

Abstract
Dominant intermediate Charcot-Marie-Tooth neuropathy type B is caused by mutations in dynamin 2. We studied the clinical, haematological, electrophysiological and sural nerve biopsy findings in 34 patients belonging to six unrelated dominant intermediate Charcot-Marie-Tooth neuropathy type B families in whom a dynamin 2 mutation had been identified: Gly358Arg (Spain); Asp551_Glu553del; Lys550fs (North America); Lys558del (Belgium); Lys558Glu (Australia, the Netherlands) and Thr855_Ile856del (Belgium). The Gly358Arg and Thr855_Ile856del mutations were novel, and in contrast to the other Charcot-Marie-Tooth-related mutations in dynamin 2, which are all located in the pleckstrin homology domain, they were situated in the middle domain and proline-rich domain of dynamin 2, respectively. We report the first disease-causing mutation in the proline-rich domain of dynamin 2. Patients with a dynamin 2 mutation presented with a classical Charcot-Marie-Tooth phenotype, which was mild to moderately severe since only 3% of the patients were wheelchair-bound. The mean age at onset was 16 years with a large variability ranging from 2 to 50 years. Interestingly, in the Australian and Belgian families, which carry two different mutations affecting the same amino acid (Lys558), Charcot-Marie-Tooth cosegregated with neutropaenia. In addition, early onset cataracts were observed in one of the Charcot-Marie-Tooth families. Our electrophysiological data indicate intermediate or axonal motor median nerve conduction velocities (NCV) ranging from 26 m/s to normal values in four families, and less pronounced reduction of motor median NCV (41–46 m/s) with normal amplitudes in two families. Sural nerve biopsy in a Dutch patient with Lys558Glu mutation showed diffuse loss of large myelinated fibres, presence of many clusters of regenerating myelinated axons and fibres with focal myelin thickenings—findings very similar to those previously reported in the Australian family. We conclude that dynamin 2 mutations should be screened in the autosomal dominant Charcot-Marie-Tooth neuropathy families with intermediate or axonal NCV, and in patients with a classical mild to moderately severe Charcot-Marie-Tooth phenotype, especially when Charcot-Marie-Tooth is associated with neutropaenia or cataracts.
Keywords: intermediate CMT, dynamin 2, neutropaenia, hereditary neuropathy, cataracts
Introduction
The hereditary motor and sensory neuropathies (HMSN), also called Charcot-Marie-Tooth (CMT) disease, are a clinically and genetically heterogeneous group of disorders of the peripheral nervous system. CMT is characterized by slowly progressive weakness and atrophy, initially obvious in the muscles of the anterior and lateral compartments of the legs, and in a later stage also affecting the intrinsic hand muscles. Sensory disturbances involving the distal parts of the limbs are often present. Deep tendon reflexes are decreased or absent. Skeletal malformations including pes cavus, hammer-toes and scoliosis are frequently observed (Harding and Thomas, 1980).
Based on the findings of Dyck and co-workers (1993) that motor nerve conduction velocities (NCV) for the median nerve in a large cohort of unrelated CMT patients showed a bimodal distribution, hereditary motor and sensory neuropathies have been divided into two main subtypes: HMSN I (or CMT1) and HMSN II (or CMT2). The first is a primarily demyelinating neuropathy with median motor NCV slowing below 38 m/s, and the latter is a primarily axonal neuropathy characterized by normal or slightly reduced NCV and usually decreased compound muscle action potential (CMAP) amplitudes. Most CMT patients can be classified as either CMT1 or CMT2 using the motor NCV cut-off value of 38 m/s for the upper limbs (Dyck et al., 1993). However, in some CMT families, patients have been difficult to classify using these strict electrophysiological criteria, leading to the concept of intermediate CMT with median motor NCV between 25 and 45 m/s (Davis et al., 1978; Nicholson and Myers, 2006). In fact, some CMT families show an even broader range of NCV with the lowest values usually around 25 m/s and the highest values within the normal range. The definition of ‘intermediate CMT’ has also been used to classify these families, although ‘overlap between CMT1 and CMT2′ would be a more accurate description.
With the advent of molecular genetic studies, it became obvious that patients and families with X-linked CMT (CMT1X), due to mutations in the gap junction beta 1 gene (GJB1, connexin 32), defy the classification into CMT1 and CMT2. Male CMT1X patients usually have median nerve motor NCV between 25 and 45 m/s, fitting exactly within the intermediate group defined by Davis et al. (1978). In female CMT1X patients, NCV show a broad range from 25 m/s to normal values. CMT1B patients with mutations in the myelin protein zero gene (MPZ) may also have NCV ranging from severely slowed to normal (De Jonghe et al., 1999; Shy et al., 2004b). A similar pattern has been observed with neurofilament light chain gene (NEFL) mutations (Jordanova et al., 2003a) and with ganglioside-induced differentiation-associated protein-1 gene (GDAP1) mutations (Senderek et al., 2003).
Three types of dominantly inherited CMT with intermediate NCV (dominant intermediate-CMT or DI-CMT) are known so far. These include a large Italian dominant intermediate-CMT family linked to chromosome 10q24.1-q25.1 (DI-CMTA; Rossi et al., 1985; Verhoeven et al., 2001; Villanova et al., 1998), and two unrelated Midwestern-American and Bulgarian families with intermediate CMT linked to a novel locus on chromosome 1p34-p35 (DI-CMTC; Jordanova et al., 2003b). In these two dominant intermediate-CMTC families, a mutation has recently been identified in the tyrosyl-tRNA synthetase gene (YARS; Jordanova et al., 2006). The dominant intermediate-CMTA gene remains currently unknown. Züchner and coworkers (2005) reported mutations in dynamin 2 (DNM2) as the cause of dominant intermediate-CMTB in three unrelated families originating from Australia, Belgium and North America. Here, we report the clinical, electrophysiological, haematological and nerve biopsy findings in these three original families and in three additional unrelated Spanish, Belgian and Dutch families with a DNM2 mutation, including two novel mutations. Remarkably, two of the families showed neutropaenia as a part of the phenotype, and in one family early-onset cataracts were additionally present.
Patients and Methods
Patients and pedigrees
The study included 34 patients belonging to six unrelated families with a DNM2 mutation (Fig. 1), originating from Australia (CMT-310), Belgium (CMT-48, CMT-72), the Netherlands (H20), North America (DUK-1118) and Spain (CMT-103). The molecular genetic results of the Australian, Belgian (CMT-48) and North American pedigrees have been described previously (Kennerson et al., 2001; Speer et al., 2002; Züchner et al., 2005). We additionally screened a cohort of 87 unrelated index patients with distinct CMT subtypes, in whom mutations in peripheral myelin protein (PMP22), MPZ and GJB1 had been excluded. Based upon clinical and electrophysiological data, 20 patients were diagnosed as CMT1 (23%), 46 as CMT2 (53%), 6 patients as intermediate CMT (7%), 2 patients as distal HMN (2%) and 13 patients as unspecified CMT (15%). For the three initially reported families, 250 clinically healthy Australian, European, and North-American individuals were screened as controls for sequence variations. For the three additional pedigrees, 192 European controls were screened. Each collaborator's institutional review board or equivalent approved the study. The patients and control persons gave informed consent, according to the Declaration of Helsinki. None of the patients experienced severe and/or frequently recurring infections. They did not take any medication that could affect the blood cells.
Figure 1.

CMT pedigrees with a DNM2 mutation. Open/filled/half-filled diamond = unaffected/affected/probably affected; ‘?’ within diamond = CMT-status unknown; slashed diamonds = deceased; n, normal allele; m, mutant allele; ‘?’ at left upper side of diamond, blood cell counts unknown; * = neutropaenia; £ = normal number of white blood cells but only one measurement; § = cataract. The arrow indicates the propositus. The gender is not shown to preserve confidentiality.
Clinical, electrophysiological and haematological studies
In the patients we performed a standard neurological examination. Motor and sensory nerve conduction studies were done in 24 patients, and needle electromyography was performed in nine CMT patients (CMT-103/I.2, II.2, II.3; CMT48/II.7, III.10; H20/II.3, III3, III.4, III.5) using standard techniques. Furthermore, an ophthalmologic examination and haematological tests including peripheral blood cell counts of erythrocytes or red blood cells, platelets and white blood cells and white blood cell differentiation were performed. We determined blood cell counts on freshly obtained samples from affected and unaffected individuals from all families using an automated cell counter in accordance with standard procedures. In patients II.2, II.4, III.4 and III.6 of family CMT-48 additional blood tests were done, including blood cell morphology, flowcytometry, platelet glycoproteins and phosphatidylinositol glycoproteins. In family CMT-48, we performed follow-up nerve conduction studies in patients II.7, III.10 and III.11, and follow-up blood cell counts in patients II.2, II.7 and III.4, within a time interval of 11 years. In patients III.2, III.3 and IV.15 of pedigree CMT-310 we counted blood cells twice within a time interval of 2 weeks. Serum creatine kinase levels were determined in seven patients (CMT-103/I.2, II.2, II.3; CMT-48/II.7, III.10; H20/II.3, III.5). In the three patients of family CMT-103, somatosensory-evoked potentials were additionally performed.
Sural nerve biopsy
In the index patient II.3 of the Dutch family, H20, a sural nerve biopsy has been performed at the age of 45 years. Sural nerve biopsy was prepared for light and electron microscopic examination using standard techniques (Joosten et al., 1974; Vos et al., 1983). The cluster ratio, defined as the number of clusters per 1000 myelinated fibres, was assessed by counting the number of clusters (three or more closely packed myelinated fibres) on the electron microscopic prints, divided by the number of myelinated fibres × 1000 in the same prints. Sural nerve biopsy findings in the Australian family CMT-310 have been reported previously (Kennerson et al., 2001).
Molecular genetic studies
The DNM2 mutation screening in pedigrees CMT-310, CMT-48 and DUK-1118 has been reported by Züchner et al. (2005). We additionally screened a cohort of 87 unrelated CMT patients for mutations in DNM2. We isolated genomic DNA from total blood samples obtained from CMT patients and control persons using standard extraction protocols. In the patients, the DNM2 mutation screening was performed by PCR amplifying all 22 coding exons of DNM2 using intronic primers (Züchner et al., 2005). PCR products were sequenced on an ABI3730 DNA Analyser (Applied Biosystems) using the BigDye Terminator Cycle Sequencing Kit 3.1 (Applied Biosystems). The DNA sequence data were collected and analysed using the ABI DNA Sequencing Analysis software 5.0 and Seqman II 5.07 (DNASTAR Inc., Madison, USA). When a mutation was detected in an index person, additional family members were analysed in order to determine co-segregation of the observed sequence variation with the disease. The numbering of the DNM2 codons was based upon the published amino acid sequence (National Centre for Biotechnology Information accession numbers NP_004936 for the protein sequence and NM_004945 for the mRNA sequence). The DNM2 mutations were defined according to the guidelines as described previously (den Dunnen and Antonarakis, 2001).
Results
Molecular genetic analysis
The North-American family DUK-1118 showed a 9-bp deletion of the 3′-end of exon 14 of DNM2, which is predicted to result in a shift of the open reading frame leading to a premature stop codon (Lys550fs) and the production of an in-frame mRNA with predicted deletion of three amino acids (Asp551_Glu553del) (for further details see Züchner et al., 2005). The Australian family CMT-310 (Züchner et al., 2005) and the Dutch family H20 carried a missense mutation in exon 15 (c.1672A>G), resulting in the amino acid substitution Lys558Glu, and the Belgian family CMT-48 showed a deletion of a single amino acid, Lys558del (c.1672_1674delAAG) in exon 15 (Züchner et al., 2005). We identified the novel missense mutation Gly358Arg (c.1072G>A) in exon 7 of DNM2 in the Spanish family CMT-103, and the novel Thr855_Ile856del (c.2564_2569delCCATTA) mutation in exon 19 in the index patient of the Belgian CMT-72 family, which were absent in 192 control individuals. The first four mutations are located in the pleckstrin homology domain of DNM2, the novel Gly358Arg mutation is situated in the middle domain whereas the novel Thr855_Ile856del mutation is localized in the proline-rich domain of DNM2.
Clinical findings
In the cohort of 87 unrelated index patients with distinct CMT subtypes, we identified a DNM2 mutation in three probands, resulting in a diagnostic yield of 3.4%. The six families with a DNM2 mutation included in total 34 examined patients, of which the clinical data are summarized in Table 1. The mean age at onset was 16 years, ranging from 2 to 50 years, even within the same family. The first symptoms in most patients were gait difficulties with frequent falls and/or foot deformities. One patient had a very early disease onset and showed delayed motor milestones without any other explanatory cause (CMT-48, patient III.4). Two individuals carried a DNM2 mutation but experienced no symptoms of a neuropathy (CMT-48/III.9; CMT-310/V.6). The former patient showed haematological abnormalities and cataracts, without any symptoms or clinical signs of a neuropathy. In the latter patient, however, clinical examination revealed some mild features of CMT. Most patients showed a rather typical CMT phenotype. Only one patient was wheelchair-bound (3%). Pyramidal signs were absent in all patients. In some patients additional features were present, in particular haematological abnormalities and/or cataracts. We observed cataracts in five affected individuals of family CMT-48. The cataracts consisted of a nuclear cataract in combination with a cortical cataract with a bluish cerulean-like appearance (Fig. 2). The cataracts were characterized by an early onset age and severely affected vision. Patients III.4 and III.6 had bilateral cataracts since the age of 12 and 15 years, respectively, for which they had undergone operation at the ages of 48 and 43 years. Patient III.9 of family CMT-48 also had cataract from an early age without any more precise data. Her vision is, however, less severely affected compared to her sisters, and at the current age of 38 years she is not visually handicapped. Patients II.4 and II.7 of family CMT-48 have been operated for cataract at the ages of 64 and 58 years, respectively. It is not known at what age cataract had initially occurred in these two patients. Cataract was not present in the other CMT patients of family CMT-48, or in the other families included in the current study.
Table 1.
Clinical features of patients harbouring a DNM2 mutation
| Patient | Amino-acid change | AAO | Symptoms at onset | ALE | Walking | Weakness dist. LL | Weakness prox. LL | Atrophy dist. LL | Pes cavus | DTR Ach/knee | Vibration LL | Weakness dist. UL | Atrophy dist. UL | DTR UL | Additional features |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CMT-103/I.2 | G358R | 9 | Foot deformities, gait difficulties, hand clumsiness | 55 | Steppage, bilateral foot drop, canes | + (0) | − (5) | + | + | −/+ | ↓ | + (2) | + | + | – |
| CMT-103/II.2 | G358R | 13 | Gait unsteadiness | 32 | Steppage, no aids | + (2,3,4) | − (5) | + | + | −/+ | ↓ | + (4) | − | + | – |
| CMT-103/II.3 | G358R | 5 | Foot deformities | 23 | Steppage, no aids | + (3,4) | − (5) | + | + | −/+ | ↓ | − (5) | − | + | − |
| DUK-1118/III.7 | D551_E553del; K550fs | Inf | Could not run fast | 71 | Steppage, no aids | + (0,4) | − (5) | + | + | −/− | ↓ | + (2,3) | + | + | − |
| DUK-1118/IV.2 | D551_E553del; K550fs | ? | ? | 54 | Steppage, no aids | + (1,4) | − (5) | + | + | −/− | ↓ | − (5) | + | + | − |
| DUK-1118/IV.11 | D551_E553del; K550fs | Inf | Weak ankles | 63 | Steppage, no aids | + (1,2) | − (5) | + | − | −/+ | ↓ | + (2,4) | + | + | − |
| DUK-1118/IV.13 | D551_E553del; K550fs | 50 | Weak ankles | 51 | Moderately impaired, no aids | + (3,4) | − (5) | + | + | −/+ | ↓ | + (4) | + | + | − |
| DUK-1118/V.1 | D551_E553del; K550fs | Inf | Always walked pigeon-toed | 35 | Moderately impaired, no aids | + (3,5) | − (5) | + | + | −/− | ↓ | + (3,4) | + | + | − |
| DUK-1118/V.4 | D551_E553del; K550fs | 12 | Frequent sprained ankles | 29 | Steppage, bilateral foot drop, no aids | + (0) | − (5) | + | + | −/− | ↓ | + (4) | + | − | − |
| DUK-1118/V.5 | D551_E553del; K550fs | 14 | Sprained ankles | 25 | Steppage, no aids | + (0,3,4) | − (5) | + | + | −/− | ↓ | − (5) | + | − | − |
| DUK-1118/V.19 | D551_E553del; K550fs | 37 | Some imbalance | 37 | Some imbalance, no aids | − (5) | − (5) | + | + | ±/+ | ↓ | + (4) | + | + | − |
| DUK-1118/V.20 | D551_E553del; K550fs | 20 | Catching toes | 33 | Steppage, no aids | + (1,2) | − (5) | + | + | −/− | ↓ | + (3,4) | + | + | − |
| DUK-1118/V.21 | D551_E553del; K550fs | 28 | Weak ankles | 29 | Slightly impaired, no aids | + (4,5) | − (5) | + | + | −/+ | ↓ | + (4) | + | + | − |
| DUK-1118/V.31 | D551_E553del; K550fs | 6 | Could not keep up with peers | 19 | Moderately impaired, no aids | + (3,4,5) | − (5) | + | + | −/− | ↓ | + (4) | + | − | − |
| CMT-48/II.2 | K558del | 14 | Gait difficulties, cramps | 68 | Steppage, bilateral foot drop, frequent falls | + (0) | − (5) | + | + | −/− | − | + (3,4) | + | − | Haematol. |
| CMT-48/II.4 | K558del | 30 | Gait difficulties, adjusted shoes | 63 | Steppage, bilateral foot drop | + (0) | − (5) | + | + | −/− | + | + (2,3,4) | + | − | Haematol., cataracts |
| CMT-48/II.7 | K558del | 19 | Gait difficulties, adjusted shoes | 56 | Steppage, bilateral foot drop | + (0) | − (5) | + | + | −/− | ↓ | + (3,4,5) | + | − | Haematol., cataracts |
| CMT-48/III.4 | K558del | 2 | Delayed motor milestones | 44 | Steppage, bilateral foot drop, walking stick | + (0) | − (5) | + | + | −/± | + | + (3,4) | + | ± | Haematol., cataracts |
| CMT-48/III.6 | K558del | 18 | Gait difficulties, foot deformities, cramps | 39 | Steppage, bilateral foot drop, walking stick | + (0) | − (5) | + | + | −/− | + | + (4,5) | + | − | Haematol., cataracts |
| CMT-48/III.9 | K558del | – | Asymptomatic | 23 | Normal | − (5) | − (5) | − | − | +/+ | + | − (5) | − | + | Haematol., cataracts |
| CMT-48/III.10 | K558del | ? | Gait difficulties | 30 | Moderately impaired, no aids | + (3,4) | − (5) | + | + | ±/± | ↓ | − (5) | + | ± | Haematol. |
| CMT-48/III.11 | K558del | ? | ? | 25 | Steppage, no aids | + (0,3,4) | − (5) | + | − | −/− | + | − (5) | − | − | Haematol. |
| CMT-310/III.2 | K558E | 14 | Foot drags | 68 | Bilateral foot drop, wheelchair at 61 years | + (0) | + (3) | + | − | −/− | ↓ | + (4) | + | + | Haematol. |
| CMT-310/IV.2 | K558E | 7 | Unable to run, falling | 38 | Bilateral foot drop, splints from age 15 years | + (0) | + (3) | + | − | −/− | ↓ | + (3) | + | − | Haematol. |
| CMT-310/IV.5 | K558E | 5 | Frequent falling | 45 | Bilateral foot drop, AFOs from early 20s | + (0) | + (4) | + | + | −/− | − | + (3) | + | + | Haematol. |
| CMT-310/IV.15 | K558E | 19 | Gait difficulties | 41 | Steppage, AFOs | + (2) | + (4) | + | + | −/− | ↓ | + (3) | − | − | Haematol. |
| CMT-310/IV.17 | K558E | 9 | Gait difficulties, drop foot | 33 | Ankles fused | + (4) | − (5) | + | − | −/− | − | + (3) | − | − | Haematol. |
| CMT-310/IV.20 | K558E | ? | Sprained ankles in sports | 23 | Weak ankles during sports | − (5) | − (5) | + | + | +/+ | + | − (5) | − | + | − |
| CMT-310/V.6 | K558E | – | asymptomatic | 18 | Normal | − (5) | − (5) | + | + | −/− | + | − (5) | − | + | ? |
| H20/II.3 | K558E | 20 | Gait difficulties | 64 | Steppage, canes | + (0,3,4) | + (4,5) | + | + | +/± | ↓ | + (4,5) | − | + | − |
| H20/III.3 | K558E | 12 | Gait difficulties, frequent falling | 38 | Steppage, AFOs | + (0,2) | − (5) | + | − | +/± | ↓ | + (4,5) | + | + | − |
| H20/III.4 | K558E | 15 | Could not keep up with peers | 36 | Steppage/bilateral footdrop, no aids | + (4,5) | − (5) | Edb only | + | −/+ | ↓ | + (4,5) | − | + | − |
| H20/III.5 | K558E | 12 | Could not keep up with peers | 31 | Steppage, no aids | + (3,4) | − (5) | + | + | −/− | ↓ | − (5) | − | ± | − |
| CMT-72/II.1 | T855_I856del | Inf | Gait difficulties, cramps LL | 67 | Steppage, bilateral foot drop | + (0,2,4,5) | − (5) | + | − | −/− | ↓ | − (5) | − | ± | − |
+ = present; − = absent; ± = weak; ↓ = decreased; ? = unknown; AAO = age at onset (years); Ach = Achilles tendon; AFOs = ankle-foot orthoses; ALE = age at last exam (years); dist. = distal; DTR = deep tendon reflexes; Edb = extensor digitorum brevis muscle; haematol. = haematological abnormalities; Inf = infancy; LL = lower limbs; () = MRC or Medical Research Council scale for muscle strength; prox. = proximal; UL = upper limbs.
Figure 2.
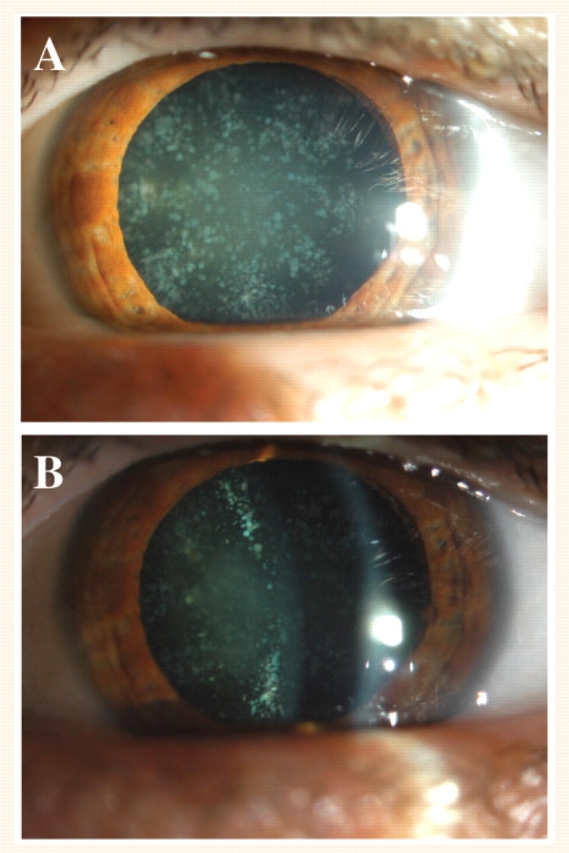
Cataract in a DNM2-mutated CMT patient. Presence of nuclear cataract in combination with cortical cataract in the left eye of patient III.4 of family CMT-48 at the age of 45 years.
Haematological findings
In all eight patients of the Belgian pedigree CMT-48 and in seven CMT patients belonging to the Australian family CMT-310, haematological abnormalities were observed (Table 1, Figs 1 and 3), which were not present in the other DNM2-mutated pedigrees including the Dutch H20 family harbouring the same mutation as CMT-310. The haematological findings in these patients are summarized in Fig. 3. Interestingly, in three patients, we counted a normal and a decreased number of white blood cells and neutrophils (Fig. 3A). The cell counts of the other white blood cell subtypes including lymphocytes, monocytes, eosinophils and basophils were normal overall. The red blood cells and platelets were inconsistently normal to decreased in both families (Fig. 3B). The additional blood tests, including blood cell morphology, flowcytometry, platelet glycoproteins and phosphatidylinositol–glycoproteins, did not show any abnormalities.
Figure 3.

Blood cell counts in patients of two DNM2-mutated CMT families with neutropaenia. For each patient of family CMT-48 and CMT-310 the number of white blood cells (WBC; black dots) and neutrophils (grey squares) is shown in (A), and the number of red blood cells (RBC; asterisks for males and triangles for females) and platelets (grey dots) is indicated in (B). The horizontal lines indicate the lower normal limit(s) for the different blood cells. Patients with normal blood cell counts are underlined.
Serum creatine kinase levels were either normal (CMT-103/I.2, II.2, II.3; CMT-48/III.10; H20/III.5) or slightly elevated (CMT-48/II.7, III.10; H20/II.3, III.5) with the highest value of 329 U/L (normal <195 U/L).
Electrophysiological results
Median nerve motor conduction velocities were available for 27 nerves in 20 affected members of all six families with a DNM2 mutation (Table 2). The median nerve motor NCV ranged overall from 26.2 to 57.0 m/s. Within four families we observed a similar range of median motor NCV; however, median motor NCV were less reduced and varied between 41.0 and 46.0 m/s in family H20, and were less slowed to 45.0 m/s in the proband of family CMT-72. Reduced motor median NCV between 38 and 49 m/s were always associated with normal CMAP amplitudes. Moderately to severely reduced CMAP amplitudes were observed in median nerves with motor NCV below 38 m/s. In some nerves both NCV and CMAP were normal, while in one individual (CMT-310/III.2), NCV was normal but the CMAP amplitude was reduced. Noteworthy, in patient I.2 of family CMT-103, motor NCV of the median nerve in the elbow–wrist segment was 33.5 m/s (CMAP amplitude at abductor pollicis brevis muscle, 0.1 mV), whereas in the axilla–elbow segment (upon stimulation at axilla and elbow, and recording from flexor digitorum superficialis muscle) NCV was 44 m/s (CMAP amplitude at flexor digitorum superficialis muscle, 0.9 mV, normal > 2 mV). The nerve conduction studies performed within a time interval of 11 years showed that NCV and CMAP remained relatively stable over this period of time. Needle electromyography was performed in nine patients. All showed chronic neurogenic alterations but no myopathic changes on needle electromyography.
Table 2.
Nerve conduction velocity studies in patients with a DNM2 mutation
| Patient | Age | R/L | Median motor |
Ulnar motor |
Peroneal motor |
Tibial motor |
Median sensory |
Ulnar sensory |
Sural sensory |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amp | CV | Amp | CV | Amp | CV | Amp | CV | Amp | CV | Amp | CV | Amp | CV | |||
| Normal values → | 4.0 | 49.0 | 4.0 | 49.0 | 3.0 | 41.0 | 3.0 | 41.0 | 17.0 | 46.0 | 17.0 | 47.0 | 12.0 | 44.0 | ||
| CMT-103/I.2 | 55 | R | 0.1 | 33.5 | 1.9 | 34.7 | A | A | A | A | 0.9 | 36.0 | 0.9 | 34.6 | A | A |
| L | – | – | 1.2 | 39.8 | A | A | – | – | – | – | – | – | – | – | ||
| CMT-103/II.2 | 32 | R | 10.1 | 50.5 | 7.5 | 51.9 | A | A | A | A | 2.5 | 46.9 | 1.4 | 45.9 | A | A |
| L | – | – | 6.5 | 50.6 | A | A | – | – | – | – | – | – | – | – | ||
| CMT-103/II.3 | 23 | R | 8.5 | 46.7 | 6.3 | 41.5 | A | A | A | A | 2.6 | 50.0 | 3.6 | 44.4 | A | A |
| L | 10.4 | 45.9 | 6.6 | 48.8 | A | A | – | – | – | – | – | – | – | – | ||
| DUK-1118/III.7 | 62 | L | 2.0 | 36.0 | – | – | – | – | – | – | – | – | – | – | – | – |
| DUK-1118/V.1 | 27 | L | 6.6 | 46.0 | – | – | – | – | A | A | 30.0 | – | – | – | – | – |
| CMT-48/II.2 | 68 | R | 1.0 | 32.8 | 2.1 | 45.5 | – | – | – | – | 5.6 | 38.7 | A | A | – | – |
| L | 0.8 | 26.2 | 4.6 | 50.0 | – | – | – | – | 6.6 | 37.1 | A | A | – | – | ||
| CMT-48/II.4 | 52 | R | A | A | 0.4 | 41.1 | – | – | – | – | A | A | A | A | – | – |
| L | A | A | 0.4 | 42.9 | – | – | – | – | A | A | A | A | – | – | ||
| CMT-48/II.7 | 45 | R | A | A | 1.9 | 47.0 | A | A | A | A | A | A | A | A | A | A |
| L | – | – | 1.9 | 52.0 | – | – | – | – | – | – | – | – | – | – | ||
| 56 | R | A | A | 1.7 | 49.0 | – | – | – | – | – | – | A | A | – | – | |
| L | A | A | 0.4 | 42.1 | – | – | – | – | – | – | A | A | – | – | ||
| CMT-48/III.4 | 44 | R | 5.8 | 41.9 | 6.5 | 42.3 | – | – | – | – | 4.1 | 41.5 | 2.1 | 37.5 | – | – |
| L | 0.9 | 37.6 | 4.4 | 41.7 | – | – | – | – | A | A | A | A | – | – | ||
| CMT-48/III.6 | 39 | R | 3.2 | 35.8 | 8.6 | 48.1 | – | – | – | – | 1.0 | 50.0 | 3.8 | 47.6 | – | – |
| L | 7.0 | 45.5 | 6.5 | 57.1 | – | – | – | – | 3.2 | 47.8 | 3.9 | 47.5 | – | – | ||
| CMT-48/III.10 | 19 | R | 6.7 | 42.0 | 10.1 | 54.0 | A | A | A | A | 10.0 | 48.0 | 8.0 | 43.0 | 4.0 | 52.0 |
| L | – | – | – | – | – | – | – | – | – | – | – | – | 5.0 | 48.0 | ||
| 30 | R | 9.6 | 42.6 | 6.0 | 54.4 | A | A | – | – | 4.0 | 42.6 | 1.3 | 36.8 | – | – | |
| L | 7.5 | 40.0 | 3.7 | 45.0 | – | – | – | – | 1.4 | 44.1 | 2.2 | 40.7 | – | – | ||
| CMT-48/III.11 | 14 | R | 8.4 | 47.0 | – | – | – | – | 1.2 | 31.0 | 5.0 | 40.0 | – | – | 4.0 | 36.0 |
| L | – | – | – | – | – | – | 1.1 | 34.0 | – | – | – | – | 4.0 | 35.0 | ||
| 25 | R | 8.7 | 43.0 | 7.1 | 50.5 | – | – | – | – | 3.2 | 45.0 | 2.4 | 43.4 | – | – | |
| L | 8.6 | 45.5 | 6.5 | 48.0 | – | – | – | – | 2.5 | 43.5 | 2.4 | 39.3 | – | – | ||
| CMT-310/III.2 | 68 | R | 1.9 | 54.0 | – | – | A | A | – | – | 3.0 | 65.0 | – | – | – | – |
| CMT-310/IV.15 | 44 | R | 0.5 | 29.0 | 6.9 | 39.0 | A | A | – | – | 8.0 | 32.0 | 2.2 | 31.0 | – | – |
| L | 0.7 | 37.0 | – | – | – | – | – | – | – | – | – | – | – | – | ||
| CMT-310/IV.17 | 44 | R | 1.0 | 33.0 | 6.0 | 41.0 | A | A | – | – | 3.0 | 31.0 | 5.9 | 28.0 | – | – |
| L | – | – | – | – | – | – | – | – | 5.3 | 30.0 | 4.7 | 32.0 | – | - | ||
| CMT-310/IV.2 | 38 | R | 0.3 | 36.0 | 2.9 | 40.0 | A | A | A | A | 2.0 | 56.0 | 1.0 | 50.0 | A | A |
| CMT-310/IV.5 | 45 | R | A | A | – | – | A | A | – | – | A | A | A | A | A | A |
| CMT-310/IV.20 | 24 | R | 6.0 | 43.0 | – | – | 0.9 | 27.0 | – | – | 7.0 | 37.0 | 7.0 | 32.0 | – | – |
| CMT-310/V.6 | 18 | R | 6.0 | 57.0 | – | – | 1.1 | 42.0 | – | – | 19.0 | 59.0 | 10.0 | 57.0 | – | – |
| H20/II.3 | 64 | R | – | – | – | – | A | A | – | – | A | A | – | – | A | A |
| L | – | – | 12.3 | 42.0 | A | A | A | A | – | – | A | A | – | – | ||
| H20/III.3 | 38 | R | 5.8 | 41.0 | – | – | A | A | – | – | A | A | – | – | A | A |
| L | – | – | 9.5 | 46.0 | A | A | A | A | – | – | A | A | – | – | ||
| H20/III.4 | 36 | R | 15.9 | 46.0 | – | – | A | A | – | – | 14.2 | 47.0 | – | – | A | A |
| L | – | – | 9.3 | 44.0 | A | A | A | A | – | – | 8.2 | 46.0 | – | – | ||
| H20/III.5 | 31 | R | 8.5 | 42.0 | – | – | A | A | A | A | A | A | – | – | – | – |
| CMT-72/II.1 | 67 | R | 10.7 | 45.0 | 3.6 | 54.0 | – | – | – | – | 8.0 | 41.0 | A | A | – | – |
Boldface represents abnormal values. A = absent response; Age = age at examination; Amp = amplitude (motor: in mV; sensory: in μV); CV = conduction velocity (in m/s); – = not measured; R/L = right/left.
Somatosensory-evoked potentials (Fig. 4) in the proband of family CMT-103 showed severely attenuated sensory nerve action potential (SNAP) amplitudes, a reduced sensory conduction velocity, an attenuated and delayed N20 response and a disorganization of parietal responses. In patient II.2, SNAP amplitudes were diminished with sensory conduction velocity at the lower limit of normal values, a normal N20 response and disorganized parietal responses. Patient II.3 had an attenuation of SNAP amplitude with normal sensory conduction velocity and normal somatosensory-evoked potentials.
Figure 4.

Somatosensory-evoked potentials in the three DNM2-mutated patients belonging to family CMT-103. Median nerve sensory potentials obtained between digitIII and wrist, and somatosensory-evoked potentials obtained after stimulation of the median nerve at the wrist, in patient I.2 (A, B), in patient II.2 (C, D) and in patient II.3 (E, F) of family CMT-103. N2 represents the N20 response.
Sural nerve biopsy
Sural nerve biopsy was performed in the index patient II.3 belonging to the Dutch family H20 at the age of 45 years. There was a complete loss of fibres larger than 8 μm and an increased number of myelinated fibres [12 573 fibres/mm2, age-matched controls 9760 (n = 5)] due to the presence of many clusters of regenerated myelinated axons (Fig. 5A). The cluster ratio was 54.8. The changes were distributed quite homogeneously throughout the fascicles. Only very rare acute axonal degeneration was observed. On semi-thin sections, axons with abnormally thick myelin sheets were found in three of the six fascicles, most probably corresponding with focal myelin thickenings observed on teased fibres (Fig. 5B). Several groups of collagenous pockets and groups of flattened Schwann cell processes were also present, representing damage to unmyelinated fibres (Fig. 5C). Fibres with abnormally broad Schmidt–Lanterman incisures (Fig. 5D) were easily found. No signs of segmental demyelination, remyelination or onion bulb formation were observed in the propositus of family H20. This contrasts the presence of rare segmental demyelination and remyelination with onion bulb formation in the sural nerve biopsy in the Australian family CMT-310, which has previously been reported by Kennerson et al. (2001). Similar to the Dutch index patient, the sural nerve biopsy in the Australian family showed axonal degeneration and loss of large diameter fibres.
Figure 5.
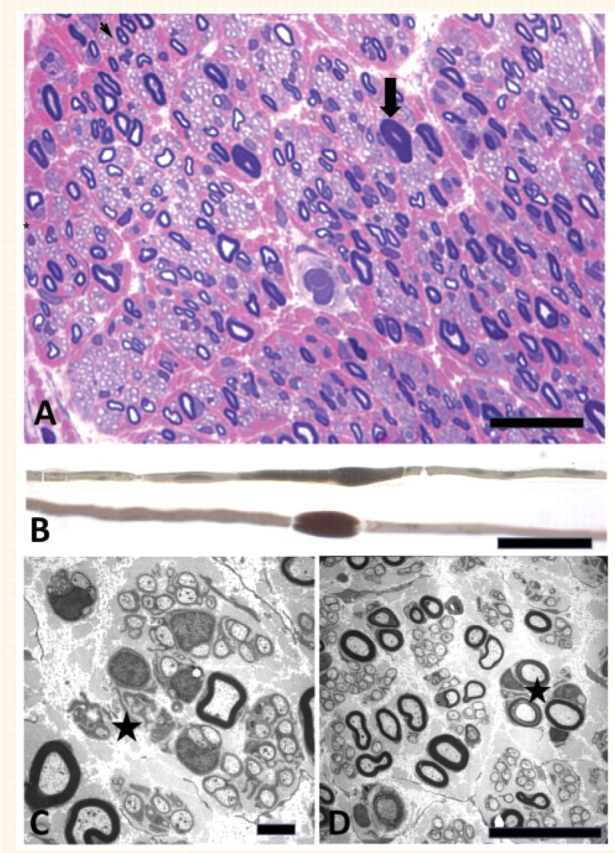
Sural nerve biopsy of index patient of CMT family H20 with DNM2 mutation. (A) Semi-thin section of sural nerve showing diffuse loss of large myelinated fibres, and many clusters of myelinated and unmyelinated fibres. A thickly myelinated fibre is indicated by an arrow. Thionin Basic Fuchsin, magnification bar = 50 μm. (B) Teased fibres with focal myelin thickenings are shown. Magnification bar = 50 μm. (C) Electronmicrograph showing clusters of unmyelinated fibres and two clusters of flattened Schwann cell processes (asterisk). Magnification bar = 1 μm. (D) Clusters of myelinated and unmyelinated fibres and two fibres with broad Schmidt–Lanterman incisures (asterisk). Magnification bar = 10 μm.
Discussion
We report the clinical, haematological, electrophysiological and histopathological data of six unrelated CMT families with a mutation in DNM2. The diagnostic yield of DNM2 mutations in our cohort comprising distinct CMT subtypes, in whom CMT1A, 1B and 1X had already been excluded, was 3.4%. Patients with a DNM2 mutation presented with a classical, mild to moderately severe CMT phenotype, with mean age at onset of 16 years. Some patients developed additional features such as neutropaenia and/or cataracts. A combined nuclear and cortical cataract with early onset was present in some CMT patients of family CMT-48 but was not observed in the other DNM2-mutated families. The presence of congenital cataracts has been reported recently in one CMT patient with a distinct DNM2 mutation (K559del; Bitoun et al., 2008). Further studies are necessary to elucidate whether cataracts are indeed part of the dominant intermediate-CMTB phenotype, and whether this finding is repeatedly associated with particular DNM2 mutations.
Interestingly, in two families CMT cosegregated with neutropaenia. CMT patients of the remaining four families had normal blood cell counts, suggesting that neutropaenia is not a general feature of dominant intermediate-CMTB, but may be associated with particular mutations. Remarkably, the Belgian (Lys558del mutation) and Australian (Lys558Glu mutation) pedigrees with neutropaenia carried two different mutations affecting the same amino acid (Lys558). This might suggest that Lys558 has an unknown function in the development of peripheral blood cells. However, in the Dutch family, the Lys558Glu mutation was not associated with haematological abnormalities. Mutations in neutrophil elastase 2 (ELA2), which is located on 19p13.3 about 9 Mb upstream to the most telomeric short tandem repeat marker for dominant intermediate-CMTB, cause congenital neutropaenia or Kostmann disease and cyclic neutropaenia (OMIM 202700; Horwitz et al., 1999; Dale et al., 2000). However, mutations in ELA2 as a cause of neutropaenia had been excluded in family CMT-48 by sequencing analysis of all coding exons (Züchner et al., 2005). The abnormal blood cell counts in our patients were not constantly present over time. The sampled time intervals were, however, too long and the number of measurements too low to evaluate a possible cyclic character of the observed haematological abnormalities. It is noteworthy that CMT patients with neutropaenia did not experience more severe or recurrent infections compared with the patients without this feature. A low neutrophil count has also been documented recently in another CMT patient harbouring a DNM2 mutation (Bitoun et al., 2008). The additional features of neutropaenia and cataracts are hard to explain as they are not even gene-specific for DNM2 but seem to be rather mutation-specific. Based on the data available in this manuscript, no further comment about possible pathogenic mechanisms can be made without becoming really speculative.
The median motor NCV in four of our DNM2-mutated CMT families varied from 26.0 m/s to normal values. In the Dutch pedigree (H20) and in the proband of the Belgian family CMT-72, median motor NCV were only mildly slowed, within the axonal NCV range, and corresponding CMAP amplitudes were not reduced. In the recent literature, the terminology of ‘intermediate CMT’ is used in two ways: to define a strictly intermediate group with median motor NCV between 25 and 45 m/s or to identify a more broadly defined overlap group showing median motor NCV ranging from 25 m/s to normal values (for review see Nicholson and Myers, 2006). Our findings in dominant intermediate-CMTB families correspond to the latter and broader definition of intermediate CMT. With the exception of the Spanish family CMT-103, the electrophysiological data in our CMT families are not typical for either CMT1 or CMT2, but rather show a combination of axonal and demyelinating features. These findings are confirmed by our pathological observations in two sural nerve biopsies, one in the Dutch and one in the Australian family (for the latter see also: Kennerson et al., 2001), both harbouring the same DNM2 mutation. In contrast, family CMT-103 was diagnosed with axonal CMT (CMT2). All three patients belonging to the Spanish CMT-103 family showed inexcitability of lower limb nerves, a fact indicative of severe axonal degeneration. Hand amyotrophy was only observed in the proband (CMT-103/I.2, aged 55 years). This woman showed severe attenuation of CMAP of median and ulnar nerves, and therefore the observed motor NCV might be accounted for by loss of larger axons. To further clarify the issue, we determined in this family motor NCV of median nerve in the axilla–elbow segment with stimulation at axilla/elbow and recording in flexor digitorum superficialis muscle, which resulted in reduced CMAP amplitude of 0.9 mV and motor NCV of 44 m/s, i.e. 10 m/s faster than in the forearm segment. Patient CMT-103/II.2, aged 32 years, showed normal motor NCV and CMAP amplitudes of median and ulnar nerves, whereas her younger sister (CMT-103/II.3, aged 23 years) exhibited minimal reduction of motor NCV with preserved CMAP amplitudes. SNAP amplitudes of median and ulnar nerves were severely reduced in the proband CMT-103/I.2, resulting in a proportionate slowing of the corresponding sensory NCV. In both the proband's daughters, we recorded a lesser reduction of SNAP amplitudes with minimal reduction of sensory NCV of the ulnar nerve only. Taking all these features into account, we concluded that family CMT-103 should rather be classified as axonal CMT (Gallardo et al., 2008). These results are similar to the findings of Fabrizi et al. (2007) and Bitoun et al. (2008) in patients with DNM2 mutations distinct from those reported here. Our data also emphasize that the term ‘intermediate’ should not be used to describe a single NCV but rather the CMT subtype at the level of the family. This is unlikely to be affected by the duration of illness at the time of the electrophysiological examination, since over a time period of 11 years, we observed that NCV and CMAP amplitudes remained relatively stable in three patients belonging to family CMT-48. The combined abnormalities of somatosensory-evoked potentials and distal SNAP might suggest that the pathological hallmark of the neuropathy caused by a mutation in DNM2 implies a degeneration of both the peripheral and central sensory axons. This is analogous with a pathological study in CMT2G patients that showed loss of spinal motor neurons and loss of ganglion cells in the posterior root ganglia with demyelination of the fasciculus gracilis and distally accentuated axonal loss in peroneal and tibial nerves. It has been proposed that CMT2G might be a type of motor and sensory neuronopathy with length-dependent axonal degeneration (Berciano et al., 1986).
Mutations in DNM2 are also known to cause autosomal dominant centronuclear myopathy (Bitoun et al., 2005, 2007; Jeub et al., 2008; Jungbluth et al., 2008). However, none of our CMT patients showed features of centronuclear myopathy, such as facial, extraocular or proximal limb weakness or myopathic changes on needle electromyography. In some patients serum creatine kinase levels were slightly elevated; however, these values can still be explained by their chronic neurogenic disease. Muscle biopsy has not been performed in any of the patients. Elaborating further on the possible concomitant myopathic abnormalities in CMT patients is certainly very interesting but beyond the scope of the current study. Furthermore, a muscle biopsy in any such patient might be difficult to interpret due to the presence of neurogenic changes resulting from their chronic neuropathy.
We identified five different DNM2 mutations in six unrelated CMT families: Asp551_Glu553del; Lys550fs in exon 14 in the American DUK-1118 family, a deletion of a single amino acid Lys558del in exon 15 in the Belgian CMT-48 family (Züchner et al., 2005), Thr855_Ile856del in exon 19 in the Belgian CMT-72 patient, three missense mutations Lys558Glu in exon 15 in the Australian CMT-310 and the Dutch H20 families and Gly358Arg in exon 7 in the Spanish CMT-103 pedigree. So far, six distinct DNM2 mutations have been reported in CMT patients (Bitoun et al., 2008; Fabrizi et al., 2007; Züchner et al., 2005). These mutations were all localized in the pleckstrin homology domain. In the current study, we additionally identified two novel mutations in CMT patients, of which Gly358Arg was located in the middle domain and Thr855_Ile856del in the proline-rich domain of DNM2. We report the first disease-causing mutation in the proline-rich domain of DNM2. This Thr855_Ile856del mutation in the proline-rich domain was not associated with a different phenotype compared to the other mutations. The Gly358Arg mutation in the middle domain was associated with an axonal CMT phenotype (see above); however, no other phenotypic differences were observed. In autosomal dominant centronuclear myopathy, mutations are mainly situated in the middle domain of DNM2 (Bitoun et al., 2005; Echaniz-Laguna et al., 2007). However, in a recent study of centronuclear myopathy patients with a more severe neonatal phenotype, mutations in DNM2 were found in the pleckstrin homology domain (Bitoun et al., 2007). The DNM2 mutations causing CMT or centronuclear myopathy are different. Recently, however, the Glu368Gln mutation has been shown to cause a combined phenotype of centronuclear myopathy and mild axonal peripheral nerve involvement (Echaniz-Laguna et al., 2007). Furthermore, intramuscular nerve fascicles found in the muscle biopsy of a centronuclear myopathy patient harbouring the Glu368Lys mutation showed alterations indicative for peripheral nerve involvement (Jeub et al., 2008).
DNM2 is a large GTPase involved in receptor-mediated endocytosis at the plasma membrane, membrane trafficking from the late endosomes and Golgi, actin assembly and centrosome cohesion (Hinshaw, 2000; McNiven, 1998; Schafer et al., 2002; Thompson et al., 2004). DNM2 is expressed in the peripheral and central nervous system, and is composed of different domains, such as an N-terminal GTPase domain, a middle domain, a pleckstrin homology domain that mediates membrane binding, a GTPase effector domain and a C-terminal proline-rich domain that interacts with numerous other proteins (McNiven, 1998; Urrutia et al., 1997). Since DNM2 is a component of the centrosome (Thompson et al., 2004), dysfunction of mutated DNM2 might lead to a destabilization of the microtubule network inducing an abnormal axonal transport and protein trafficking, which has been described as a pathophysiological mechanism in different subtypes of CMT (Shy, 2004a).
To conclude, DNM2 mutations should be part of the screening programme in autosomal dominant CMT families with intermediate or axonal NCV ranging from 25 m/s to normal values, after exclusion of CMT1A, 1B (and 1X), and in patients with a classical, mild to moderately severe CMT phenotype, especially when CMT is associated with neutropaenia or cataracts.
Funding
Fund for Scientific Research (FWO-Flanders); the Medical Foundation Queen Elisabeth (GSKE); the University of Antwerp (UA); the Interuniversity Attraction Poles program of the Belgian Federal Science Policy Office (BELSPO P6/43); the Association Belge contre les Maladies Neuromusculaires (ABMM); the CMT Association of North America (to S.Z.); the National Institute of Neurological Disorders and Stroke (5R01NS052767) (to S.Z.); the National Health and Medical Research Council of Australia; the CMT Association of Australia. PhD fellowship of the FWO-Flanders, Belgium (to J.B.).
Acknowledgements
We gratefully acknowledge the participation of all patients and their relatives in this study. We appreciated the contribution of the VIB Genetic Service Facility (http://www.vibgeneticservicefacility.be/) for genotyping and sequencing. We thank Dr A. Gabreëls-Festen for advice and Mrs L. Eshuis for technical assistance with the nerve biopsy.
Glossary
Abbreviations
- CMAP
compound muscle action potential
- CMT
Charcot-Marie-Tooth
- DI-CMTB
dominant intermediate Charcot-Marie-Tooth neuropathy type B
- DNM2
dynamin 2
- NCV
nerve conduction velocities
- SNAP
sensory nerve action potential
References
- Berciano J, Combarros O, Figols J, Calleja J, Cabello A, Silos I, et al. Hereditary motor and sensory neuropathy type II. Clinicopathological study of a family. Brain. 1986;109:897–914. doi: 10.1093/brain/109.5.897. [DOI] [PubMed] [Google Scholar]
- Bitoun M, Bevilacqua JA, Prudhon B, Maugenre S, Taratuto AL, Monges S, et al. Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann Neurol. 2007;62:666–70. doi: 10.1002/ana.21235. [DOI] [PubMed] [Google Scholar]
- Bitoun M, Maugenre S, Jeannet PY, Lacène E, Ferrer X, Laforêt P, et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. 2005;37:1207–9. doi: 10.1038/ng1657. [DOI] [PubMed] [Google Scholar]
- Bitoun M, Stojkovic T, Prudhon B, Maurage CA, Latour P, Vermersch P, et al. A novel mutation in the dynamin 2 gene in a Charcot-Marie-Tooth type 2 patient: clinical and pathological findings. Neuromuscul Disord. 2008;18:334–8. doi: 10.1016/j.nmd.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, et al. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000;96:2317–22. [PubMed] [Google Scholar]
- Davis CJF, Bradley W, Madrid R. The peroneal muscular atrophy syndrome. Clinical, genetic, electrophysiological and nerve biopsy studies. J Genet Hum. 1978;26:311–49. [PubMed] [Google Scholar]
- De Jonghe P, Timmerman V, Ceuterick C, Nelis E, De Vriendt E, Lofgren A, et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain. 1999;122:281–90. doi: 10.1093/brain/122.2.281. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet. 2001;109:121–4. doi: 10.1007/s004390100505. [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Chance P, Lebo R, Carney JA. Hereditary motor and sensory neuropathies. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF, editors. Peripheral neuropathy. Philadelphia: W.B. Saunders Company; 1993. pp. 1094–136. [Google Scholar]
- Echaniz-Laguna A, Nicot AS, Carré S, Franques J, Tranchant C, Dondaine N, et al. Subtle central and peripheral nervous system abnormalities in a family with centronuclear myopathy and a novel dynamin 2 gene mutation. Neuromuscul Disord. 2007;17:955–9. doi: 10.1016/j.nmd.2007.06.467. [DOI] [PubMed] [Google Scholar]
- Fabrizi GM, Ferrarini M, Cavallaro T, Cabrini I, Cerini R, Bertolasi L, et al. Two novel mutations in dynamin-2 cause axonal Charcot-Marie-Tooth disease. Neurology. 2007;69:291–5. doi: 10.1212/01.wnl.0000265820.51075.61. [DOI] [PubMed] [Google Scholar]
- Gallardo E, Claeys KG, Nelis E, García A, Canga A, Combarros O, et al. Magnetic resonance imaging findings of leg musculature in Charcot-Marie-Tooth disease type 2 due to dynamin 2 mutation. J Neurol. 2008;255:986–92. doi: 10.1007/s00415-008-0808-8. [DOI] [PubMed] [Google Scholar]
- Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103:259–80. doi: 10.1093/brain/103.2.259. [DOI] [PubMed] [Google Scholar]
- Hinshaw JE. Dynamin and its role in membrane fission. Annu Rev Cell Dev Biol. 2000;16:483–519. doi: 10.1146/annurev.cellbio.16.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz M, Benson KF, Person RE, Aprikyan AG, Dale DC. Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nat Genet. 1999;23:433–6. doi: 10.1038/70544. [DOI] [PubMed] [Google Scholar]
- Jeub M, Bitoun M, Guicheney P, Kappes-Horn K, Strach K, Druschky KF, et al. Dynamin 2-related centronuclear myopathy: clinical, histological and genetic aspects of further patients and review of the literature. Clin Neuropathol. 2008;27:430–8. doi: 10.5414/npp27430. [DOI] [PubMed] [Google Scholar]
- Joosten EM, Krijgsman JB, Gabreëls-Festen AA, Gabreëls FJ, Baars PE. Infantile globoid cell leucodystrophy (Krabbe's disease). Some remarks on clinical, biochemical and sural nerve biopsy findings. Neuropediatrie. 1974;5:191–209. doi: 10.1055/s-0028-1091702. [DOI] [PubMed] [Google Scholar]
- Jordanova A, De Jonghe P, Boerkoel CF, Takashima H, De Vriendt E, Ceuterick C, et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain. 2003a;126:590–7. doi: 10.1093/brain/awg059. [DOI] [PubMed] [Google Scholar]
- Jordanova A, Irobi J, Thomas FP, Van Dijck P, Meerschaert K, Dewil M, et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- Jordanova A, Thomas FP, Guergueltcheva V, Tournev I, Gondim FA, Ishpekova B, et al. Dominant intermediate Charcot-Marie-Tooth type C maps to chromosome 1p34-p35. Am J Hum Genet. 2003b;73:1423–30. doi: 10.1086/379792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbluth H, Wallgren-Pettersson C, Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis. 2008;3:26. doi: 10.1186/1750-1172-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennerson ML, Zhu D, Gardner RJ, Storey E, Merory J, Robertson SP, et al. Dominant intermediate Charcot-Marie-Tooth neuropathy maps to chromosome 19p12-p13.2. Am J Hum Genet. 2001;69:883–8. doi: 10.1086/323743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNiven MA. Dynamin: a molecular motor with pinchase action. Cell. 1998;94:151–4. doi: 10.1016/s0092-8674(00)81414-2. [DOI] [PubMed] [Google Scholar]
- Nicholson G, Myers S. Intermediate forms of Charcot-Marie-Tooth neuropathy: a review. Neuromolecular Med. 2006;8:123–30. doi: 10.1385/nmm:8:1-2:123. [DOI] [PubMed] [Google Scholar]
- Rossi A, Paradiso C, Cioni R, Rizzuto N, Guazzi G. Charcot-Marie-Tooth disease: study of a large kinship with an intermediate form. J Neurol. 1985;232:91–8. doi: 10.1007/BF00313907. [DOI] [PubMed] [Google Scholar]
- Schafer DA, Weed SA, Binns D, Karginov AV, Parsons JT, Cooper JA. Dynamin2 and cortactin regulate actin assembly and filament organization. Curr Biol. 2002;12:1852–7. doi: 10.1016/s0960-9822(02)01228-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senderek J, Bergmann C, Ramaekers VT, Nelis E, Bernert G, Makowski A, et al. Mutations in the ganglioside-induced differentiation-associated protein-1 (GDAP1) gene in intermediate type autosomal recessive Charcot-Marie-Tooth neuropathy. Brain. 2003;126:642–9. doi: 10.1093/brain/awg068. [DOI] [PubMed] [Google Scholar]
- Shy ME. Charcot-Marie-Tooth disease: an update. Curr Opin Neurol. 2004a;17:579–85. doi: 10.1097/00019052-200410000-00008. [DOI] [PubMed] [Google Scholar]
- Shy ME, Jani A, Krajewski K, Grandis M, Lewis RA, Li J, et al. Phenotypic clustering in MPZ mutations. Brain. 2004b;127:371–84. doi: 10.1093/brain/awh048. [DOI] [PubMed] [Google Scholar]
- Speer MC, Graham FL, Bonner E, Collier K, Stajich JM, Gaskell PC, et al. Reduction in the minimum candidate interval in the dominant-intermediate form of Charcot-Marie-Tooth neuropathy to D19S586 to D19S432. Neurogenetics. 2002;4:83–5. doi: 10.1007/s10048-002-0139-3. [DOI] [PubMed] [Google Scholar]
- Thompson HM, Cao H, Chen J, Euteneuer U, McNiven MA. Dynamin 2 binds gamma-tubulin and participates in centrosome cohesion. Nat Cell Biol. 2004;6:335–42. doi: 10.1038/ncb1112. [DOI] [PubMed] [Google Scholar]
- Urrutia R, Henley JR, Cook T, McNiven MA. The dynamins: redundant or distinct functions for an expanding family of related GTPases? Proc Natl Acad Sci USA. 1997;94:377–84. doi: 10.1073/pnas.94.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven K, Villanova M, Rossi A, Malandrini A, De Jonghe P, Timmerman V. Localization of the gene for the intermediate form of Charcot-Marie-Tooth to chromosome 10q24.1-q25.1. Am J Hum Genet. 2001;69:889–94. doi: 10.1086/323742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanova M, Timmerman V, De Jonghe P, Malandrini A, Rizzuto N, Van Broeckhoven C, et al. Charcot-Marie-Tooth disease: an intermediate form. Neuromuscul Disord. 1998;8:392–3. doi: 10.1016/s0960-8966(98)00044-3. [DOI] [PubMed] [Google Scholar]
- Vos AJ, Joosten EM, Gabreëls-Festen AA. Adult polyglucosan body disease: clinical and nerve biopsy findings in two cases. Ann Neurol. 1983;13:440–4. doi: 10.1002/ana.410130411. [DOI] [PubMed] [Google Scholar]
- Züchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, De Jonghe P, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 2005;37:289–94. doi: 10.1038/ng1514. [DOI] [PubMed] [Google Scholar]