

Abstract
Recessive Charcot-Marie-Tooth disease type-4J (CMT4J) and its animal model, the pale tremor mouse (plt), are caused by mutations of the FIG4 gene encoding a PI(3,5)P2 5-phosphatase. We describe the 9-year clinical course of CMT4J, including asymmetric, rapidly progressive paralysis, in two siblings. Sensory symptoms were absent despite reduced numbers of sensory axons. Thus, the phenotypic presentation of CMT4J clinically resembles motor neuron disease. Time-lapse imaging of fibroblasts from CMT4J patients demonstrates impaired trafficking of intracellular organelles because of obstruction by vacuoles. Further characterization of plt mice identified axonal degeneration in motor and sensory neurons, limited segmental demyelination, lack of TUNEL staining and lack of accumulation of ubiquitinated protein in vacuoles of motor and sensory neurons. This study represents the first documentation of the natural history of CMT4J. Physical obstruction of organelle trafficking by vacuoles is a potential novel cellular mechanism of neurodegeneration.
Keywords: FIG4 or SAC3 gene; PI(3,5)P2-5-phosphatase; neuronopathy; axonal degeneration; vacuoles; amyotrophic lateral sclerosis; motor neuron disease; segmental demyelination; Schwann cells
Introduction
Phosphoinositides regulate intracellular membrane trafficking by recruiting effector proteins to the surface of specific organelles (Bolis et al., 2007; Volpicelli-Daley et al., 2007). The levels of phosphoinositides are regulated by specific kinases and phosphatases (Di Paolo et al., 2006; Volpicelli-Daley et al., 2007). Phosphoinositide signalling is particularly important for the integrity of the nervous system. Mutations in MTMR2 and MTMR13, which dephosphorylate PI3P and PI(3,5)P2 at the 3rd position of inositol, cause the recessive inherited neuropathies Charcot-Marie-Tooth type 4B1 (CMT4B1) and CMT4B2, with a distinctive pathology of excessive myelin out-folding. Mutations of FIG4/SAC3 were recently shown to cause Charcot-Marie-Tooth type 4J (CMT4J), as discussed below.
FIG4/SAC3 is a phosphatase that dephosphorylates PI(3,5)P2 at the 5th position of inositol (Duex et al., 2006b; Chow et al., 2007). FIG4 interacts with FAB1 and VAC14 in a protein complex that regulates the overall concentration of PI(3,5)P2 (Di Paolo et al., 2006; Sbrissa et al., 2007). Although the detailed molecular mechanism of this regulation remains to be clarified, loss of function of FIG4 reduces the levels of PI(3,5)P2 (Duex et al., 2006a, b; Chow et al., 2007). We recently described a mouse mutant, pale tremor (plt), with a spontaneous mutation that eliminates the expression of FIG4. Homozygous plt mice have a neuronopathy with extensive neuronal degeneration in the central and peripheral nervous systems, including loss of dorsal root ganglion (DRG) neurons and large myelinated axons in the sciatic nerve. A screen of 95 patients with CMT identified four patients with mutations in FIG4 (Chow et al., 2007). The natural history and pathogenesis of CMT4J has not been well characterized. In this report, we provide evidence that CMT4J is a primary neuronopathy. Patients develop rapidly progressive, asymmetric motor neuron degeneration without symptoms of sensory loss, although both sensory and motor neurons exhibit morphological abnormalities. Abnormal transport of intracellular organelles was observed by time-lapse imaging in fibroblasts from CMT4J patients. The data provide insights into the pathogenesis of CMJ4J and the pattern of disease progression in motor neuron disease (MND).
Methods
Plt mice and genotyping
The plt mutation was genotyped by PCR as previously described (Chow et al., 2007). Eleven mice were examined at 6 weeks of age and two mice at 3 weeks of age.
Semithin section and EM
Mice were transcardially perfused with 4% paraformaldhyde and 3.5% glutaraldhyde. Sciatic nerve and dorsal/ventral roots were dissected and post-fixed for 24 h. Nerves were then osmicated for 1.5 h, dehydrated and embedded in Epon. Tissue blocks were sectioned with 1 μm thickness and stained with methylene blue for light microscopic examination. The blocks were then trimmed and sectioned into ultrathin sections for EM examination (Zeiss EM 900). The same methods were applied to patients’ sural nerve.
Morphometric analysis
Semithin sections of dorsal and ventral roots of plt mice. (thickness = 1 µM) were examined under the 63X objective. We imaged the entire field of transverse sections of each lumbar root for analysis. Images were imported into morphometric software (ImagePro Plus). Areas of each field were counted to obtain nerve fibre density, myelin thickness, g-ratio and histograms.
Teased nerve fibre immunohistochemistry (IHC)
Nerves were fixed in 4% paraformaldehyde for 30 min after dissection from perfused mice. Sciatic nerves were teased into individual fibres on glass slides. The slides were dried overnight, reacted with primary antibodies, and stored at 4°C overnight, then incubated for 2 h with secondary antibodies. The stained slides were examined under a Leica fluorescent microscope.
Patients
We evaluated two siblings, a male of current age 44 years (patient 1) and his sister of 46 years (patient 2). These patients, designated BAB1372 and BAB1373 in reference Chow et al., are compound heterozygotes for mutations of FIG4, R183X in exon 6 and I41T in exon 2. Detailed medical histories were obtained and serial neurological examinations were performed, followed by electrophysiological studies below.
Electromyogram/nerve conduction studies
Nerve conduction studies (NCS) of median, ulnar, peroneal, tibial and sural nerves were performed using conventional methods. The distal stimulation distances for motor conduction studies were 7 cm in the arms and 9 cm in the legs. A concentric electromyogram (EMG) needle was used and recordings were made at standard settings (Li et al., 2002).
Skin biopsy and human fibroblast culture
Punch skin biopsies were obtained from patients, rinsed with PBS, minced into coarse slurry using an iris scissor and transferred into a 15 ml plastic tube containing 4 mg/ml collagenase II in basal medium eagle with 20% foetal bovine serum and 100 IU/ml penicillin–streptomycin. The tube was placed in incubator at 37°C for 24 h and centrifuged at 1000 rpm for 5 min next day. Supernatants were removed using a pipette. Pellets were washed twice with PBS, suspended in 1 ml basal medium eagle medium and transferred into a flask to be distributed over the bottom surface. The flask was placed in a humidified 5% CO2 incubator at 37°C. Cells were passaged by removing the media, washing with calcium- and magnesium-free PBS and incubated with 0.25% trypsin-EDTA in PBS for 3–5 min. The action of trypsin was terminated by adding medium containing 20% foetal bovine serum. After centrifugation, pelleted cells were washed with PBS and resuspended. The suspension was split into thirds for seeding. Cultures were passaged approximately every other week. Cultures were examined under light microscopy. All experiments were conducted between passages 3 and 8. Control and mutant cells were compared at the same passage.
IHC, TUNEL staining and proliferation assay
IHC
Mice were transcardially perfused with 4% paraformaldhyde. Dissected nerves were fixed overnight at 4°C in fresh 4% paraformaldehyde and cryoprotected in PBS containing 20% sucrose. Tissues were embedded in OCT, sectioned into 10 μm thickness slices in a cryostat, and thaw-mounted onto Superfrost Plus slides (Fisher Scientific, PA). After drying, sections were washed in PBS, permeabilized with cold acetone in −20°C, blocked for 1 h in blocking solution (0.5% Triton-X-100 fish gelatin solution in PBS) and incubated overnight at 4°C with primary antibodies (Table 1); followed by secondary antibody for 2 h at room temperature. After washing with PBS, sections were dried and coversliped. For teased nerve fibre IHC, dissected nerves were post-fixed in 4% paraformaldehyde for 30 min and teased into individual fibres on slides. Teased fibres were dried overnight and immunostained as described (Bai et al., 2006; Li et al., 2006). The stained slides were examined using a fluorescent microscope (Leica Microsystems, Germany).
Table 1.
Primary antibodies
| Antibody | Source | Species raised in | Specific antigen | Type | Reference |
|---|---|---|---|---|---|
| α-Ubiquitin | Vector, CA Cat # VP-U576 | Rabbit | Ubiquitin cross linked to limpet haemocyanin | Polyclonal IgG | Li et al. (2006) |
| SMI31 | COVANCE # SMI31R | Mouse | NFP | Monoclonal IgG1 | Giasson et al. (1996) |
| VCP | BioReagents # MA3-004 | Mouse | A synthetic peptide corresponding to residues 792–806 | Monoclonal IgG2a | Vij et al. (2006) |
| MAG | Zymed Laboratories # 34-6200 | Rabbit | The shared C-terminal region of the small and large MAG | Polyclonal Antiserum | Yamamoto et al. (1994) |
| CD68 | AbD Serotec # MCA1957 | Rat | Macrosialin | Monoclonal IgG2a | da Silva et al. (1999) |
| Lamp2 | USBiological # L9190-14A | Mouse | D107b | Monoclonal IgG1 | Fanin et al. (2006) |
| EEA1 | USBiological # E2211-01 | Rabbit | EEA1 | Polyclonal IgG | Mills et al. (1998) |
| NF-L | COVANCE # PRB-574C | Rabbit | Full length recombinant NF-L | Polyclonal Antiserum | Goldstain et al. (1991) |
| S-100 | CHEMICON # AB941 | Rabbit | Keyhole limpet hemocyanin (KLH) with carbodiimide | Polyclonal Antiserum | Gonzalez-Martinez et al. (2003) |
| ChAT | CHEMICON # AB144P | Goat | Choline acetyltransferase | Polyclonal Affinity purified immunoglobulin | Barber et al. (1984) |
| Ki67 | DakoCytomation | Mouse | Bacterially expressed parts of hunan ki67 cDNA | Monoclonal IgG1 | Muskhelishvili et al. (2003) |
TUNEL staining and proliferation assay
Apoptosis was evaluated by TUNEL using a ApopTag® Plus Fluorescein In-Situ Apoptosis Detection Kit (Chemicon, Illinois, USA) according to the manufacturer's protocol. Briefly, cryostat sections, 10 µM thickness, were washed in PBS for 5 min and post-fix in pre-cooled ethanol: acetic acid 2 : 1 for 5 min at −20°C. Slides were first incubated with equilibration buffer, and then replaced with Working Strength TdT enzyme for 1 h at 37°C in a humidified chamber. The reaction was terminated by adding working strength stop/wash buffer. For the negative control, the TdT enzyme was replaced by equilibration buffer. Tissues were further incubated with warmed (RT) working strength antidigoxigenin conjugates for 0.5 h at RT. The nucleuses were counterstained by DAPI. For the proliferation assessment, antibody against Ki67 was used for IHC as described above. Neurons in the DRG, spinal cord anterior horn and Schwann cells in sciatic nerve were identified by antibodies against neurofilament (NF), ChAT and S-100, respectively. Numbers of TUNEL-positive and total cells (neurons or Schwann cells) were counted under 40× lens in each section for apoptotic cells. Apoptotic index was expressed as the number of TUNEL-positive cells in total cell of 1000 for each mouse. The nucleus localization of TUNEL-positive signal was verified with double-stained of DAPI (Thompson et al., 2000).
Time-lapse imaging
Time-lapse recording was made under a phase contrast optic using a computer-assisted invert microscope (Olympus IX70). Temperature in the culture chamber was maintained at 37°C with a thermostat controller (MATS-U55R30, Tokai Hit Co., Japan). Images under a 40× or 60× lens were captured every 5 s through a CCD camera over 30−60 min. To stabilize CO2 concentration during recording, culture medium was replaced with Leibovitz's L-15 medium containing 20% foetal bovine serum 2 h prior to imaging (Yan et al., 2007).
Results
Rapidly progressive and asymmetric paralysis in two patients with FIG4 mutations
To characterize the natural history of patients with FIG4 mutations, we followed two siblings with adult onset CMT4J for 9 years. Both patients are compound heterozygotes carrying the FIG4 protein truncation mutation R183X and the missense mutation I41T. Two patients with early-onset of CMT4J were not seen by us and were not included (Chow et al., 2007).
Patient 1
When first seen in 1998, the proband was a 37-year-old woman who had developed progressive right arm weakness over the course of 1 year following a fall from a desk in 1993. She was the product of a normal pregnancy and delivery, and walked at 9 months of age. She was a dancer as a child until the age of 13 when she was no longer able to dance on her toes. During several clinical visits, no obvious cognitive dysfunction was observed. She was a high school teacher until the severe paralysis developed. Her brain MRI was unremarkable. An examination in 2002 revealed no voluntary movements in the right upper extremity, which was diffusely atrophied. She also had mild weakness of her left upper limb (intrinsic hand muscles 1/5, deltoid 4+/5) and asymmetric, mild, weakness of her legs, with the right worse than the left (Supplementary Video 1). By June 2007, she had no voluntary movement of either arm or right leg (Supplementary Video 2). She had 4−/5 strength throughout the left leg. Her sensory examination was normal for pin prick and position sense. She had mild loss of vibration sense in the right hand. She was diffusely areflexic. The patient died of presumed respiratory compromise in February 2008.
Patient 2
The proband's brother (a computer programmer) was healthy until 1995 when his left leg gave way and he fell down the stairs. He walked by 1 year of age and participated in sports, at a varsity level, in high school. We evaluated him in 1998 at age 35 and then again in 2002 (Supplementary Video 3). In 2002, he had <3/5 strength in iliopsoas and quadriceps muscles bilaterally, but was able to walk with assistance. Anterior tibialis strength was 1/5 bilaterally. Gastrocnemius strength was nearly normal (4+/5) of the right but only 1/5 on the left. Upper extremity strength was entirely normal. By 2007, he was confined to a wheel chair with no voluntary movement of any muscle in either leg (Supplementary Video 4). Strength was now reduced (4/5) in his left deltoid although distal muscles of this arm and all muscles of the right arm were still 5/5. On sensory examination, he had a mild decrease to vibration in the toes of both feet. Proprioception and pin prick sensation were normal in all limbs. He was diffusely areflexic. Like the proband, we found no obvious cognitive dysfunction. Neither patient had abnormalities of skin or hair colour.
NCS in both patients demonstrated that compound motor and sensory responses were asymmetric, severely reduced and progressed in severity between 1998 and 2007 (Table 2). There were prolonged distal latencies, significantly slowed conduction velocities and prolonged F-wave latencies in both patients. The NCS features suggest a demyelinating polyneuropathy in addition to severe axonal loss. Needle EMG showed abundant abnormal spontaneous activities (fibrillations and positive waves) in all limbs. Motor unit action potentials were of long duration and polyphasic with large amplitudes. The recruitment of these Motor unit action potentials was severely reduced. Taken together, the abnormalities suggested severe and wide-spread denervation, similar to that observed in patients with MND (Eisen, 2001). The denervation was severe in the proximal muscles, and, like the weakness, was asymmetric, suggestive of a neuronopathy but not length dependent axonal polyneuropathy.
Table 2.
NCS/EMGa in patients with CMT4J
| Nerves | CMAP/SNAP |
DL (M/S) |
CV (M/S) |
F-wave Latency |
||||
|---|---|---|---|---|---|---|---|---|
| 1998 | 2007 | 1998 | 2007 | 1998 | 2007 | 1998 | 2007 | |
| Patient 1 | ||||||||
| Median | 2.0/8.0 | nr/nr | 10.3/3.9 | nr/nr | 19.8/34.8 | nr/nr | 67.9 | nr |
| Ulnar | 6.0/12.0 | nr/nr | 5.6/2.3 | nr/nr | 23.8/50.0 | nr/nr | 64.9 | nr |
| Per EDB | 0.8 | nr | 13.2 | nr | 8.1 | nr | nr | nr |
| Tibial | 2.0 | nr | 2.1 | nr | 8.3 | nr | nr | nr |
| Sural | nr | nr | nr | nr | nr | nr | ||
| Patient 2 | ||||||||
| Median | 11.2/26.6 | 7.3/3.8 | 4.7/2.8 | 6.5/2.8 | 46.7/50.0 | 36.0/39.0 | 37.0 | 40.4 |
| Ulnar | 11.7/22.3 | 9.7/nr | 3.7/3.1 | 4.3/nr | 50.0/45.1 | 35.0/nr | 35.9 | 29.6 |
| Per EDB | nr | nr | nr | nr | nr | nr | nr | nr |
| Per AT | 3.3 | nr | 6.3 | nr | 46 | nr | nr | nr |
| Sural | nr | nr | nr | nr | nr | nr | ||
aNeedle EMG: abnormal spontaneous activities and reduced recruitment in all limbs.
CMAP = compound muscle action potential (mV); SNAP = sensory nerve action potential (µV); DL = distal latency (ms); M/S = motor nerve distal latency (ms)/sensory nerve distal latency (ms); CV = conduction velocity (m/s); nr = non-responsive; Per EDB = motor peroneal nerve recorded at the extensor digitorium brevis muscle; Per AT = motor peroneal nerve recorded at the anterior tibialis muscle.
A sural nerve biopsy was obtained from patient 2. Semithin sections and EM revealed severely reduced numbers of myelinated nerve fibres with a large amount of collagen in the extracellular matrix (Fig. 1). Onion bulbs and thinly myelinated nerve fibres were observed. Osmicated teased nerve fibres showed evidence of de- and re-myelination in 60% of the myelinated nerve fibres.
Fig. 1.

Loss of myelinated nerve fibres in sural biopsy of a patient with CMT4J. A semithin section with toluidine blue staining was performed on the sural nerve biopsy of the patient 2. The number of myelinated nerve fibres is severely reduced, and there is a large amount of collagen in the extracellular matrix. Arrowhead: onion bulbs and thinly myelinated nerve fibres.
Taken together, the patients’ clinical presentation demonstrates a novel CMT phenotype with two unusual features: (i) aggressive widespread denervation accompanied by concomitant demyelination; and (ii) rapidly progressive and asymmetric weakness without sensory complaints. The clinical presentation and needle EMG imitated MND, although NCS and sural nerve biopsy suggest a demyelinating sensorimotor neuropathy.
Vacuoles interfere with intracellular organelle trafficking in human fibroblasts with FIG4 deficiency
To determine whether FIG4 deficiency is accompanied by vacuolization in patient fibroblasts, as observed in plt mice (Chow et al., 2007), we cultured skin fibroblasts from the two CMT4J-patients and an age-matched control. Although control fibroblasts rarely contained vacuoles, many fibroblasts with multiple vacuoles were present in cultures from both patients (Fig. 2A and B). We manually assessed the vacuolization of 529 cells from patient 1 and 201 cells from patient 2, as well as 270 cells from the control culture. The percentage of cells with vacuoles was significantly higher in FIG4-deficient cultures (Fig. 2C).
Fig. 2.

Vacuolation in FIG4-deficient human fibroblasts. Fibroblasts were isolated from skin biopsies of patients with CMT4J and normal control. Cells were imaged under phase-contrast lens. (A) Cultured fibroblasts from the control contained occasional vacuoles. Intracellular organelles appear as ‘black-dots’ (Waterman-Storer et al., 1999) and are scattered in the cytoplasm (arrows). (B) Excessive vacuoles in fibroblasts from patients (arrows). (C) Nineteen to twenty two fields from cultures of each human subject were randomly selected under 40× magnification, and the cell number and number of cells with vacuoles were counted. Means ± SD values: 7% ± 6 vacuolated control cells; 39% ± 10 vacuolated cells from patient 1; 45% ± 22 vacuolated cells from patient 2. (D) Time lapse imaging was carried out on regions of fibroblasts containing many vacuoles (vacuolated) or lacking vacuoles (non-vacuolated). The percentage of intracellular organelles exhibiting vectorial movement was measured as described in Methods section (Schrader, 2001) (p = 0.01 for filled circles; p = 0.02 for open circles; compared with non-vacuolated area).
To investigate the nature and contents of the vacuoles, we labelled cells with antibodies against the late-endosome–lysosome membrane marker LAMP-2 and the early endosome marker EEA1. LAMP-2 was visible on the vacuolar membrane, but EEA1 did not label the vacuoles (Fig. 3C and D). These findings agree with our observations on plt mice (Chow et al., 2007), and indicate that the vacuoles are derived from dysfunctioned late-endosomes or lysosomes.
Fig. 3.

Vacuoles contain markers for late- endosomes and lysosomes. (A) Fibroblasts from human skin biopsies were cultured on glass slides. IHC was then performed on these slides. (B) Numerous vacuoles were visualized under phase contrast imaging. (C) Cells were stained with antibodies against late-endosome-lysosome marker LAMP-2. (D) Cells were stained with antibodies against early endosome marker EEA1. Vacuoles were co-localized with LAMP-2, but not EEA1. This finding is consistent with the observation in mouse fibroblasts (Chow et al., 2007) and suggests that the vacuoles are derived from dysfunctional late-endosomes or lysosomes, and not from early endosomes.
Ubiquitinated inclusions containing excessive phosphorylated neurofilaments (NFp) are a common feature in many neurodegenerative disease (Ballatore et al., 2007). Immunoreactivity against ubiquitin was widespread and evenly distributed in the cytoplasm of motor and sensory neurons in spinal cord and DRG from plt mice (Fig. 4A–D). There were no ubiquitinated inclusions within the vacuoles (Fig. 4D, inset). Antibodies against NFp strongly labelled axons but not neuronal cytoplasm or vacuoles (Fig. 4B, inset 4D) There was profound loss of large motor neurons in anterior horns of plt mice (4D), consistent with the loss of large diameter motor axons shown in Fig. 5 below.
Fig. 4.

NF and ubiquitin do not accumulate in neuronal vacuoles. Sections of mouse spinal cord and DRG were examined with IHC technique. (A–D) Immunoreactivity against ubiquitin was evenly distributed in the cytoplasm of motor and sensory neurons. Ubiquitinated inclusions were not present in vacuoles. (A–B) Antibodies against NFp produced a strong staining in the axons but not in neuronal cytoplasm. Some axons appeared irregular and enlarged (B, arrows). Large motor neurons in the anterior horn were reduced in number (5D). Inset: A spinal motor neuron at high magnification lacking NFp and ubiquitin in vacuoles (arrows).
Fig. 5.
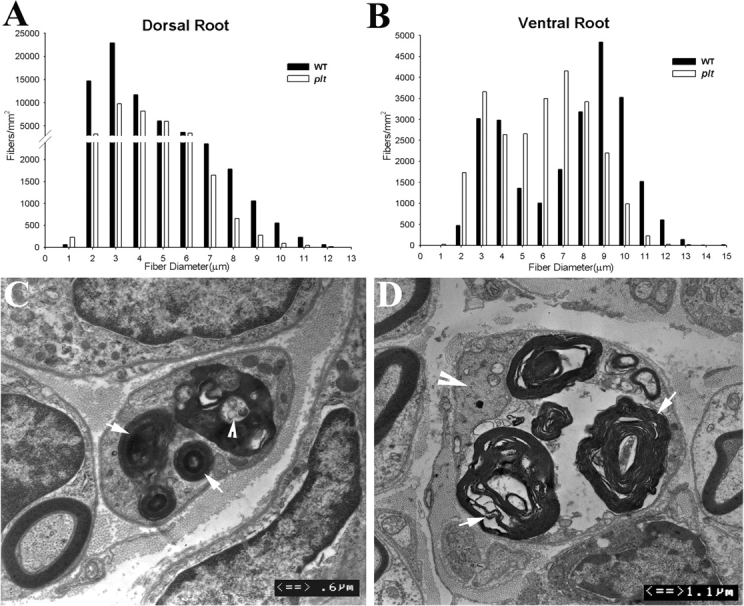
Axonal degeneration in plt mice. Morphometric analysis was performed on the semithin sections of dorsal and ventral roots of 6-week-old plt (n = 4) and wild-type (n = 4) mice (total number of counted myelinated nerve fibres = 3679 for dorsal roots and 2480 for ventral roots in 4 plt mice and 4300 for dorsal roots and 2611 for ventral roots in wild-type mice). Sensory nerve fibre loss in the dorsal roots involved both small and large diameter myelinated fibres (A). In contrast, axonal loss in the ventral roots mainly affected the large diameter fibres. Small diameter myelinated nerve fibre numbers were increased (B). We have also examined femoral, saphenous, tibial and sural nerves under light microscopy (semithin sections) and EM. Active axonal degeneration was conspicuous. Examples are shown in Figs C and D. In (C), a degenerated nerve fibre is in the centre of the picture. Myelin debris is localized in the Schwann cell cytoplasm (arrows). An axon-like residue is visible (arrowhead), presumably from a degenerated axon. A similar nerve fibre is shown in (D). Again, myelin debris is prominent (arrows in D). The axon is still preserved, but appears irregular with condensed cytoskeletons and intra-axonal organelles (arrowhead).
Mutations of VCP (valosin-containing protein), a regulator of ER protein degradation, result in neuronal vacuolation and degeneration (Ballar et al., 2006). No VCP immunoreactivity was detected in vacuoles in DRG or motor neurons for plt mice (arrows in Supplementary Fig. S1).
Vacuoles in plt mice were not stained by Filipin (Ko et al., 2001) or Nile red (Schrader, 2001), which detect cholesterol and other lipids (Supplementary Fig. S2). The failure to demonstrate accumulation of lipid or protein within the vacuoles suggests that although these vacuoles originate from late endosomes–lysosomes, they fail to incorporate materials for processing within these organelles.
The abundance of vacuoles in the cell bodies of FIG4-deficient cells suggested that they might physically disrupt trafficking of other intracellular organelles. We performed time-lapse imaging on cultured fibroblasts isolated from patient skin biopsies over 30 min and quantified movement during a 5 min period of observation (Schrader, 2001). In control fibroblasts vacuoles were rarely visible. Many organelles exhibited fast movement in long trajectories towards or away from the nucleus. In contrast, FIG4-deficient fibroblasts contain many stationary vacuoles, often clustered in the perinuclear region of the cell. Organelle movement was normal in regions of mutant cells lacking vacuoles, but was severely limited around and between vacuoles (Supplementary Video 5).
To quantify the organelle motility defect, we randomly placed three 4 × 4 µM ‘interest boxes’ in each fibroblast captured by time-lapse imaging. Intracellular organelles were manually counted and categorized by their motility features as has been previously described (Ko et al., 2001). Stationary organelles lacked any movements over a 5 min period; vectorial organelles were observed to move in long paths either toward or away from nucleus; Brownian organelles, displayed short, continuous back-and-forth movements (Ko et al., 2001). Stationary organelles accounted for ∼20% of the total organelles in control cells and in the non-vacuolated regions of mutant cells. In the areas that were heavily vacuolated, 60–80% of organelles were stationary (Supplementary Table S1). There was a decrease in organelles exhibiting vectorial movement from ∼30% in control cells to 5% in vacuolated regions of mutant cells (Fig. 2D). In the non-vacuolated areas, there was no difference in the percentages of any movements between FIG4-deficient fibroblasts and control fibroblasts (Supplementary Table S1). These findings suggest that the trafficking of intracellular organelles is blocked or prevented by the clustered vacuoles in FIG4-deficient cells (a regional impairment) since there were no trafficking abnormalities in non-vacuolated areas. Since there is extensive vacuolation in plt neurons as well (Chow et al., 2007), obstruction of intracellular organelle trafficking may contribute to neuronal degeneration in mice and CMT4J patients.
Neuronal degeneration in plt mice
plt mice exhibit severe neuronal degeneration and neuronal vacuolization in brain, spinal cord and sensory ganglia (Chow et al., 2007). The disease is progressive over a 6-week period resulting in juvenile death. To better characterize the degeneration in the PNS we performed morphometric analysis of dorsal and ventral roots. At 6 weeks of age, sensory nerve fibre loss in the dorsal roots affected both small and large diameter myelinated fibres (Fig. 5A). Axonal loss in the ventral roots mainly affected the large diameter fibres (Fig. 5B). The number of small diameter nerve fibres (≤4 μm) was increased by 25% in the ventral roots, possibly due to collateral branch regeneration. At 3 weeks of age, degenerating axons were visible in femoral (primarily motor), saphenous (primarily sensory) and tibial (mixed) nerves (Fig. 5C and D). The data demonstrate degeneration of both motor and sensory axons in plt mice.
Evaluation of apoptosis and cell proliferation in plt mice
To determine whether apoptosis contributes to neuronal loss in FIG4 deficiency, we performed TUNEL staining on sections from DRG, spinal cord, sciatic nerve and dorsal root of 6-week-old plt mice. TUNEL staining was readily detected in control tumour cells, but there was no or negligible staining in mutant tissues and no significant difference from wild-type mice (Supplementary Fig. S3). There thus was no increase in apoptosis of neurons and Schwann cells of plt mice at this age.
The same tissues were stained with antibodies to Ki67 to assess cell proliferation (Muskhelishvili et al., 2003). No significant difference was observed between wild-type and plt tissues (Supplementary Fig. S4). As a positive control, strong Ki67 immunoreactivity was detected in hair follicles of the skin biopsies, an anatomical region with abundant proliferating cells (Morris et al., 2004).
Segmental demyelination in plt mice
The reduced conduction velocity in nerves of patients (Table 2) and plt mice (Chow et al., 2007) was suggestive of segmental demyelination. Therefore, we examined teased nerve fibres from sciatic nerves of plt mice. Schwann cell-specific MAG (myelin-associated glycoprotein) was present in myelenated neurons and was strongly expressed in Schmidt-Lantermann incisures (Fig. 6A). In some segments of myelinated nerve fibres there was an absence of MAG staining, demonstrating segmental demyelination (Fig. 6B). Segmental demyelination was observed in 11% of a random selection of 100 myelinated nerve fibres from plt mice, but was not detected in wild-type fibres.
Fig. 6.

Segmental demyelination in teased sciatic nerve fibres from plt mice. Mouse sciatic nerves were teased into individual glass slides and stained with IHC techniques. (A) Myelin specific protein MAG was strongly expressed in Schmidt-Lantemann incisures. MAG staining was absent in some segments of myelinated nerve fibres (e.g. between arrowheads in A, B). Labelling for NFp was intact in the same segment (C), demonstrate segmental demyelination. Many nuclei were lined up along this segment of the axon (D) derived from invading macrophages or remyelinating Schwann cells. Demyelination was not observed in wild-type nerve fibres.
Macrophages containing debris, presumably from engulfed myelin, were visible in dorsal roots of mutant mice (arrowheads in Fig. 7B). The identity of macrophages was confirmed using CD68 antibodies (Supplementary Fig. S5). Myelinated nerve fibres from the mutant mice exhibited thin myelin, absence of myelin or basal lamina redundancy (Fig. 7C and D). Myelin debris was occasionally seen in Schwann cells (7E). Vacuoles were abundant in the perinuclear region of neuronal bodies, but were rarely observed in Schwann cells and never in axons. Excessive myelin out-folding was not observed in plt mice, in contrast to MTMR2/13 deficient patients and mice (Bolino et al., 2004; Robinson et al., 2005; Tersar et al., 2007). Taken together, the ultrastructural changes and the IHC findings in Fig. 6 demonstrate segmental demyelination in peripheral nerves. Consistent with this result, morphometric analysis of the dorsal and ventral roots revealed an increased g-ratio and decreased myelin thickness in the large diameter (≥4 µM) myelinated nerve fibres (Table 3).
Fig. 7.
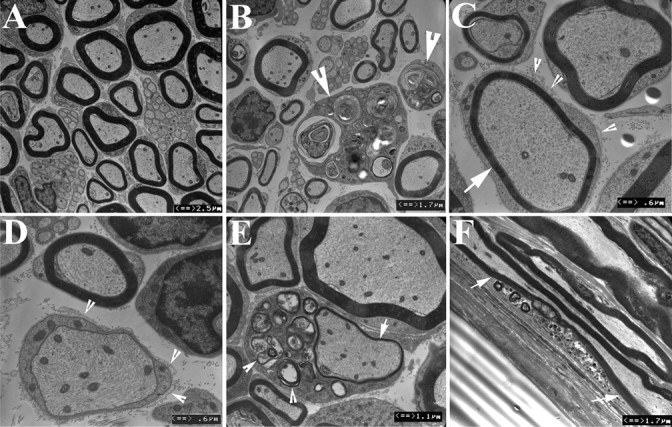
Demyelination confirmed by ultrastructure. Peripheral nerves were prepared into ultrathin sections and examined under EM. (A) Sciatic nerves from wild-type mice exhibited myelinated nerve fibres of various diameter as well as non-myelinated fibres. (B) Sciatic nerves from plt mice showed significantly reduced density of myelinated nerve fibres. Macrophages were present in dorsal roots but less frequently in other nerves (arrowheads). Macrophage cytoplasm contained debris, presumably from engulfed myelin. Nerve fibres with thin myelin (arrow in C) or absence of myelin (D) were observed. Many had basal lamina redundancy (arrowheads in C and D), a pathological change suggesting remyelination. (E) Myelin debris was occasionally observed in Schwann cell cytoplasm (arrowheads), suggesting active demyelination/remyelination. (F) Myelination debris in a segment of myelinated nerve fibre (between arrows) can also be seen in longitudinal sections of nerves. Vacuoles were restricted to neuronal soma and not present in axons or Schwann cells. The ultrastructural changes are consistent with the IHC findings in Fig. 6 supporting segmental demyelination in plt mice.
Table 3.
Morphometric analysis of myelinated nerve fibre myelin thickness in the dorsal and ventral roots of plt and wild-type mice (mean ± SD, n = 4 mice for each genotype)
| Ventral Root |
Dorsal Root |
|||||
|---|---|---|---|---|---|---|
| Diameter | g-ratio | Myelin thickness | P-value | g-ratio | Myelin thickness | P-value |
| wt < 4 μm | 0.45 ± 0.05 | 1.93 ± 0.39 | 0.068g | 0.51 ± 0.01 | 1.18 ± 0.03 | 0.058g |
| Plt < 4 μm | 0.52 ± 0.04 | 1.47 ± 0.28 | 0.104m | 0.55 ± 0.03 | 1.47 ± 0.12 | 0.049m |
| wt ≥ 4 μm | 0.53 ± 0.05 | 2.28 ± 0.32 | 0.048g | 0.59 ± 0.03 | 1.77 ± 0.19 | 0.019g |
| Plt ≥ 4 μm | 0.60 ± 0.03 | 1.60 ± 0.28 | 0.019m | 0.65 ± 0.03 | 1.31 ± 0.23 | 0.023m |
g, P-value for g-ratio comparison between wt and plt mice. G-ratio = the numerical ratio between the diameter of the axon proper and the outer diameter of the myelinated fibre; m, P-value for myelin thickness comparison between wt and plt mice.
Discussion
We describe a novel phenotype of rapidly progressive and asymmetric paralysis in two patients with mutations in FIG4. The pattern of regional progression is distinct from typical CMT, which exhibits slowly progressive, symmetric, length-dependent polyneuropathy. CMT4J more closely resembles MND, a disorder also characterized by rapidly progressive asymmetric weakness in which symptoms move from one limb to the contralateral limb before progressing to the other extremities (Caroscio et al., 1987; Brooks et al., 1995). CMT4J patients exhibit no sensory symptoms and minimal signs of sensory loss on neurological examination. The reasons for this are unclear since there is evidence of physiological and morphological sensory nerve involvement in both patients and mice. Taken together, the clinical progression and electrophysiological finding of active denervation with massive fibrillations and positive waves are consistent with an aggressive neuronopathy, as distinguished from polyneuropathy characterized by chronic, symmetric, length-dependent denervation.
The mechanism of rapid and asymmetric progression in CMT4J is not understood. In both CMT4J patients, the initially affected limb experienced minor trauma that preceded the development of marked weakness. Over several years the weakness rapidly progressed to the other limbs. Although the trauma could be coincidental in the two patients, trauma has been hypothesized to influence neurodegeneration in a number of disorders (Chio et al., 2005; Hachiya et al., 2007). Alternatively, the falls taken by both patients near the onset of progression may have resulted from the prior development of subclinical weakness. Uneven distribution of vacuolated motor neurons in the spinal cord could contribute to the asymmetric progression in our patients. Whatever the explanation, FIG4 deficiency may provide a unique opportunity to investigate the molecular mechanisms underlying asymmetric progression in MND.
Time-lapse imaging of cultured CMT4J fibroblasts suggested a potential cellular mechanism for the clinical disorder: defective trafficking of intracellular organelles due to obstruction by vacuoles. Trafficking of intracellular organelles is essential for cell survival. Maintenance of an energy supply, for example, requires mitochondrial transport within cells. Impaired trafficking could affect survival, particularly for polarized neurons with long axons (Baloh et al., 2007). Vacuoles were observed only in the neuronal cell body, and not in the axons, indicating that there is impaired organelle trafficking in the cell body. This is consistent with the fact that weakness was not length dependent in our patients. The possibility of a direct or secondary effect on axonal transport has not been investigated. Impaired organelle trafficking has been implicated in the pathogenesis of other MNDs. For example, mutations of the vesicle-associated membrane protein-associated protein-B (VAPB) gene are responsible for an inherited form of amyotrophic lateral sclerosis and spinal muscular atrophy [Nishimura et al., 2004; Marques et al., 2006]. VAPB is localized on endoplasmic reticulum and Golgi apparatus, where it regulates organelle transport and interacts with lipid-binding proteins. VAPB levels are also reduced in patients with sporadic amyotrophic lateral sclerosis (Nishimura et al., 2004; Marques et al., 2006).
Vacuole formation in FIG4 deficient cells appears to involve the endocytic pathway. Endocytic trafficking requires coordinated signalling by PI3P, which plays a crucial role in the formation of early endosomes (Volpicelli-Daley et al., 2007), and PI(3,5)P2, which may trigger budding of vesicles from late endosomes (Efe et al., 2005; Di Paolo et al., 2006; Rutherford et al., 2006). Reduced PI(3,5)P2 due to loss of function of FIG4 could inhibit budding, leading to the accumulation of large vesicles in the endocytic compartment (Marks, 2008). This model is supported by the presence of LAMP2, a marker of late endosomes and early lysosomes, on the enlarged vacuoles in cultured fibroblasts from human patients (this article) and the plt mouse (Chow et al., 2007).
It is intriguing that vacuoles are rarely observed in Schwann cells although they are abundant in neurons. Differential effects on neurons and Schwann cells have been observed for mutations of other genes in the phosphoinositide pathway. Mtmr2 encodes a phosphatase that dephosphorylates the 3-position of PI(3,5)P2. Mutations of human MTMR2 result in CMT4B1 (Bolino et al., 2000; Suter 2007). Conditional inactivation of Mtmr2 in Schwann cells in the mouse recapitulates the pathology of CMT4B1, while inactivation in motor neurons had no effect (Bolis et al., 2005). There thus appear to be differences in phosphoinositide signalling in the two cell types.
Despite their low level of vacuolization, Schwann cells are likely to be responsible for the observed segmental demyelination. However, this mild demyelination is unlikely to be a major cause of disability, since the pattern of rapid asymmetric progress is characteristic of neuronopathy. Abnormal Schwann cell-axonal interactions may also contribute to the axonal loss. Altered interactions between Schwann cells and axons occur in many demyelinating neuropathies. For instance, trembler mice with a Pmp22 missense mutation (Suter et al., 1992) have decreased NF-phosphorylation and axonal transport in addition to demyelination (de Waegh et al., 1990; de Waegh et al., 1992; Watson et al., 1994). Similarly, the myelin protein zero mutations T95M and H10P produced axonal neuropathy with minimal demyelination (De Jonghe et al., 1999; Bai et al., 2006; Li et al., 2006). Axonal degeneration in all of these models is chronic, symmetric and length dependent. None of these features are characteristic of mice or patients with FIG4 deficiency.
The pathological evidence of segmental demyelination in the two patients and in plt mice is consistent with the reduced nerve conduction velocities. The striking difference in conduction velocity between the two patients may reflect a different extent of segmental demyelination, which could result from genetic differences in modifying genes. In spite of the difference in conduction velocity, the clinical deficits in the two patients are very similar.
In contrast to the severe neuronal degeneration in the CNS of plt mice (Chow et al., 2007), we found no upper motor neuron signs in the patients with CMT4J. One patient had an unremarkable brain MRI. Detailed psychometric tests were not performed but, there was no obvious cognitive dysfunction in either patient, both of whom had successful professional careers. Thus, any CNS abnormalities were subtle. The partial function of the I41T variant of FIG4 may protect the CNS of patients from the degeneration seen in the null plt mice, and may also explain the normal pigmentation in patients with CMT4J. It is possible that differences in PI(3,5)P2 signalling between human and mouse also contribute to the phenotypic differences.
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Acknowledgements
Authors thank Dr Istvan Katonia and Ms Yanmei Yuan for technical assistance. This research is supported by grants from the NINDS (K08 NS048204), NIGMS (R01 GM24872), MDA (MDA 4029) and the Hiller ALS foundation. We would like to especially thank the family involved for their contributions to this study.
Glossary
Abbreviations:
- CMT4B1
Charcot-Marie-Tooth type 4B1
- CMT4J
Charcot-Marie-Tooth disease type-4J
- DRG
dorsal root ganglion
- EMG
electromyogram
- IHC
immunohistochemistry
- MAG
myelin-associated glycoprotein
- MND
motor neuron disease
- NCS
nerve conduction studies
- NF
neurofilament
- NFp
phosphorylated neurofilaments
- VAPB
vesicle-associated membrane protein-associated protein-B
- VCP
valosin-containing protein
References
- Bai YH, Ianokova E, Pu Q, Ghandour K, Levinson R, Martin JJ, et al. R69C Mutation in P0 Gene alters myelination and ion channel subtypes. Arch Neurol. 2006;63:1787–94. doi: 10.1001/archneur.63.12.1787. [DOI] [PubMed] [Google Scholar]
- Ballar P, Shen Y, Yang H, Fang S. The role of a novel p97/valosin-containing protein-interacting motif of gp78 in endoplasmic reticulum-associated degradation. J Biol Chem. 2006;281:35359–68. doi: 10.1074/jbc.M603355200. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–72. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci. 2007;27:422–30. doi: 10.1523/JNEUROSCI.4798-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber RP, Phelps PE, Houser CR, Crawford GD, Salvaterra PM, Vaughn JE. The morphology and distribution of neurons containing choline acetyltransferase in the adult rat spinal cord: an immunocytochemical study. J Comp Neurol. 1984;229:329–46. doi: 10.1002/cne.902290305. [DOI] [PubMed] [Google Scholar]
- Bolino A, Bolis A, Previtali SC, Dina G, Bussini S, Dati G, et al. Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J Cell Biol. 2004;167:711–21. doi: 10.1083/jcb.200407010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolino A, Muglia M, Conforti FL, LeGuern E, Salih MA, Georgiou DM, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet. 2000;25:17–9. doi: 10.1038/75542. [DOI] [PubMed] [Google Scholar]
- Bolis A, Coviello S, Bussini S, Dina G, Pardini C, Previtali SC, et al. Loss of Mtmr2 phosphatase in Schwann cells but not in motor neurons causes Charcot-Marie-Tooth type 4B1 neuropathy with myelin outfoldings. J Neurosci. 2005;25:8567–77. doi: 10.1523/JNEUROSCI.2493-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolis A, Zordan P, Coviello S, Bolino A. Myotubularin-related (MTMR) phospholipid phosphatase proteins in the peripheral nervous system. Mol Neurobiol. 2007;35:308–16. doi: 10.1007/s12035-007-0031-0. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Shodis KA, Lewis DH, Rawling JD, Sanjak M, Belden DS, et al. Natural history of amyotrophic lateral sclerosis. Quantification of symptoms, signs, strength, and function. Adv Neurol. 1995;68:163–84. [PubMed] [Google Scholar]
- Caroscio JT, Mulvihill MN, Sterling R, Abrams B. Amyotrophic lateral sclerosis. Its natural history. Neurol Clin. 1987;5:1–8. [PubMed] [Google Scholar]
- Chio A, Benzi G, Dossena M, Mutani R, Mora G. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain. 2005;128:472–6. doi: 10.1093/brain/awh373. [DOI] [PubMed] [Google Scholar]
- Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva RP, Gordon S. Phagocytosis stimulates alternative glycosylation of macrosialin (mouse CD68), a macrophage-specific endosomal protein. Biochem J. 1999;338:687–94. [PMC free article] [PubMed] [Google Scholar]
- De Jonghe P, Timmerman V, Ceuterick C, Nelis E, De Vriendt E, Löfgren A, et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain. 1999;122:281–90. doi: 10.1093/brain/122.2.281. [DOI] [PubMed] [Google Scholar]
- de Waegh S, Brady ST. Altered slow axonal transport and regeneration in a myelin-deficient mutant mouse: the trembler as an in vivo model for Schwann cell-axon interactions. J Neurosci. 1990;10:1855–65. doi: 10.1523/JNEUROSCI.10-06-01855.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–63. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–7. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Duex JE, Nau JJ, Kauffman EJ, Weisman LS. Phosphoinositide 5-phosphatase Fig 4p is required for both acute rise and subsequent fall in stress-induced phosphatidylinositol 3,5-bisphosphate levels. Eukaryot Cell. 2006a;5:723–31. doi: 10.1128/EC.5.4.723-731.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duex JE, Tang F, Weisman LS. The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J Cell Biol. 2006b;172:693–704. doi: 10.1083/jcb.200512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efe JA, Botelho RJ, Emr SD. The Fab1 phosphatidylinositol kinase pathway in the regulation of vacuole morphology. Curr Opin Cell Biol. 2005;17:402–8. doi: 10.1016/j.ceb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Eisen A. Clinical electrophysiology of the upper and lower motor neuron in amyotrophic lateral sclerosis. Semin Neurol. 2001;21:141–54. doi: 10.1055/s-2001-15261. [DOI] [PubMed] [Google Scholar]
- Fanin M, Nascimbeni AC, Fulizio L, Spinazzi M, Melacini P, Angelini C. Generalized lysosome-associated membrane protein-2 defect explains multisystem clinical involvement and allows leukocyte diagnostic screening in Danon disease. Am J Pathol. 2006;168:1309–20. doi: 10.2353/ajpath.2006.050646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Mushynski WE. Aberrant stress-induced phosphorylation of perikaryal neurofilaments. J Biol Chem. 1996;271:30404–9. doi: 10.1074/jbc.271.48.30404. [DOI] [PubMed] [Google Scholar]
- Goldstein ME, House SB, Gainer H. NF-L and peripherin immunoreactivities define distinct classes of rat sensory ganglion cells. J Neurosci Res. 1991;30:92–104. doi: 10.1002/jnr.490300111. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Martinez T, Perez-Piñera P, Díaz-Esnal B, Vega JA. S-100 proteins in the human peripheral nervous system. Microsc Res Tech. 2003;60:633–8. doi: 10.1002/jemt.10304. [DOI] [PubMed] [Google Scholar]
- Hachiya NS, Kozuka Y, Kaneko K. Mechanical stress and formation of protein aggregates in neurodegenerative disorders. Med Hypotheses. 2008;70:1034–7. doi: 10.1016/j.mehy.2007.06.043. [DOI] [PubMed] [Google Scholar]
- Ko DC, Gordon MD, Jin JY, Scott MP. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol Biol Cell. 2001;12:601–14. doi: 10.1091/mbc.12.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Bai YH, Anokova E, Grandis M, Uchwat F, Trostinskaia A, et al. Major myelin protein gene (P0) mutation causes a novel form of axonal degeneration. J Comp Neurol. 2006;498:252–65. doi: 10.1002/cne.21051. [DOI] [PubMed] [Google Scholar]
- Li J, Petajan J, Smith G, Bromberg M. Electromyography of sternocleidomastoid muscle in ALS: a prospective study. Muscle Nerve. 2002;25:725–8. doi: 10.1002/mus.10115. [DOI] [PubMed] [Google Scholar]
- Marks MS. FIG4, Charcot-Marie-Tooth disease, and hypopigmentation: a role for phosphoinositides in melanosome biogenesis? Pigment Cell Melanoma Res. 2008;21:11–4. doi: 10.1111/j.1755-148X.2007.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques VD, Barreira AA, Davis MB, Abou-Sleiman PM, Silva WA Jr, Zago MA, et al. Expanding the phenotypes of the Pro56Ser VAPB mutation: proximal SMA with dysautonomia. Muscle Nerve. 2006;34:731–9. doi: 10.1002/mus.20657. [DOI] [PubMed] [Google Scholar]
- Mills IG, Jones AT, Clague MJ. Involvement of the endosomal autoantigen EEA1 in homotypic fusion of early endosomes. Curr Biol. 1998;8:881–4. doi: 10.1016/s0960-9822(07)00351-x. [DOI] [PubMed] [Google Scholar]
- Morris RJ, Liu Y, Marles L, Yang Z, Trempus C, Li S, et al. Capturing and profiling adult hair follicle stem cells. Nat Biotechnol. 2004;22:411–7. doi: 10.1038/nbt950. [DOI] [PubMed] [Google Scholar]
- Muskhelishvili L, Latendresse JR, Kodell RL, Henderson EB. Evaluation of cell proliferation in rat tissues with BrdU, PCNA, Ki-67(MIB-5) immunohistochemistry and in situ hybridization for histone mRNA. J Histochem Cytochem. 2003;51:1681–8. doi: 10.1177/002215540305101212. [DOI] [PubMed] [Google Scholar]
- Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–31. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson FL, Dixon JE. The phosphoinositide-3-phosphatase MTMR2 associates with MTMR13, a membrane-associated pseudophosphatase also mutated in type 4B Charcot-Marie-Tooth disease. J Biol Chem. 2005;280:31699–707. doi: 10.1074/jbc.M505159200. [DOI] [PubMed] [Google Scholar]
- Rutherford AC, Traer C, Wassmer T, Pattni K, Bujny MV, Carlton JG, et al. The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci. 2006;119:3944–57. doi: 10.1242/jcs.03153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbrissa D, Ikonomov OC, Fu Z, Ijuin T, Gruenberg J, Takenawa T, et al. Core protein machinery for mammalian phosphatidylinositol 3,5-bisphosphate synthesis and turnover that regulates the progression of endosomal transport. Novel Sac phosphatase joins the ArPIKfyve-PIKfyve complex. J Biol Chem. 2007;282:23878–91. doi: 10.1074/jbc.M611678200. [DOI] [PubMed] [Google Scholar]
- Schrader M. Tubulo-reticular clusters of peroxisomes in living COS-7 cells: dynamic behavior and association with lipid droplets. J Histochem Cytochem. 2001;49:1421–9. doi: 10.1177/002215540104901110. [DOI] [PubMed] [Google Scholar]
- Suter U. Phosphoinositides and Charcot-Marie-Tooth disease: new keys to old questions. Cell Mol Life Sci. 2007;64:3261–5. doi: 10.1007/s00018-007-7381-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL, et al. A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc Natl Acad Sci USA. 1992;89:4382–6. doi: 10.1073/pnas.89.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersar K, Boentert M, Berger P, Bonneick S, Wessig C, Toyka KV, et al. Mtmr13/Sbf2-deficient mice: an animal model for CMT4B2. Hum Mol Genet. 2007;16:2991–3001. doi: 10.1093/hmg/ddm257. [DOI] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. HSV latency-associated transcript and neuronal apoptosis. Science. 2000;289:1651. [PubMed] [Google Scholar]
- Vij N, Fang S, Zeitlin PL. Selective inhibition of endoplasmic reticulum-associated degradation rescues DeltaF508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels: therapeutic implications. J Biol Chem. 2006;281:17369–78. doi: 10.1074/jbc.M600509200. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley L, De Camilli P. Phosphoinositides’ link to neurodegeneration. Nat Med. 2007;13:784–6. doi: 10.1038/nm0707-784. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Worthylake RA, Liu BP, Burridge K, Salmon ED. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol. 1999;1:45–50. doi: 10.1038/9018. [DOI] [PubMed] [Google Scholar]
- Watson DF, Nachtman FN, Kuncl RW, Griffin JW. Altered neurofilament phosphorylation and beta tubulin isotypes in Charcot-Marie-Tooth disease type 1. Neurology. 1994;44:2383–7. doi: 10.1212/wnl.44.12.2383. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Mizuno R, Nishimura T, Ogawa Y, Yoshikawa H, Fujimura H, et al. Cloning and expression of myelin-associated oligodendrocytic basic protein. A novel basic protein constituting the central nervous system myelin. J Biol Chem. 1994;269:31725–30. [PubMed] [Google Scholar]
- Yan Y, Jensen K, Brown A. The polypeptide composition of moving and stationary neurofilaments in cultured sympathetic neurons. Cell Motil Cytoskeleton. 2007;64:299–309. doi: 10.1002/cm.20184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.