

Abstract
An excess of extracellular glutamate in the hippocampus has been linked to the generation of recurrent seizures and brain pathology in patients with medically intractable mesial temporal lobe epilepsy (MTLE). However, the mechanism which results in glutamate excess in MTLE remains unknown. We recently reported that the glutamate-metabolizing enzyme glutamine synthetase is deficient in the hippocampus in patients with MTLE, and we postulated that this deficiency is critically involved in the pathophysiology of the disease. To further explore the role of glutamine synthetase in MTLE we created a novel animal model of hippocampal glutamine synthetase deficiency by continuous (∼28 days) microinfusion of methionine sulfoximine (MSO: 0.625 to 2.5 μg/h) unilaterally into the hippocampus in rats. This treatment led to a deficiency in hippocampal glutamine synthetase activity by 82–97% versus saline. The majority (>95%) of the MSO-treated animals exhibited recurrent seizures that continued for several weeks. Some of the MSO-treated animals exhibited neuropathological features that were similar to mesial temporal sclerosis, such as hippocampal atrophy and patterned loss of hippocampal neurons. However, many MSO-treated animals displayed only minimal injury to the hippocampus, with no clear evidence of mesial temporal sclerosis. These findings support the hypothesis that a deficiency in hippocampal glutamine synthetase causes recurrent seizures, even in the absence of classical mesial temporal sclerosis, and that restoration of glutamine synthetase may represent a novel approach to therapeutic intervention in this disease.
Keywords: astrocyte, epileptiform EEG discharges, animal models, glutamate, temporal lobes
Introduction
MTLE is one of the most common types of medically intractable epilepsies, and improved therapies for this disorder are requisite. However, the discovery of new therapies has been lagging, partly due to an incomplete understanding of the pathophysiology of MTLE and partly due to a lack of animal models that recapitulate key features of the disease.
Several studies have suggested a critical role for glutamate in the pathophysiology of MTLE. First, glutamate and glutamate analogues cause seizures and brain damage in laboratory animals (Olney et al., 1972; McNamara, 1994). Second, patients with MTLE have sustained elevations of extracellular glutamate in the epileptogenic hippocampus (Cavus et al., 2005), which is the site of seizure onset in this disease, and hippocampal glutamate increases markedly during seizures (During and Spencer, 1993). Moreover, the increased glutamate in MTLE is cleared very slowly from the extracellular space after the cessation of a seizure (During and Spencer, 1993). Key questions pertinent to these observations are as follows: What causes the increased concentration and slowed clearance of hippocampal glutamate in MTLE, and what role does the excess glutamate play in the development of epilepsy and triggering of seizures?
We recently reported that the enzyme glutamine synthetase is deficient in astrocytes of the epileptogenic hippocampus in patients surgically treated for MTLE (Eid et al., 2004). Glutamine synthetase is pivotal for the metabolism of the neurotransmitter glutamate, and a deficiency in this enzyme has been postulated to cause accumulation of astrocytic and extracellular glutamate, recurrent seizures and neuropathological changes typical for MTLE (Eid et al., 2004). However, because it can be argued that the downregulation of glutamine synthetase in MTLE is an epiphenomenon and not a causative factor for the disease, we tested the hypothesis that a chronic deficiency in hippocampal glutamine synthetase leads to recurrent seizures and neuropathological features typical of MTLE. Part of this work has been presented in abstract's form (Wang et al., 2006).
Materials and Methods
Chemicals and animals
All chemicals were obtained from Sigma Chemical Co. (St Louis, MO) unless otherwise noted. Male Sprague Dawley rats were used in this study (180–220 g, Charles River Laboratories, Wilmington, MA). The rats had free access to food and water and were housed on a 12 h light/dark cycle with lights on from 7 am to 7 pm. The animal care and use procedures were approved by the Institutional Animal Care and Use Committee of Yale University. All experiments were performed in accordance with current guidelines.
Surgery
The rats were anaesthetized with 2–3% Isoflurane (Baxter, Deerfield, IL) in O2 and placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). A 28 gauge stainless steel cannula attached to a plastic pedestal (Brain infusion kit II, Plastics One Inc., Roanoke, VA) was introduced through a burr hole in the skull and into the right hippocampus using the following coordinates with bregma as the reference: AP = –5.6 mm, ML = 5.3 mm, DV = –6.5 mm. The cannula was cemented to the skull using cyanoacrylate and connected via plastic tubing to a subcutaneously implanted Alzet osmotic pump (Model 2004, Durect Corp., Cupertino, CA). This pump holds a total volume of 200 μl, and delivers a continuous flow of 0.25 μl/h for ∼28 days (as per manufacturer's specifications). The pumps were filled with MSO [dissolved in 0.9% NaCl (saline)] to achieve the following drug delivery rates: 2.5, 1.25 and 0.625 µg/h. Separate pumps were filled with saline (0.9% NaCl) as controls.
In one set of animals, two unipolar stainless-steel depth electrodes (E363-1-SPC stainless steel electrode, bare diameter 0.25 mm, insulated diameter 0.28 mm, Plastics One, Roanoke, VA) were introduced into the brain parenchyma to record intrahippocampal EEG activity. The coordinates used were (AP = –3.3 mm, ML = 2.5 mm, DV = –3.9 mm, one into each hippocampus). A third depth electrode was positioned in the white matter of the cerebellum (AP = –11 mm, ML = 5.3 mm, DV = –5.6 mm) to serve as the reference. A fourth screw electrode was positioned in the occipital bone to serve as the ground. In a separate set of animals, two epidural, stainless-steel screw electrodes (Plastics One Inc., Roanoke, VA) were positioned in the skull to record cortical EEG activity. The approximate coordinates used were 4 mm lateral to the midline and 2 mm anterior to the lambda (one over each hemisphere). A third epidural screw electrode was positioned as far posterior over the skull as possible to serve as the reference. A fourth screw electrode was positioned in the occipital bone to serve as the ground. The female socket contacts on the other end of the electrodes were inserted into a plastic pedestal (Plastics One) and the entire setup was secured by acrylic adhesive (Loctite 3106 light cure adhesive, Henkel Loctite Corporation, Rocky Hill, CT). The wound was closed with surgical clips and Meloxicam was given for postoperative analgesia as follows: one dose 30 min prior to surgery (0.5 mg/kg s.c.) followed by oral administration (∼1 mg/kg/day in the drinking water) for 2 days.
Behavioural observations and video-intracranial (ic) EEG-monitoring
During the first phase of this study, the MSO and saline-infused animals were observed visually for the occurrence of behavioural seizures. The rats were monitored in real-time or by video recording for 6–12 h/day, starting on the day of surgery and continuing for a total of 1–3 weeks. The Racine criteria (Racine et al., 1973) were modified and used to identify and classify the seizures as follows: Stage 1, immobilization, eye blinking, twitching of vibrissae and mouth movements; Stage 2, head nodding, often accompanied by severe facial clonus; Stage 3, forelimb clonus; Stage 4, rearing, Stage 5, rearing, falling and generalized convulsions. However, due to difficulties in distinguishing seizures from normal activity, behaviours of stages 1 and 2 were not counted as seizure activity in the absence of confirmatory EEG.
In order to more accurately identify and characterize the seizures, a separate set of animals infused with the lowest dose of MSO (0.625 µg/h, n = 13 with depth electrodes, n = 9 with surface screw electrodes) or saline (n = 6 with depth electrodes, n = 4 with surface screw electrodes) were subjected to video and intracranial (ic)-EEG-monitoring. The experimental set-up for recording video-icEEGs was adapted from Bertram et al. (1997). The rats were placed, individually, in custom-made Plexiglas cages. A spring covered, 6-channel cable was connected to the electrode pedestal on one end and to a commutator (Plastics One) on the other. A second cable was employed to connect the commutator and the digital video and EEG recording unit (CEEGraph 4, Bio-logic Systems Corp., Mundelein, IL). Digital cameras with infrared light detection capacity were used to record animal behaviour (two cages per camera). The digital video signal is encoded and synchronized to the digital EEG signals. Seizures were detected through visual inspection of the video and the EEG record. The Teager energy of the icEEGs was calculated to aid in the review of these signals. Teager energy is a weighted measure of signal energy such that high-frequency signals contribute greater energy than low-frequency signals (E ∝ w2A2, where E is the energy, w is the frequency and A is the amplitude of the signal). Energy estimates were obtained at a 1 s resolution.
Glutamine synthetase assay
The glutamine synthetase enzyme assay method was adapted from that of Shapiro and Stadtman (1970) and Stanimirovic et al. (1999). The assay mixture contained: 80 mM imidazole–HCl buffer (pH 7.0), 30 mM glutamine, 3 mM MnCl2, 30 mM hydroxylamine–HCl (pH 7.0), 20 mM sodium arsenate (pH 7.0), 0.4 mM ADP and tissue homogenate. The assay was stopped after 30 min at 37°C by adding a stopping mixture containing 4/1/0.5/6.5 (v/v/v/v) of 10% (w/v) ferric chloride, 24% (w/v) trichloroacetic acid, 6 M HCl, and water. The colour of the reaction product, γ-glutamyl hydroxamate, was measured at 540 nm using a Bio-Tek Synergy plate-reader and converted to the amount of product formed by comparison to a standard curve. Enzymatic reactivity was expressed as unit(s) per milligram of sample protein.
Glutathione assay
Rats infused with MSO (0.625 µg/h, n = 6, 2.5 µg/h, n = 4) or saline (n = 18) were anaesthetized with Nembutal (60 mg/kg) 10 days after surgery. The animals were perfused transcardially with ice cold saline (50 ml/min for <30 s) to remove intravascular blood. The brains were rapidly removed from the skull and both hippocampi were isolated on ice, transferred to a Lysing Matrix D tube (Q-Biogene, Morgan Irvine, CA) and weighed. Ice cold 5% (w/v) metaphosphoric acid (20 ml/g tissue) was added to the tube and the tissue was homogenized using the Fast Prep system (Q-Biogene, Morgan Irvine, CA), followed by centrifugation at 3000 × g for 10 min at 4°C. The supernatant was collected and stored at –80°C until analysis. At the day of analysis, the homogenate was thawed on ice and assayed for total glutathione using the Glutathione Assay Kit (Trevigen, Gaithersburg, MD). The kit uses glutathione reductase to quantify glutathione. The sulfhydryl group of glutathione reacts colorimetrically with 5,5′-dithionbis-2-nitrobenzoic acid to produce 5-thio-2-nitrobenzoic acid (TNB). The mixed disulfide between glutathione and TNB that is concomitantly formed is reduced by glutathione reductase to recycle glutathione and produce more TNB. The intensity of the reaction product, measured spectrophotometrically at 415 nm, is directly proportional to the concentration of glutathione in the tissue and the exact concentration is determined by comparison to a standard curve.
Histology
For Nissl staining, rats were anaesthetized with pentobarbital 60 mg/kg (Nembutal, Ovation Pharmaceuticals, Deerfield, IL) and perfused transcardially with saline followed by Zamboni fixative [4% formaldehyde and 15% (v/v) saturated picric acid in 0.1 M PB, pH 7.4]. The brains were removed and post-fixed in the same fixative overnight at 4°C and then sectioned on a Vibratome at 50 μm. Every fifth section was mounted on gelatin-coated slides and stained with cresyl violet.
For silver staining, animals were anaesthetized with Nembutal (60 mg/kg) and perfused transcardially with 0.9% saline followed by 4% formaldehyde in 0.1 M PB, pH 7.4. The brains were left in situ for at least 48 h before they were removed from the skull and post-fixed for another 1–2 days in the same fixative. Eighty micrometre thick horizontal sections were cut on a Vibratome and collected in 4% formaldehyde. Every fifth section was processed for silver staining of degenerating neurons and fibres using the FD NeuroSilver Kit (FD NeuroTechnologies, Catonsville, MD). All stained sections were mounted onto gelatin-coated slides, coverslipped and examined by light microscopy.
Results
We first determined whether chronic intrahippocampal infusion of the irreversible glutamine synthetase antagonist methionine sulfoximine (MSO), results in reduced activity of glutamine synthetase in the hippocampus. The activity of glutamine synthetase was measured in the hippocampus 10 days after the onset of MSO infusion. The activity of glutamine synthetase in animals injected with either 0.625 or 2.5 µg/h MSO was reduced by 82% (n = 7, P < 0.0001, Student's t-test) or 97% (n = 5, P < 0.0001, Student's t-test), respectively, when compared to saline-injected sham-controls (Fig. 1). A smaller reduction in glutamine synthetase of 46% (n = 5, P < 0.05, Student's t-test) was observed in the contralateral hippocampus in animals injected with 2.5 µg/h MSO, whereas no change was seen in the contralateral hippocampus in animals injected with 0.625 µg/h MSO (Fig. 1). While the mechanism of the contralateral reduction in glutamine synthetase in the higher-dosed animals is unclear, diffusion of the drug via the ventricular-subarachnoid space to other brain areas, due to the higher drug concentration, is a possibility.
Fig. 1.

Intracranial infusion of methionine sulfoximine (MSO) inhibits hippocampal glutamine synthetase. MSO (0.625 μg/h, n = 7; 2.5 μg/h, n = 5), or saline (0.9% NaCl, n = 11), were continuously infused into the right hippocampus (R Hipp) of rats. The activity of glutamine synthetase in the right and left (L Hipp) hippocampus were assayed 10 days after the onset of infusion. (*P < 0.05 and **P < 0.0001, Student's t-test). Abbreviations, GS: glutamine synthetase.
Because MSO can cause depletion of the free radical scavenger glutathione (Shaw and Bains, 2002), we assayed the concentrations of hippocampal glutathione at each of the two doses of MSO given earlier (0.625 µg/h; n = 6, and 2.5 µg/h; n = 4) 10 days after the onset of MSO infusion. No decrease in hippocampal glutathione was observed in the lower-dosed animals when compared to saline infused controls (n = 6), whereas a 21% decrease (Student's t-test; P < 0.05) was observed in the higher-dosed animals (saline controls, n = 12; Fig. 2). Thus, glutathione depletion with subsequent free radical injury might exist in the animals treated with the higher dose of MSO, but is less likely in the animals treated with the lower dose of the drug.
Fig. 2.

MSO infused at 0.625 μg/h does not cause depletion of hippocampal glutathione. MSO (0.625 μg/h, n = 6; 2.5 μg/h, n = 4), or saline (0.9% NaCl, n = 18), were infused into the right hippocampus (R Hipp) of rats. Glutathione was measured in the right and left (L Hipp) hippocampus 10 days after the onset of infusion. (*P < 0.05, Student's t-test).
We next assessed whether chronic intrahippocampal infusion of MSO results in recurrent seizures (Table 1). Animals treated with increasing doses of MSO (0.625 µg/h, n = 19; 1.25 µg/h, n = 8; 2.5 µg/h, n = 28) or saline (n = 32) were monitored for behavioural seizures and classified according to established criteria (Racine et al., 1973) (see ‘Materials and methods’ section for details). Recurrent behavioural seizures [defined as ≥2 behavioural seizures of stage 3 (Racine et al., 1973) (forelimb clonus) or higher and separated at least 1 h apart] occurred with all doses of MSO and in 54/55 (98%) of the animals (Table 1). None of the saline-treated animals exhibited recurrent seizures.
Table 1.
The distribution of animals exhibiting recurrent seizures and different grades of hippocampal pathologies after continuous intrahippocampal infusion with saline (control) and increasing doses of MSO
| Dose | Animals with recurrent seizures |
Histopathology |
||||
|---|---|---|---|---|---|---|
| Video only | Continuous intracranial video-EEG | Grade I (‘minimal’) | Grade II (‘moderate’) | Grade III (‘necrosis’) | Grade IV (‘sclerosis’) | |
| NaCl 0.9% | 0/32 | 0/10 | 10/10 | 0/10 | 0/10 | 0/10 |
| MSO 0.625 µg/h | 19/19 | 22/22 | 9/22 | 4/22 | 6/22 | 3/22 |
| MSO 1.25 µg/h | 8/8 | 1/8 | 1/8 | 6/8 | 0/8 | |
| MSO 2.5 µg/h | 27/28* | 0/8 | 0/8 | 5/8 | 3/8 | |
None of the saline-treated animals exhibited recurrent seizures or more than minimal neuronal loss in the hippocampus. *All but one of the MSO-treated rats exhibited recurrent seizures. The single exception amongst the MSO-treated animals was observed by video only for 6–12 hrs/day for a maximum of 7 days. It is possible that the presence of recurrent seizures may have been missed in this animal due to the use of intermittent video observation only, which is less sensitive than continuous video-EEG. The histopathology was assessed by light microscopy using Nissl- and silver-stained sections and each animal was scored and classified into one of the following grades. I: Minimal neuronal loss (<100 μm diameter) around the infusion cannula track by Nissl stain; no patterned involvement of other hippocampal subfields by silver or Nissl stain (e.g. Fig. 6A and D). II: More extensive (‘moderate’) neuronal loss (>100 μm diameter) around the infusion cannula track by Nissl stain, often spreading into nearby hippocampal and cortical subfields. Necrosis is not present. III: Large necrotic lesions involving and usually extending beyond the hippocampus (Fig. 6C). IV: Hippocampal sclerosis-like picture as evident by patterned neuronal injury (silver stain, e.g. Fig. 7H, J) or neuronal loss (Nissl stain, e.g. Fig. 7B, D and F) in selected hippocampal subfields.
To further characterize the seizures, separate groups of animals infused with the lowest dose of MSO (0.625 µg/h; n = 22) or saline (n = 10) were subjected to periods of continuous video and intracranial (ic)-EEG-monitoring (Table 1, Figs 3–5). One set of animals was monitored by depth electrodes in both hippocampi (MSO, n = 13; saline, n = 6) beginning immediately after surgery and lasting for 8–27 days (Fig. 4). Another set of animals were monitored by epidural screw electrodes (MSO, n = 9; saline, n = 4) for one or more continuous periods, extending as far as 83 days after surgery (Fig. 5). Epidural recordings were chosen to detect seizures that had spread beyond the hippocampus. Moreover, because screw electrodes attach more securely to the skull than depth electrodes, the use of screws allowed us to perform longer recordings with lesser risk of electrode failure. The lowest dose of MSO was chosen to avoid the potential confounding effects of glutathione depletion and contralateral glutamine synthetase inhibition seen with the higher dose of the drug (Figs 1 and 2).
Fig. 3.

MSO precipitates recurrent seizures. (A) Continuous three-day-record (days 24–26 after surgery) of Teager energy (see ‘Methods’ section for details) of a single ic (intracranial) EEG time-series from a representative animal infused continuously with intrahippocampal MSO (0.625 μg/h). Energy estimates were obtained at 1 s resolution and averaged over a 5 s running window (x-axis). A 24-h-period (midnight to midnight) is displayed in each trace. Two seizures were recorded during the 72-h-period. As in human epilepsy, seizures in the MSO model are readily identified by the difference in energy (y-axis) from background icEEG activity. (B) Continuous 3.5 min icEEG (from day 24: 18: 11: 50 to 18: 15: 20) displaying the first seizure identified in A.
Fig. 4.

Continuous intrahippocampal EEG recordings of MSO-treated rats—an overview. Thirteen animals were treated with intrahippocampal MSO (0.625 μg/h) and subjected to continuous video-intracranial (ic)EEG monitoring for several days (d), beginning immediately after surgical implantation of the MSO-infusion system. Unipolar depth electrodes were placed into each hippocampus for EEG recordings using an electrode in the cerebellar white matter as the reference. The duration of continuous recordings per animal in days (d), total number of seizures per animal (sz), and mean number of seizures per animal per day (/d) are indicated. The mean numbers of these parameters for the entire group are given below the dashed line. Error bar: standard deviation.
Fig. 5.

Epidural (neocortical) EEG recordings of MSO-treated rats—an overview. Nine animals were treated with intrahippocampal MSO (0.625 μg/h) and subjected to discrete periods of continuous video-intracranial (ic)EEG monitoring for up to 82 days after surgical implantation of the MSO-infusion system. Epidural screw electrodes were placed over the dorsal neocortex of each hemisphere for EEG recordings. The duration of continuous recordings per animal in days (d) and mean number of seizures per animal per day (/d) are indicated.
Electrographic seizures occurred in all (22/22) of the MSO-treated (Figs 3–5) and in none (0/10) of the saline-treated animals. The first evidence of electrographic seizures by depth electrode recordings occurred in the MSO-treated animals at 4.4 ± 2 h (mean ± SD) after implantation of the MSO-infusion pump. The seizures in both the depth and epidural screw electrode recorded animals recurred for several weeks after surgery (Figs 4 and 5), and there was a tendency for the seizures to occur in clusters throughout the monitoring period (not shown). There was also a trend towards a reduction in seizure frequency over time as the two animals with the longest recordings (epidural recordings of #16 and 18; Fig 5) completely stopped seizing at approximately 45–55 days after implantation of the MSO-infusion pump. The average number of seizures per day was 5.3 ± 4.2 (mean ± SD) in the depth electrode recorded group (Fig. 4), whereas the same number for the epidural screw electrode group was 1.0 ± 0.9 [mean ± SD; after excluding the two periods of no seizures in animals #16 and 18 (Fig. 5)]. The electrographic seizures in both groups were characterized by a wide range of behaviours such as wet-dog shakes and immobilization, chewing and whisker twitching, head nodding, forelimb clonus, rearing, and rearing and falling.
One of the hallmarks of MTLE is the presence of mesial temporal sclerosis, which is a collective term for several distinct pathologies affecting the limbic structures of the brain [reviewed in (Gloor, 1991)]. One of these pathologies is hippocampal sclerosis, which is characterized by atrophy and loss of neurons in CA1, CA3, and the dentate hilus of the hippocampus. The subiculum, CA2, and granule cell layer of the dentate gyrus are relatively spared (Sommer, 1880). Some patients with mesial temporal sclerosis show more extensive lesions of the hippocampus, also involving CA2 and the dentate granule cell layer (Babb et al., 1984a; Kim et al., 1990), whereas others reveal very restricted lesions, involving CA1 only (de Lanerolle et al., 2003), or the hilus (end-folium) of the dentate gyrus (Margerison and Corsellis, 1966). Neuronal loss has also been reported in other limbic regions such as the entorhinal cortex (Du et al., 1993) and amygdala (Gloor, 1992).
To explore the association between hippocampal glutamine synthetase deficiency and neuronal loss, we analysed MSO- and saline-infused rats histologically, using Nissl-staining and a highly sensitive silver impregnation technique for injured neurons (Gallyas et al., 1980). Intrahippocampal infusion of MSO was associated with a dose-dependent loss of neurons in the hippocampal formation and nearby brain areas (Table 1, Fig. 6). The majority of animals treated with the highest dose of MSO (2.5 µg/h) developed large necrotic lesions around the infusion site, typically involving the entire hippocampus and nearby cortical areas (Fig. 6C). The lowest dose of MSO (0.625 µg/h) resulted in much smaller lesions; in fact, many animals exhibited only minimal neuronal loss (i.e. an area of neuronal loss <100 µm diameter) that in Nissl-stained sections were present around the track of the drug infusion cannula (Fig. 6B and E). The animals treated with 1.25 µg/h of MSO exhibited a loss of neurons intermediate between the highest and lowest dose of MSO (not shown).
Fig. 6.
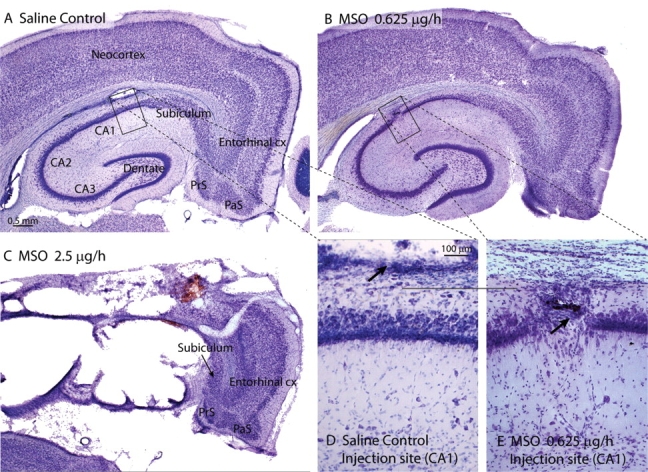
MSO is associated with dose-dependent neuronal loss in the hippocampus and nearby structures. (A–E) Nissl-stains of the hippocampal formation, entorhinal cortex and nearby neocortex of representative rats infused continuously into the hippocampus with 0.9% NaCl (A, D), 0.625 µg/h MSO (B, E) and 2.5 µg/h MSO (C). No significant neuronal loss is seen in the rat treated with 0.9% NaCl (A), and there is only a small vacuolar lesion at the site of the infusion cannula (arrow in D). There is only minimal neuronal loss in CA1 of the rat treated with the lowest dose (0.625 µg/h) of MSO (B, E), and the damage is restricted to the track of the infusion cannula (arrow in E). The highest dose of MSO (C, 2.5 µg/h) is associated with extensive necrosis of the hippocampus and adjacent neocortex, whereas the entorhinal cortex, pre- (PrS), parasubiculum (PaS), and part of the subiculum are relatively spared in this animal. All MSO-treated rats depicted here exhibited recurrent seizures. Animals were euthanized 2 weeks after onset of saline or MSO infusion. Abbreviations: cx, cortex; CA1–3, hippocampal subfields cornu ammonis 1–3; PaS, parasubiculum; PrS, presubiculum. Magnification: A = B = C; D = E.
Interestingly, some animals developed lesions that were reminiscent of hippocampal sclerosis (Fig. 7). Shrinkage of the hippocampus along with loss of neurons in CA1-CA3 and the dentate hilus were observed in a small subset of animals in Nissl-stained sections (Fig. 7B, D and F). In these animals neurons in the subiculum and the dentate granule cell layer were typically spared (Fig. 7B, D and F). Even if minimal neuronal loss was present by Nissl-staining in many animals treated with MSO, the more sensitive silver-impregnation technique often revealed injury to subpopulations of neurons distant from the infusion site, such as the CA1 pyramidal cells (Fig. 7H and J), which are known to be exquisitely sensitive to seizures (Sommer, 1880; Meldrum, 1986) and hypoxic-ischaemic insults (Pulsinelli et al., 1982; Petito et al., 1987). Silver-stained neurons were not observed in the saline-treated animals (Fig. 7G and I).
Fig. 7.
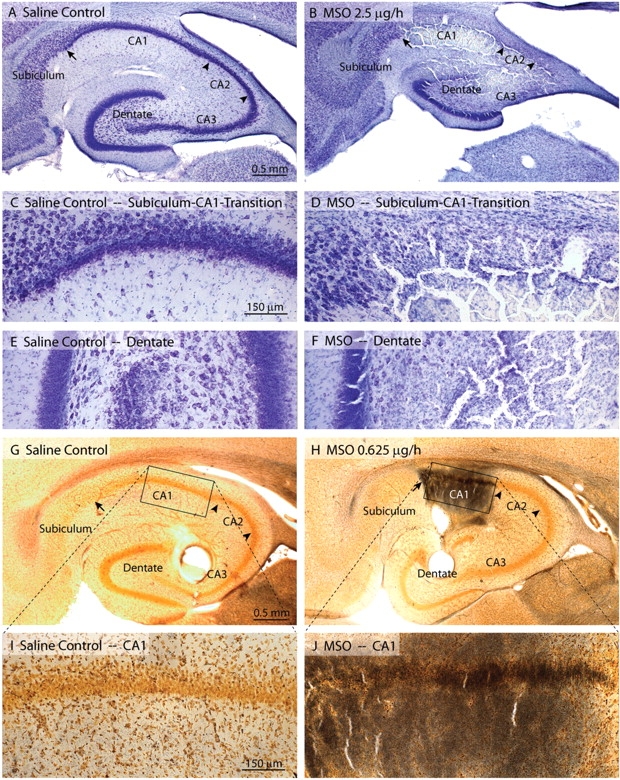
MSO is in a subpopulation of animals associated with neuropathological features that are reminiscent of hippocampal sclerosis. (A–F) Nissl stains of the hippocampal formation of selected animals infused with intrahippocampal saline (left column: A, C, E) or MSO (2.5 μg/h; right column: B, D, F). There is hippocampal atrophy (B) and extensive loss of neurons in the CA-fields (B, D) of the MSO-treated rat. Subiculum (B, D) and portions of the dentate granule cell layer (B, F) are spared in the MSO-treated animal. (G–J) A sensitive silver staining technique for degenerating neurons and fibre tracts reveals numerous stained neurons and fibres preferentially in area CA1 of the hippocampus in a rat infused with a lower dose of MSO (0.625 μg/h, right column: H, J). A saline-infused control rat (left column: G, I) is unstained. The MSO-treated rats depicted here exhibited recurrent seizures. Animals were euthanized 2 weeks after onset of saline or MSO infusion. Magnification: A = B; C = D = E = F; G = H; I = J.
Discussion
We used, for the first time, continuous intrahippocampal infusion of MSO to suppress the activity of glutamine synthetase locally in the brain in laboratory rats. MSO (in the presence of ATP) is phosphorylated by glutamine synthetase, resulting in an irreversible inhibition of the enzyme (Ronzio and Meister, 1968). MSO has other biological effects, one of the most notable being its ability to inhibit gamma-glutamylcysteine synthetase, thus reducing the cellular levels of glutathione (Shaw and Bains, 2002). Importantly, we did not observe an effect on glutathione at the lowest dose of MSO (0.625 µg/h).
MSO has been used extensively in prior studies of glutamine synthetase. For example, in vitro experiments with organotypical hippocampal cultures have shown that inhibition of glutamine synthetase by MSO causes depletion of glutamine and accumulation of glutamate in astrocytes, as well as glutamate-induced excitotoxicity (Laake et al., 1995). MSO also precipitates an episode of acute (i.e. non-recurrent) convulsive seizures when given to rodents as a single systemic injection (Folbergrova et al., 1969; Szegedy, 1978); however, the seizures are often very severe, with only a small proportion of animals surviving the initial convulsive episode (Subbalakshmi and Murthy, 1981; Bernard-Helary et al., 2000). Because human MTLE is characterized by a highly localized and chronic deficiency in glutamine synthetase (Eid et al., 2004), a single systemic dose of MSO would not reproduce these features of the disease. Moreover, patients with MTLE exhibit recurrent seizures that are frequently non-convulsive and rarely lethal. Thus, the novel approach of continuous intrahippocampal infusion of MSO used here is most reminiscent of human MTLE.
How does intrahippocampal MSO cause seizures? Glutamine synthetase is critical for the conversion of glutamate to glutamine, and its deficiency has been postulated to cause an abnormal increase in astrocytic and extracellular glutamate (Laake et al., 1995; Eid et al., 2004). This postulate is based on the known stoichiometry of glutamate transport across the glial plasma membrane. The stoichiometry dictates that rapid metabolism of intracellular glutamate is a prerequisite for efficient glutamate clearance from the extracellular space (Otis and Jahr, 1998). Glutamate is a potent excitotoxin, and excessive concentrations of extracellular glutamate can cause seizures and brain damage in laboratory animals (Olney et al., 1972, 1986). Thus, the hypothesis is that intrahippocampal MSO leads to inhibition of glutamine synthetase, accumulation of extracellular glutamate and seizures. However, this scenario has not been established experimentally, and further studies are required to address this issue. Another possible explanation is that low levels of glutamine synthetase may cause loss of inhibition by neurons due to impaired vesicular release of GABA (Liang et al., 2006). After blocking glutamine synthetase by MSO in slice preparations of the hippocampus, Liang and colleagues found a reduction in evoked IPSC (inhibitory post-synaptic currents) in area CA1. Thus, a disruption in the glutamate–glutamine cycle by MSO may not only increase the extracellular glutamate but also diminish the vesicular release of GABA.
The trend towards a reduction in seizure frequency over time in the MSO-treated animals is interesting and raises two pertinent questions. First, the infusion pump is manufactured to deliver MSO for ∼28 days. At 45–55 days (the time of zero seizures in two of the MSO-infused animals) the pump should no longer deliver MSO, and the activity of hippocampal glutamine synthetase is presumably normal, suggesting that recovery of glutamine synthetase leads to cessation of seizures. Thus, can recovery (or re-expression) of hippocampal glutamine synthetase be used therapeutically to prevent seizures in patients with established epilepsy? Secondly, the two MSO-treated animals which exhibited long-term seizure cessation were recorded with epidural screw electrodes. Intracranial depth electrodes may be more sensitive than epidural electrodes for detection of focal hippocampal seizures. Thus, is it possible that the two MSO-treated animals had highly focal and/or subclinical seizures which were not detected by the epidural electrodes? Further investigations are required to properly address these questions.
The deficiency in glutamine synthetase caused by intrahippocampal MSO infusion is associated, in some animals, with neuropathological changes similar to mesial temporal sclerosis, such as unilateral atrophy of the hippocampus with injury and/or loss of neurons in vulnerable areas of the hippocampus such as CA1. Two questions are of particular relevance to this pathology. First, what is the mechanism of the hippocampal injury, and second, is mesial temporal sclerosis a key factor in the pathophysiology of MTLE? One of the possible mechanisms of hippocampal injury after MSO infusion is that chronic high levels of extracellular glutamate in the hippocampus lead to excitotoxic damage of vulnerable neuronal populations. This concept is supported by animal studies in which administration of glutamate analogues results in loss of neurons, whereas co-injection of glutamate-receptor antagonists prevents the neuronal damage (Clifford et al., 1989; Turski et al., 1990). Another possible scenario is that accumulation of reactive oxygen species may be responsible for a large portion of the hippocampal damage [reviewed in (Patel, 2002)]. This is because the highest dose of MSO (2.5 µg/h) resulted in depletion of glutathione and the most extensive damage to the brain.
Two observations suggest that the sclerotic mesial temporal lobe is important in the pathophysiology of MTLE. First, depth electrode recordings from patients with MTLE indicate that the seizures originate from the sclerotic mesial temporal lobe, particularly the hippocampus (Babb et al., 1984b; Mathern et al., 1997). Second, surgical removal of the sclerotic hippocampus is associated with an excellent clinical outcome, resulting in cessation of the seizures in about 85% of patients (Engel Class I) (de Lanerolle et al., 2003). The sclerotic mesial temporal lobe is characterized by a large number of structural, molecular and chemical alterations other than neuronal loss [reviewed in (de Lanerolle and Lee, 2005; Binder and Steinhauser, 2006)], and a major challenge has been to identify which of these changes are pivotal for the generation of recurrent seizures. Even though many of these alterations can be reproduced throughout the brain by chemical alteration or transgenic approaches, it has been very difficult to restrict the pathology to the mesial temporal lobe. For example, systemic administration of kainic acid or pilocarpine causes a sclerosis-like pathology in the hippocampus; however, there is widespread loss of neurons in many other brain areas after such treatments (Olney, 1978). Transgenic animals with alterations in dystrophin (Bulfield et al., 1984) and aquaporin 4 (Ma et al., 1997; Adams et al., 2000; Amiry-Moghaddam et al., 2003) are indeed available; however, the alterations in these animals are not restricted to the mesial temporal lobe, whereas in human MTLE they appear to be (Eid et al., 2005).
In summary, we were able to successfully inhibit glutamine synthetase in the hippocampus in laboratory rats. A deficiency in this enzyme in the hippocampus is one of the defining features of mesial temporal sclerosis and human MTLE (Eid et al., 2004); thus, our approach of continuous intrahippocampal MSO infusion appears to replicate this feature very well. We have shown that inhibition of hippocampal glutamine synthetase leads to recurrent seizures, which is one of the hallmarks of epilepsy. Furthermore, only a minority of animals develops a pattern of neuronal injury and/or loss that is reminiscent of mesial temporal sclerosis. Thus, our data strongly suggest that a deficiency in hippocampal glutamine synthetase is critically important for the generation of recurrent seizures in MTLE. The observation that recurrent seizures occurs independently of extensive neuronal loss also raises the possibility that a deficiency in glutamine synthetase may be more important in the pathogenesis of MTLE than the patterned loss of hippocampal neurons characteristic of mesial temporal sclerosis.
Acknowledgements
We thank Ms Ilona Kovacs and Ms Maria B. Lai for excellent technical assistance and Ms Anne Sommer for editorial advice. This study was supported by a grant from the National Institutes of Health (NIH/NINDS, NS054801 to T.E.).
Glossary
Abbreviations:
- CA1-3
cornu ammonis subfields 1–3 of the hippocampus
- MSO
methionine sulfoximine
- MTLE
mesial temporal lobe epilepsy
References
- Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, et al. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol. 2000;150:1385–98. doi: 10.1083/jcb.150.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, et al. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci USA. 2003;100:13615–20. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babb TL, Brown WJ, Pretorius J, Davenport C, Lieb JP, Crandall PH. Temporal lobe volumetric cell densities in temporal lobe epilepsy. Epilepsia. 1984a;25:729–40. doi: 10.1111/j.1528-1157.1984.tb03484.x. [DOI] [PubMed] [Google Scholar]
- Babb TL, Lieb JP, Brown WJ, Pretorius J, Crandall PH. Distribution of pyramidal cell density and hyperexcitability in the epileptic human hippocampal formation. Epilepsia. 1984b;25:721–8. doi: 10.1111/j.1528-1157.1984.tb03483.x. [DOI] [PubMed] [Google Scholar]
- Bernard-Helary K, Lapouble E, Ardourel M, Hevor T, Cloix JF. Correlation between brain glycogen and convulsive state in mice submitted to methionine sulfoximine. Life Sci. 2000;67:1773–81. doi: 10.1016/s0024-3205(00)00756-6. [DOI] [PubMed] [Google Scholar]
- Bertram EH, Williamson JM, Cornett JF, Chen ZF. Design and construction of a long-term continuous video-EEG monitoring unit for simultaneous recording of multiple small animals. Brain Res Brain Res Protoc. 1997;1997:85–97. doi: 10.1016/s1385-299x(97)00033-0. [DOI] [PubMed] [Google Scholar]
- Binder DK, Steinhauser C. Functional changes in astroglial cells in epilepsy. Glia. 2006;54:358–68. doi: 10.1002/glia.20394. [DOI] [PubMed] [Google Scholar]
- Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–92. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. 2005;57:226–35. doi: 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- Clifford DB, Zorumski CF, Olney JW. Ketamine and MK-801 prevent degeneration of thalamic neurons induced by focal cortical seizures. Exp Neurol. 1989;105:272–9. doi: 10.1016/0014-4886(89)90130-1. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Williamson A, Spencer SS, Zaveri HP, Eid T, et al. A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: evidence for distinctive patient subcategories. Epilepsia. 2003;44:677–87. doi: 10.1046/j.1528-1157.2003.32701.x. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Lee TS. New facets of the neuropathology and molecular profile of human temporal lobe epilepsy. Epilepsy Behav. 2005;7:190–203. doi: 10.1016/j.yebeh.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Du F, Whetsell WO, Jr, Abou-Khalil B, Blumenkopf B, Lothman EW, Schwarcz R. Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Res. 1993;16:223–33. doi: 10.1016/0920-1211(93)90083-j. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341:1607–10. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjornsen LP, Spencer DD, et al. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci USA. 2005;102:1193–8. doi: 10.1073/pnas.0409308102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet. 2004;363:28–37. doi: 10.1016/s0140-6736(03)15166-5. [DOI] [PubMed] [Google Scholar]
- Folbergrova J, Passonneau JV, Lowry OH, Schulz DW. Glycogen, ammonia and related metabolities in the brain during seizures evoked by methionine sulphoximine. J Neurochem. 1969;16:191–203. doi: 10.1111/j.1471-4159.1969.tb05937.x. [DOI] [PubMed] [Google Scholar]
- Gallyas F, Wolff JR, Bottcher H, Zaborsky L. A reliable and sensitive method to localize terminal degeneration and lysosomes in the central nervous system. Stain Technol. 1980;55:299–306. doi: 10.3109/10520298009067258. [DOI] [PubMed] [Google Scholar]
- Gloor P. Mesial temporal sclerosis: historical background and an overview from a modern perspective. In: Luders H, editor. Epilepsy surgery. New York: Raven Press; 1991. pp. 6889–703. [Google Scholar]
- Gloor P. The role of the amygdala in temporal lobe epilepsy. In: Aggleton JP, editor. The amygdala. Neurobiological aspects of emotion, memory, and mental dysfunction. New York: Wiley-Liss; 1992. pp. 505–38. [Google Scholar]
- Kim JH, Guimaraes PO, Shen M-Y, Masukawa LM, Spencer DD. Hippocampal neuronal density in temporal lobe epilepsy with and without gliomas. Acta Neuropathol. 1990;80:41–5. doi: 10.1007/BF00294220. [DOI] [PubMed] [Google Scholar]
- Laake JH, Slyngstad TA, Haug FM, Ottersen OP. Glutamine from glial cells is essential for the maintenance of the nerve terminal pool of glutamate: immunogold evidence from hippocampal slice cultures. J Neurochem. 1995;65:871–81. doi: 10.1046/j.1471-4159.1995.65020871.x. [DOI] [PubMed] [Google Scholar]
- Liang SL, Carlson GC, Coulter DA. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J Neurosci. 2006;26:8537–48. doi: 10.1523/JNEUROSCI.0329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest. 1997;100:957–62. doi: 10.1172/JCI231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margerison JH, Corsellis JAN. Epilepsy and the temporal lobes. Brain. 1966;89:499–530. doi: 10.1093/brain/89.3.499. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Armstrong DL. Hippocampal sclerosis. In: Engel J, Pedley TA, editors. Epilepsy: a comprehensive textbook. Philadelphia: Lippincott-Raven; 1997. pp. 133–55. [Google Scholar]
- McNamara JO. Cellular and molecular basis of epilepsy. J Neurosci. 1994;14:3413–25. doi: 10.1523/JNEUROSCI.14-06-03413.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum BS. Cell damage in epilepsy and the role of calcium in cytotoxicity. Adv Neurol. 1986;44:849–55. [PubMed] [Google Scholar]
- Olney JW. Neurotoxicity of excitatory amino acids. In: McGeer EG, Olney JW, McGeer PL, editors. Kainic acid as a tool in neurobiology. New York: Raven Press; 1978. pp. 37–70. [Google Scholar]
- Olney JW, Collins RC, Sloviter RS. Excitotoxic mechanims of epileptic brain damage. In: Delgado-Escueta AV, Ward AA Jr, Woodbury DM, Porter RJ, editors. Advances in neurology. New York: Raven Press; 1986. pp. 857–77. [PubMed] [Google Scholar]
- Olney JW, Sharpe LG, Feigin RD. Glutamate-induced brain damage in infant primates. J Neuropathol Exp Neurol. 1972;31:464–88. doi: 10.1097/00005072-197207000-00006. [DOI] [PubMed] [Google Scholar]
- Otis TS, Jahr CE. Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J Neurosci. 1998;18:7099–110. doi: 10.1523/JNEUROSCI.18-18-07099.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MN. Oxidative stress, mitochondrial dysfunction, and epilepsy. Free Radic Res. 2002;36:1139–46. doi: 10.1080/1071576021000016391. [DOI] [PubMed] [Google Scholar]
- Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–6. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–8. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Racine RJ, Burnham WM, Gartner JG, Levitan D. Rates of motor seizure development in rats subjected to electrical brain stimulation: strain and inter-stimulation interval effects. Electroencephalogr Clin Neurophysiol. 1973;35:553–6. doi: 10.1016/0013-4694(73)90033-3. [DOI] [PubMed] [Google Scholar]
- Ronzio RA, Meister A. Phosphorylation of methionine sulfoximine by glutamine synthetase. Proc Natl Acad Sci USA. 1968;59:164–70. doi: 10.1073/pnas.59.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro BM, Stadtman ER. The regulation of glutamine synthesis in microorganisms. Annu Rev Microbiol. 1970;24:501–24. doi: 10.1146/annurev.mi.24.100170.002441. [DOI] [PubMed] [Google Scholar]
- Shaw CA, Bains JS. Synergistic versus antagonistic actions of glutamate and glutathione: the role of excitotoxicity and oxidative stress in neuronal disease. Cell Mol Biol (Noisy-le-grand) 2002;48:127–36. [PubMed] [Google Scholar]
- Sommer W. Erkrankung des Ammonshorns als aetiologisches Moment der Epilepsie. Arch Psychiatr Nervenkr. 1880;10:631–75. [Google Scholar]
- Stanimirovic DB, Ball R, Small DL, Muruganandam A. Developmental regulation of glutamate transporters and glutamine synthetase activity in astrocyte cultures differentiated in vitro. Int J Dev Neurosci. 1999;17:173–84. doi: 10.1016/s0736-5748(99)00028-3. [DOI] [PubMed] [Google Scholar]
- Subbalakshmi GY, Murthy CR. Effects of methionine sulfoximine on cerebral ATPase. Biochem Pharmacol. 1981;30:2127–30. doi: 10.1016/0006-2952(81)90232-x. [DOI] [PubMed] [Google Scholar]
- Szegedy L. Enzyme histochemical studies in the rat hippocampus, cerebellar cortex and brainstem motor nuclei during methionine sulphoximine convulsions. Cell Mol Biol. 1978;23:43–53. [PubMed] [Google Scholar]
- Turski L, Niemann W, Stephens DN. Differential effects of antiepileptic drugs and beta-carbolines on seizures induced by excitatory amino acids. Neuroscience. 1990;39:799–807. doi: 10.1016/0306-4522(90)90262-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zaveri HP, Ghosh A, Beckstrom H, De Lanerolle NC, Eid T. Inhibition of hippocampal glutamine synthetase causes spontaneously recurrent seizures in rats. Epilepsia. 2006;47:328–329. [Google Scholar]