

Abstract
Aim: Presented is the neuroradiological signature of acute Wernicke's encephalopathy (WE), derived from different types of magnetic resonance imaging (MRI) sequences. WE results from thiamine depletion, and its most typical antecedent is chronic alcohol dependence. Brain regions observed with in vivo MRI affected in acute WE include the mammillary bodies, periaqueductal and periventricular gray matter, collicular bodies and thalamus. These affected areas are usually edematous and are best visualized and quantified with MRI sequences that highlight such tissue. Following the acute WE phase and resolution of edema and inflammation of affected brain tissue, WE, if not adequately treated with thiamine repletion, can herald Korsakoff's syndrome (KS), with its symptomatic hallmark of global amnesia, that is, the inability to commit newly encountered (episodic) information to memory for later recall or recognition. Methods: Neuropathology of KS detectable with MRI has a different neuroradiological signature from the acute stage and can be observed as tissue shrinkage or atrophy of selective brain structures, including the mammillary bodies and thalamus and ventricular expansion, probably indicative of atrophy of surrounding gray matter nuclei. Quantification of these and additional gray matter structures known to underlie global amnesia reveal substantial bilateral volume deficits in the hippocampus, in addition to the mammillary bodies and thalamus, and modest deficits in the medial septum/diagonal band of Broca. The infratentorium is also affected, exhibiting volume deficits in cerebellar hemispheres, anterior superior vermis and pons, contributing to ataxia of gait and stance. Results: Consideration of WKS structural brain changes in the context of the neuropathology of non-WKS alcoholism revealed a graded pattern of volume deficits, from mild in non-WKS alcoholics to moderate or severe in WKS, in the mammillary bodies, hippocampus, thalamus, cerebellum and pons. The development and resolution of brain structures affected in acute, chronic and treated WE was verified in longitudinal MRI study of rats that modeled of the interaction of extensive alcohol consumption and thiamine depletion and repletion. Conclusions: Thus, neuroradiological examination with MRI is valuable in the diagnosis of acute WE and enables in vivo tracking of the progression of the brain pathology of WE from the acute pathological phase to resolution with thiamine treatment or to progression to KS without treatment. Further, in vivo MRI facilitates translational studies to model antecedent conditions contributing to the development, sequelae and treatment of WE.
Introduction
This report focuses on in vivo characterization of the acute and chronic brain lesions associated with Wernicke's encephalopathy (WE) detected with different forms of structural MRI, with particular emphasis on sequences with sensitivity to the edematous lesions marking WE. Although studies of MR spectroscopy (MRS) and functional MRI (fMRI) have been conducted in WE, these modalities are less often available than structural MRI in clinical settings and so have been less useful diagnostically. Further, task-based fMRI would be difficult to impossible to conduct in the acute, confusional phase of WE. Therefore, neither MRS nor fMRI studies are reviewed herein. Because of WE's close relationship with Korsakoff's syndrome (KS), examples of MR images and new quantitative MRI analysis of KS are featured. Also presented is a hypothesis about the etiology of the heterogeneity of neuroradiological and behavioral features of the WE–KS complex.
Acute We: Aetiologies and Clinical Signs
Classically, acute thiamine deficiency-associated WE is marked neuropathologically by lesions of periventricular nuclei, hypothalamic nuclei, tectal plate and thalamus, which are caused by thiamine (vitamin B1) deficiency and result in WE's cardinal signs of ophthalmoplegia, nystagmus, ataxia and confusional state (as noted in Victor et al., 1971, 1989). The acute WE condition, when left untreated with thiamine or if treated incompletely or too late, can herald profound, debilitating global amnesia marking KS. Another form of WE, beriberi, is also caused by a thiamine-deficient diet but usually has a more insidious progression; beriberi causes damage to central and peripheral nervous systems, musculature and cardiovascular system (Victor et al., 1989).
Clinically relevant thiamine deficiency (Bender and Schilder, 1933; Jolliffe et al., 1941) and evolving WE can result from conditions that restrict eating, such as orofacial cancers, or limit adequate vitamin absorption, such as gastric bypass surgery, gastric and colon cancer, hyperemesis gravidarum or starvation by choice or associated with sepsis, surgical complications and comma (for review of physiological mechanisms of thiamine deficiency, see Martin et al., 2003; Thomson and Marshall, 2006; Sechi and Serra, 2007). Infarction of the mammillothalamic tracts is another antecedent of WE (Yoneoka et al., 2004). By far the most common cause of thiamine deficiency throughout the world is alcoholism (Thomson, 2000), as described in landmark studies of alcoholic WE–KS (Victor et al., 1971; Jarho, 1973; Harper et al., 1986, 1995). Alcoholics are at special risk for thiamine deficiency because of the poor diet associated with their lifestyle and because chronic alcoholism compromises thiamine absorption from the gastrointestinal tract, impairs thiamine storage and may reduce thiamine phosphorylation, essential for cellular function (Thomson et al., 1987; Todd and Butterworth, 1999; Thomson, 2000; Lieber, 2003; Martin et al., 2003). Severe or insufficiently treated cases may show the enduring memory impairment and ataxia defining KS (Victor et al., 1959; Talland, 1965; Butters and Cermak, 1980; Kopelman, 1995). Although WE does not necessarily evolve into KS if adequately treated (Victor et al., 1971, 1989; Caine et al., 1997; Thomson et al., 2002), it remains controversial whether KS can develop without WE and whether mild forms of neuropsychological sequelae arise from repeated bouts of thiamine deficiency or inadequate treatment of such episodes.
Acute We: Neuroradiological Signs
Noninvasive neuroradiological examination of WE dates to the 1970s. Early studies used computed tomography (CT) scanning and revealed ventricular enlargement, especially of the third ventricle (Escobar et al., 1983; McDowell and LeBlanc, 1984; Mensing et al., 1984; Shimamura et al., 1988) but were largely unable to detect edema or focal damage (Gotze et al., 1978; Gallucci et al., 1990). The introduction of MR imaging with its exquisite sensitivity to tissue water content and mobility enabled visualization of acute and chronic radiological signs of neuropathology not visible on CT (Antunez et al., 1998). In some sequences, the MR signal of selective brain structures is hyperintense, indicative of high water content, present in WE because of the edematous nature of the lesions. A direct comparison of CT and MRI in the detection of WE-related neuropathology identified low-density signal abnormalities on CT in the paraventricular regions of thalamus in only 2 of 15 WE patients examined, whereas MRI identified abnormality in this thalamic region in 7 patients of this WE group. Additional affected areas included periaqueductal gray matter in 6 patients and mammillary body shrinkage in 6 WE and 4 of 15 non-WE alcoholic patients. Thus, in vivo neuroimaging has been instrumental in revealing WE-like neuropathology in alcoholics who do not present with the obvious signs of WE (Sullivan, 2003; Sullivan and Pfefferbaum, 2005). Overall, although the sensitivity of MRI in detecting WE was only 53%, the specificity was 93% (Antunez et al., 1998). Fig. 1 presents an exemplary comparison of CT and different MRI sequences.
Fig. 1.

CT and MR images of an acute 35-year-old man with schizophrenia and acute nutritional deficiency-induced WE. (A) Axial CT at the level of the lateral ventricles. (B–E) Axial MR images at a similar level to the CT. (B) A proton density-weighted image. (C) A T2-weighted late-echo fast spin echo (FSE) image. (D) A fluid-attenuated inversion recovery (FLAIR) image. (E) A diffusion-weighted image (DWI). Note the hyperintensity of the fornix and thalamus, especially in D and E, less so in C, and lack of lesion conspicuity in A and B.
MR techniques used to enhance visualization of edematous lesions were initially based on T2-weighted late-echo sequences, which are acquired after the majority of the tissue signal has decayed but while the signal of unbound water remains robust (for review, see Bigler, 1996). The most obvious neuroradiological sign of acute WE, regardless of etiology, is bilateral hyperintensity on late-echo MRI, generally occurring in gray matter tissue of the mammillary bodies, anterior and medial nuclei of the thalamus, periventricular gray matter, inferior and superior colliculi (e.g. nonalcoholics: Doraiswamy et al., 1994; Chu et al., 2002; Unlu et al., 2006; Zhong et al., 2005) (alcoholics: Schroth et al., 1991) and occasionally cererbellum (Shear et al., 1996; Nicolas et al., 2000; Sullivan et al., 2000; Bae et al., 2001). These observations are consistent with postmortem reports (e.g. Torvik et al., 1982, 1986; Harper and Kril, 1988, 1990; Kril et al., 1997; Baker et al., 1999). The bilateral distribution of the neuropathology may contribute to the severity of the clinical signs and symptoms. Although the pons is not usually implicated in WE, an MR study examining T2 relaxation time, a measure of interstitial fluid reflecting axonal and myelin integrity, provided evidence of excessive fluid in the central pons of patients with alcoholic WKS (Sullivan and Pfefferbaum, 2001). Predictors of prolonged relaxation time in non-KS alcoholics in this study were hematological measures of nutritional status, e.g. macrocytic anemia and cognitive fluency.
The development of the MR fluid-attenuated inversion recovery (FLAIR) sequence provided significant improvement over the conventional T2 approach by incorporating additional T1 contrast mechanisms. An advantage of the FLAIR approach is that it essentially eliminates only signal with T1 characteristics of CSF, including in regions of non-tissue CSF, such as sulci and ventricles, and therefore enhances the conspicuity of the signal in boggy, edematous tissue. Several case studies have published in vivo FLAIR images of acute WE (Maeda et al., 1995; Ashikaga et al., 1997). An early study of a woman with hyperemesis gravidarum revealed high signal intensity of the mammillary bodies and hypothalamus; following thiamine treatment, although the high signal intensity resolved, the mammillary bodies shrank (Maeda et al., 1995). A series of six cases of nonalcoholic WE studied with FLAIR revealed hyperintense signal in the tissue around the aqueduct, third ventricle, floor of the fourth ventricle, anterior ventricular caps and medial thalami; follow-up examination noted recovery in the four cases without cortical damage but not in the two cases with such damage (Zhong et al., 2005).
MR diffusion-weighted imaging (DWI), in which signal from freely diffusing water is suppressed, is another MRI method that has proved sensitive to the detection of WE brain pathology. An example of WE lesions with DWI is presented in Fig. 1. Paradoxically, the edematous lesions of WE, which would be expected to have high levels of diffusivity and have their signal suppressed with DWI, are instead hyperintense. This is an example of the ‘T2 shine-through effect’, in which tissue with long T2 value is bright (Koch and Norris, 2005); thus, the bright signal, rather than representing low diffusivity, reflects the opposite. In addition to the periventricular and thalamic tissue abnormalities typically identified with WE (Halavaara et al., 2003; Unlu et al., 2006), one case study concluded that bright signal on DWI was caused by abnormally low diffusivity in the cerebellum. Even though the diffusivity abnormality in the cerebellum resolved with thiamine repletion, noted at a 3-month follow-up study, associated motor impairment persisted (Lapergue et al., 2006). DWI-increased signal intensity, confirmed as decreased diffusivity with apparent diffusion coefficient (ADC) images, in affected brain regions has been reported in two studies of acute WE (Halavaara et al., 2003; Lapergue et al., 2006). Because interpretation of DWI can be confounded by the T2 shine-through effect, DWI is probably of greatest value when accompanied by ADC imaging for quantitative assessment of the water diffusion. Together, the two techniques provide a method for characterizing the evolution of WE lesions from early edematous high diffusivity through later atrophic low diffusivity.
Figures 1 and 2 present FLAIR images of an acute WE case, a 35-year-old man with schizophrenia, found lethargic and confused in his apartment. He had suffered weight loss from inadequate nutrition. Examination revealed failure of horizontal gaze and ataxia of gait; management included daily doses of intravenous thiamine 100 mg. This case is striking because all neuropathological indices are present, and the lesions are bilateral and visible as signal hyperintensities. The structures affected in this case of WE are the mammillary bodies, periventricular gray matter, thalamus, inferior colliculi and fornix.
Fig. 2.

Three contiguous FLAIR images (5 mm thick with a 2.5 mm skip) of the acute WE case in Fig. 1. Note the hyperintense signal in the mammillary bodies and colliculi (left), periventricular gray matter (middle), and fornix and thalamus (right).
Sequelae of We: Neuroradiological Signs of Alcoholic Wernicke–Korsakoff Syndrome
Despite the utility of neuroimaging in diagnosing WE and in identifying the loci and extent of damage, MRI examination often post-dates the acute phase marked by edema and inflammation of affected brain tissue. In this later phase, targeted structures, notably the mammillary bodies, become atrophic (Charness and DeLaPaz, 1987; Sheedy et al., 1999) and assume a different neuroradiological signature from the acute stage. This acute to chronic progression can be tracked with in vivo MRI. A gross morphological view of alcoholism-related WKS reveals cortical thinning, sulcal widening and ventriculomegaly (Fig. 3). More detailed examination reveals regional structural volume deficits, consistent with postmortem-identified atrophy and prominent in thalamus and mammillary bodies (Fig. 4).
Fig. 3.

Surface rendered brains (top) and rendered ventricular system (bottom, green) of a 59-year-old healthy man (A and C) and a 53-year-old man with WKS (B and D). Note the shrinking of the cortical gyri and widening of the sulci (B) and expansion of the ventricles (D) of the WKS compared with the control (A and C).
Fig. 4.

T1-weighted SPoiled GRadient echo (SPGR) images of the healthy (left panel) and WKS (right panel) men in Fig. 3. Note the shrunken mammillary bodies (arrows) in the WKS (B and D) compared with the control (A and C).
Uncomplicated alcoholism to WKS: a graded effect of brain structural volume deficits
The WKS structural brain changes noted above must be considered in the context of the neuropathology of non-WKS alcoholism. In Fig. 5 we present an examination of the potential compounded effects of chronic alcoholism, WE and KS on brain structure and function in individuals with alcohol-related KS, ‘uncomplicated’ alcoholism and healthy controls. Selective brain structures were manually identified on MRI to permit volumetric quantification. Description of the MRI sequences used and MRI quantification procedures have been published: volumetric SPGR for mammillary bodies (Sullivan et al., 1999), thalamus (Sullivan et al., 2003, 2004), medial septum/diagonal band (MD/DB) (Sullivan et al., 2005), cerebellum (Sullivan et al., 2000) and pons (Sullivan et al., 2003, 2004); and coronal, dual-echo spin-echo images for the hippocampus (Sullivan et al., 1995). Regional brain volumes were adjusted for normal variation in intracranial volume and age and expressed as standardized Z-scores, where the expected mean of the controls was 0 (standard deviation = 1); thus, low scores reflect smaller volumes than would be expected for a particular intracranial volume and age, and the mean Z-scores also reflect the effect size. All KS patients had been alcohol dependent, were abstinent from alcohol at examination and met retrospective, chart review criteria for WE (Caine et al., 1997). Neuropsychological tests assessed multiple functional domains, targeting executive functions, declarative and procedural memory, visuospatial abilities and postural stability, and revealed severe to profound deficits in the KS group in memory for new material and gait and balance with sparing of general intelligence, short-term memory and visuoperceptual implicit learning (Fama et al., 2004, 2006; Sullivan et al., 2000). This pattern of functional sparing and impairment is consistent with KS (also see paper in this issue byes Kopelman).
Fig. 5.

(a) Mean ± SE of volumes of brain structures considered to subserve memory for new information: healthy controls (white), uncomplicated alcoholics (gray) and alcoholic WKS (black). All volumes are expressed as standardized Z-scores, adjusted for normal variation in intracranial volume and age. The expected value of the controls is 0 (standard deviation = 1); low values of volume in the alcoholic and WKS groups reflect volume deficits. A graded effect of uncomplicated alcoholism to WKS is present for each of these brain structures but is statistically significant for the mammillary bodies and hippocampus. These four brain structures are considered to contribute to the support of declarative memory, that is, memory for new information; profound impairment in declarative memory is characteristic of WKS. (b) Mean ± SE of volumes of brain stem structures. The pons, cerebellar hemispheres and vermis each demonstrate a trend toward a graded effect from uncomplicated alcoholism to WKS.
MRI indicated graded regional brain volume shrinkage (Fig. 5a and b), where deficits of uncomplicated alcoholics were significant (generally about 0.5 standard deviation deficit) but less severe than those of KS (generally ∼1–2 standard deviation deficit) in the mammillary bodies (Shear et al., 1996; Sullivan et al., 1999), thalamus (this report), pons (this report), cerebellar hemispheres and anterior superior vermis (V1 of Fig. 5b) (Sullivan et al., 2000). Contrary to traditional belief and evidence (Squire et al., 1990; Visser et al., 1999; Reed et al., 2003), we observed bilateral deficits in the anterior hippocampus of alcoholics with WKS (Fig. 5a). As an additional neuropathological context, we compared the hippocampal volumes of KS with those of Alzheimer's disease (AD) patients, whose neuroradiological hallmark is severe hippocampal volume loss (Sullivan and Marsh, 2003). We found that the KS group exhibited a deficit in hippocampal volumes bilaterally equivalent to that observed in the patients with AD and more than twice that we previously observed in nonamnesic alcoholic patients (Sullivan et al., 1995). Relations between the amnesia index and hippocampal volumes but not volumes of the mammillary bodies or the temporal cortex, despite tissue deficits in both structures, support the relevance of the hippocampal volume to the amnesia of KS (Sullivan and Marsh, 2003); further, performance on tests requiring historical event naming and sequencing (i.e. non-declarative memory) related to frontoparietal white matter volumes (Fama et al., 2004). Volume of the medial septum/diagonal band of Broca was modestly reduced in KS (Fig. 5a), suggesting that volume loss and potential cholinergic compromise due to damage in this nuclear complex may also contribute to the KS amnesic syndrome (cf., Butters and Stuss, 1989; De Rosa and Sullivan, 2003; Sullivan et al., 2005).
The graded deficits in regional brain volumes from uncomplicated alcoholics without KS are substantially less than in alcoholics with WE or KS (cf., Blansjaar et al., 1992; Charness, 1993, 1999; Sullivan, 2000; Mulholland et al., 2005) or even Alzheimer's disease (Charness and DeLaPaz, 1987). This pattern, however, implicates nutritional deficiency as a mechanism of alcoholic regional brain volume shrinkage and ventricular expansion, which can be examined in humans only with naturalistic observational methods.
Factors contributing to brain abnormalities in alcoholic WKS
That brain regions outside of those traditionally associated with thiamine depletion were affected in both uncomplicated and KS alcoholics suggests a role for alcoholism alone or nutritional deficiencies in interaction with continued drinking as mechanisms for the brain abnormalities. Indeed, multiple subclinical bouts of thiamine or other nutritional deficiencies in alcoholism may contribute to the graded effect of brain regional volume deficits and to heterogeneity of presenting signs and neuroradiological profile (cf., Blansjaar and Van Dijk, 1992). Because sustained heavy drinking frequently occurs at the expense of eating (Santolaria et al., 2000), alcoholics prone to intermittent binge drinking are at risk for nutritional depletion. A recent report on the association between alcohol drinking pattern and diet quality in a representative sample of the US population found diet quality was poorest among the highest-quantity–lowest-frequency drinkers and best among the lowest-quantity–higher-frequency drinkers. When alcohol consumption was expressed as average drinks per day, no association with diet was seen (Breslow et al., 2006). A study of recent nutritional intake in a small sample of heavy drinkers found that ∼60% of energy intake came from alcohol and intake of vitamins fell below recommended norms (Manari et al., 2003). Nutritionally based anemia (probably folate deficiency) in alcoholics has been associated with deficits in cortical white matter volume (Pfefferbaum et al., 2004), whereas hematological indices improved with short-term sobriety and nutritional supplementation related to concurrent reduction of ventricular enlargement. Hematocrit and hemoglobin levels at discharge from a 28-day inpatient rehabilitation program discriminated patients who maintained sobriety from those who relapsed over the ensuing months. This finding may reflect the common sense observation that patients who are better fortified physically at the end of treatment will be better equipped for maintaining sobriety.
Because alcoholism is a chronic disorder, often spanning decades, the interaction of aging-related involutional brain changes must be considered when interpreting the effects of alcoholism. Cortical gray and white matter may sustain long-term volume shrinkage and even loss (Jernigan et al., 1991; Pfefferbaum et al., 1992), especially in the prefrontal cortex (De Bellis et al., 2005) of older alcoholics (Pfefferbaum et al., 1997; Cardenas et al., 2005). Although alcohol-related brain abnormalities are partially reversible with prolonged sobriety (Carlen et al., 1978; Schroth et al., 1988; Pfefferbaum et al., 1995, 1998; Mann et al., 1999; O’Neill et al., 2001; Parks et al., 2002; Gazdzinski et al., 2005), the extent to which KS can recover is controversial. Incomplete recovery of affected neural structures and systems are candidate substrates for the enduring cognitive and motor impairments defining alcoholic WE and KS as well as ‘uncomplicated’ alcoholism. Whether thiamine deficiency is a necessary or sufficient cause of alcoholism-related brain volume and neuropsychological deficits remains a question in humans but is amenable to a systematic study in animals. Conversely, whether alcoholics who, while continuing to drink, achieve good physical condition with exercise and maintain adequate nutrition can escape or at least reduce the neurological throes of alcoholism remains a consideration.
Animal Model of We: Neuroradiological Confirmation of Acute and Chronic Phases
Animal models of the brain lesions marking the thiamine deficiency syndrome have typically revealed subsets of the lesions noted in humans [reviewed by Witt (1985), Martin et al. (2003)), involving the thalamus, superior and inferior colliculi, and hypothalamic nuclei. Because dietary deficiency alone requires a month or longer to deplete thiamine stores, a standard approach to shorten experimental time is administration of the thiamine antagonist, pyrithiamine (Langlais, 1995). Studies using pyrithiamine-induced thiamine deficiency (Langlais and Savage, 1995; Langlais and Zhang, 1997; Savage et al., 1999; Pitkin and Savage, 2001, 2004) have demonstrated characteristic neuropathology and behavioral deficits in animal models. For example, significant shrinkage of the corpus callosum was induced several months after only a single bout of pyrithiamine-induced thiamine deficiency (Langlais and Savage, 1995). Ultrastructural studies show splitting of myelin sheaths and swelling of periaxonal spaces within the cerebral cortex of pyrithiamine-treated rats (Takahashi et al., 1988). Some have demonstrated an enhancement of thiamine deficiency pathology by alcohol (Zimitat et al., 1990; He et al., 2007), and others have shown that the thiamine deficiency does not need to be complete to have untoward effects (Pires et al., 2001; Bruce et al., 2003).
Neuroimaging with MR methods provides the means for repeated, longitudinal in vivo high-resolution surveys of the whole brain and its structures that can be analyzed with a variety of approaches, including longitudinal examinations before and after treatment. The earliest neuroimaging experiments using rodent models of thiamine deficiency to produce WE were conducted at 1.5T (Pentney et al., 1993) and reported volume enlargement of lateral ventricles followed by normalization with a thiamine-enriched diet (Acara et al., 1995). Glucose administration to thiamine-deficient rats produced the untoward effect of blood-brain barrier impairment, observed with contrast-enhanced T1-weighted MR images (Zelaya et al., 1995). Using T2-weighted MR imaging to emphasize the tissue free-water concentration, hyperintensities in the hippocampus as well as in the thalamus, hypothalamus and collicular bodies were visible and exacerbated by glucose infusion; some lesions noted as hyperintensities endured for a month (Jordan et al., 1998).
Neuropathological and MRI studies of pyrithiamine-induced thiamine deficiency yield inconsistent results with regard to the timing of lesion development and resolution. Langlais and colleagues report white matter and cortical lesions without mammillothalamic involvement at the time of ataxia and righting reflex impairment, with mammillothalamic lesions emerging only after seizure development (Langlais and Zhang, 1997). Nixon and colleagues (Jordan et al., 1998) describe MRI evidence of mammillothalamic and hippocampal involvement at the time of ataxia and righting reflex impairment. The contradiction may reside in differential sensitivity of the two methods. It may be that the histopathology was insensitive to the initial edematous mammillothalamic process, whereas the T2-weighted structural MRI was insensitive to the white matter pathology. Implementation of MR diffusion tensor imaging for the assessment of regional white matter microstructural integrity in combination with high resolution MRI for macrostructural measurement in longitudinal study may serve to resolve inconsistent in vivo and postmortem results.
We conducted a longitudinal MRI study of thiamine deficiency in alcohol-preferring rats (Pfefferbaum et al., 2007). Half the sample had voluntarily consumed large amounts of alcohol prior to thiamine manipulation, and the other half had never been exposed to alcohol (‘water’ control), but both groups had had free access to rat chow, which was adequately enriched with thiamine and other required vitamins and minerals (Pfefferbaum et al., 2006; Sullivan et al., 2006). Following the alcohol exposure arm of the experiment, both samples of rats were given a thiamine-depleted diet for 14 days, and all received daily intraperitoneal injections of either thiamine or pyrithiamine, resulting in a four-group design depending on historical exposure to alcohol and current exposure to pyrithiamine: alcohol + pyrithiamine, alcohol + thiamine, water + pyrithiamine and water + thiamine. Serial MRI examinations identified significant ventricular enlargement and increase in signal intensities in thalamus, inferior colliculi and mammillary nuclei of pyrithiamine-treated rats compared with thiamine-treated rats from baseline to 18 days after thiamine repletion (Fig. 6). Comparison of MR results from 18 to 35 days revealed significant normalization in thalamus and inferior colliculi, but neither in the mammillary nuclei nor in ventricles (Fig. 6). Postmortem examination of white matter with electron microscopy of these rats revealed increased density of small diameter fibers in the corpus callosum and compromised myelin (He et al., 2007). Whether the structural abnormalities noted in the corpus callosum and hippocampus require combined alcohol exposure and thiamine depletion remains incompletely answered.
Fig. 6.
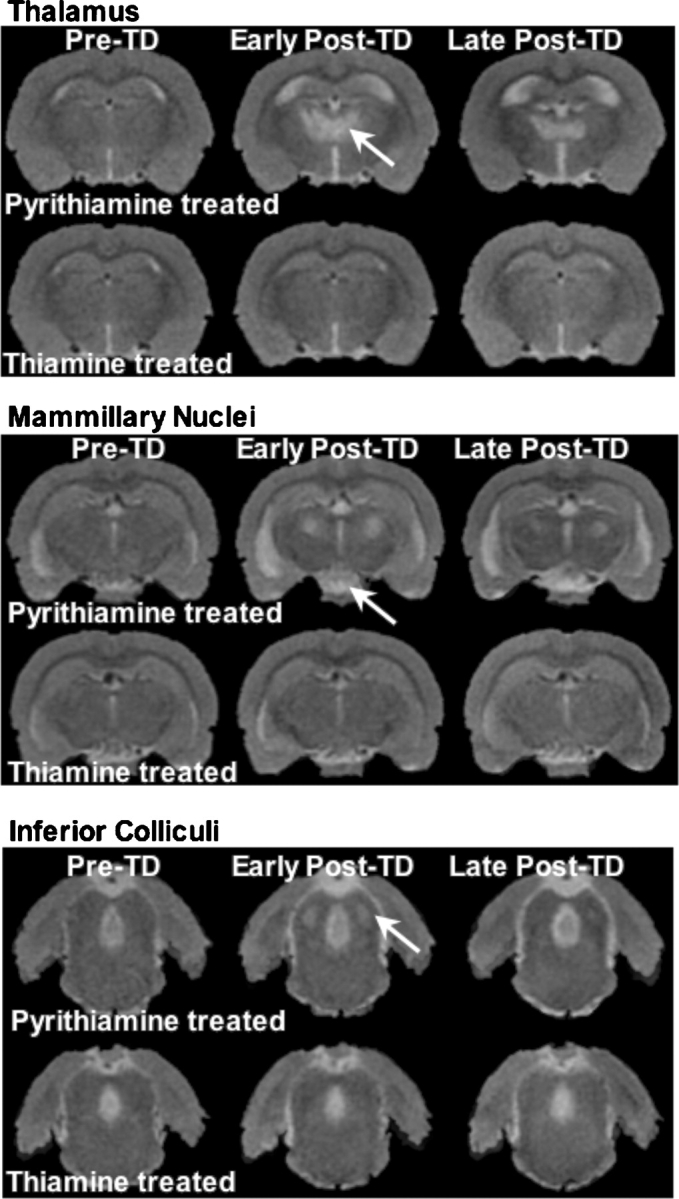
Left: T2-weighted late-echo fast spin echo (FSE) images of a thiamine-deficient (TD) WE model in rats. Animals were treated with pyrithiamine (top images of each panel) or thiamine supplementation (bottom images of each panel). Images on the left of each panel are pre-treatment (pre-TD); images in the middle are early post-treatment (post-TD); and images on the right are late post-treatment (late post-TD) after thiamine repletion. Early post-TD, hyperintense lesions are prominent in the thiamine-deficient (pyrithiamine-treated) rats in the thalamus, mammillary nuclei and inferior colliculi. Late post-TD after thiamine repletion, the thalamus showed some recovery, the mammillary nuclei remained affected and the inferior colliculi showed complete recovery.
Our longitudinal in vivo experiment coupled with postmortem examination provides neuroradiological and histological confirmation of the cause, progression and resolve of thiamine deficiency through an animal model of WE. These experiments also support an alcohol–thiamine deficiency interaction, which suggests an added risk of WE in individuals with alcohol dependence. The enduring macrostructural abnormalities involving critical nodes of the Papez circuit carry liabilities for the development of amnesia (Talland, 1968; Zola-Morgan and Squire, 1993; Eichenbaum and Cohen, 2001) and incomplete recovery from other cognitive and motor functions subserved by the affected neural systems.
Conclusions: Implications for Prognosis and Neuropathological Liability
The confusional state marking the presentation of WE can impede accurate diagnosis. Incoherence of thought and communication in cases of alcohol intoxication and protracted withdrawal can further mask the seriousness of acute WE. MRI has been shown to improve and expedite accurate diagnosis, and therefore adequate treatment of WE (Sechi and Serra, 2007). As further noted by Sechi and Serra (2007), ‘…MRI is currently considered the most valuable method to confirm a diagnosis of Wernicke's encephalopathy. MRI has a sensitivity of only 53%, but its high specificity of 93% means that it can be used to rule out the disorder’.
Not all neurological or neuroradiological signs are necessarily present in all WE cases, and the severity of the signs is likely related to the degree of the underlying pathology. This speculation was supported in a retrospective study of 25 WE–KS patients, whose clinical outcome was more favorable with fewer radiological signs detected at onset (Varnet et al., 2002). The bilateral distribution of pathology typically observed probably also contributes to the severity of the signs and symptoms at all stages of encephalopathy. Because only about a third of WE cases exhibit all three signs of the classical triad (Harper, 1983; Zuccoli et al., 2007), diagnosis needs supplementation with more reliable information than signs and symptoms alone. Chart review can help determine whether a symptomatic patient has had bouts of WE (e.g. Caine et al., 1997). MR imaging has also become an indispensable diagnostic tool for encephalopathy, especially in light of the reversibility of the otherwise devastating cognitive and motor features of untreated WE. MRI is sensitive to detection of WE-related or -induced lesions in brain regions classically identified with this encephalopathy—mammillary bodies, periaqueductal gray matter, thalamus and colliculi—but, as noted by Victor (Victor, 1990), can also be helpful in diagnosis and prognosis in cases with atypical symptomatic or neuroradiological presentation, which can include involvement of pons, cerebellum and hippocampus.
The most common antecedent of WE is chronic alcoholism. A possible consequence of clinical or subclinical thiamine deficiency in alcoholism may contribute to the considerable variability in the extent of alcoholism-related brain abnormalities reported, with some individuals having massive brain shrinkage and others little observed effect. Possible explanations for this variability include individual differences in susceptibility, alcohol use pattern (quantity, frequency, duration) and nutrition. Non-treatment-seeking alcoholics tend to have less brain pathology than those seeking treatment (Fein and Landman, 2005) and may represent individuals with lesser predisposing susceptibility or better nutrition when drinking. Subclinical bouts of thiamine deficiency in alcoholism may, therefore, account for the graded effect from uncomplicated alcoholism to KS we noted (also see Blansjaar et al., 1992; Jauhar and Montaldi, 2000). Indeed, the incidence of undetected WE-associated lesions among alcoholics at autopsy suggests greater influence of nutritional deficiency (especially thiamine) than generally appreciated clinically (Victor et al., 1989; Harper and Butterworth, 1997; Thomson, 2000; Harper, 2006), and the neuroradiological traces of WE may linger in the brains of ‘uncomplicated’ alcoholics yet remain a liability for interaction with other neurodegenerative conditions and the ineluctable regression of brain tissue integrity with aging.
Acknowledgments
We would like to thank Barton Lane, M.D., for identifying the acute Wernicke encephalopathy case reported herein and for clinical radiological readings of MRI studies, and Elfar Adalsteinsson, Ph.D., for oversight on MR descriptions. We also thank Anjali Deshmukh, M.D., Kathleen Serventi, M.D., and Eve De Rosa, Ph.D., for careful manual delineation of selective brain structures on MRI. This work was supported by grants from the U.S. National Institute on Alcohol Abuse and Alcoholism (AA005965, AA012388, AA010723, AA017168).
References
- Acara M, Alletto JJ, Dlugos C, et al. Small animal MRI. Alcohol Health Res World. 1995;19:321–4. [PMC free article] [PubMed] [Google Scholar]
- Antunez E, Estruch R, Cardenal C, et al. Usefulness of CT and MR imaging in the diagnosis of acute Wernicke's encephalopathy. AJR Am J Roentgenol. 1998;171:1131–7. doi: 10.2214/ajr.171.4.9763009. [DOI] [PubMed] [Google Scholar]
- Ashikaga R, Araki Y, Ono Y, et al. FLAIR appearance of Wernicke encephalopathy. Radiat Med. 1997;15:251–3. [PubMed] [Google Scholar]
- Bae SJ, Lee HK, Lee JH, et al. Wernicke's encephalopathy: atypical manifestation at MR imaging. AJNR Am J Neuroradiol. 2001;22:1480–2. [PMC free article] [PubMed] [Google Scholar]
- Baker K, Harding A, Halliday G, et al. Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke's encephalopathy. Neuroscience. 1999;91:429–38. doi: 10.1016/s0306-4522(98)90664-9. [DOI] [PubMed] [Google Scholar]
- Bender L, Schilder P. Encephalopathia alcoholica: polioencephalitis haemorrhagica superior of Wernicke. Arch Neurol Psychiatry. 1933;29:990–1053. [Google Scholar]
- Bigler ED. Neuroimaging I: Basic Science. I. New York:: Plenum; 1996. [Google Scholar]
- Blansjaar B, Van Dijk J. Korsakoff minus Wernicke syndrome. Alcohol Alcohol. 1992;27:435–7. [PubMed] [Google Scholar]
- Blansjaar B, Vielvoye G, van Dijk J, et al. Similar brain lesions in alcoholics and Korsakoff patients: MRI, psychometric and clinical findings. Clin Neurol Neurosurg. 1992;93:197–203. doi: 10.1016/0303-8467(92)90089-l. [DOI] [PubMed] [Google Scholar]
- Breslow RA, Guenther PM, Smothers BA. Alcohol drinking patterns and diet quality: the 1999–2000 National Health and Nutrition Examination Survey. Am J Epidemiol. 2006;163:359–66. doi: 10.1093/aje/kwj050. [DOI] [PubMed] [Google Scholar]
- Bruce WR, Furrer R, Shangari N, et al. Marginal dietary thiamin deficiency induces the formation of colonic aberrant crypt foci (ACF) in rats. Cancer Lett. 2003;202:125–9. doi: 10.1016/j.canlet.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Butters N, Cermak LS. Alcoholic Korsakoff's Syndrome: An Information Processing Approach to Amnesia. New York: Academic Press; 1980. [Google Scholar]
- Butters N, Stuss DT. Diencephalic amnesia. In: Boller F, Grafman J, editors. Handbook of Neuropsychology. Vol. 3. Amsterdam: Elsevier; 1989. pp. 107–48. Vols. 4. [Google Scholar]
- Caine D, Halliday GM, Kril JJ, et al. Operational criteria for the classification of chronic alcoholics: identification of Wernicke's encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62:51–60. doi: 10.1136/jnnp.62.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas VA, Studholme C, Meyerhoff DJ, et al. Chronic active heavy drinking and family history of problem drinking modulate regional brain tissue volumes. Psychiatry Res. 2005;138:115–30. doi: 10.1016/j.pscychresns.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Carlen PL, Wortzman G, Holgate RC, et al. Reversible cerebral atrophy in recently abstinent chronic alcoholics measured by computed tomography scans. Science. 1978;200:1076–8. doi: 10.1126/science.653357. [DOI] [PubMed] [Google Scholar]
- Charness ME. Brain lesions in alcoholics. Alcohol Clin Exp Res. 1993;17:2–11. doi: 10.1111/j.1530-0277.1993.tb00718.x. [DOI] [PubMed] [Google Scholar]
- Charness ME. Intracranial voyeurism: revealing the mammillary bodies in alcoholism. Alcohol Clin Exp Res. 1999;23:1941–4. doi: 10.1111/j.1530-0277.1999.tb04095.x. [DOI] [PubMed] [Google Scholar]
- Charness ME, DeLaPaz RL. Mammillary body atrophy in Wernicke's encephalopathy: antemortem identification using magnetic resonance imaging. Ann Neurol. 1987;22:595–600. doi: 10.1002/ana.410220506. [DOI] [PubMed] [Google Scholar]
- Chu K, Kang DW, Kim HJ, et al. Diffusion-weighted imaging abnormalities in wernicke encephalopathy: reversible cytotoxic edema? Arch Neurol. 2002;59:123–7. doi: 10.1001/archneur.59.1.123. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Narasimhan A, Thatcher DL, et al. Prefrontal cortex, thalamus, and cerebellar volumes in adolescents and young adults with adolescent-onset alcohol use disorders and comorbid mental disorders. Alcohol Clin Exp Res. 2005;29:1590–600. doi: 10.1097/01.alc.0000179368.87886.76. [DOI] [PubMed] [Google Scholar]
- De Rosa E, Sullivan EV. Enhanced release from proactive interference in nonamnesic alcoholic individuals: implications for impaired associative binding. Neuropsychology. 2003;17:469–81. doi: 10.1037/0894-4105.17.3.469. [DOI] [PubMed] [Google Scholar]
- Doraiswamy PM, Massey EW, Enright K, et al. Wernicke-Korsakoff syndrome caused by psychogenic food refusal—MR findings. Am J Neuroradiol. 1994;15:594–6. [PMC free article] [PubMed] [Google Scholar]
- Eichenbaum H, Cohen NJ. From Conditioning to Conscious Recollection. Oxford: Oxford University Press; 2001. [Google Scholar]
- Escobar A, Aruffo C, Rodriguez-Carbajal J. Wernicke's encephalopathy. A case report with neurophysiologic and CT-scan studies. Acta Vitaminol Enzymol. 1983;5:125–31. [PubMed] [Google Scholar]
- Fama R, Marsh L, Sullivan EV. Dissociation of remote and anterograde memory impairment and neural correlates in alcoholic Korsakoff syndrome. J Int Neuropsychol Assoc. 2004;10:427–41. doi: 10.1017/S135561770410310X. [DOI] [PubMed] [Google Scholar]
- Fama R, Pfefferbaum A, Sullivan EV. Visuoperceptual priming in alcoholic Korsakoff syndrome. Alcohol Clin Exp Res. 2006;30:680–7. doi: 10.1111/j.1530-0277.2006.00085.x. [DOI] [PubMed] [Google Scholar]
- Fein G, Landman B. Treated and treatment-naive alcoholics come from different populations. Alcohol. 2005;35:19–26. doi: 10.1016/j.alcohol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Gallucci M, Bozzao A, Splendiani A, et al. Wernicke encephalopathy: MR findings in five patients. AJR Am J Roentgenol. 1990;155:1309–14. doi: 10.2214/ajr.155.6.2122685. [DOI] [PubMed] [Google Scholar]
- Gazdzinski S, Durazzo TC, Meyerhoff DJ. Temporal dynamics and determinants of whole brain tissue volume changes during recovery from alcohol dependence. Drug Alcohol Depend. 2005;78:263–73. doi: 10.1016/j.drugalcdep.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Gotze P, Kuhne D, Hansen J, et al. Brain atrophy in chronic alcoholism. Clinical and computer tomographic study. Arch Psychiatr Nervenkr. 1978;226:137–56. doi: 10.1007/BF00345948. [DOI] [PubMed] [Google Scholar]
- Halavaara J, Brander A, Lyytinen J, et al. Wernicke's encephalopathy: is diffusion-weighted MRI useful? Neuroradiology. 2003;45:519–23. doi: 10.1007/s00234-003-1043-8. [DOI] [PubMed] [Google Scholar]
- Harper C. Thiamine (vitamin B1) deficiency and associated brain damage is still common throughout the world and prevention is simple and safe! Eur J Neurol. 2006;13:1078–82. doi: 10.1111/j.1468-1331.2006.01530.x. [DOI] [PubMed] [Google Scholar]
- Harper C, Butterworth R. Nutritional and metabolic disorders. In: Graham DI, Lantos PL, editors. Greenfield's Neuropathology. London: Arnold; 1997. pp. 601–42. [Google Scholar]
- Harper C, Fornes P, Duyckaerts C, et al. An international perspective on the prevalence of the Wernicke–Korsakoff syndrome. Metab Brain Dis. 1995;10:17–24. doi: 10.1007/BF01991779. [DOI] [PubMed] [Google Scholar]
- Harper CG. The incidence of Wernicke's encephalopathy in Australia: a neuropathological study of 131 cases. J Neurol, Neurosurg, Psychiatry. 1983;46:593–8. doi: 10.1136/jnnp.46.7.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke–Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry. 1986;49:341–5. doi: 10.1136/jnnp.49.4.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CG, Kril JJ. Corpus callosal thickness in alcoholics. Br J Addict. 1988;83:577–80. doi: 10.1111/j.1360-0443.1988.tb02577.x. [DOI] [PubMed] [Google Scholar]
- Harper CG, Kril JJ. Neuropathology of alcoholism. Alcohol Alcohol. 1990;25:207–16. doi: 10.1093/oxfordjournals.alcalc.a044994. [DOI] [PubMed] [Google Scholar]
- He X, Sullivan EV, Stankovic RK, et al. Interaction of thiamine deficiency and voluntary alcohol consumption disrupts rat corpus callosum ultrastructure. Neuropsychopharmacology. 2007;32:2207–16. doi: 10.1038/sj.npp.1301332. [DOI] [PubMed] [Google Scholar]
- Jarho L. Korsakoff-like amnesic syndrome in penetrating brain injury. A study of Finnish war veterans. Acta Neurol Scand Suppl. 1973;54:3–156. [PubMed] [Google Scholar]
- Jauhar P, Montaldi D. Wernicke–Korsakoff syndrome and the use of brain imaging. Alcohol Alcohol. 2000;35:21–3. doi: 10.1093/alcalc/35.supplement_1.21. [DOI] [PubMed] [Google Scholar]
- Jernigan TL, Butters N, DiTraglia G, et al. Reduced cerebral grey matter observed in alcoholics using magnetic resonance imaging. Alcohol Clin Exp Res. 1991;15:418–27. doi: 10.1111/j.1530-0277.1991.tb00540.x. [DOI] [PubMed] [Google Scholar]
- Jolliffe N, Wortis H, Fein HD. The Wernicke syndrome. Arch Neurol Psychiatry. 1941;46:569–97. [Google Scholar]
- Jordan LR, Zelaya FO, Rose SE, et al. Changes in the hippocampus induced by glucose in thiamin deficient rats detected by MRI. Brain Res. 1998;791:347–51. doi: 10.1016/s0006-8993(98)00203-0. [DOI] [PubMed] [Google Scholar]
- Koch MA, Norris DG. Artifacts and pitfalls in diffusion MR imaging: diffusion, perfusion and spectroscopy. In: Gillard J, Waldman A, Barker P, editors. Clinical MR Neuroimaging. Cambridge: Cambridge University Press; 2005. pp. 99–108. [Google Scholar]
- Kopelman MD. The Korsakoff syndrome. Br J Psychiatry. 1995;166:154–73. doi: 10.1192/bjp.166.2.154. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Halliday GM, Svoboda MD, et al. The cerebral cortex is damaged in chronic alcoholics. Neuroscience. 1997;79:983–98. doi: 10.1016/s0306-4522(97)00083-3. [DOI] [PubMed] [Google Scholar]
- Langlais PJ. Pathogenesis of diencephalic lesions in an experimental model of Wernicke's encephalopathy. Metab Brain Dis. 1995;10:31–44. doi: 10.1007/BF01991781. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Savage LM. Thiamine deficiency in rats produces cognitive and memory deficits on spatial tasks that correlate with tissue loss in diencephalon, cortex and white matter. Behav Brain Res. 1995;68:75–89. doi: 10.1016/0166-4328(94)00162-9. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang SX. Cortical and subcortical white matter damage without Wernicke's encephalopathy after recovery from thiamine deficiency in the rat. Alcohol Clin Exp Res. 1997;21:434–43. doi: 10.1111/j.1530-0277.1997.tb03788.x. [DOI] [PubMed] [Google Scholar]
- Lapergue B, Klein I, Olivot JM, et al. Diffusion weighted imaging of cerebellar lesions in Wernicke's encephalopathy. J Neuroradiol. 2006;33:126–8. doi: 10.1016/s0150-9861(06)77243-1. [DOI] [PubMed] [Google Scholar]
- Lieber CS. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health. 2003;27:220–31. [PMC free article] [PubMed] [Google Scholar]
- Maeda M, Tsuchida C, Handa Y, et al. Fluid attenuated inversion recovery (FLAIR) imaging in acute Wernicke encephalopathy. Radiat Med. 1995;13:311–3. [PubMed] [Google Scholar]
- Manari AP, Preedy VR, Peters TJ. Nutritional intake of hazardous drinkers and dependent alcoholics in the UK. Addict Biol. 2003;8:201–10. doi: 10.1080/1355621031000117437. [DOI] [PubMed] [Google Scholar]
- Mann K, Gunther A, Stetter F, et al. Rapid recovery from cognitive deficits in abstinent alcoholics: a controlled test-retest study. Alcohol Alcohol. 1999;34:567–74. doi: 10.1093/alcalc/34.4.567. [DOI] [PubMed] [Google Scholar]
- Martin PR, Singleton CK, Hiller-Sturmhofel S. The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health. 2003;27:134–42. [PMC free article] [PubMed] [Google Scholar]
- McDowell JR, LeBlanc HJ. Computed tomographic findings in Wernicke–Korsakoff syndrome. Arch Neurol. 1984;41:453–4. doi: 10.1001/archneur.1984.04050160119026. [DOI] [PubMed] [Google Scholar]
- Mensing JW, Hoogland PH, Slooff JL. Computed tomography in the diagnosis of Wernicke's encephalopathy: a radiological-neuropathological correlation. Ann Neurol. 1984;16:363–5. doi: 10.1002/ana.410160316. [DOI] [PubMed] [Google Scholar]
- Mulholland PJ, Self RL, Stepanyan TD, et al. Thiamine deficiency in the pathogenesis of chronic ethanol-associated cerebellar damage in vitro. Neuroscience. 2005;135:1129–39. doi: 10.1016/j.neuroscience.2005.06.077. [DOI] [PubMed] [Google Scholar]
- Nicolas J, Fernandez-Sola J, Robert J, et al. High ethanol intake and malnutrition in alcoholic cerebellar shrinkage. Q J Med. 2000;93:449–56. doi: 10.1093/qjmed/93.7.449. [DOI] [PubMed] [Google Scholar]
- O’Neill J, Cardenas VA, Meyerhoff DJ. Effects of abstinence on the brain: quantitative magnetic resonance imaging and magnetic resonance spectroscopic imaging in chronic alcohol abuse. Alcohol Clin Exp Res. 2001;25:1673–82. [PubMed] [Google Scholar]
- Parks MH, Dawant BM, Riddle WR, et al. Longitudinal brain metabolic characterization of chronic alcoholics with proton magnetic resonance spectroscopy. Alcohol Clin Exp Res. 2002;26:1368–80. doi: 10.1097/01.ALC.0000029598.07833.2D. [DOI] [PubMed] [Google Scholar]
- Pentney RJ, Alletto JJ, Acara MA, et al. Small animal magnetic resonance imaging: a means of studying the development of structural pathologies in the rat brain. Alcohol Clin Exp Res. 1993;17:1301–08. doi: 10.1111/j.1530-0277.1993.tb05245.x. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Adalsteinsson E, Bell RL, et al. Development and resolution of brain lesions caused by pyrithiamine and dietary induced thiamine deficiency and alcohol exposure in the alcohol-preferring (P) rat: a longitudinal MR imaging and spectroscopy study. Neuropsychopharmacology. 2007;30:1159–77. doi: 10.1038/sj.npp.1301107. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Adalsteinsson E, Sood R, et al. Part II: longitudinal brain MRI study of the alcohol-preferring (P) rat: effects of voluntary chronic alcohol consumption. Alcohol Clin Exp Res. 2006;30:1248–61. doi: 10.1111/j.1530-0277.2006.00146.x. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Lim KO., Zipursky RB, et al. Brain gray and white matter volume loss accelerates with aging in chronic alcoholics: a quantitative MRI study. Alcohol Clin Exp Res. 1992;16:1078–89. doi: 10.1111/j.1530-0277.1992.tb00702.x. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Rosenbloom MJ, Serventi KL, et al. Brain volumes, RBC status, and hepatic function in alcoholics after 1 and 4 weeks of sobriety: predictors of outcome. Am J Psychiatry. 2004;161:1190–96. doi: 10.1176/appi.ajp.161.7.1190. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Mathalon DH, et al. Longitudinal changes in magnetic resonance imaging brain volumes in abstinent and relapsed alcoholics. Alcohol Clin Exp Res. 1995;19:1177–91. doi: 10.1111/j.1530-0277.1995.tb01598.x. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Mathalon DH, et al. Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcohol Clin Exp Res. 1997;21:521–9. doi: 10.1111/j.1530-0277.1997.tb03798.x. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Rosenbloom MJ, et al. A controlled study of cortical gray matter and ventricular changes in alcoholic men over a five year interval. Arch Gen Psychiatry. 1998;55:905–12. doi: 10.1001/archpsyc.55.10.905. [DOI] [PubMed] [Google Scholar]
- Pires RG, Pereira SR, Pittella JE, et al. The contribution of mild thiamine deficiency and ethanol consumption to central cholinergic parameter dysfunction and rats’ open-field performance impairment. Pharmacol Biochem Behav. 2001;70:227–35. doi: 10.1016/s0091-3057(01)00593-7. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, Savage LM. Aging potentiates the acute and chronic neurological symptoms of pyrithiamine-induced thiamine deficiency in the rodent. Behav Brain Res. 2001;119:167–77. doi: 10.1016/s0166-4328(00)00350-8. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, Savage LM. Age-related vulnerability to diencephalic amnesia produced by thiamine deficiency: the role of time of insult. Behav Brain Res. 2004;148:93–105. doi: 10.1016/s0166-4328(03)00208-0. [DOI] [PubMed] [Google Scholar]
- Reed LJ, Lasserson D, Marsden P, et al. FDG-PET findings in the Wernicke–Korsakoff syndrome. Cortex. 2003;39:1027–45. doi: 10.1016/s0010-9452(08)70876-1. [DOI] [PubMed] [Google Scholar]
- Santolaria F, Perez-Manzano JL, Milena A, et al. Nutritional assessment in alcoholic patients. Its relationship with alcoholic intake, feeding habits, organic complications and social problems. Drug Alcohol Depend. 2000;59:295–304. doi: 10.1016/s0376-8716(99)00129-5. [DOI] [PubMed] [Google Scholar]
- Savage LM, Pitkin SR, Knitowski KM. Rats exposed to acute pyrithiamine-induced thiamine deficiency are more sensitive to the amnestic effects of scopolamine and MK-801: examination of working memory, response selection, and reinforcement contingencies. Behav Brain Res. 1999;104:13–26. doi: 10.1016/s0166-4328(99)00049-2. [DOI] [PubMed] [Google Scholar]
- Schroth G, Naegele T, Klose U, et al. Reversible brain shrinkage in abstinent alcoholics, measured by MRI. Neuroradiology. 1988;30:385–9. doi: 10.1007/BF00404102. [DOI] [PubMed] [Google Scholar]
- Schroth G, Wichmann W, Valavanis A. Blood-brain-barrier disruption in acute Wernicke encephalopathy: MR findings. J Comput Assist Tomogr. 1991;15:1059–61. doi: 10.1097/00004728-199111000-00034. [DOI] [PubMed] [Google Scholar]
- Sechi G, Serra A. Wernicke's encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6:442–55. doi: 10.1016/S1474-4422(07)70104-7. [DOI] [PubMed] [Google Scholar]
- Shear PK, Sullivan EV, Lane B, et al. Mammillary body and cerebellar shrinkage in chronic alcoholics with and without amnesia. Alcohol Clin Exp Res. 1996;20:1489–95. doi: 10.1111/j.1530-0277.1996.tb01153.x. [DOI] [PubMed] [Google Scholar]
- Sheedy D, Lara A, Garrick T, et al. Size of mamillary bodies in health and disease: useful measurements in neuroradiological diagnosis of Wernicke's encephalopathy. Alcohol Clin Exp Res. 1999;23:1624–8. [PMC free article] [PubMed] [Google Scholar]
- Shimamura AP, Jernigan TL, Squire LR. Korsakoff's syndrome: radiological (CT) findings and neuropsychological correlates. J Neurosci. 1988;8:4400–10. doi: 10.1523/JNEUROSCI.08-11-04400.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR, Amaral DG, Press GA. Magnetic resonance imaging of the hippocampal formation and mammillary nuclei distinguish medial temporal lobe and diencephalic amnesia. J Neurosci. 1990;10:3106–17. doi: 10.1523/JNEUROSCI.10-09-03106.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV. Human brain vulnerability to alcoholism: evidence from neuroimaging studies. In: Noronha A, Eckardt M, Warren K, editors. Review of NIAAA's Neuroscience and Behavioral Research Portfolio, NIAAA Research Monograph No. 34. Bethesda, MD: National Institutes of Health; 2000. pp. 473–508. [Google Scholar]
- Sullivan EV. Compromised pontocerebellar and cerebellothalamocortical systems: speculations on their contributions to cognitive and motor impairment in nonamnesic alcoholism. Alcohol Clin Exp Res. 2003;27:1409–19. doi: 10.1097/01.ALC.0000085586.91726.46. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Adalsteinsson E, Sood R, et al. Part I: longitudinal brain MRI brain study of the alcohol-preferring (P) rat: adult brain growth. Alcohol Clin Exp Res. 2006;30:1234–47. doi: 10.1111/j.1530-0277.2006.00145.x. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Deshmukh A, De Rosa E, et al. Striatal and forebrain nuclei volumes: contribution to motor function and working memory deficits in alcoholism. Biol Psychiatry. 2005;57:768–76. doi: 10.1016/j.biopsych.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Deshmukh A, Desmond JE, et al. Cerebellar volume decline in normal aging, alcoholism, and Korsakoff's syndrome: relation to ataxia. Neuropsychology. 2000;14:341–52. doi: 10.1037//0894-4105.14.3.341. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Lane B, Rosenbloom MJ, et al. In vivo mammillary body volume deficits in amnesic and nonamnesic alcoholics. Alcohol Clin Exp Res. 1999;23:1629–36. [PubMed] [Google Scholar]
- Sullivan EV, Marsh L. Hippocampal volume deficits in alcoholic Korsakoff's syndrome. Neurology. 2003;61:1716–19. doi: 10.1212/01.wnl.0000098940.31882.bb. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Marsh L, Mathalon DH, et al. Anterior hippocampal volume deficits in nonamnesic, aging chronic alcoholics. Alcohol Clin Exp Res. 1995;19:110–22. doi: 10.1111/j.1530-0277.1995.tb01478.x. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. Magnetic resonance relaxometry reveals central pontine abnormalities in clinically asymptomatic alcoholic men. Alcohol Clin Exp Res. 2001;25:1206–12. [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. Neurocircuitry in alcoholism: a substrate of disruption and repair. Psychopharmacology (Berlin) 2005;180:583–94. doi: 10.1007/s00213-005-2267-6. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Rosenbloom MJ, Serventi KL, et al. The effects of alcohol dependence comorbidity and anti-psychotic medication on volumes of the thalamus and pons in schizophrenia. Am J Psychiatry. 2003;160:1110–6. doi: 10.1176/appi.ajp.160.6.1110. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Rosenbloom MJ, Serventi KL, et al. Effects of age and sex on volumes of the thalamus, pons, and cortex. Neurobiol Aging. 2004;25:185–92. doi: 10.1016/s0197-4580(03)00044-7. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Nakazawa S, Yoshino Y, et al. Metabolic studies of the edematous cerebral cortex of the pyrithiamine-treated thiamine-deficient rat. Brain Res. 1988;441:202–8. doi: 10.1016/0006-8993(88)91399-6. [DOI] [PubMed] [Google Scholar]
- Talland GA. Deranged Memory: A Psychonomic Study of the Amnesic Syndrome. New York: Academic Press; 1965. [Google Scholar]
- Talland GA. Disorders of Memory and Learning. Harmondsworth: Penguin; 1968. [Google Scholar]
- Thomson AD. Mechanisms of vitamin deficiency in chronic alcohol misusers and the development of the Wernicke–Korsakoff syndrome. Alcohol Alcohol Suppl. 2000;35:2–7. doi: 10.1093/alcalc/35.supplement_1.2. [DOI] [PubMed] [Google Scholar]
- Thomson AD, Cook CC, Touquet R, et al. The Royal College of Physicians report on alcohol: guidelines for managing Wernicke's encephalopathy in the accident and emergency department. Alcohol Alcohol. 2002;37:513–21. doi: 10.1093/alcalc/37.6.513. [DOI] [PubMed] [Google Scholar]
- Thomson AD, Jeyasingham MD, Pratt OE, et al. Nutrition and alcoholic encephalopathies. Acta Med Scand. 1987;717(Suppl):55–65. doi: 10.1111/j.0954-6820.1987.tb13042.x. [DOI] [PubMed] [Google Scholar]
- Thomson AD, Marshall EJ. The natural history and pathophysiology of Wernicke's encephalopathy and Korsakoff's psychosis. Alcohol Alcohol. 2006;41:151–8. doi: 10.1093/alcalc/agh249. [DOI] [PubMed] [Google Scholar]
- Todd K, Butterworth RF. Mechanisms of selective neuronal cell death due to thiamine deficiency. Ann N Y Acad Sci. 1999;893:404–11. doi: 10.1111/j.1749-6632.1999.tb07866.x. [DOI] [PubMed] [Google Scholar]
- Torvik A, Lindboe CF, Rodge S. Brain lesions in alcoholics: a neuropathological study with clinical correlations. J Neurol Sci. 1982;56:233–48. doi: 10.1016/0022-510x(82)90145-9. [DOI] [PubMed] [Google Scholar]
- Torvik A, Torp S, Lindboe CF. Atrophy of the cerebellar vermis in aging: a morphometric and histologic study. J Neurol Sci. 1986;76:283–94. doi: 10.1016/0022-510x(86)90176-0. [DOI] [PubMed] [Google Scholar]
- Unlu E, Cakir B, Asil T. MRI findings of Wernicke encephalopathy revisited due to hunger strike. Eur J Radiol. 2006;57:43–53. doi: 10.1016/j.ejrad.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Varnet O, de Seze J, Soto-Ares G, et al. Wernicke–Korsakoff syndrome: diagnostic contribution of magnetic resonance imaging. Rev Neurol (Paris) 2002;158:1181–5. [PubMed] [Google Scholar]
- Victor M. MR in the diagnosis of Wernicke–Korsakoff syndrome. AJR Am J Roentgenol. 1990;155:1315–6. doi: 10.2214/ajr.155.6.2122686. [DOI] [PubMed] [Google Scholar]
- Victor M, Adams RD, Collins GH. The Wernicke–Korsakoff Syndrome. Philadelphia, PA: F.A. Davis; 1971. [PubMed] [Google Scholar]
- Victor M, Adams RD, Collins GH. The Wernicke–Korsakoff Syndrome and Related Neurologic Disorders Due to Alcoholism and Malnutrition. 2nd edn. Philadelphia, PA: F.A. Davis; 1989. [Google Scholar]
- Victor M, Herman K, White EE. A psychological study of the Wernicke–Korsakoff syndrome. Results of Wechsler–Bellevue Intelligence Scale and Wechsler Memory Scale testing at different stages in the disease. Q J Stud Alcohol. 1959;20:467–79. [PubMed] [Google Scholar]
- Visser P, Krabbendam L, Verhey F, et al. Brain correlates of memory dysfunction in alcoholic Korsakoff's syndrome. J Neurol Neurosurg Psychiatry. 1999;67:774–8. doi: 10.1136/jnnp.67.6.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt E. Neuroanatomical consequences of thiamine deficiency: a comparative analysis. Alcohol Alcohol. 1985;20:201–21. [PubMed] [Google Scholar]
- Yoneoka Y, Takeda N, Inoue A, et al. Acute Korsakoff syndrome following mammillothalamic tract infarction. Am J Neuroradiol. 2004;25:964–8. [PMC free article] [PubMed] [Google Scholar]
- Zelaya FO, Rose SE, Nixon PF, et al. MRI demonstration of impairment of the blood-CSF barrier by glucose administration to the thiamin-deficient rat brain. Magn Reson Imaging. 1995;13:555–61. doi: 10.1016/0730-725x(95)00020-h. [DOI] [PubMed] [Google Scholar]
- Zhong C, Jin L, Fei G. MR imaging of nonalcoholic Wernicke encephalopathy: a follow-up study. AJNR Am J Neuroradiol. 2005;26:2301–5. [PMC free article] [PubMed] [Google Scholar]
- Zimitat C, Kril J, Harper CG, et al. Progression of neurological disease in thiamin-deficient rats is enhanced by ethanol. Alcohol. 1990;7:493–501. doi: 10.1016/0741-8329(90)90038-e. [DOI] [PubMed] [Google Scholar]
- Zola-Morgan S, Squire LR. Neuroanatomy of memory. Annu Rev Neurosci. 1993;16:547–63. doi: 10.1146/annurev.ne.16.030193.002555. [DOI] [PubMed] [Google Scholar]
- Zuccoli G, Gallucci M, Capellades J, et al. Wernicke encephalopathy: MR findings at clinical presentation in twenty-six alcoholic and nonalcoholic patients. AJNR Am J Neuroradiol. 2007;28:1328–31. doi: 10.3174/ajnr.A0544. [DOI] [PMC free article] [PubMed] [Google Scholar]