancreatic tumor xenografts was suppressed compared with control mice (at 6 weeks, mean tumor volume of control vs BITC-treated mice = 334 vs 172 mm3, difference =162 mm3, 95% confidence interval = 118 to 204 mm3; P = .008) and tumors had increased apoptosis and reduced STAT-3 protein expression.
Conclusion
BITC induces apoptosis in some types of pancreatic cancer cells by inhibiting the STAT-3 signaling pathway.
CONTEXT AND CAVEATS
Prior knowledge
Benzyl isothiocyanate (BITC), a compound found in cruciferous vegetables, has been reported to have anticancer properties. The mechanism by which it inhibits proliferation of human pancreatic cancer cells in culture was not well understood.
Study design
Human pancreatic cancer cell lines and a human nonmalignant, but immortalized, pancreatic cell line were used to examine the effects of BITC on proliferation and survival and on STAT-3 expression and activity in vitro. A mouse model of pancreatic cancer was used to study the effects of BITC on tumor growth in vivo.
Contribution
BITC treatment increased cell death in the pancreatic cancer cell lines tested compared with the nonmalignant cell line. BITC-sensitive cells also showed reduced levels of total and activated STAT-3 protein. Overexpression of STAT-3 eliminated BITC-induced apoptosis. Tumors grew more slowly in mice fed BITC than in untreated control mice.
Implications
BITC induces apoptosis by a STAT-3–dependent mechanism in several human pancreatic cancer cell lines.
Limitations
BITC promoted cell death and inhibited STAT-3 activation to varying degrees in several pancreatic cancer cell lines. Only one nonmalignant pancreatic cell line was studied. The in vivo experiments were performed in a small number of mice that were continuously fed BITC from the time of tumor cell implantation, and it is not clear whether the protective dose would be practical in terms of human consumption of vegetables.
From the Editors
Pancreatic cancer is one of the most common invasive malignancies and is the fourth leading cause of cancer-related deaths in the United States (1). The high mortality can be attributed to late diagnosis, rapid disease progression, poor response to systemic remedies, and resistance to chemotherapy and radiotherapy (2,3). Therefore, the development of novel approaches to prevent and treat pancreatic cancer is an important mission.
Evidence from epidemiological, pharmacological, and case– control studies has indicated that isothiocyanates present in cruciferous vegetables may have substantial chemopreventive activity against human malignancies including pancreatic cancer (4–7). The accumulated data from several in vitro models suggest that isothiocyanates induce cell death by multiple signaling pathways (8–13). Benzyl isothiocyanate (BITC), an agent that is present in cruciferous vegetables such as broccoli, watercress, cabbage, cauliflower, mustard, and horseradish, is widely consumed as part of a routine diet and has been reported to inhibit the initiation, growth, and metastasis of chemically induced human cancers in rodents (14–17). In previous studies, we demonstrated that BITC suppressed the proliferation of human pancreatic cancer cells by inducing DNA damage that caused G2/M cell cycle arrest and apoptosis (18) and by inhibiting the activation of nuclear factor kappa B (10). However, the mechanism by which BITC might inhibit human pancreatic carcinogenesis was not fully understood.
Members of the signal transducer and activator of transcription (STAT) family of transcription factors have been identified to play a crucial role in the expression of genes that are involved in cell survival, proliferation, chemoresistance, and angiogenesis (19–26). The constitutive activation of STAT-3 has been reported in different human tumors and cell lines and to some extent in pancreatic cancer (27–38), which makes STAT-3 an attractive molecular target for cancer therapy (39,40). Interleukin 6 (IL-6), Janus-activated kinases, epidermal growth factor receptor, and Src family kinases are among the activators of STAT-3, all of which phosphorylate critical tyrosine and serine residues, leading to STAT-3 dimerization, nuclear translocation of dimers, and initiation of transcription through their specific binding to DNA response elements in the promoter region of target genes (26,39–41). A few recent studies have shown that several naturally occurring anticancer agents induce apoptosis in human cancer cells via inhibition of STAT-3, although in these cases the exact role of the STAT-3 molecule is not clear (27,36,38).
This study aimed to investigate the molecular mechanism by which BITC inhibited the growth of pancreatic cancer cells. We first tested the BxPC-3 pancreatic cancer cell line for the effect of BITC on apoptosis and on STAT-3 activation, protein and mRNA levels, and DNA-binding and transcriptional activity. We examined whether overexpression of STAT-3α could prevent each of these effects of BITC. We then extended these findings to four other pancreatic cancer cell lines, AsPC-1, Capan-2, MiaPaCa-2, and Panc-1, and one relatively normal pancreatic cell line, HPDE-6. Last, we looked at the effect of BITC on the growth of BxPC-3 cells as xenografts in nude mice. Our results further elucidate the mechanism of BITC as an anticancer agent.
Materials and Methods
Cell Culture and Cell Survival Assays
The well-differentiated epithelial human pancreatic adenocarcinoma cell lines BxPC-3, Capan-2, AsPC-1, and MiaPaCa-2 were obtained from American Type Cell Culture (ATCC, Manassas, VA), and Panc-1 cells were a kind gift from Dr Thomas L. Brown (Wright State University, Dayton, OH). The normal human pancreatic duct epithelial cell line, HPDE-6, which was derived from normal (benign) adult human pancreata immortalized by infection with a retrovirus containing the E6 and E7 genes of the human papillomavirus 16, was a generous gift from Dr Ming-Sound Tsao (University of Toronto, Toronto, Ontario, Canada). Cultures of HPDE-6 cells were maintained as described previously (42,43). Monolayer cultures of BxPC-3 and AsPC-1 cells were maintained in RPMI-1640 medium (Sigma-Aldrich, St Louis, MO), adjusted to contain 10% heat-inactivated fetal bovine serum (FBS) (Mediatech Inc, Herndon, VA), supplemented with 2 mM l-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM HEPES, 1.0 mM sodium pyruvate, and 1% (vol/vol) penicillin and streptomycin (GIBCO-BRL, Carlsbad, CA) in a humidified incubator at 37°C and 5% CO2. Capan-2 cells were maintained in McCoy’s 5A medium (Mediatech Inc) supplemented with 2 mM l-glutamine and adjusted to contain 10% FBS and 1% (vol/vol) penicillin/streptomycin. MiaPaCa-2 cells were maintained in Dulbecco's Modified Eagle medium (DMEM; ATCC) that was supplemented with 4 mM l-glutamine and adjusted to contain 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, 10% FBS, 2.5% horse serum (Mediatech Inc), and 1% (vol/vol) penicillin/streptomycin. Monolayer cultures of Panc-1 cells were maintained in DMEM supplemented with 4 mM l-glutamine and adjusted to contain 10% FBS and 1% (vol/vol) penicillin/streptomycin. HPDE-6 cells were cultured in Keratinocyte serum-free medium (GIBCO-BRL) supplemented with 4 mM l-glutamine and adjusted to contain 0.2 ng/mL epidermal growth factor (GIBCO-BRL), 30 μg/mL bovine pituitary extract (GIBCO-BRL), and 1% (vol/vol) penicillin/streptomycin.
A 100 mM stock solution of BITC (Sigma-Aldrich) was prepared in 100% dimethyl sulfoxide (DMSO) and subsequently diluted in the cell culture medium so that the final concentration of DMSO was 0.1% in the medium. Cells were treated with BITC at 5, 10, or 20 μM concentration for 24 hours. The effect of BITC on proliferation of BxPC-3 cells was determined by sulforhodamine B assay (Sigma-Aldrich), as described previously (44). The plates were read at 590 nm using the Bio Kinetics plate reader EL-800 (BioTek Instrument Inc, Winooski, VA).
Western Blot Analysis
BxPC-3, AsPC-1, Capan-2, MiaPaCa-2, Panc-1, and HPDE-6 cells were treated with varying concentrations of BITC (0, 5, 10, and 20 μM) for 24 hours. For time-dependent experiments, cells were treated with 10 μM BITC for 0, 1, 8, and 24 hours. Whole-cell extracts were prepared as we described previously (18). To examine whether degradation of STAT-3 protein was mediated by the ubiquitin-proteasome pathway, BxPC-3 cells were treated with BITC and with the proteasome inhibitor MG-132 (Calbiochem EMD Chemicals, Gibbstown, NJ) at 10 μM for 2 hours. In a separate experiment, cells were pretreated with 10 μg/mL cycloheximide, a protein synthesis inhibitor (Sigma-Aldrich), for 4 hours followed by treatment with 10 μM BITC. Cell lysates containing 20–40 μg of protein were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and proteins were transferred onto polyvinylidene fluoride membranes. After blocking with 5% nonfat dry milk, membranes were incubated overnight with the desired primary antibody, followed by an appropriate secondary antibody, and the immunoreactive bands were visualized using the enhanced chemiluminescence kit from Perkin-Elmer (Waltham, MA) according to the manufacturer's instructions. Two mouse monoclonal antibodies (anti-pp38 [Thr180/Tyr182] [#9261] and anti-ubiquitin Ub [#3936]) and all of the following rabbit antibodies were diluted 1:1000 in 50 mM Tris–HCl (pH 7.5) and 150 mM NaCl Tris-buffered saline (TBS) containing 0.1% Tween-20 (TBST) before use on blots: anti–caspase-3 (#9664), anti-PARP (#9541), anti–pSTAT-3 (Tyr705) (#9131), anti–pSTAT-3 (Ser727) (#9134), anti–STAT-3 (total) (#9139), anti–Mcl-1 (#4572), anti–Bcl-2 (#2872), anti-pJNK (Thr183/Tyr185) (#9251), anti-JNK (#9252), anti-p38 (#9212), anti-pERK (Thr202/Tyr204) (#9101), and anti-ERK (#9102) (all from Cell Signaling Technologies, Danvers, MA). The same membranes were reprobed with a 1:50 000 dilution of mouse monoclonal anti–β-actin antibody (#A5441; Sigma-Aldrich) as a control for equal protein loading. The intensity of immunoreactive bands on western blots was determined using a densitometer (Molecular Dynamics, Sunnyvale, CA) equipped with Image QuaNT software. Unless otherwise stated, each experiment was repeated independently at least two to three times and expressed as mean values with 95% confidence intervals (CIs).
Immunoprecipitation
Briefly, 1 × 106 BxPC-3 cells were seeded and treated with different concentrations of BITC for 24 hours. The medium was removed and cells were rinsed with ice-cold 10 mM phosphate buffer (pH 7.4) containing 137 mM NaCl and 2.7 mM KCl phosphate-buffered saline (PBS) and then lysed with 0.5 mL of cell lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μg/mL leupeptin for 5 minutes on ice. Cells were scraped, transferred to microcentrifuge tubes, and sonicated three times for 5 seconds. Cell extracts were centrifuged for 10 minutes at 14 000g at 4°C, and the supernatant was isolated. About 250 μg of total protein from control or BITC-treated cells were each incubated with anti–STAT-3 antibody overnight at 4°C with gentle rocking. Protein A agarose beads (20 μL of a 50% slurry) were added to each sample and incubated for 3 hours at 4°C. Lysates were centrifuged for 30 seconds at 1000g, and pellets were washed five times with 0.5 mL cell lysis buffer. STAT-3 protein from each sample was eluted with 40 μL of 1% SDS and quantified. About 3 μg of pure STAT-3 protein was resolved on 10% SDS–PAGE and blotted with anti–pSTAT-3 (Tyr705) antibody. The same membrane was stripped and reprobed with anti–STAT-3 antibody. In another experiment, cells were treated with 10 μM BITC for 24 hours and with 10 μM MG-132, a specific proteasome inhibitor, for 2 hours. The cells were lysed and STAT-3 protein was immunoprecipitated as described above. However, in this experiment, instead of quantifying and loading equal amounts of STAT-3 protein, total immunoprecipitated STAT-3 protein was resolved by SDS–PAGE and blotted, and the blots were probed with anti–STAT-3 and anti-Ub antibodies.
Determination of STAT-3 mRNA Transcripts
To determine the effects of BITC on STAT-3 gene transcription, BxPC-3, AsPC-1, Capan-2, MiaPaCa-2, Panc-1, or HPDE-6 cells were treated with varying concentrations of BITC (0, 5, 10, and 20 μM) for 24 hours. Total RNA was extracted from control and treated cells of each cell lines using Trizol RNA extraction reagent (Sigma-Aldrich), and RNA samples were prepared for reverse transcription–polymerase chain reaction (RT-PCR) by incubation with DNase I, then RT-PCR of STAT-3 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA was performed using the Verso 1-Step RT-PCR kit from Thermo-Fisher Scientific (Surrey, UK), according to the manufacturer's protocol. The following primer sets were used: STAT-3 forward primer, 5′-ATCCTGAAGCTGACCCAGGTA-3′; STAT-3 reverse primer, 5′- AGGTCGTTGGTGTCACACAGA-3′; GAPDH forward primer, 5′- ACCACAGTCCATGCCATCAC-3′; and GAPDH reverse primer, 5′-TCCACCACCCTGTTGCTGTA -3′. Reactions were carried out in 50-μL volumes using 100 ng of total template RNA, 200 nM each of forward and reverse primers, 25 μL polymerase chain reaction (PCR) master mix, and 1 μL of Verso enzyme mixture. Reaction conditions for initial complementary DNA synthesis were 50°C for 15 minutes followed by 40 cycles of denaturation at 94°C for 30 seconds, annealing at 63–61°C for STAT-3 or at 55°C for GAPDH for 30 seconds, extension at 72°C for 45 seconds, and then final extension at 72°C for 5 minutes on a 7300 real time PCR system AB1 (Perkin-Elmer-Applied Biosystems, Foster City, CA). The products were separated on 2% agarose gels and visualized by staining with ethidium bromide.
STAT-3 DNA-Binding Assay
DNA binding of the STAT-3 transcription factor to the promoters of its target genes in pancreatic cancer cells was measured by Universal EZ-TFA transcription factor assay colorimetric kit (Upstate Biotechnology, Inc, Lake Placid, NY) according to the manufacturer's protocol. In these experiments, BxPC-3 cells were treated either with 0.1% DMSO or 10 μM BITC for 24 hours. Then, nuclear extracts were prepared and incubated with 2 μL of “capture probe,” a biotinylated double-stranded oligonucleotide that contained the consensus sequence for STAT-3. To determine the specificity of STAT-3 DNA binding, a mixture of capture probe and “competitor probe,” that contained the exact same sequence as the capture probe but did not include any biotin modifications, was also incubated in streptavidin-coated plates. The biotinylated oligonucleotide, along with any bound STAT-3, was then immobilized on the streptavidin-coated plate, and the inactive, unbound material was washed away. The bound transcription factor was then detected by incubation with a 1:500 dilution of rabbit anti–STAT-3 antibody and a 1:500 dilution of horseradish peroxidase (HRP)–conjugated secondary antibody. A negative control containing binding buffer and free probe without cell lysate was used in each assay. HRP activity was colorimetrically detected at 450 nm using an EL-800 ELISA plate reader (BioTek Instruments Inc).
STAT-3 Luciferase Reporter Assay
STAT-3 luciferase transcriptional activity was first determined in BxPC-3 cells cotransfected, using lipofectamine reagent (Invitrogen, Carlsbad, CA), with 2 μg of pLuc-TK/STAT3 (a generous gift from Dr J. F. Bromberg, Rockefeller University, New York), which encoded firefly luciferase under the control of the STAT-3 promoter, and with 0.2 μg of a pRL-TK (Promega Corp, Madison, WI), which constitutively expressed Renilla luciferase, the latter as a transfection efficiency control. Twenty-four hours after transfection, BxPC-3 cells were treated with 10 μM BITC or with 0.1% DMSO for 24 hours or pretreated with 10 μg/mL cycloheximide for 4 hours and then treated with 10 μM BITC for 24 hours. Whole-cell lysates were collected according to the dual luciferase reporter kit assay protocol, and the light output of lysates was measured with a luminometer. Firefly luciferase activities were corrected for Renilla values and then normalized relative to the DMSO control, which was considered as 1.0. A value less than 1 in this assay indicated attenuation of STAT-3–directed transcription by BITC.
In other experiments, human pancreatic cancer cells AsPC-1, MiaPaCa-2, Capan-2, Panc-1, and normal HPDE-6 cells were cotransfected for 24 hours with 2 μg pLuc-TK/STAT3 and with 0.2 μg pRL-TK plasmid as a transfection efficiency control. Cells were again treated with 0.1% DMSO or with 10 μM BITC for 24 hours. In additional experiments, cells were 1) pretreated with 5 ng/mL IL-6 for 15 minutes, 2) pretreated with 10 μg/mL cycloheximide for 4 hours, or 3) transfected for 48 hours with 2 μg STAT-3α, cloned in the pBabe Puro mammalian expression vector (a generous gift from Dr J. F. Bromberg, Rockefeller University, New York) followed by cotransfection for 24 hours with the pLuc-TK/STAT3 and pRL-TK plasmids. Transfected cells were then treated with or without 10 μM BITC for 24 hours, and lysates were analyzed for luciferase activity as above.
Induction of STAT-3 by IL-6
Dishes containing 1 × 106 subconfluent BxPC-3 cells were seeded 24 hours before stimulation with 5, 10, or 20 ng/mL human recombinant IL-6 (Sigma-Aldrich) for 0.25, 0.50, or 24 hours. Whole-cell extracts were prepared and analyzed for activated STAT-3 (pTyr705) and for total STAT-3 by western blotting with phospho-specific or anti–STAT-3 (total) antibodies. Substantial STAT-3 activation was observed after 15 minutes of exposure to 5 ng/mL IL-6. Therefore, these conditions were used in subsequent experiments to activate STAT-3.
Overexpression of STAT-3α
A plasmid that contained STAT-3α (Gene Accession number U06922) cloned into the pBabe Puro mammalian expression vector (a generous gift from Dr J. F. Bromberg, Rockefeller University, New York) was transfected to overexpress STAT-3 in BxPC-3 cells. Briefly 3 × 105 cells were transfected with 2 μg of the STAT-3α plasmid diluted in Opti-MEM serum-free medium to which lipofectamine reagent (Invitrogen) was added before the mixture was added to cells. Cells were incubated with plasmid–lipofectamine mixture for 5 hours and then replenished with normal growth medium for 48 hours. Transfected cells were treated with 0.1% DMSO or with 10 μM BITC for 24 hours. Cell lysates were prepared, and 20 μg of protein was analyzed by western blotting. The same blots were stripped and reprobed with anti-actin antibody for equal loading.
Apoptosis Detection by Cell Death Enzyme-Linked Immunosorbent Assay Method
In addition to caspase and PARP cleavage on western blots, the Cell Death Detection Enzyme-linked ImmunoSorbent Assay (ELISA) kit (Roche Applied Science, Mannheim, Germany) was used, following the manufacturer's instructions, to measure apoptotic cell death. Briefly, 1 × 104 BxPC-3 cells were seeded in 96-well plates and treated with either 10 μM BITC for 24 hours or with 0.1% DMSO as a control. In other experiments, similarly grown BxPC-3 cells were either stimulated with IL-6 or transfected with STAT-3α and then treated with BITC. Cell lysates were mixed with biotinylated anti-histone antibodies and peroxide POD conjugated anti-DNA antibodies in streptavidin-coated multiwell dishes, so that histone-bound DNA fragments that are characteristic of apoptosis could be detected and quantified. The plates were read at 405 nm and at 490 nm for background on EL-800 ELISA plate reader (BioTek Instruments Inc). Each sample was analyzed in triplicate, and the average values were subtracted from the background values.
In Vivo Xenograft Experiment
To assess the mechanism of BITC and its efficacy in vivo, BxPC-3 human pancreatic cancer cells were grown as xenografts in mice. The use of athymic nude mice and their treatment was approved by the Institutional Animal Care and Use Committee (IACUC), Texas Tech University Health Sciences Center, and all the experiments were carried out in strict compliance with their regulations. Six-week-old female athymic nude mice (NCR nu/nu, n = 10) were purchased from Tacomic (Germantown, NY). Mice were put on an antioxidant-free AIN-76A special diet (ICN Biomedicals, Aurora, OH) 1 week before starting the experiment. Tumor xenografts were implanted in athymic nude mice as described previously (8). Briefly, 1 × 106 BxPC-3 tumor cells in 0.1 mL PBS were injected subcutaneously in both left and right flanks of the mice. Mice were divided randomly into two groups with five mice in each group. Because each mouse had two tumors, every group contained 10 tumors. In our past experience and in this experiment, the two tumors from the same mouse typically showed different growth patterns; however, a permutation-based Wilcoxon rank test was used to sum the two log-transformed measurements on each mouse. Treatment with BITC started the same day after tumor cell implantation. Group 1 served as controls and received 0.1 mL PBS as vehicle, whereas group 2 received 12 μmol BITC in 0.1 mL PBS five times a week (Monday to Friday) by oral gavage. Starting 8 days after tumor cell implantation and after each mouse started to develop palpable tumors, tumors were measured three times a week (Monday, Wednesday, and Friday) using vernier calipers. Each mouse was also weighed twice a week (Monday and Friday) from the day of tumor cell implantation until 42 days after that time. At day 42, at which time the tumors typically started to show signs of necrosis, mice were killed by CO2 asphyxiation followed by cervical dislocation in accordance with IACUC guidelines. The tumors were removed aseptically from each mouse, and half of each tumor was snap frozen in liquid nitrogen for western blotting while the remaining other half was fixed in 10% neutral buffered formalin overnight. For western blotting, tumors from control and BITC-treated mice were washed with ice-cold PBS, minced, and homogenized in lysis buffer containing 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin, and 1 mM phenylmethane sulfonyl fluoride. The tumor lysate was cleared by centrifugation at 14 000g for 30 minutes. Lysate containing 60 μg of protein was resolved by 10% SDS–PAGE, and immunoblots were probed with anti–pSTAT-3 (Tyr705) and anti–STAT-3 antibody.
Apoptosis Measurements from Human Tumor Xenografts
Tumor tissues fixed in 10% neutral buffered formalin were dehydrated and embedded in paraffin, and sections that were 4 μm in thickness were prepared at every 100-μm interval. Paraffin-embedded tumor tissues were stained using hematoxylin and eosin (Anatech Ltd, Battle Creek, MI). Apoptosis was measured using a TdT-mediated dUTP-biotin nick end labeling (TUNEL)-based In Situ Apoptosis detection kit (#4828-30-DK; Trevigen, Inc, Gaithersburg, MD) according to the manufacturer's instructions. Briefly, tissue sections were deparaffinized and hydrated by sequential incubation in xylene, absolute alcohol, 70% alcohol, deionized water, and PBS at room temperature. Sections on slides were then incubated in proteinase K (20 μg/mL in 10 mM Tris–HCl, pH 7.4) for 15 minutes at 37°C, quenched in 3% hydrogen peroxide, and washed with PBS. Slides were incubated with TdT labeling buffer in a humidifying chamber at 37°C for 30 minutes followed by immersion in TdT stop buffer for 5 minutes at room temperature, incubation with 50 μL of biotinylated bromodeoxyuridine antibody at 37°C for 1 hour, and washed with PBS. Finally, apoptotic staining was developed with (diluted 1:50 in PBS) streptavidin-conjugated HRP and (diluted 1:200 in PBS) 3,3′-diaminobenzidene, an HRP substrate. After washing, sections were counterstained with methyl green and analyzed under a phase-contrast Olympus microscope (Olympus America Inc, Central Valley, PA).
Immunohistochemistry for STAT-3 Localization
Paraffin-embedded tissue sections for immunohistochemistry were deparaffinized and rehydrated by incubating sections in three washes of xylene for 5 minutes each, two washes of 100% ethanol for 10 minutes each followed by two washes of 95% ethanol for 10 minutes, and the sections were given two 5-minute washes in double-distilled water (dH2O). Antigens were unmasked by boiling the sections in 10 mM sodium citrate buffer (pH 6.0) and then reducing the temperature to below the boiling point at around 95°C for 10 minutes. The slides were cooled on the bench top for 30 minutes and washed in dH2O three times for 5 minutes each, then incubated in 3% hydrogen peroxide for 10 minutes followed by two washes in dH2O for 5 minutes each. Then, tumor sections were washed twice in wash buffer (PBS with 0.1% Tween-20) for 5 minutes each, blocked in 200 μL of blocking solution (5% horse serum diluted in TBST) for 1 hour at room temperature, and incubated with anti–STAT-3 antibody (1:300 in TBST) overnight at 4°C. After removal of the primary antibody, sections were washed three times in wash buffer for 5 minutes each followed by incubation with 200 μL of HRP-conjugated secondary antibody diluted 1:5000 in blocking solution for 30 minutes. Subsequently, sections were washed with wash buffer and incubated with 200 μL of avidin-biotin conjugate (ABC) reagent containing avidin and biotinylated HRP for 30 minutes at room temperature using ABC staining kit according to the manufacturer's instructions (Santa Cruz Biotechnology Inc, Santa Cruz, CA). Three drops of peroxidase substrate was added to each section and incubated until the desired color developed. The sections were counterstained with hematoxylin and mounted and analyzed under a phase-contrast Olympus microscope (Olympus America Inc).
Statistical Analysis
All statistical calculations were performed using InStat software and GraphPad Prizm 4.0. Nonparametric analysis of variance followed by Bonferroni or Newman–Keuls post hoc multiple comparison tests were used to test the statistical significance of STAT-3 DNA-binding assay, luciferase reporter assay, apoptosis and cell survival assays between multiple control and treated groups. The Student t test was used to compare the control and treated groups in the STAT-3 luciferase reporter assay. Experiments were usually repeated three times unless otherwise indicated with three replicates each. The data represent mean values with 95% confidence intervals. Differences were considered statistically significant when the P value was less than .05. To analyze the mouse tumor size, we computed the geometric mean of the right and left flank tumor size measurements for each mouse. The control and treatment groups were compared at each time point using a two-sided exact Wilcoxon rank sum test (no corrections for multiple comparisons were made because measurements at successive time points are highly correlated). Differences were considered statistically significant when the P value was less than .05. Mean tumor sizes were based on the within-group means of the geometric means for each of the five mice, and 95% confidence intervals were computed.
Results
Effect of BITC on Constitutively Activated STAT-3 in Human Pancreatic Cancer Cells
In our previous studies, we demonstrated that BITC strongly suppressed the growth of human pancreatic cancer cells by causing cell cycle arrest and apoptosis, but the exact mechanism by which BITC induced apoptosis was not clear (18). Recent studies have implicated STAT-3 in the promotion of cell survival and resistance to apoptosis in a wide variety of human tumor cell lines (22,30,31,45,46). We hypothesized that BITC-induced apoptosis in pancreatic cancer cells could be mediated by inhibiting STAT-3 activity; however, a role for STAT-3 in pancreatic cancer had not yet been clearly determined. To test our hypothesis and to establish a role for STAT-3 in pancreatic tumorigenesis, the effects of BITC treatment were evaluated in BxPC-3 human pancreatic cancer cells, in which STAT-3 is normally constitutively active.
Treatment of BxPC-3 cells with 0–40 μM BITC for a 24-hour period substantially reduced their survival in a concentration-dependent manner, with an IC50 of about 10 μM (Figure 1, A). In support of this finding, BxPC-3 cells that were treated with 0–20 μM BITC showed cleavage of caspase-3 and PARP, both indicative of apoptosis, when their lysates were separated on western blots (Figure 1, B). Treatment of BxPC-3 cells with 0–20 μM BITC for 24 hours not only substantially decreased STAT-3 activation, as reflected by reduced phosphorylation of tyrosine 705 (Tyr705) and serine 727 (Ser727), but also reduced the total levels of STAT-3 protein in a dose-dependent manner (Figure 1, C). Therefore, the same concentrations of BITC that effectively reduced STAT-3 expression and activation also substantially induced apoptosis in this cell line (Figure 1, C vs B).
Figure 1.

Effect of benzyl isothiocyanate (BITC) on the constitutive phosphorylation and expression of STAT-3 in BxPC-3 cells. A) Cytotoxicity of BITC in BxPC-3 cells as measured by the sulforhodamine B cell survival assay. Cells were treated with varying concentrations of BITC or with dimethyl sulfoxide (DMSO) as a control and surviving cells were quantified spectrophotometrically. The experiment was repeated four times, each time with eight replicates, and the data are expressed as the means of all experiments with 95% confidence intervals (error bars). B) Induction of apoptosis by BITC as assayed by caspase-3 and PARP cleavage. Whole-cell lysates were prepared, and samples containing 40 μg of protein were resolved by 10% SDS–PAGE and analyzed for the induction of apoptosis as assessed by caspase-3 and PARP cleavage (PARP-CF). β-Actin was used as a control for loading and transfer. The experiment was repeated three times with similar results. C) Effect of BITC on STAT-3 protein levels and phosphorylation. BxPC-3 cells were treated for 24 hours with 0–20 μM BITC, and cell lysates were examined on western blots for STAT-3 phosphorylation at Tyr705 and Ser727 and for STAT-3 protein expression. β-Actin was used as a control for loading and transfer. The experiment was repeated three times with similar results. D) Further examination of STAT-3 phosphorylation. Total STAT-3 was immunoprecipitated from control and BITC-treated BxPC-3 cells, and an equal amount of STAT-3 protein was resolved on 10% SDS–PAGE and analyzed for pSTAT-3 (Tyr705). The experiment was repeated three times and the mean of the three experiments was represented as the ratio of pSTAT-3 (Tyr705)/STAT-3 protein with 95% confidence intervals (error bars). E and F) Effect of BITC on STAT-3 mRNA levels as a function of concentration (E) and time (F). Cells were treated with 0–20 μM BITC for 24 hours (E) or with 10 μM BITC for 0–24 hours (F) before total RNA was extracted by the Trizol method and analyzed by reverse transcription– polymerase chain reaction. In each experiment the same RNA samples were analyzed for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal control for loading and transfer. The experiment in (E) was repeated four times, the experiment in (F) twice, and similar results were obtained. G) Effect of BITC on JNK, ERK, and p38 phosphorylation. BxPC-3 cells were treated with varying concentrations of BITC for 24 hours and cell lysates were examined on western blots using antibodies specific for the activated pJNK (Thr183/Tyr185), pERK (Thr202/Tyr204), and pp38 (Thr180/Tyr182) proteins and for total protein of each type. β-Actin was used as a control for loading and transfer. The experiment was repeated twice and similar results were obtained.
To test whether BITC treatment was associated with reduced STAT-3 phosphorylation in the absence of decreased STAT-3 protein levels, total STAT-3 protein was immunoprecipitated from both 0.1% DMSO-treated and BITC-treated (10 μM, 24 hours) BxPC-3 cells, using anti–STAT-3 antibody. Equal amounts of STAT-3 protein were resolved by SDS–PAGE, blotted, and probed with an anti–pSTAT-3 (Tyr705) antibody. Interestingly, on a molar basis, no substantial change was observed in STAT-3 phosphorylation in response to BITC treatment (Figure 1, D). These results suggested that the decreased STAT-3 phosphorylation that we observed in the presence of BITC is likely to be due to reduced STAT-3 protein levels.
Because we observed substantial reductions in STAT-3 protein expression in response to BITC treatment, we also tested whether BITC might affect STAT-3 mRNA levels. When STAT-3 mRNA transcripts were amplified by RT-PCR from lysates of BxPC-3 cells that had been treated with BITC or 0.1% DMSO, BITC substantially decreased STAT-3 mRNA levels in a dose- and time-dependent manner as compared with controls (Figure 1, E and F). To establish whether BITC specifically affected STAT-3 activation, the effect of BITC on MAPK signaling pathways was also evaluated. We found that BITC treatment was not associated with changes in the activation (phosphorylation) nor expression of JNK, ERK, or p38 in BxPC-3 cells (Figure 1, G). Therefore, among the pathways tested, BITC specifically affected synthesis and/or stability of STAT-3.
Effect of BITC on DNA-Binding and Transcriptional Activity of STAT-3
To perform its role in cell survival, activated STAT-3 translocates to the nucleus and binds to specific response elements in the promoters of its target genes. Because we observed substantially reduced levels of STAT-3 in the nuclear fraction of BITC-treated cells (data not shown), we expected that, in treated cells, decreased amounts of STAT-3 would be able to bind to DNA response elements and promote transcription.
We determined the DNA-binding activity of STAT-3 using the Universal EZ-TFA transcription factor colorimetric assay, which combined the DNA-binding principle of the electrophoretic mobility shift assay with the 96-well format of an enzyme-linked immunosorbent assay. Compared with extracts from DMSO-treated control cells, extracts from BxPC-3 cells treated with 10 μM BITC for 24 hours exhibited an approximately 48% decrease in the STAT-3 DNA-binding activity (A450 of STAT-3–DNA binding, control cells vs BITC-treated cells: 0.089 vs 0.049, difference = 0.040, 95% CI = 0.033 to 0.046; P = .017; Figure 2, A). We also expected that if BITC inhibited the activation and expression of STAT-3, it might inhibit transcription of STAT-3–responsive target genes. To examine this question, BxPC-3 cells were treated with 0–20 μM BITC for 24 hours and the whole-cell lysate was resolved on 10% SDS–PAGE followed by western blotting. As shown in Figure 2, B, BITC treatment was associated with substantial and dose-dependent reductions in the expression of Bcl-2 and Mcl-1 proteins, the products of two STAT-3–responsive genes that have been shown to play roles in apoptosis and cell growth (27,36).
Figure 2.

Effect of benzyl isothiocyanate (BITC) on STAT-3 DNA-binding and transcriptional activity in BxPC-3 cells. A) Effect of BITC on STAT-3 binding to its cognate DNA sequence. BxPC-3 cells were treated for 24 hours with or without 10 μM BITC, and cell extracts were tested for STAT-3 DNA-binding activity as measured by the Universal EZ-TFA transcription factor colorimetric assay, in which a biotinylated double-stranded oligonucleotide containing the consensus sequence for STAT-3 was used to link bound transcription factors to streptavidin-coated wells. The negative control contained only binding buffer and capture probe. Control wells contained non–BITC-treated cell lysates, with or without a competitor probe (CP) that was unbiotinylated. Means and 95% confidence intervals of two independent experiments performed in triplicate are shown. Differences between all the groups were compared by nonparametric analysis of variance with Bonferroni post hoc multiple comparison test. Statistical tests were two-sided. B) Effect of BITC on the expression of the STAT-3–regulated genes Mcl-1 and Bcl-2. Cells were treated with either dimethyl sulfoxide (DMSO) or 0–20 μM BITC for 24 hours. Whole-cell lysates were prepared, and 40 μg of protein was resolved by 10% SDS–PAGE, blotted, and analyzed for Mcl-1 and Bcl-2 proteins. β-Actin was used as a control for loading and transfer. The experiment was repeated twice and similar results were obtained. C) Effect of the proteosome inhibitor MG-132 on BITC-mediated reduction of STAT-3 protein levels. BxPC-3 cells were treated either with 10 μM BITC or DMSO alone (as a control) for 24 hours and with or without 10 μM MG-132 for 2 hours, and the expression of STAT-3 was determined by western blotting of whole-cell lysates. β-Actin was used as a control for loading and transfer. The experiment was repeated twice and similar results were obtained. D) Effect of the proteosome inhibitor MG-132 on BITC-mediated STAT-3 degradation. BxPC-3 cells were treated with or without BITC and MG-132 as described in (C). Cell lysates were immunoprecipitated overnight with anti–STAT-3 antibody and whole immunoprecipitates were resolved by 10% SDS–PAGE and immunoblotted with anti–STAT-3 and anti-ubiquitin antibodies. E) Effect of BITC on STAT-3–regulated luciferase reporter activity. STAT-3 luciferase transcriptional activity was determined in BxPC-3 cells cotransfected with 2 μg of a plasmid encoding firefly luciferase under the control of the STAT-3 promoter and with 0.2 μg of a plasmid expressing Renilla luciferase as a transfection efficiency control. At 24 hours after transfection, cells were treated with or without 10 μM BITC for 24 hours or pretreated with 10 μg/mL cycloheximide for 4 hours and then treated with or without 10 μM BITC for 24 hours. Whole-cell lysates were collected, and firefly luciferase activities were corrected for Renilla luciferase levels and then normalized relative to the DMSO control, which was considered as 1. A value less than 1.0 in this assay indicates attenuation of STAT-3–directed transcription by BITC. Means and 95% confidence intervals of two independent experiments performed in triplicate are shown. The differences between all the groups were compared by nonparametric analysis of variance with Bonferroni post hoc comparisons. Statistical tests were two-sided. F) Effect of cycloheximide on BITC-mediated reduction of STAT-3 mRNA content. Cells were pretreated with 10 μg/mL cycloheximide for 4 hours followed by treatment with 10 μM BITC for 24 hours. Total RNA was extracted by Trizol method and probed for STAT-3 expression. The same RNA samples were probed for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal loading control. The experiments were repeated three times and similar results were obtained.
Because we had observed dramatic reductions of both STAT-3 mRNA and protein levels, we next asked whether STAT-3 protein might have been inhibited through BITC-mediated activation of the ubiquitin-proteasome pathway. To address this question, we treated BxPC-3 cells with or without 10 μM BITC for 24 hours and with or without 10 μM MG-132, a specific proteasome inhibitor, for 2 hours. In the blotted whole-cell lysates shown in Figure 2, C, the BITC-mediated decline in the STAT-3 protein expression could be almost completely prevented by MG-132 treatment. Furthermore, when STAT-3 was first immunoprecipitated from BITC- and/or MG-132–treated and untreated cells, then blotted, probed with anti–STAT-3 antibody, and then reprobed with anti-ubiquitin antibodies, high–molecular weight polyubiquitin conjugates were observed in response to BITC treatment suggesting that STAT-3 is degraded by the ubiquitin-proteasome pathway (Figure 2, D).
To delineate whether the mRNA levels for STAT-3 were reduced due to decreased STAT-3–mediated transcription after BITC treatment, we transfected BxPC-3 cells with a plasmid that encoded the STAT-3 promoter upstream of a luciferase reporter gene, treated the cells with or without 10 μM BITC, and performed luciferase assays on extracts made 24 hours later. We observed that BITC treatment statistically significantly inhibited STAT-3–regulated luciferase reporter gene activity (luciferase activity in control vs BITC-treated cells: 1.08 vs 0.245, difference = 0.835, 95% CI = 0.415 to 1.264; P = .001) (Figure 2, E).
The transcription of the STAT-3 gene has been shown to be autoregulated by STAT-3 protein through a composite IL-6 response element in its promoter that contains both a STAT-3–binding element and a cyclic AMP-responsive element (47–49). To examine whether the results in Figure 2, E, which showed that BITC treatment inhibited STAT-3 transcription, might be due to transcriptional autoregulation by STAT-3, we pretreated the BxPC-3 cells for 4 hours with 10 μg/mL cycloheximide, a protein synthesis inhibitor, before treating them with 10 μM BITC for 24 hours and then evaluated STAT-3–mediated luciferase reporter activity in one experiment and STAT-3 mRNA levels in another. Cycloheximide pretreatment blocked STAT-3 transcriptional activity in the reporter assay and also blocked the BITC-mediated decrease in STAT-3 mRNA levels, albeit incompletely (Figure 2, E and F). However, STAT-3 mRNA levels were also consistently reduced by cycloheximide treatment alone (Figure 2, F). The explanation for this paradox is not clear at this time and warrants further investigation. Taken together, our results consistently show that BITC reduces STAT-3 mRNA and protein levels, attenuates STAT-3 activation as demonstrated by reduced phosphorylation at Tyr705 and Ser727, and reduces DNA-binding and STAT-3–directed promoter activity.
Rescue of BITC-Induced Apoptosis by IL-6–Induced STAT-3 Phosphorylation
Because IL-6 is a growth factor that has been shown to activate STAT-3 (50–52), we next sought to determine whether IL-6 can increase STAT-3 activity to higher levels than the constitutive levels normally seen in BxPC-3 cells and whether the effect of BITC on apoptosis is rescued under these conditions. BxPC-3 cells were stimulated with 5 ng/mL of IL-6 for 15 minutes to 24 hours. In lysates from cells treated with IL-6 for 15 minutes, we observed 20-fold increased phosphorylation of STAT-3 at Tyr705 compared with constitutive levels, without any change in STAT-3 protein levels. In lysates from cells treated with IL-6 for 24 hours, STAT-3 phosphorylation was increased 11-fold as compared with DMSO-treated controls (data not shown).
Next, we determined whether BITC could inhibit IL-6–induced STAT-3 phosphorylation. BxPC-3 cells were first stimulated with 5 ng/mL IL-6 for 15 minutes and then treated with either DMSO or 10 μM BITC for 24 hours. As shown in Figure 3, A, in the presence of IL-6 pretreatment, STAT-3 protein levels still appeared to be reduced in association with BITC treatment. Moreover, IL-6–induced increases in STAT-3 phosphorylation at Tyr705 and Ser727 (that were still visible 24 hours after IL-6 stimulation) were markedly lessened after BITC treatment. Similar results were obtained when BxPC-3 cells were first treated for 24 hours with BITC and then treated for 15 minutes with IL-6 (data not shown). Furthermore, IL-6–mediated enhancement of STAT-3 activation was accompanied with a modest decrease in BITC-induced apoptosis, as indicated by reduced levels of caspase-3 and PARP cleavage in lysates from BxPC-3 cells treated with both IL-6 and BITC compared with those from cells treated with BITC alone (Figure 3, A).
Figure 3.

Effects of benzyl isothiocyanate (BITC) in BxPC-3 cells in which STAT-3 has been activated by interleukin 6 (IL-6) or in which STAT-3 has been overexpressed. A) Effect of BITC on STAT-3 activation, STAT-3 protein expression, and apoptosis in IL-6–treated BxPC-3 cells. Cells were stimulated with 5 ng/mL of IL-6 for 15 minutes followed by treatment with 10 μM of BITC for 24 hours. Whole-cell lysates were resolved on 10% SDS–PAGE for the analysis of STAT-3 phosphorylation at Tyr705 and Ser727, STAT-3 expression, and cleavage of caspase-3 and PARP. β-Actin was used as a control for loading and transfer. The experiment was repeated three times and similar results were obtained. B) Effect of BITC on STAT-3 activation, STAT-3 expression, and apoptosis in BxPC-3 cells overexpressing STAT-3α. Cells were transfected with an empty vector or a plasmid expressing STAT-3α and 48 hours later were treated with 10 μM of BITC for 24 hours. Whole-cell lysates were analyzed as in (A). The experiment was repeated three times and similar results were obtained. C) Effect of STAT-3α overexpression on BITC-induced apoptosis in BxPC-3 cells. Apoptosis was further confirmed by the cell death detection assay, which uses monoclonal antibodies directed against DNA and histones to quantify apoptosis. Nontransfected BxPC-3 cells were treated with or without 10 μM BITC for 24 hours and analyzed for apoptosis. Separately, cells were transfected with empty vector or STAT-3α–expressing vector as described above and then treated with or without 10 μM BITC for 24 hours. Means and 95% confidence intervals of two independent experiments performed in triplicate are shown. Differences between all the groups were compared by nonparametric analysis of variance (ANOVA) with Bonferroni post hoc multiple comparison test. Statistical tests were two-sided. D) Effect of STAT-3α overexpression on BITC-associated reductions in cell survival. BxPC-3 cells were transfected with a STAT-3α−expressing vector as described above and treated with 10 μM BITC or 0.1% dimethyl sulfoxide (control) for 24 hours and the cell survival was evaluated by sulforhodamine B assay. Means and 95% confidence intervals of two independent experiments performed in triplicate are shown. Differences between all the groups were compared by nonparametric ANOVA with the Newman–Keuls post hoc multiple comparison test. Statistical tests were two-sided. E) Effect of IL-6 or STAT-3α overexpression on BITC-associated reductions in STAT-3 DNA binding. The Universal EZ-TFA transcription factor colorimetric assay was used to determine the DNA-binding activity of STAT-3 in BxPC-3 cells that were stimulated with 5 ng/mL IL-6 for 15 minutes or that overexpressed STAT-3α with and without treatment with 10 μM BITC for 24 hours. A negative control contained binding buffer and free probe without cell lysate, whereas free probe contained neither binding buffer nor cell lysate. Empty vector and control are defined above in (C). Means and 95% confidence intervals of two independent experiments performed in triplicate are shown. Differences between all the groups were compared by nonparametric ANOVA with the Newman–Keuls post hoc multiple comparison test. Statistical tests were two-sided. F) Effect of IL-6 or STAT-3α overexpression on BITC-associated reductions in STAT-3 transcriptional activity. Transcriptional activity of STAT-3 in BxPC-3 cells that were stimulated with 5 ng/ml IL-6 for 15 minutes or that overexpressed STAT-3α with and without 24-hour treatment with 10 μM BITC was determined by luciferase assays. Control cells were neither stimulated with IL-6 nor transfected with empty vector. Means and 95% confidence intervals of two independent experiments performed in triplicate are shown. Differences between all the groups were compared by nonparametric ANOVA with Newman–Keuls post hoc multiple comparison test. Statistical tests were two-sided. G) Effect of STAT-3α overexpression on BITC-associated reductions in Mcl-1 and Bcl-2 expression. At 48 hours after transfection with empty vector or a STAT-3α–expressing vector, BxPC-3 cells were treated with 10 μM BITC for 24 hours. Cell lysates were separated on 10% SDS–PAGE and blots were probed using Mcl-1– and Bcl-2–specific antibodies, then stripped and reprobed with β-actin antibody to ensure equal loading. The experiment was repeated twice and similar results were obtained.
Rescue of BITC-Induced Apoptosis by Overexpression of STAT-3α
Two isoforms of STAT-3, STAT-3α (p92) and STAT-3β (p83), are derived from a single gene by alternative mRNA splicing and demonstrate both distinct and overlapping functions in gene transcription (53,54). STAT-3α is the predominant isoform expressed in the cell lines used in this study (or, it is possible that the STAT-3 antibody that we used was more specific to STAT-3α, because we detected a single band in our blots). To further confirm the role of STAT-3α in BITC-induced apoptosis, we overexpressed STAT-3α in BxPC-3 cells by transient transfection with a STAT-3α–expressing plasmid. STAT-3α overexpression (2.6-fold over control) completely protected BxPC-3 cells from BITC-induced apoptosis as assessed by caspase-3 and PARP cleavage (Figure 3, B). Furthermore, STAT-3α overexpression protected BxPC-3 cells from BITC-induced apoptosis as shown by quantitative apoptosis (Figure 3, C), and cell survival assays (Figure 3, D). STAT-3α overexpression (and, to a lesser extent, IL-6–mediated activation of STAT-3) also abrogated BITC-associated reductions in STAT-3 DNA-binding activity (Figure 3, E) and BITC-associated reductions in STAT-3 transcriptional activity as measured by luciferase reporter assay (Figure 3, F) in BxPC-3 cells. The ability of STAT-3α overexpression to reverse BITC-mediated inhibition of STAT-3 transcriptional activity and cell survival was also reflected by the restoration of Mcl-1 and Bcl-2 protein expression, levels of both of which were otherwise reduced in response to BITC treatment (Figure 3, G).
Effect of BITC on Apoptosis, STAT-3 Activity, and STAT-3 mRNA and Protein Levels in Other Human Pancreatic Cancer Cell Lines
To rule out the possibility that the observed effects of BITC were specific to BxPC-3 cells, we also evaluated the effects of BITC on the AsPC-1, Capan-2, MiaPaCa-2, and Panc-1 human pancreatic cancer cell lines, which have varying degrees of baseline STAT-3 expression and activation. Treatment of AsPC-1, Capan-2, or Mia-PaCa-2 cells for 24 hours with increasing concentrations of BITC was associated with substantially reduced cell survival (Figure 4, A) and increased apoptosis as determined by caspase-3 and PARP cleavage (Figure 4, B) for each of these cell lines. AsPC-1, Capan-2, and Mia-PaCa-2 cells treated with BITC also showed substantially decreased levels of STAT-3 protein and its phosphorylation at Tyr705 and Ser727 (Figure 4, C). Accordingly, the DNA-binding activity of STAT-3 was substantially reduced in all three cell lines after treatment with BITC for 24 hours (Figure 4, D). The transcriptional activity of STAT-3 was decreased in AsPC-1 cells but not in Capan-2 or MiaPaCa-2 cells in response to BITC treatment, as tested by the luciferase reporter assay (Figure 4, E). Nevertheless, in contrast to what was seen in BxPC-3 cells, STAT-3 mRNA levels were not altered in any of these cell lines following treatment with up to 20 μM BITC (Figure 4, F). We also tested the effects of BITC treatment on Panc-1 pancreatic adenocarcinoma cells and HPDE-6–immortalized “normal” pancreatic cells. Exposure of Panc-1 cells to 0–40 μM BITC for 24 hours resulted in decreased survival of the cells, with an IC50 of about 8 μM (Figure 5, A). BITC also induced apoptosis in Panc-1 cells (Figure 5, B). However, to our surprise, BITC treatment had no effect on the STAT-3 pathway in these cells. BITC failed to reduce the phosphorylation or protein level of STAT-3 (Figure 5, C). Similarly, BITC had no effect on the DNA-binding, transcriptional activity or mRNA level of STAT-3 (Figure 5, D–F), suggesting that in Panc-1 cells, BITC reduces cell survival by affecting pathways other than STAT-3.
Figure 4.

Effect of benzyl isothiocyanate (BITC) on STAT-3 signaling in AsPC-1, Capan-2, and MiaPaCa-2 cells. AsPC-1, Capan-2, and MiaPaCa-2 cells were treated with either dimethyl sulfoxide or with varying concentrations of BITC. A) Cytotoxicity of BITC in these cell types, measured by the sulforhodamine B cell survival assay. The experiment was performed as described in Figure 1, A. It was repeated two times, each with eight replicates, and similar results were obtained. Means and 95% confidence intervals are shown. B) Apoptosis of these cell types in response to BITC measured by caspase-3 and PARP cleavage (caspase-3-CF and PARP-CF, respectively). The experiment was performed as described in Figure 1, B. It was repeated twice and similar results were obtained. C) Phosphorylation and expression of STAT-3 protein in these cell types in response to BITC. Western blots were performed as described in Figure 1, C. These experiments were repeated twice and similar results were obtained. D) Effect of BITC on STAT-3 DNA binding in these cell lines. DNA binding was measured by the Universal EZ-TFA transcription factor colorimetric assay as described in Figure 2, A. Means and 95% confidence intervals of two independent experiments performed in triplicate are shown. Differences between all the groups were compared by nonparametric analysis of variance with the Bonferroni post hoc multiple comparison test. Statistical tests were two-sided. E) Effect of BITC on STAT-3 transcriptional activity in these cell lines. Transcriptional activity was measured by luciferase assay as described in Figure 2, E. Means and 95% confidence intervals of two independent experiments performed in triplicate are shown. Differences between groups were compared using the Student t test (two-sided; ns denotes “not significant”). F) Effect of BITC on STAT-3 mRNA expression in these cell lines. Cells were treated for 24 hours with 0–20 μM BITC, and total RNA was analyzed for STAT-3 content by reverse transcription–polymerase chain reaction. The same RNA samples were analyzed for glyceraldehyde 3-phosphate (GAPDH) as an internal loading control. The experiment was repeated twice and similar results were obtained.
Figure 5.
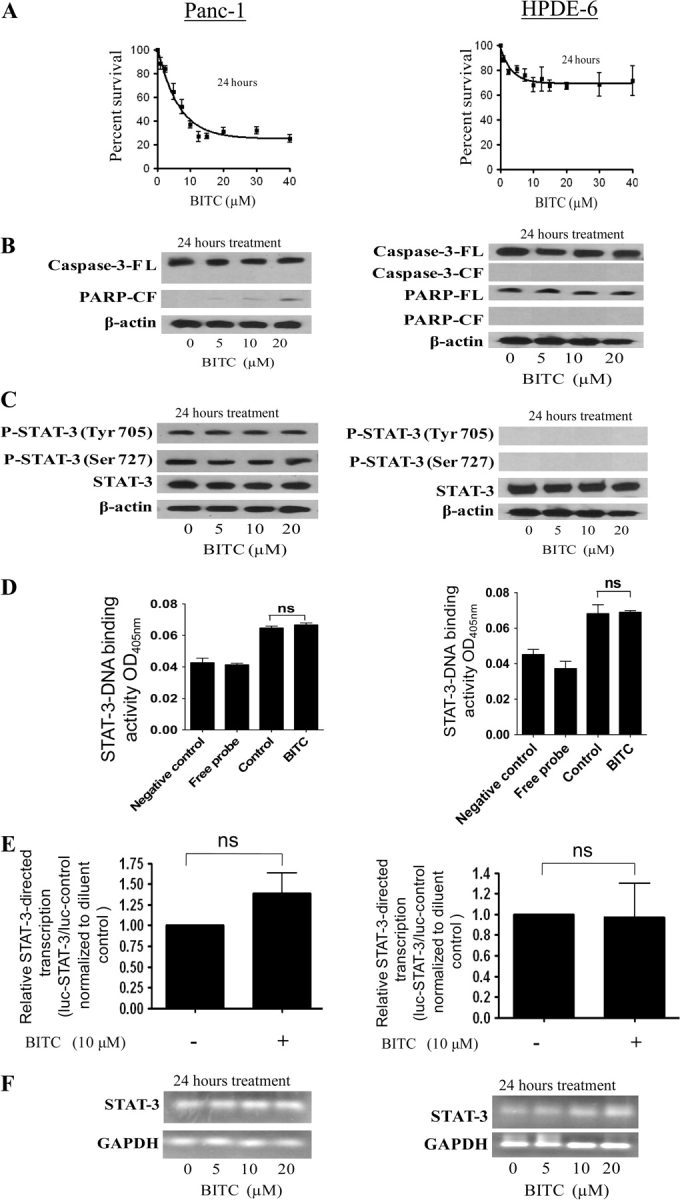
Effect of benzyl isothiocyanate (BITC) on STAT-3 signaling in Panc-1 and immortalized normal pancreatic ductal epithelial cells. The effects of BITC on STAT-3 in Panc-1 and HPDE-6 cells were determined as described in Figure 4. A–F) Each experiment was repeated twice and similar results were obtained. The same blots were stripped and reprobed with β-actin antibody as a control for equal loading and transfer. The same RNA samples were analyzed for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression, as an internal control for loading and transfer. Means and 95% confidence intervals of two independent experiments performed in triplicate are shown. Differences between groups in (D) were compared by nonparametric analysis of variance with the Bonferroni post hoc multiple comparison test and in (E) were compared by the Student t test. Statistical tests were two-sided. Nonsignificance is denoted as “ns.”
The effects of BITC were then evaluated in HPDE-6 cells, a line of immortalized human pancreatic ductal epithelial cells that has been characterized extensively and is considered to be similar to normal (nonmalignant) pancreatic cells in character (42,43). The cells were exposed to 0–40 μM BITC for 24 hours. As shown in Figure 5, A, survival of HPDE-6 cells was not affected by BITC treatment, even at concentrations that were very toxic to cancer cells. Next, we determined the induction of apoptosis by cleavage of caspase-3 and PARP. In contrast to what we found in pancreatic cancer cells, BITC failed to induce apoptosis in HPDE-6 cells (Figure 5, B). We also observed no constitutive activation of STAT-3, as indicated by the absence of phosphorylation at Tyr705 or Ser727, in HPDE-6 cells (Figure 5, C), consistent with the general observation in the literature that STAT-3 is activated in transformed cells and not in normal cells (37). Furthermore, BITC treatment did not alter the protein level of STAT-3 in HPDE-6 cells (Figure 5, C). Similarly, BITC failed to inhibit DNA binding, the transcriptional activity, or the mRNA level of STAT-3 (Figure 5, D–F), suggesting that HPDE-6 cells are altogether resistant to the cytotoxic effects of BITC.
Effect of BITC Treatment on Apoptosis and STAT-3 Activity on BxPC-3 Pancreatic Cancer Cell Xenografts in Nude Mice
We next aimed to determine whether BITC could suppress the growth of pancreatic tumors in vivo and if so, to examine the mechanism. Based on our in vitro results, we hypothesized that BITC treatment would inhibit in vivo tumor growth by reducing STAT-3 expression in the tumor cells. To test our hypothesis, human pancreatic tumor xenografts were implanted in athymic nude mice by injecting 1 × 106 BxPC-3 cells subcutaneously in both the left and right flanks of each mouse, so that each mouse had two independent tumors. Mice were randomly divided into two groups with five mice per group, and because each mouse had two implanted tumors, each group had 10 tumors. Starting the same day, after tumor cell implantation, one group of mice was fed 12 μmol BITC dissolved 0.1 mL of PBS per day, 5 days a week, and control mice received PBS alone. Tumors were measured three times a week, and each mouse was weighed twice a week as we described previously (8). By day 42, the growth of the tumors in BITC-fed mice was substantially retarded compared with tumor growth in control mice, and tumors also appeared to grow more slowly in BITC-fed mice compared with control mice at all earlier stages (Figure 6, A). For example, 6 weeks after treatment with 12 μmol BITC (60 μmol/wk), the average tumor volume in control mice was about 1.92-fold higher than that in BITC-treated mice (mean tumor volume, control vs BITC treated: 334 vs 172 mm3, difference = 162 mm3, 95% CI = 118 to 204 mm3; P = .008) (Figure 6, A).
Figure 6.

Effects of benzyl isothiocyanate (BITC) on growth, apoptosis, and STAT-3 expression in BxPC-3 human pancreatic tumor xenografts in nude mice. Tumor cells were implanted into athymic nude mice, and each mouse received 12 μmol BITC by oral gavage starting the day of tumor implantation and continuing 5 days a week for 6 weeks before the mice were killed. A) Effect of BITC on tumor volumes. Tumors were measured by vernier calipers and sizes were calculated as follows: volume (mm3) = length × (width2)/2. B) Effect on tumor weights. Tumors were excised on day 42 after implantation and weighed before use in immunohistochemical and western blotting experiments. C) Effect on average mouse weight. Tumor-bearing mice were weighed over the course of the experiment as a measure of possible BITC toxicity. Values in (A) are means of 10 (five sets of two) observations in each group with 95% confidence interval. Differences between control and BITC-treated groups in (A) were compared with exact two-sided Wilcoxon tests after summing the two log-transformed measurements on each mouse (P = .008). The weight of BITC-treated tumors was found to be statistically different from controls (P = .025). Differences between groups in (B) were compared by Student t test (two-sided) with 95% confidence intervals. D) Apoptosis and STAT-3 expression in tumor sections from BITC-treated and control mice. TdT-mediated dUTP-biotin nick end labeling (TUNEL), hematoxylin and eosin, and STAT-3 immunostaining were performed on representative tumor sections and photographs were taken at high (×100) magnification. E) STAT-3 expression and activation in lysates from the tumors of BITC-treated and control mice. Tumor lysates from tumor samples in (D) were further analyzed for phospho-Tyr705 STAT-3 and total STAT-3 protein by immunoblotting. Blots were stripped and reprobed with β-actin antibody to verify equal protein loading. Each lane represents a different tumor sample.
At 42 days after the implantation of the tumors, all mice were killed, and the tumors were excised, weighed, and processed for immunohistochemistry and western blotting. In accordance with the tumor volumes that were estimated from external measurements, the weight of the tumors from BITC-treated mice was approximately 53% less than the weight of tumors from control mice (mean weight, control vs BITC treated: 0.425 vs 0.225 g, difference = 0.2 g, 95% CI = 0.181 to 0.219; P = .025) (Figure 6, B). The average body weight of control and BITC-treated mice did not change throughout the experiment (Figure 6, C), which suggested that BITC was not associated with any discernible toxicity to the mice.
To investigate the mechanism by which BITC reduced tumor growth, tumors from control and BITC-treated mice were evaluated by immunohistochemistry. As analyzed by TUNEL assay, substantially increased numbers of apoptotic bodies were observed in the tumor sections obtained from BITC-treated mice compared with control mice (Figure 6, D), suggesting that the suppression of tumor growth in the BITC-treated mice was due to increased apoptosis of the tumor cells. Furthermore, immunostaining of similar tumor sections with antibodies to STAT-3 revealed substantially reduced STAT-3 staining in the tumors of BITC-treated mice as compared with controls (Figure 6, D) These observations were further confirmed by western blotting of protein lysates prepared from tumors obtained from control and BITC-fed mice. BITC-treated BxPC-3 tumors grown in mice contained lower levels of STAT-3 total protein and of STAT-3 phosphorylation than untreated tumors as shown on blots using anti–STAT-3 anti–pSTAT-3 (Tyr705) antibodies (Figure 6, E). Taken together, these in vivo results are in agreement with our in vitro results and suggest that STAT-3 is a critical signaling molecule responsible for BITC-induced apoptosis both in vitro and in vivo.
Discussion
STAT family proteins are cytoplasmic transcription factors that are activated by receptor tyrosine kinases and intracellular kinases through the phosphorylation of critical tyrosine and serine residues that regulate gene expression in response to cytokine and growth factor receptor signaling (19–24). These proteins mediate many diverse biological processes including cell survival, differentiation, inflammation, immune response, and apoptosis. Among the STAT proteins, STAT-3 has been extensively studied due to its constitutive expression in a large proportion of human cancers (25,27,29–32,35–37,55–57) and its role in neoplastic development and transformation (22,23,28,29,48) through the transcriptional control of its regulated genes (19,20,24,58). Inhibition of STAT-3 activation has been shown to suppress the growth of human malignant cells in experimental systems both in vitro and in vivo (25,27,30,32,35,36,55–57), and thus, targeted disruption of STAT-3 could be one potential approach to treat human cancers (26,39,40,59).
It is well established that various cytokines and growth factors can induce the activation of STAT-3 (44,60,61). Several lines of evidence have suggested that IL-6 stimulates cancer cell growth by the phosphorylation and activation of STAT-3 (50–53). Recent studies have consistently demonstrated that phospho–STAT-3 levels are elevated in malignant prostate cells in vivo, further supporting a role for activated STAT-3 in cancer (32). In agreement with these findings, in this study we find differential levels of constitutive STAT-3 phosphorylation in various pancreatic cancer cell lines but no activating phosphorylation of STAT-3 in normal HPDE-6 cells.
Our findings clearly demonstrate that apoptotic death of human pancreatic cancer cells in the presence of BITC, a naturally occurring anticancer agent, is associated with substantial reductions in the levels of both activated STAT-3 (as represented by phosphorylation at Tyr705 and Ser727) and total STAT-3 protein. The loss of STAT-3 expression in response to BITC treatment is both dose and time dependent. In BxPC-3 pancreatic cancer cells, STAT-3 mRNA levels are similarly reduced. The inhibition of STAT-3 by BITC was associated with decreased amounts of STAT-3–mediated DNA-binding activity and decreased transcription of the genes for Mcl-1 and Bcl-2, which are both known to be downstream of STAT-3 activation (26,30,37,56,62,63). In addition, our results show that overexpression of STAT-3 by gene transfection completely protected BxPC-3 pancreatic cancer cells from BITC-induced apoptosis, confirming the role of STAT-3 in BITC-induced apoptotic cell death. BITC treatment not only blocked constitutive STAT-3 activation but also was able to block tyrosine phosphorylation of STAT-3 induced by IL-6. Moreover, BITC failed to cause any cytotoxic effects in HPDE-6 cells, which are considered to be similar to normal human pancreatic cells. Last, we provided evidence that orally feeding BITC to athymic nude mice substantially suppressed the growth of BxPC-3 pancreatic tumor xenografts and that suppressed tumor growth was associated with increased apoptosis and decreased STAT-3 expression. Thus, we establish STAT-3 as a molecular target of BITC in pancreatic cancer cells. Our results are in agreement with recent studies that showed that the naturally occurring agents capsaicin and silibinin suppress the growth of multiple myeloma and prostate cancer cells, respectively, by blocking STAT-3 activation (27,36). In another study, resveratrol was reported to cause cell cycle arrest and apoptosis in breast and prostate cancer cells by inhibiting constitutive STAT-3 activation and suppressing STAT-3–regulated cyclin D1, Bcl-xL, and Mcl-1 gene expression (38). It is noteworthy that our study demonstrates the degradation of STAT-3 protein by BITC, in contrast to some previously published reports in which only phosphorylation was inhibited (27,36). In agreement with other previous studies, degradation of STAT-3 protein in our model involves the ubiquitin-proteasome pathway (64,65).
The identified targets of STAT-3 transcriptional activation include Bcl-2, Mcl-1, Bcl-xL, and cyclin D1 and reflect the roles of STAT-3 to promote cell survival and cell cycle progression (26,30,37,51,62,63). Moreover, constitutively active STAT-3 contributes to resistance to apoptosis in colorectal tumors and multiple myeloma cells, possibly through Bcl-2 and cyclin D1 expression (26,34,60). In our previous study, we showed that BITC treatment decreased the expression of Bcl-2 and cyclin D1 in pancreatic cancer cells (10). In this study, inhibition of STAT-3 activation together with reduced expression of Bcl-2 and Mcl-1 was associated with the ability of BITC to induce apoptosis in pancreatic cancer cells. These results are consistent with previous observations which showed that transfection with dominant-negative STAT-3 induced apoptosis in HeLa and SiHa cells that contained constitutively active STAT-3 (59). Our results also showed that overexpression of STAT-3α in BxPC-3 cells completely abrogated the apoptosis-inducing effects of BITC. Expression of Bcl-2 and Mcl-1 was not decreased after BITC treatment in cells that overexpressed STAT-3α. These results support the notion that STAT-3α is a critical target of BITC.
To rule out the possibility that BITC affects BxPC-3 cells specifically, the effects of BITC on apoptosis and STAT-3 activation were also evaluated in AsPC-1, Capan-2, MiaPaCa-2, and Panc-1 pancreatic cancer cells and compared with normal HPDE-6 pancreatic cells. BITC inhibited the STAT-3 signaling pathway in AsPC-1, Capan-2, and MiaPaCa-2 cells but not in Panc-1 cells. Furthermore, normal HPDE-6 cells were totally resistant to the deleterious effects of BITC. These results are in agreement with our previous studies (18), in which we demonstrated that BITC was least effective against acinar cells isolated from normal human pancreas. Taken together, these results suggest that BITC may not be as toxic to normal cells as to cancer cells. To extend the observations made in cultured cells and to evaluate the effect of BITC in vivo, we determined the effect of BITC on the growth of BxPC-3 pancreatic tumor xenografts in athymic nude mice. The growth rate of BxPC-3 tumor xenografts was substantially retarded in mice that were administered 12 μmol BITC orally for each of 5 days a week for 6 weeks without causing detectable side effects. The tumors obtained from BITC-treated mice exhibited substantially enhanced apoptosis and decreased STAT-3 staining as assessed by immunohistochemistry and immunoblotting. Our results in mice complement our in vitro results confirming STAT-3 as a potential therapeutic target of BITC.
BITC is a dietary agent that is abundant in many cruciferous vegetables that are consumed by humans on a daily basis. Epidemiological studies continue to support the notion that dietary intake of cruciferous vegetables may reduce the risk of different types of malignancies, including pancreatic cancer (4–7). Our data suggest that BITC is relatively safe to normal pancreatic cells and also safe to mice as established previously (18) and in this study. These data are consistent with previous in vitro studies of other isothiocyanates such as allyl isothiocyanate, phenethyl isothiocyanate (PEITC), and sulforaphane, in which cell growth arrest and induction of apoptosis were observed at concentrations lower than 40 μM (8,9,66,67). Because the pharmacokinetics of BITC in humans have not been determined, it is difficult to predict how much cruciferous vegetable would need to be consumed to clinically achieve a serum concentration of 10 μM BITC, the concentration that was most effective in inhibiting STAT-3 activation in our model. However, a very recent study suggested that orally feeding male Sprague-Dawley rats with PEITC (an analog of BITC) resulted in rapid absorption that reached a maximal plasma concentration of 9.2 ± 0.6 μM after 0.44 ± 0.1 hours of feeding 10 μmol/kg PEITC and of 42.1 ± 11.4 μM after 2.0 ± 1 hours of feeding 100 μmol/kg PEITC, suggesting that micromolar concentrations of isothiocyanates may be achieved in vivo (68). In another pharmacokinetics study in which four human volunteers were fed with a single dose of myrosinase-hydrolyzed extract from 3-day-old broccoli sprouts (which contained about 200 μmol of total isothiocyanates), a peak concentration of 2.27 μM isothiocyanates was reached in the plasma at 1 hour after broccoli extract ingestion (69). Nevertheless, detailed pharmacokinetic studies of BITC are required before conducting clinical testing of BITC as a cancer chemopreventive agent.
There are some limitations to our study. Although we have established the efficacy of BITC in terms of limiting the growth of human pancreatic cancer cells in vitro and in vivo and have demonstrated that STAT-3 is a critical target, it remains to be determined whether BITC modulates signals upstream of STAT-3 to inhibit STAT-3 transcription and protein levels. Furthermore, the number of mice used in our animal studies was limited, and we did not explore the growth of BxPC-3 cells with either stable overexpression of STAT-3 or with deletion of STAT-3 as tumor xenografts in mice. Such studies could better establish the role of STAT-3 in pancreatic tumorigenesis. The effect of BITC treatment under these conditions is not known and will be the focus of our future studies.
In conclusion, our results demonstrate that BITC can suppress both constitutive and inducible STAT-3 activation in human pancreatic cancer cells, block the DNA-binding and transcriptional activity of STAT-3, reduce the expression of Bcl-2 and Mcl-1, induce apoptosis, and inhibit cell proliferation. These effects of BITC were not observed in normal pancreatic epithelial cells. BITC-stimulated apoptosis was blocked when STAT-3α was overexpressed in BxPC-3 cells. Taken together, these findings may provide the basis for further preclinical and clinical investigation of BITC for the chemoprevention and/or chemotherapy of pancreatic cancer.
Funding
This investigation was supported in part by United States Public Health Service RO1 grant CA106953 (to SKS) awarded by the National Cancer Institute. Funding from Texas Tech University Health Sciences Center, School of Pharmacy (to SKS), and an instrument grant from Turner Biosystems Inc, Sunnyvale, CA (to RPS), are also acknowledged.
Footnotes
The authors wish to thank Ruifen Zhang and Jeffery Richards for help in performing animal experiments; Dr J. F. Bromberg, Rockefeller University, New York, NY, for providing the STAT-3 expression plasmid and pLuc-TK/STAT3 construct; Dr Ming-Sound Tsao, University of Toronto, Toronto, Ontario, Canada, for providing HPDE-6 cells; and Dr Thomas L. Brown, Wright State University, Dayton, OH, for providing Panc-1 cells. The authors also wish to thank Dr Doug Potter (University of Pittsburgh) and Dr David Fike for statistical help; Dr Kalkunte Srivenugopal and Dr Jayarama Gunaje, Texas Tech University Health Sciences Center, for constructive suggestions during the revision of the manuscript; and Christopher Adkins for technical help in performing fluorescent microscopy.
The authors take sole responsibility for the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication.
References
- 1.Jemal A, Murray T, Ward E, et al. Cancer statistics 2005 [erratum appears in: CA Cancer J Clin. 2005;55(4):259] CA Cancer J Clin. 2005;55(1):10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.DiGiuseppe JA, Yeo CJ, Hruban RH. Molecular biology and the diagnosis and treatment of adenocarcinoma of the pancreas. Adv Anat Pathol. 1996;3:139–155. [Google Scholar]
- 3.DiMagno EP, Reber HA, Tempero MA. AGA technical review on the epidemiology, diagnosis, and treatment of pancreatic ductal adenocarcinoma. American Gastroenterological Association. Gastroenterology. 1999;117(6):464–484. doi: 10.1016/s0016-5085(99)70298-2. [DOI] [PubMed] [Google Scholar]
- 4.Bueno de Mesquita HB, Maisonneuve P, Runia S, Moerman CJ. Intake of foods and nutrients and cancer of the exocrine pancreas: a population-based case-control study in The Netherlands. Int J Cancer. 1991;48(4):540–549. doi: 10.1002/ijc.2910480411. [DOI] [PubMed] [Google Scholar]
- 5.Olsen GW, Mandel JS, Gibson RW, Wattenberg LW, Schuman LM. Nutrients and pancreatic cancer: a population-based case-control study. Cancer Causes Control. 1991;2(5):291–297. doi: 10.1007/BF00051668. [DOI] [PubMed] [Google Scholar]
- 6.Block G, Patterson B, Subar A. Fruit, vegetables and cancer prevention: a review of the epidemiological evidence. Nutr Cancer. 1992;18(1):1–29. doi: 10.1080/01635589209514201. [DOI] [PubMed] [Google Scholar]
- 7.Ji BT, Chow WH, Gridley G, et al. Dietary factors and risk of pancreatic cancer: a case control study in Shanghai, China. Cancer Epidemiol Biomarkers Prev. 1995;4(8):885–893. [PubMed] [Google Scholar]
- 8.Srivastava SK, Xiao D, Lew KL, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits growth of PC-3 human prostate cancer xenografts in vivo. Carcinogenesis. 2003;24(10):1665–1670. doi: 10.1093/carcin/bgg123. [DOI] [PubMed] [Google Scholar]
- 9.Xiao D, Srivastava SK, Lew KL, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis. 2003;24(5):891–897. doi: 10.1093/carcin/bgg023. [DOI] [PubMed] [Google Scholar]
- 10.Srivastava SK, Singh SV. Cell cycle arrest, apoptosis induction and inhibition of nuclear factor kappa B activation in anti-proliferative activity of benzyl isothiocyanate against human pancreatic cancer cells. Carcinogenesis. 2004;25(9):1701–1709. doi: 10.1093/carcin/bgh179. [DOI] [PubMed] [Google Scholar]
- 11.Miyoshi N, Uchida K, Osawa T, Nakamura Y. A link between benzyl isothiocyanate-induced cell cycle arrest and apoptosis: involvement of mitogen-activated protein kinases in the Bcl-2 phosphorylation. Cancer Res. 2004;64(6):2134–2142. doi: 10.1158/0008-5472.can-03-2296. [DOI] [PubMed] [Google Scholar]
- 12.Miyoshi N, Uchida K, Osawa T, Nakamura Y. Benzyl isothiocyanate modifies expression of the G2/M arrest-related genes. Biofactors. 2004;21(1–4):23–26. doi: 10.1002/biof.552210106. [DOI] [PubMed] [Google Scholar]
- 13.Trachootham D, Zhou Y, Zhang H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10(3):241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci USA. 1992;89(6):2399–2403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang E-S, Lee HJ. Benzyl isothiocyanate inhibits metalloproteinase-2/9 expression by suppressing the mitogen-activated protein kinase in SK-Hep1 human hepatoma cells. Food Chem Toxicol. 2008;46(7):2358–2364. doi: 10.1016/j.fct.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 16.Stoner GD, Morse MA. Isothiocyanates and plant polyphenols as inhibitors of lung and esophageal cancer. Cancer Lett. 1997;114(1–2):113–119. doi: 10.1016/s0304-3835(97)04639-9. [DOI] [PubMed] [Google Scholar]
- 17.Hecht SS. Chemoprevention of cancer by isothiocyanates, modifiers of carcinogen metabolism. J Nutr. 1999;129(3):S768–S774. doi: 10.1093/jn/129.3.768S. [DOI] [PubMed] [Google Scholar]
- 18.Zhang R, Loganathan S, Humphreys I, Srivastava SK. Benzyl isothiocyanate-induced DNA damage causes G2/M cell cycle arrest and apoptosis in human pancreatic cancer cells. J Nutr. 2006;136(11):2728–2734. doi: 10.1093/jn/136.11.2728. [DOI] [PubMed] [Google Scholar]
- 19.Ihle JN. STATs: signal transducers and activators of transcription. Cell. 1996;84(3):331–334. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 20.Darnell JE., Jr STATs and gene regulation. Science. 1997;277(5332):1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 21.Horvath CM, Darnell JE. The state of the STATs: recent developments in the study of signal transduction to the nucleus. Curr Opin Cell Biol. 1997;9(2):233–239. doi: 10.1016/s0955-0674(97)80067-1. [DOI] [PubMed] [Google Scholar]
- 22.Duncan SA, Zhong Z, Wen Z, Darnell JE., Jr STAT signaling is active during early mammalian development. Dev Dyn. 1997;208(2):190–198. doi: 10.1002/(SICI)1097-0177(199702)208:2<190::AID-AJA6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 23.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98(3):295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 24.Brivanlou AH, Darnell JE., Jr Signal transduction and the control of gene expression. Science. 2002;295(5556):813–818. doi: 10.1126/science.1066355. [DOI] [PubMed] [Google Scholar]
- 25.Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101(8):2940–2954. doi: 10.1182/blood-2002-04-1204. [DOI] [PubMed] [Google Scholar]
- 26.Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res. 2007;13(19):5665–5669. doi: 10.1158/1078-0432.CCR-06-2491. [DOI] [PubMed] [Google Scholar]
- 27.Agarwal C, Tyagi A, Kaur M, Agarwal R. Silibinin inhibits constitutive activation of Stat3, and causes caspase activation and apoptotic death of human prostate carcinoma DU145 cells. Carcinogenesis. 2007;28(7):1463–1470. doi: 10.1093/carcin/bgm042. [DOI] [PubMed] [Google Scholar]
- 28.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19(21):2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 29.Coffer PJ, Koenderman L, de Groot RP. The role of STATs in myeloid differentiation and leukemia. Oncogene. 2000;19(21):2511–2522. doi: 10.1038/sj.onc.1203479. [DOI] [PubMed] [Google Scholar]
- 30.Huang M, Page C, Reynolds RK, Lin J. Constitutive activation of stat 3 oncogene product in human ovarian carcinoma cells. Gynecol Oncol. 2000;79(1):67–73. doi: 10.1006/gyno.2000.5931. [DOI] [PubMed] [Google Scholar]
- 31.Song JI, Grandis JR. STAT signaling in head and neck cancer. Oncogene. 2000;19(21):2489–2495. doi: 10.1038/sj.onc.1203483. [DOI] [PubMed] [Google Scholar]
- 32.Mora LB, Buettner R, Seigne J, et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002;62(22):6659–6666. [PubMed] [Google Scholar]
- 33.Nagpal JK, Mishra R, Das BR. Activation of Stat-3 as one of the early events in tobacco chewing-mediated oral carcinogenesis. Cancer. 2002;94(9):2393–2400. doi: 10.1002/cncr.10499. [DOI] [PubMed] [Google Scholar]
- 34.Quintanilla-Martinez L, Kremer M, Specht K, et al. Analysis of signal transducer and activator of transcription 3 (Stat 3) pathway in multiple myeloma: Stat 3 activation and cyclin D1 dysregulation are mutually exclusive events. Am J Pathol. 2003;162(5):1449–1461. doi: 10.1016/S0002-9440(10)64278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sánchez A, Nagy P, Thorgeirsson SS. STAT-3 activity in chemically-induced hepatocellular carcinoma. Eur J Cancer. 2003;39(14):2093–2098. doi: 10.1016/s0959-8049(03)00393-9. [DOI] [PubMed] [Google Scholar]
- 36.Bhutani M, Pathak AK, Nair AS, et al. Capsaicin is a novel blocker of constitutive and interleukin-6-inducible STAT3 activation. Clin Cancer Res. 2007;13(10):3024–3032. doi: 10.1158/1078-0432.CCR-06-2575. [DOI] [PubMed] [Google Scholar]
- 37.Chen CL, Hsieh FC, Lieblein JC, et al. Stat3 activation in human endometrial and cervical cancers. Br J Cancer. 2007;96(4):591–599. doi: 10.1038/sj.bjc.6603597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotha A, Sekharam M, Cilenti L, et al. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther. 2006;5(3):621–629. doi: 10.1158/1535-7163.MCT-05-0268. [DOI] [PubMed] [Google Scholar]
- 39.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2(10):740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 40.Darnell JE. Validating Stat3 in cancer therapy. Nat Med. 2005;11(6):595–596. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- 41.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 42.Furukawa T, Duguid WP, Rosenberg L, Viallet J, Galloway DA, Tsao MS. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am J Pathol. 1996;148(6):1763–1770. [PMC free article] [PubMed] [Google Scholar]
- 43.Ouyang H, Lj Mou, Luk C, et al. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am J Pathol. 2000;157(5):1623–1631. doi: 10.1016/S0002-9440(10)64800-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13):1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 45.Grandis JR, Drenning SD, Chakraborty A, et al. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J Clin Invest. 1998;102(7):1385–1392. doi: 10.1172/JCI3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terui K, Enosawa S, Haga S, et al. Stat3 confers resistance against hypoxia/reoxygenation-induced oxidative injury in hepatocytes through upregulation of Mn-SOD. J Hepatol. 2004;41(6):957–965. doi: 10.1016/j.jhep.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 47.Narimatsu M, Maeda H, Itoh S, et al. Tissue-specific autoregulation of the stat3 gene and its role in interleukin-6-induced survival signals in T cells. Mol Cell Biol. 2001;21(19):6615–6625. doi: 10.1128/MCB.21.19.6615-6625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ichiba M, Nakajima K, Yamanaka Y, Kiuchi N, Hirano T. Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J Biol Chem. 1998;273(11):6132–6138. doi: 10.1074/jbc.273.11.6132. [DOI] [PubMed] [Google Scholar]
- 49.Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18(4):443–451. doi: 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- 50.Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE., Jr Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol. 1998;18(5):2553–2558. doi: 10.1128/mcb.18.5.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Isomoto H, Mott JL, Kobayashi S, et al. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology. 2007;132(1):384–396. doi: 10.1053/j.gastro.2006.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moran DM, Mattocks MA, Cahill PA, Koniaris LG, McKillop IH. Interleukin-6 mediates G(0)/G(1) growth arrest in hepatocellular carcinoma through a STAT 3-dependent pathway. J Surg Res. 2007;147(1):23–33. doi: 10.1016/j.jss.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang X, Darnell JE., Jr Functional importance of Stat3 tetramerization in activation of the alpha 2-macroglobulin gene. J Biol Chem. 2001;276(36):33576–33581. doi: 10.1074/jbc.M104978200. [DOI] [PubMed] [Google Scholar]
- 54.Schaefer TS, Sanders LK, Nathans D. Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc Natl Acad Sci USA. 1995;92(20):9097–9101. doi: 10.1073/pnas.92.20.9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alas S, Bonavida B. Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin's lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clin Cancer Res. 2003;9(1):316–326. [PubMed] [Google Scholar]
- 56.Amit-Vazina M, Shishodia S, Harris D, et al. Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells. Br J Cancer. 2005;93(1):70–80. doi: 10.1038/sj.bjc.6602637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hussain SF, Kong LY, Jordan J, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67(20):9630–9636. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- 58.Bromberg J, Darnell JE., Jr The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19(21):2468–2473. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 59.Lui VW, Boehm AL, Koppikar P, et al. Antiproliferative mechanisms of a transcription factor decoy targeting signal transducer and activator of transcription (STAT) 3: the role of STAT1. Mol Pharmacol. 2007;71(5):1435–1443. doi: 10.1124/mol.106.032284. [DOI] [PubMed] [Google Scholar]
- 60.Lassmann S, Schuster I, Walch A, et al. STAT3 mRNA and protein expression in colorectal cancer: effects on STAT3-inducible targets linked to cell survival and proliferation. J Clin Pathol. 2007;60(2):173–179. doi: 10.1136/jcp.2005.035113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Albrecht U, Yang X, Asselta R, et al. Activation of NF-kappaB by IL-1beta blocks IL-6-induced sustained STAT3 activation and STAT3-dependent gene expression of the human gamma-fibrinogen gene. Cell Signal. 2007;19(9):1866–1878. doi: 10.1016/j.cellsig.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 62.Zhang C, Li B, Gaikwad AS, et al. Avicin D selectively induces apoptosis and downregulates p-STAT-3, bcl-2, and survivin in cutaneous T-cell lymphoma cells. J Invest Dermatol. 2008;28(11):2728–2735. doi: 10.1038/jid.2008.138. [DOI] [PubMed] [Google Scholar]
- 63.Zhang X, Zhang J, Wei H, Tian Z. STAT3-decoy oligodeoxynucleotide inhibits the growth of human lung cancer via down-regulating its target genes. Oncol Rep. 2007;17(6):1377–1382. [PubMed] [Google Scholar]
- 64.Daino H, Matsumura I, Takada K, et al. Induction of apoptosis by extracellular ubiquitin in human hematopoietic cells: possible involvement of STAT3 degradation by proteasome pathway in interleukin 6-dependent hematopoietic cells. Blood. 2000;95(8):2577–2585. [PubMed] [Google Scholar]
- 65.Yeh HH, Wu CH, Giri R, et al. Oncogenic Ras-induced morphologic change is through MEK/ERK signaling pathway to downregulate Stat3 at a posttranslational level in NIH3T3 cells. Neoplasia. 2008;10(1):52–60. doi: 10.1593/neo.07691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nishikawa A, Furukawa F, Uneyama C, et al. Chemopreventive effects of phenethyl isothiocyanate on lung and pancreatic tumorigenesis in N-nitrosobis(2-oxopropyl)amine-treated hamsters. Carcinogenesis. 1996;17(6):1381–1384. doi: 10.1093/carcin/17.6.1381. [DOI] [PubMed] [Google Scholar]
- 67.Singh SV, Srivastava SK, Choi S, et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J Biol Chem. 2005;280(20):19911–19924. doi: 10.1074/jbc.M412443200. [DOI] [PubMed] [Google Scholar]
- 68.Ji Y, Kuo Y, Morris ME. Pharmacokinetics of dietary phenethyl isothiocyanate in rats. Pharm Res. 2005;22(10):1658–1666. doi: 10.1007/s11095-005-7097-z. [DOI] [PubMed] [Google Scholar]
- 69.Ye L, Dinkova-Kostova AT, Wade KL, Zhang Y, Shapiro TA, Talalay P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin Chim Acta. 2002;316(1–2):43–53. doi: 10.1016/s0009-8981(01)00727-6. [DOI] [PubMed] [Google Scholar]