

Abstract
We recently identified the X-chromosomal four and a half LIM domain gene FHL1 as the causative gene for reducing body myopathy, a disorder characterized by progressive weakness and intracytoplasmic aggregates in muscle that exert reducing activity on menadione nitro-blue-tetrazolium (NBT). The mutations detected in FHL1 affected highly conserved zinc coordinating residues within the second LIM domain and lead to the formation of aggregates when transfected into cells. Our aim was to define the clinical and morphological phenotype of this myopathy and to assess the mutational spectrum of FHL1 mutations in reducing body myopathy in a larger cohort of patients. Patients were ascertained via the detection of reducing bodies in muscle biopsy sections stained with menadione-NBT followed by clinical, histological, ultrastructural and molecular genetic analysis. A total of 11 patients from nine families were included in this study, including seven sporadic patients with early childhood onset disease and four familial cases with later onset. Weakness in all patients was progressive, sometimes rapidly so. Respiratory failure was common and scoliosis and spinal rigidity were significant in some of the patients. Analysis of muscle biopsies confirmed the presence of aggregates of FHL1 positive material in all biopsies. In two patients in whom sequential biopsies were available the aggregate load in muscle sections appeared to increase over time. Ultrastructural analysis revealed that cytoplasmic bodies were regularly seen in conjunction with the reducing bodies. The mutations detected were exclusive to the second LIM domain of FHL1 and were found in both sporadic as well as familial cases of reducing body myopathy. Six of the nine mutations affected the crucial zinc coordinating residue histidine 123. All mutations in this residue were de novo and were associated with a severe clinical course, in particular in one male patient (H123Q). Mutations in the zinc coordinating residue cysteine 153 were associated with a milder phenotype and were seen in the familial cases in which the boys were still more severely affected compared to their mothers. We expect the mild end of the spectrum to significantly expand in the future. On the severe end of the spectrum we define reducing body myopathy as a progressive disease with early, but not necessarily congenital onset, distinguishing this condition from the classic essentially non-progressive congenital myopathies.
Keywords: reducing body myopathy, FHL1, aggresomes, LIM, X-linked
Introduction
Reducing body myopathy (RBM) is a rare disorder of muscle, first described more than 35 years ago (Brooke and Neville, 1972). The typical muscle histopathological finding that gives the disease its name consist of intracytoplasmic aggregates that reduce nitro-blue-tetrazolium (NBT) and thus stain strongly with the menadione-NBT stain, presumably because of their high content of sulfhydryl groups. The inclusions were later also noted to have features of aggresomes (Liewluck et al., 2007; Schessl et al., 2008). The clinical presentation of patients with reducing body myopathy has ranged from early onset fatal, through childhood onset to adult onset cases, thus presenting a wide clinical spectrum (Brooke and Neville, 1972; Tome and Fardeau, 1975; Dubowitz, 1978; Hubner and Pongratz, 1982; Oh et al., 1983; Carpenter et al., 1985; Kobayashi et al., 1992; Nomizu et al., 1992; Bertini et al., 1994; Kiyomoto et al., 1995b; Reichmann et al., 1997; Figarella-Branger et al., 1999; Schessl et al., 2008).
Using laser microdissection of the aggregates out of biopsy material followed by proteomic analysis, we recently established the four and a half LIM domain gene FHL1 as the causative gene for RBM (Schessl et al., 2008). We detected dominantly acting mutations in highly conserved zinc coordinating histidine and cysteine residues within the second LIM domain of the gene FHL1 in two sporadic female patients and in two familial mother/son pairs. FHL1 (also known as SLIM1) is a 32 kDa protein containing an N-terminal zinc finger/half LIM domain, followed by four complete LIM domains, leading to the designation of this class of proteins four and a half LIM domain proteins. LIM domains are cysteine-rich tandem-zinc finger protein interaction motifs, first recognized in three homeodomain transcription factors (Lin-11, Isl-1, Mec-3) (Freyd et al., 1990; Dawid et al., 1995; Curtiss and Heilig, 1998; Jurata and Gill, 1998; Kadrmas and Beckerle, 2004). FHL proteins in general appear to be involved in cytoskeletal scaffolding as well as in regulation of transcription factors. FHL1 is encoded on Xq26.3 and is highly expressed in skeletal and cardiac muscle (McGrath et al., 2006). In skeletal muscle FHL1 localizes to the sarcomere and also to the sarcolemma and is believed to participate in muscle differentiation and growth as well as in sarcomeric assembly (McGrath et al., 2006). In addition to full length FHL1 (FHL1A), there are two additional splice variants referred to as FHL1B, and C, which also contain the second LIM domain in which the initial mutations occurred (Brown et al., 1999; Ng et al., 2001; Schessl et al., 2008).
Our initial report had focused on the biochemical and molecular genetic features of the disease in the four families originally identified. We now report an expanded series of 11 patients from nine families with RBM caused by mutations in FHL1 to define the clinical and histological phenotypes in relation to the emerging spectrum of FHL1 mutations. Missense mutations of histidine 123 emerge as the most common molecular mechanism of severe RBM.
Patients and Methods
Patients
We ascertained a total of 11 patients from nine unrelated families (Table 1). Sporadic Patients 1 and 7 and the two mother–son pairs were part of the original gene identification report (Schessl et al., 2008). Patients were included for mutation screening if menadione-NBT positive reducing bodies in muscle biopsy sections were present or if a mutation had been identified in an also affected relative with RBM. Informed consent from all subjects was obtained (IRB approval 2002-6-2846, The Children's Hospital of Philadelphia, PA, USA and IRB approvals at the collaborating institutions).
Table 1.
Clinical characteristics
| Patient | Sex | Age | Age at onset | Clinical presentation at onset | Progression and current clinical status | Major pattern of muscle involvement | Contractures and spinal involvement | Cardiac involvement | Respiratory involvement | CK U/l | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 8.3 yrs | <2 yrs | Delayed motor development, poor head control, stands with hyperlordosis. | Never able to run, never able to stand up from sitting position. Wheelchair dependent at age 3 yrs. | Proximally pronounced diffuse weakness. | Spinal rigidity, scoliosis surgery at age 6 yrs 10 mo. | None | BiPAP at age 4 yrs, tracheotomy at age 7 yrs. | 337 | c.367C>T/ p.H123Y |
| 2 | F | 2.5 yrs | <1.5 yrs | Delayed motor development, lordotic gait. | Unable to climb stairs, able to walk 50 m. | Predominant proximal muscle weakness, neck weakness, Gowers’ sign. | None | None | None | 330 | c.367C>T/ p.H123Y |
| 3 | F | 7 yrs | 4 yrs | Frequent falls, difficulties walking, running and getting up from the floor/Gowers’ sign, scapular and pelvic girdle muscular weakness. | Progressive diffuse weakness. Increasing difficulties walking. | Generalized hypotrophy, predominant in upper limbs, scapular winging. | Scoliosis | None | None | 114 | c.368A>T/ p.H123L |
| 4 | F | 10 yrs | 4 yrs | Frequent falls. | Since age 8 yrs. progressive weakness of proximal and axial muscles, including neck. Scapular winging. Wheelchair dependent at age 10 yrs. | Proximally pronounced weakness of upper limbs and neck, axial muscles. | Contractures of elbows, hand and Achilles tendons. Severe scoliosis. | None | None | 513 | c.369C>G/ p.H123Q |
| 5 | F | 11 yrs | 3 yrs 8 mo | Frequent falls and abnormal gait. | 5 mo after onset increased limb girdle weakness and scapular winging. Dysphagia (gastrostomy at age 10 yrs). Wheelchair dependent at age 8 yrs. | Progressive weakness, pronounced in neck and trunk. Gowers’ sign. | Knee and Achilles contractures, scoliosis. | Slight mitral insufficiency. | Restrictive pulmonary disease, atelectasis at age 8 yrs, BiPAP at night | 1080 | c.369C>G/ p.H123Q |
| 6 | M | 3.5 yrs | Between 13 and 17 mo | Head drop. Not able to climb stairs and roll over when lying flat. Scapular muscle hypotrophy, abolished reflexes. | Progressive loss of ambulation and generalized muscle hypotrophy. Unable to walk at current age, Wheelchair dependent at 31 mo of age. | Proximally pronounced weakness, generalized muscle hypotrophy. | Progressive severe contractures at the elbows, knees and Achilles tendons. Mild Scoliosis, no rigid spine. | Cardiac arrest at age 3 yrs. | Non-invasive ventilatory support | 2000 | c.369C>A/ p.H123Q |
| 7 | F | Died at age 6.5 yrs | 2 yrs | Frequent falls and mild leg weakness. | Initially treated with i.v. IG and daily prednisone for presumed myositis without improvement. At age 3.5 yrs not able to climb stairs, followed by severe progression and at age 5 yrs inability to sit or ambulate. | Generalized muscle hypotrophy with severe truncal and limb weakness; eventual flaccid paralysis. | No contractures; exam particularly noted for absence of spinal rigidity (very lax thorax). | None | Respiratory insufficiency leading to death. | 537 | c.395G>T/ p.C132F |
| 8 | M | 18 yrs | 7 yrs | Rigid spine, no weakness nor muscle fatigability, rigidity and contractures at 10 yrs involving neck, elbows and wrists (> right). | Wheelchair dependent at age 14 yrs, sitting only with sacral support. | Predominantly proximal muscle weakness, generalized muscle hypotrophy. | Progressive joint contractures in the neck, the elbows (>left), wrists, knees and lower limbs (>>right). Upper and lower limbs are in flexion. Kyphoscoliosis, Rigid spine. | None | Moderate respiratory difficulties appeared at 17 yrs. Now on BiPAP | 1770 | c.457T>C/ p.C153R |
| 9 | F | 34 yrs | 30 yrs | Muscle fatigability, difficulties with stairs. | Slowly progressive muscle weakness. | Moderate weakness to the upper limbs. | None | None | None | Normal | c.457T>C/ p.C153R |
| 10 | M | 21 yrs | 5 yrs | Poor walking endurance; frequent falls. | Wheelchair bound at 8 yrs of age. | Predominantly proximal muscle weakness. | Mild scoliosis; tightness of the Achilles tendons but no other severe contractures until the age of 18 yrs (when assessed). | Dilated Cardiomyopathy at 18 yrs (reduced fractional shortening). | Ventilatory support at 11 yrs of age (severe restrictive respiratory insufficiency) | 880 | c.458G>A/ p.C153Y |
| 11 | F | 40s | 30s | Difficulties with stairs and walking uphill. | Requires wheelchair for longer distances. | Predominantly proximal muscle weakness. Significant asymmetrical wasting of trapezii and shoulder muscles. | None | Normal Echo (6 years ago). | None | n.k. | c.458G>A/ p.C153Y |
Patients are listed by their localization of the mutation on the FHL1 gene.
Yrs = years; mo = months; BiPAP = bilevel positive airways pressure. CK = Serum creatine kinase level; < = some motor developmental delay noticed before clear onset of weakness; i.v. = intravenous, IG = immunoglobulin; n.k. = not known.
FHL1 sequencing
Exon 4 (third coding exon) of FHL1 of the genomic DNA of the patients and family members was amplified using Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA) with the following cycling parameters (primers for this and other FHL1 exons available on request) (Schessl et al., 2008): 3 min denaturation at 94°C, followed by 35 cycles of 94°C (30 s), 58°C (30 s), 72°C (1 min) and a final 72°C extension for 7 min. Purified PCR products were analysed in both strands by unidirectional sequencing with ABI PRISM BigDye Terminator cycle sequencing kit 3.1 and run on a 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA). Sequences were analysed using Sequencing Analysis 5.2 software (Applied Biosystems). The sequences were blasted against the human FHL1 clone MGC:15297 (GI 33872192, BC010998).
Morphological studies
Standard histochemical stains included haematoxylin and eosin, nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR), Congo red, adenosine triphosphatase (ATPase) 9.4 and Gomori trichrome were performed on frozen muscle samples (Fig. 2).
Fig. 2.
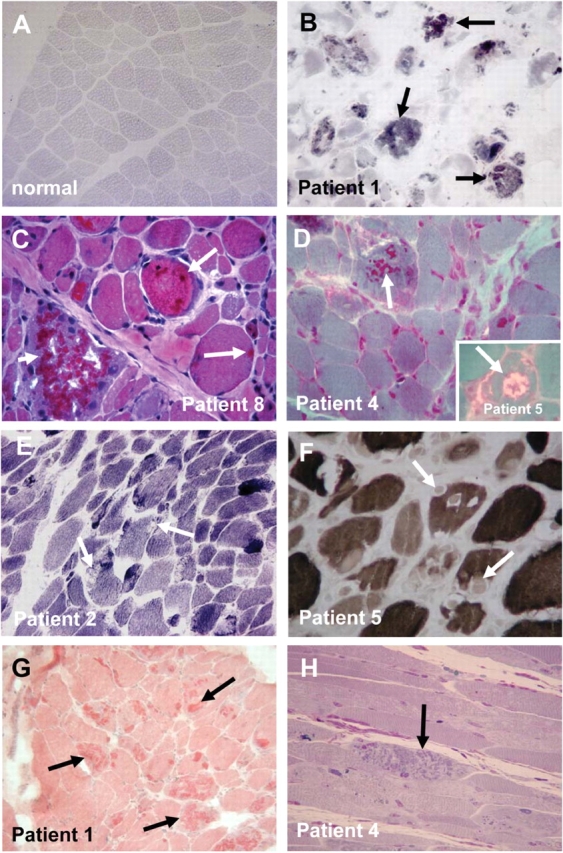
Histological appearance. (A and B) Menadione-NBT staining of normal muscle (A) and the muscle biopsy of Patient 1 with evident reactive intracytoplasmic aggregates (arrows) (B). (C–H) Haematoxylin and eosin staining demonstrates darkly staining aggregates (arrows), note degenerating fibre loaded with inclusions (short arrows) (C); modified Gomori trichrome demonstrates the dark red staining aggregates (arrows) contained in a subset of fibres (inset demonstrates a fibre with rimmed vacuole, arrows) (D). NADH (E), and ATPase at pH 9.4 (F), demonstrate that the aggregates are negative on these stains (arrows), but are positive on Congo red stain (arrows) (G). Semi thin section (H) shows characteristic aggregates (arrows) in a longitudinal section.
Menadione-NBT staining
Muscle sections were incubated at 37°C for 1 h in menadione-NBT solution (8 mg menadione (Sigma M5625, St. Louis, MO, USA), 1.3 ml acetone, 20 mg nitro-blue-tetrazolium, 20 ml Gomori–Tris–HCl buffer at pH 7.4) and followed by a series of washes in acetone (30–60–90–60–30%).
Immunohistochemistry
Methanol (room temperature) fixed frozen muscle tissue was processed as described before with goat polyclonal FHL1 (1:100, Abcam 23937, Cambridge, MA, USA) or rabbit polyclonal FHL1 (1:5, directed against a unique amino acid sequence located in the fourth LIM domain of the human FHL1, Monash University, Australia) (Schessl et al., 2008).
Electron microscopy
A small sample of the muscle biopsy was fixed in 2.5% glutaraldehyde and embedded in Polybed 812. Semithin sections were stained with methylene blue and ultrathin sections were stained with uranyl acetate and lead citrate (Reynolds) and examined under a Zeiss 109 electron microscope.
Immuno electron microscopy
Frozen tissue was thawed and then fixed in 4% paraformaldehyde with 0.2% glutaraldehyde, in 0.1 M sodium cacodylate buffer. After dehydration in graded alcohol at −20°C, the tissue was infiltrated and embedded in LRWhite resin, and polmerized at −20°C under long wave UV lamp. Using a Leica FCS ultramicrotome, 90-nm sections were collected on nickel grids. Non-specific binding was blocked with 1% ovalbumin + 0.2% cold water fish skin gelatin in 10 mM sodium phosphate buffer with 0.9% NaCl, pH 7.4. Sections were then incubated with primary antibody overnight at 4°C, washed in tris buffer multiple times and then transferred to gold labelled secondary IgG (10 nm). After 3 h of incubation at room temperature, the grids were rinsed in copious amounts of buffer, stained with aqueous uranyl acetate, and observed in a JEOL JEM 1010 Electron Microscope. Images were collected with Hamamatsu CCD Camera aided by AMT HR-12 software.
Results
Clinical presentation
Patients 1, 2, 3, 4, 5 and 7 were sporadic patients representing the severe early childhood onset form of RBM in females (Table 1). Patient 6 was a severely affected sporadic male and Patients 8–11 were members of two families with more significantly affected sons while the two mothers were less affected. In the patients with the earliest disease onset (<3 years) there was evidence for some degree of motor developmental delay during the first year of life in two of the four patients (Patients 1 and 2) (Table 1). Following a more static phase it was usually possible to define an age at which it became apparent that there was the beginning of a more progressive disorder. The earliest age of onset of this progressive phase was towards the end of the second year of life (Patients 1, 6 and 7). Only Patient 2 still appears to be stable at 2.5 years of age and may not yet have entered such a more progressive phase. In patients who lost ambulation, the time from perceived onset to loss of ambulation ranged from 18 months in the sporadic male patient (Patient 6) to 7 years in one of the familial male patients (Patient 8). Six patients developed respiratory insufficiency requiring ventilatory support either at night or permanently, and Patient 7 died from respiratory complications. Cardiomyopathy was present in one of the familial male patients (Patient 10). Scoliosis was present in seven patients, and was associated with significant spinal rigidity in Patients 1 and 8 (Fig. 1A, C and D). In the familial male Patient 8, the spinal rigidity was the presenting feature. Weakness of the neck muscles was noted in several patients. Patient 2 habitually hyperextended her neck when bending forward in order to prevent her head from dropping forward. Weakness in the upper extremity was present in a proximal and periscapular distribution leading to scapular winging (Fig. 1E). In the lower extremities there was evidence for proximal weakness, although some patients were noted to trip early, suggesting an element of foot drop or early Achilles tendon contractures. These contractures were a common and prominent feature of the more advanced disease phase with prominent pes adductus, consistent with underlying peroneal weakness. Contractures also developed in other joints to varying degree. With progression of the disease antigravity strength was lost and swallowing difficulties developed. Facial involvement with asymmetric ptosis was evident late in the course in Patient 1.
Fig. 1.

Clinical picture. (A) Patient 1, at 8 years of age is permanently wheelchair dependent, she is ventilated via tracheostomy and BiPAP and all nutrition and fluids are provided via gastrostomy tube. Her antigravity strength is limited to her finger extensors and toes. There is facial weakness and asymmetrical ptosis. (B) The male Patient 6 at age 2 years is completely paralysed requiring ventilation via tracheotomy and nutrition via G-tube. Note foot contractures. (C and D) Familial Patient 8 demonstrating scoliosis, spinal rigidity and contractures of knees and elbows in a pattern reminiscent of the Emery-Dreifuss phenotype. (E) Patient 3 showing early prominent periscapular involvement leading to scapular winging.
Illustrative case histories
Sporadic female patient
Patient 1 is a girl with initially mild delays in motor development (Fig. 1A). She achieved walking at 18 months of age but was never able to run nor able to stand up from a sitting position. CK was initially normal and was slightly elevated at 337 U/l (normal 75–230 U/l) at 2 years of age when poor head control and neck weakness were noted and accelerated progression of the disease began, leading to loss of ambulation and wheelchair dependency at 3 years of age. At that age she was found to have generalized muscle weakness and hypotonia, which appeared to be more pronounced proximally. She developed progressive spinal rigidity as well as scoliosis, requiring spinal fusion at age 6 years and 10 months. Respiratory insufficiency developed and required non-invasive ventilatory support at age 4 years. At age 6 years 10 months, at the time of her spinal surgery she had a tracheostomy and gastrostomy tube (G-tube) placed. Currently at age 8 years she is ventilated continuously via BiPAP with all nutrition and fluids provided via G-tube. She has lost all antigravity strength except for her finger extensors. There is facial weakness and asymmetrical ptosis, but extraocular movements are full. Up to now, no cardiac involvement has been diagnosed.
Sporadic male patient
This boy (Patient 6) at a routine medical examination at age 13 months was found to have had apparently normal development and no difficulties walking (Fig. 1B). When seen again at age 17 months he in the interim had developed significant weakness in a more proximal distribution. He was not able to rise from the floor, climb stairs or roll over in bed. His walking distance was limited to 50 m with assistance. There was significant axial and cervical muscle weakness with associated head drop but no evidence of frank facial or extraocular involvement. There was no spinal rigidity at that point. Reflexes were abolished. CK was elevated at 2000 U/l. Motor and sensory nerve conduction velocities were normal. He was still able to handle secretions and was able to swallow. However, 9 months later his weakness had progressed further and he could not walk >5 m without assistance and was falling frequently. He had no antigravity strength in his quadriceps muscle and he was not able to raise his arms above his shoulders. Cough and respiratory effort were diminishing. Four months later (at 30 months of age) he was no longer able to ambulate and had lost all proximal antigravity strength, and required continuous ventilatory support.
Familial case
Patient 8 is a boy born after a normal pregnancy and delivery followed by normal psychomotor development (Fig. 1C and D). He presented with thoracolumbar spinal rigidity at 7 years of age. At that point no overt muscle weakness or fatigability was noted. His CK value was elevated at 1770 U/l. At 10 years of age the spinal rigidity had spread to involve the cervical spine, while contractures were now also evident in the elbows and wrists. He then started to develop progressive weakness leading to loss of ambulation at the age of 14 years. Kyphoscoliosis had developed in addition to the spinal rigidity. Contractures of the neck, the elbows, wrists, knees and lower limbs had also progressed significantly. Moderate respiratory difficulties manifested at age 17 years and he is now ventilated via nocturnal BiPAP. No cardiac involvement was evident at this point. His mother (Patient 9) demonstrated first symptoms at age 30 years with muscle fatigue and difficulties climbing stairs. She had mild proximal weakness, but no contractures. She now can walk not more than 2 h. No cardiac or respiratory involvements are evident in her as of yet.
Morphological studies
All patients except for Patient 9 (mother of Patient 8) had at least one muscle biopsy demonstrating the presence of reducing bodies staining positively with the menadione-NBT stain (Fig. 2A and B), thus fulfilling the main histochemical criterion for RBM. The bodies appeared purple red on haematoxylin and eosin (Fig. 2C), and intensely red on the modified Gomori trichrome stain (Fig. 2D). They displaced both NADH as well ATPase stainings (Fig. 2E and F) and were positively stained using Congo red. The aggregates were easily recognizable on semi-thin sections (Fig. 2H). Muscle biopsies displayed variable myopathic findings including variability in fibre diameter and evidence of muscle fibre degeneration in some sections. Muscle fibre degeneration was more severe in biopsies with a high number of reducing bodies, in which individual fibres with the highest load of aggregates seem to undergo degeneration (Fig. 2C, short arrow). Occasional rimmed vacuoles were evident in the modified Gomori trichrome stain (Fig. 2D inset). Immunohistochemistry using antibodies directed against FHL1 confirmed that the intracytoplasmic aggregates were highly enriched for FHL1 immunoreactivity (Fig. 3A). Analysis of serial sections demonstrated that the FHL1 positive structures were identical to the menadione-NBT positive reducing bodies (Fig. 3A); although we cannot yet conclude that absolutely all FHL1 positive deposits will also be menadione-NBT positive or that all menadione-NBT positive structures will be FHL1 positive. FHL1 positive aggregates were often situated in close association with myonuclei (Fig. 3C–F). In fibres without clear aggregates FHL1 was normally localized to the I-band/Z-disc, which was also evident in a male patient, expressing mutant FHL1 only (Patient 6). In contrast, in fibres with prominent aggregates there was a marked reduction of FHL1 immunolabelling of the myofibrils in the vicinity of the aggregates as noted by us before (Schessl et al., 2008). Three of the patients had two biopsies obtained at different times during the course of the disease. In two of these (Patients 6 and 7) there appeared to be an increase in the number of FHL1 positive aggregates between the first and second biopsy specimen examined, suggesting that the increased formation of aggregates may correlate with the clinical progression of the disease, although the potential of muscle-to-muscle variability of aggregate formation has to be taken into account (Fig. 3B and C).
Fig. 3.

Immunohistochemical analysis. (A) Serial sections stained with menadione-NBT and immunostained with anti-FHL1 antibody (directed against a unique amino acid sequence located in the fourth LIM domain of the human FHL1, Monash University, Australia) demonstrating that the menadione-NBT positive aggregates are also FHL1 immunoreactive (arrows). (B and C) FHL1 immunostaining on sequential biopsies from the quadriceps from Patient 6, taken at 2 years of age (B) and 2¼ years of age (C), demonstrate an increase of FHL1 positive aggregates (arrows) (even allowing for the relatively poor preservation of the first biopsy material). (D–F) FHL1 positive aggregates in RBM are also immunoreactive for the centrosomal component pericentrin (D) as well as for gamma-tubulin (F). Gamma-tubulin and pericentrin co-localize for most, but not all aggregates (E).
It has been suggested that reducing bodies have some features of aggresomes (Liewluck et al., 2007). To further investigate proteins known to be associated with aggresomes, we confirmed that gamma-tubulin was included in the aggregates as has been reported previously (Fig. 3E) (Liewluck et al., 2007). We performed immunolabelling for the centrosomal marker pericentrin as typical aggresomes tend to assume a position close to a microtubular organizing center, thereby incorporating centrosomal components including pericentrin (Johnston et al., 2002). We found the aggregates to be strongly positive for pericentrin immunoreactivity, although like gamma-tubulin in a diffuse pattern (Fig. 3D and F) and not as a distinct centrosomal structure.
Ultrastructural analysis confirmed the typical electron-dense appearance of the aggregates in all biopsies (Fig. 4). As observed previously the aggregates were present throughout the muscle fibre, but were often seen in association with myonuclei (Fig. 4A–C) (Brooke and Neville, 1972; Tome and Fardeau, 1975; Hubner and Pongratz, 1981; Oh et al., 1983; Carpenter et al., 1985; Kiyomoto et al., 1995a,b; Reichmann et al., 1997; Figarella-Branger et al., 1999; Ikezoe et al., 2004; Shinde et al., 2004; Ohsawa et al., 2006). Although the electron-dense material was very close to the myonuclear membrane, sometimes even appearing to indent the nucleus, it did not appear to be in continuity with the nuclear membrane (Fig. 4A–C). In some instances there was a thin ribbon of electron-dense material around the nucleus, but again separated from the outer nuclear membrane by a visible gap (Fig. 4B). In addition, ultrastructural analysis revealed less dense bodies with a radial halo of filaments in all the patient biopsies analysed. These structures most closely resembled cytoplasmic bodies (Fig. 4D and E) (Brooke and Neville, 1972; Tome and Fardeau, 1975; Hubner and Pongratz, 1981; Carpenter et al., 1985; Bertini et al., 1994; Reichmann et al., 1997; Goebel et al., 2001; Shinde et al., 2004). They were often seen interspersed in between the darker appearing reducing bodies. We also detected occasional vacuolar structures possibly resembling rimmed vacuoles/autophagosomes (Fig. 4F). Occasional dilated sarcotubular systems (not shown) were seen in some of the sections.
Fig. 4.
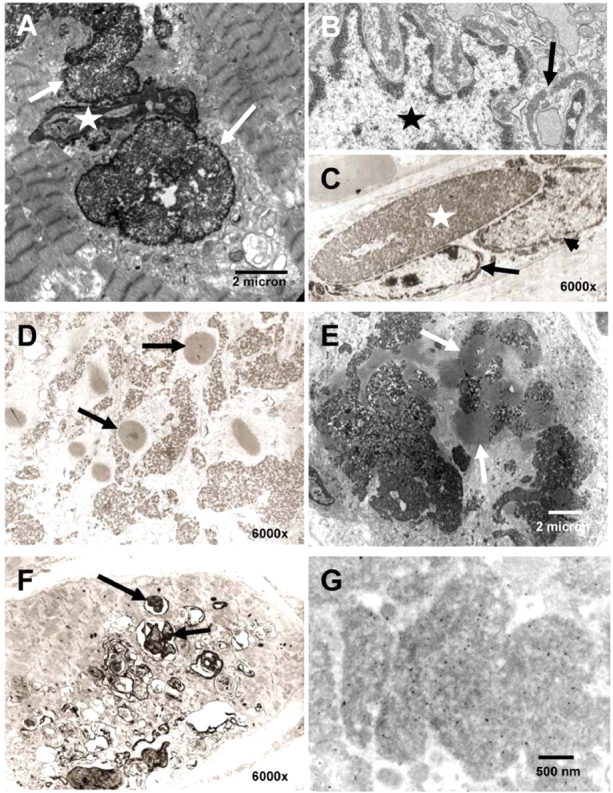
Ultrastructural analysis. (A–C) Localization of aggregates (arrows) in close proximity to a nucleus (star), appearing to indent the nucleus (A). Dense material is seen surrounding the myonucleus (star) as a thin band (B). (D and E) Cytoplasmic bodies (arrows) were found in frequent coexistence with the reducing bodies in all specimens examined. (F) Rimmed vacuoles (arrows) occur in some fibres in RBM (compare Fig. 1D inset). (G) Immuno EM: gold-particles indicate binding of the FHL1 antibody to the major electron-dense aggregates.
Immuno electron microscopy (EM) was difficult to perform since none of the EM material had been properly embedded for immuno EM at the time of the biopsy, thus requiring re-embedding of material from the frozen biopsies. Even though the ultrastructure was not optimal, gold-labelled FHL1 antibody was strongly enriched in the electron-dense inclusions (Fig. 4G). Some binding was also seen adjacent to the Z-line in the expected normal location for FHL1 in skeletal muscle (McGrath et al., 2006).
Identification of FHL1 mutations in sporadic and familial patients
All mutations identified were located in the second LIM domain of FHL1 encoded on the third coding exon (exon 4 overall, as exon 1 is noncoding). In the six sporadic female patients heterozygous mutations were detected (c.367C > T resulting in p.H123Y in Patients 1 and 2, c.368A > T resulting in p.H123L in Patient 3, c.369C > G resulting in p.H123Q in Patients 4 and 5 and c.395G > T resulting in p.C132F in Patient 7) (Table 1; Fig. 5A and B). None of the mutations detected in the girls were present in the parents or unaffected siblings and had thus arisen de novo. Patients 1 and 7 were part of our original report (Schessl et al., 2008).
Fig. 5.
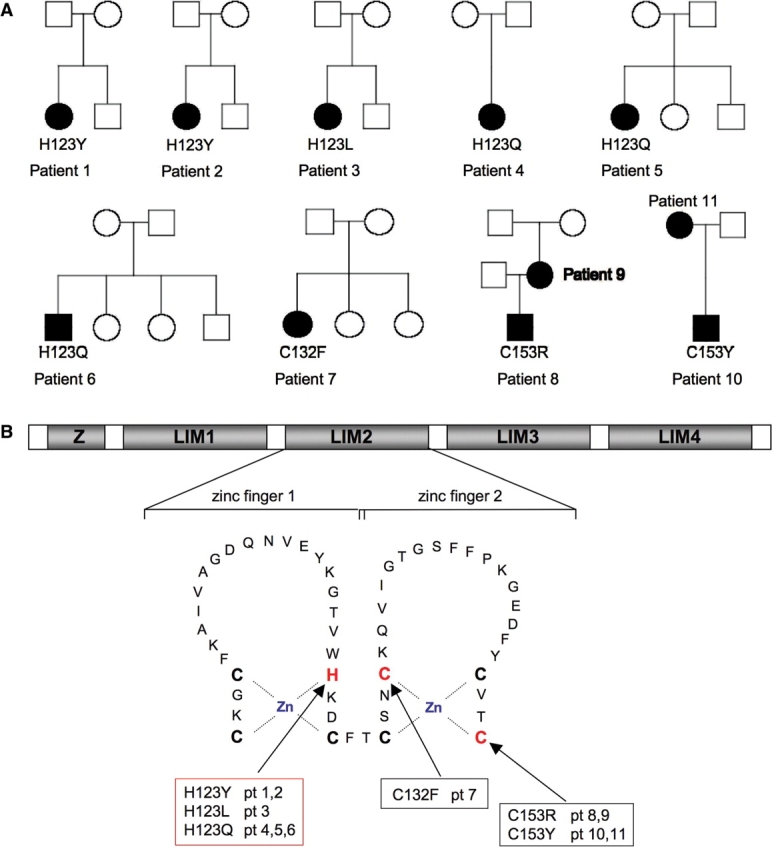
Genetic analysis. (A) Pedigrees of families with affected members, indicating mutations in FHL1. Solid symbols designate affected individuals carrying the mutation indicated. Open symbols indicate unaffected individuals not carrying the mutation. (B) Schematic representation of the domain structure of FHL1, comprised of four LIM domains (LIM1-4) with an additional N-terminal half-LIM domain (Z). The secondary structure of the second LIM domain is indicated with the Zn coordinating residues highlighted in bold, and mutated residues in addition in red. Histidine 123 is mutated independently in six patients.
Sequencing in the most severely affected sporadic male (Patient 6) identified homozygous c.369C > A resulting in p.H123Q. The mutation was not present in the boy's mother and thus arose de novo. The mutations in the two familial cases with comparatively milder manifestations (c.457T > C resulting in p.C153R in Patient 8 and his mother and c.458G > A resulting in the p.C153Y change in the same amino acid in Patient 10 and his mother) were part of our original report (Table 1, Fig. 5A and B). All of the mutations affect invariant consensus cysteine or histidine residues involved in coordinating zinc binding in the second LIM domain of FHL1 (Fig. 5C). None of the mutations were detected in 100 control X chromosomes.
Discussion
Using laser microdissection of reducing bodies followed by proteomic analysis to identify FHL1 as the major component within the aggregates, we recently identified mutations in the X-chromosomal gene FHL1 as the cause of sporadic as well as familial RBM (Schessl et al., 2008). This extended series of 11 patients now defines the clinical and morphological phenotypes in relation to RBM-associated mutations in FHL1.
Clinical analysis
The clinical severity observed in this series ranged from early childhood onset with severe progression in the sporadic patients to later childhood and adult onset cases in our two familial observations. The seven sporadic patients (six girls and one boy) presented in early childhood followed by a rather rapidly progressive course in most, which may lead to loss of ambulation at a young age and continued progression to respiratory failure. This early onset phenotype is consistent with the original description of the disease by Brooke and Neville (1972), reporting two girls with an even more severe and progressive course leading to death at respectively 9 months and 2.5 years of age due to respiratory failure. In our series two of the four patients with onset <3 years had some delay in motor development before recognition of clinically evident weakness. Strikingly, Patient 6 had normal motor development until onset of an extremely rapidly progressive course at age 13 months leading to loss of ambulation within a few months. With its at times rapidly progressive weakness and its sometimes symptom-free period in infancy, RBM thus behaves differently from a prototypical non-progressive congenital myopathy with which it is commonly classified (Goebel et al., 2001). Distribution of muscle weakness overall was more proximal with evidence for early scapuloperoneal involvement. Spinal rigidity may be prominent and a presenting features (Patient 8). Thus RBM should be considered in the differential diagnosis of patients with rigid spine syndrome and in patients with scapuloperoneal muscle involvement, i.e. in cases that may be reminiscent of Emery–Dreifuss muscular dystrophy. One patient (Patient 10) had cardiomyopathy, which was not felt to be secondary to respiratory failure. Possible cardiomyopathy may not develop until later in the course but should be screened for in patients with RBM given the cardiac expression of FHL1. In particular for the later onset cases of FHL1-associated RBM there may be considerable clinical and morphological overlap with the myofibrillar myoapathies (MFM), including the desminopathies, in which cardiac involvement may also occur (Selcen, 2008). It thus seems reasonable to recommend menadione-NBT staining and mutation analysis in FHL1 in cases of MFM that have remained unexplained.
Twenty-one patients with RBM have been documented in the literature so far, obviously at this point without molecular confirmation (Supplementary Table). Six of these cases (including the original two patients reported by Brooke and Neville) were severely affected girls with onset at birth and early childhood (birth to 3¾ years), and a rapidly progressive course leading to death in five patients between the age of 9 months and 6¾ years mostly due to respiratory failure (Brooke and Neville, 1972; Dubowitz, 1978; Carpenter et al., 1985; Kobayashi et al., 1992; Kiyomoto et al., 1995b; Goebel et al., 2001). Four girls were reported to have later onset between age 9 and 13 years with progression to loss of ambulation between the age of 13 and 27 years (Hubner and Pongratz, 1981; Hubner and Pongratz, 1982; Nomizu et al., 1992; Bertini et al., 1994). Clinical symptoms of the disease in adulthood are reported in six additional female patients, with onset between 22 and 50 years of age (Keith and Brownell, 1990; Kiyomoto et al., 1995a; Figarella-Branger et al., 1999; Goebel et al., 2001; Shinde et al., 2004; Ohsawa et al., 2006). Five of these patients progressed slowly, but one patient had a rapidly progressive course leading to loss of ambulation within 5 years after onset. Five reported male cases presented with onset of the disease between age 17 months and 10.5 years followed by variable progression of the disease (Tome and Fardeau, 1975; Oh et al., 1983; Reichmann et al., 1997; Goebel et al., 2001; Ohsawa et al., 2006). Prior to our report four familial cases of RBM have been reported, notably without any male-to-male transmission, consistent with the X-chromosomal location of FHL1 (Hubner and Pongratz, 1981; Hubner and Pongratz, 1982; Reichmann et al., 1997; Goebel et al., 2001; Ohsawa et al., 2006). In all familial reports to date the disease was always more severe in affected male offspring compared to affected mothers and grandmothers, as is the case in our two familial occurrences (Patients 8–11). The striking spinal rigidity seen in one of the familial male cases (Patient 8) is highly similar to the clinical picture in the familial observations of Goebel et al. (2001). In this latter family we have now also identified a missense mutation in the second LIM domain affecting cysteine 150, thus localized adjacent to the familial mutation in Patient 8 (Schessl et al., unpublished result).
Histological analysis
The degree of myopathic features in the biopsies appeared to reflect the number of intracytoplasmic aggregates so that in the most severely affected biopsies there was evidence for a widespread destructive myopathy with ubiquitous aggregates, correlating with an elevated CK level of 2000 U/l in the most progressive patient (Patient 6). However, at this point we would be hesitant to use the term dystrophic for this myopathy as the mechanisms of cell damage remain to be fully worked out. The morphological suggestion that the aggregates in muscle build up over time as the basis of progression of the myopathy as evidenced in the repeat biopsies of two (Patients 6 and 7) is also consistent with our observation of rapidly progressive formation of inclusions after transfection of mutant FHL1 constructs into C2C12 cells (Schessl et al., 2008). Given the possibility that a single biopsy in this disease may not show obvious aggregates (as a result of an early biopsy or because there may be different involvement in different muscles), a diagnosis of RBM may have to be considered even in the absence of specific pathological evidence in the appropriate clinical context. Our observation of normally localized FHL1 immunoreactivity in fibres without aggregates in a male patient suggests that the mutant FHL1 is able to localize normally in the muscle fibre, as only mutant FHL1 is expressed in affected males. This would imply that part of the pathomechanism of this disorder relates to the formation of the aggregates rather than solely due to a primary dysfunction of FHL1, although FHL1 will become depleted as it is progressively trapped in the aggregates. It is unclear whether the aggregates themselves are toxic, or whether the depletion of a number of important cellular proteins that are trapped in the aggregates (Liewluck et al., 2007; Schessl et al., 2008) contributes to the demise of the cell.
The aggregates in RBM have been noticed to have morphological and immunohistochemical features reminiscent of aggresomes (Liewluck et al., 2007). Aggresomes are aggregates of abnormal protein tagged for degradation that form after the functions of the proteasome have been exhausted (Kopito, 2000). Aggresomes form at microtubule organizing centers and therefore are usually found in a juxtanuclear position, a feature that has also been pointed out for reducing bodies (Brooke and Neville, 1972; Tome and Fardeau, 1975; Hubner and Pongratz, 1981; Oh et al., 1983; Carpenter et al., 1985; Kiyomoto et al., 1995a, b; Reichmann et al., 1997; Figarella-Branger et al., 1999; Kopito, 2000; Ikezoe et al., 2004; Shinde et al., 2004; Ohsawa et al., 2006). Gamma-tubulin is associated with microtubule organizing centers, centrosomes and aggresomes (Oakley and Oakley, 1989; Joshi et al., 1992) and has been shown to be associated with reducing bodies, which we were now able to confirm in our FHL1 mutation proven cases (Liewluck et al., 2007). Expanding on this observation we find that the aggregates in RBM also contain pericentrin, a crucial centrosomal component, which is also contained in aggresomes in other pathological systems, such as Lewy bodies (Doxsey et al., 1994; Johnston et al., 2002; McNaught et al., 2002). Although reducing bodies have at least some of the features of aggresomes more analysis in model systems will have to be done before a final conclusion about the relationship of aggresomes and reducing bodies can be arrived at.
Ultrastructural analysis
Ultrastructural analysis in our cases also confirms the frequent but not exclusive juxtanuclear position of the intracytoplasmic aggregates. In some samples there also was a band of material surrounding the myonuclei, which was of similar electron density as the typical aggregates. However, the exact composition of this material and its relationship to the endoplasmatic reticulum in particular will have to remain open at this point. It has been pointed out that the reducing bodies may be part of a ‘mixed myopathy’ because they have been observed in conjunction with cytoplasmic bodies (Goebel et al., 2001). Analysis of EM material from our patients confirms that cytoplasmic bodies is found in frequent coexistence with the reducing bodies, confirming that this mixed myopathy can also be caused by FHL1 mutations. In fact, in our series the presence of cytoplasmic bodies appears to be the rule and not an exception and should therefore be viewed as an integral part of RBM. It will be interesting to investigate whether myopathies subsumed under the label ‘cytoplasmic body myopathies’ can be caused by FHL1 mutations.
Genetic analysis
This series expands the mutation spectrum in RBM to 7 distinct FHL1 mutations in 11 patients. These extended observations support our initial finding that mutations in RBM were restricted to the second LIM domain of FHL1, and thereby affects all three splice forms of FHL1 (FHL1A, B, C) (Brown et al., 1999; Ng et al., 2001; Schessl et al., 2008). Furthermore, we define histidine 123 as a crucial residue mutated in severe RBM, as all of our additional patients carried de novo mutations in this residue (H123Y in 2, H123L in 1, H123Q in 3 patients), bringing the total of patients with independent de novo mutations in this residue to six (Patients 1–6, Fig. 5). Histidine 123 is evolutionarily conserved in all species down to mosquitoes and is involved in coordinating zinc in the first zinc finger motif in the second LIM domain (Schessl et al., 2008). Mutations in this residue likely severely disrupt the conformation of the domain, presumably thereby starting the process of misfolding and aggregation. This is supported by our previously reported data in which we transfected constructs of the mutation H123Y into COS7 and C2C12 cells (Schessl et al., 2008). As pointed out by us before, the somewhat milder phenotype associated with mutations in C153 likely is related to the location of that residue as the very last of the zinc coordinating residues of the second LIM domain already within an alpha-helical structure, leading to less severe disruption of the structure (Schessl et al., 2008). Mutations in this X-linked disease act in a dominant way, however, a female patient with a given mutation will be less affected compared to a male patient with the same mutation as lyonization results in a mix of mutant and wild-type nuclear domains in female muscle. The nuclear domains expressing wild-type FHL1 will thereby dilute the aggregation effect of the mutant FHL1. Thus, the male patients in our series were more severely affected compared with female patients with the same mutation as is evident in the familial observation and in the sporadic male patient with the mutation in H123Q, showing much more rapid progression of the disease compared with the female Patients 4 and 5 also carrying H123Q. Skewed X-inactivation in a female patient could obviously modify this scenario considerably, leading to milder or more severe disease (Schessl et al., 2008).
Independent from our initial observation of FHL1 mutations in RBM, the mutation W122S in the FHL1 gene was reported as the cause for an adult onset X-linked dominant scapuloperoneal myopathy in which males were more severely and earlier affected than female patients (Quinzii et al., 2008) (see also Supplementary Table). Following our report, muscle biopsies from patients from this family were reexamined and found to contain menadione-NBT positive reducing bodies, suggesting that this family in fact represents the milder end of RBM (M. Hirano, personal communication). The mutation segregating in this family is located in the second LIM domain adjacent to the frequently mutated residue H123, but does not directly change any of the zinc coordinating residues, thus likely accounting for the comparatively milder phenotype in this family. In parallel Windpassinger et al. described a novel form of X-linked recessive myopathy with postural muscle atrophy and generalized hypertrophy (XMPMA), caused by a missense mutation within the fourth LIM domain of FHL1 in a large family and by an insertion mutation within the second LIM domain in another family with an identical phenotype (Windpassinger et al., 2008) (see also Supplementary Table). Only males were affected and onset of the disease in these patients was in their 30s. No reducing bodies were described, although there were core-like lesions and subsarcolemmal foci of desmin positivity. This phenotype does appear to be different from RBM, suggesting a broader clinical spectrum of myopathies due to FHL1 mutations.
In summary, we have confirmed that mutations in the second LIM domain of FHL1 are associated with RBM and are refining the clinical and morphological spectrum of this disease. This information should provide the necessary basis for further analysis and diagnosis of patients with appropriate clinical and histological phenotypes. With ongoing mutation analysis in FHL1 we expect the phenotype to broaden even further, shedding additional light on the role of FHL1 and aggregate formation in disorders of muscle. A recent report (Shalaby et al. Neuromuscul Disord 2008; 18: 959–61) describes a male patient with a three amino acid deletion (p.151–153delVTC) in the second LIM domain presenting with spinal rigidity and reducing bodies in the biopsy, consistent with the clinical and molecular observations we report here.
Supplementary material
Supplementary material is available at Brain online.
Funding
C.G.B. is supported by grants from NIH/NIAMS (RO1AR051999) and from MDA USA (MDA3896). The MRC Translational research grant and the MDA USA grant (MDA on congenital myopathies) to F.M. are also gratefully acknowledged. J.S., H.H.G. and C.G.B are also members of the German network on muscular dystrophies (MD-NET) funded by the German Ministry of Education and Research (BMBF, Bonn, Germany). MD-NET is a partner of TREAT-NMD.
Acknowledgements
Foremost we thank the patients and their families reported here for their participation and encouragement. We thank Qian-Chun Yu, MB, PhD and Raymond Meade (Biomedical Imaging Core Facility, University of Pennsylvania, Philadelphia, PA, USA) for processing the Immuno EM samples. We thank Tom Crawford, MD (The Johns Hopkins Hospital, Baltimore, MD, USA), Daniel Galizzi, MD, Alejandro Teper, MD, M. Elisa Gurmandi, MD (R. Gutierrez Children's Hospital, Buenos Aires, Argentina), Carlo Minetti, MD (G. Gaslini's Institute, Genua, Italy) for clinical information and/or patient specimen. We thank Michio Hirano, MD, (Columbia University, New York, NY, USA) for communicating unpublished findings.
Glossary
Abbreviations
- BiPAP
bilevel positive airways pressure
- CK
creatine kinase
- EM
electron microscopy
- FHL1
four and a half LIM domain 1
- LIM
Lin-11, Isl-1, Mec-3
- NADH-TR
nicotinamide adenine dinucleotide tetrazolium reductase
- NBT
nitro-blue-tetrazolium
- RBM
reducing body myopathy
References
- Bertini E, Salviati G, Apollo F, Ricci E, Servidei S, Broccolini A, et al. Reducing body myopathy and desmin storage in skeletal muscle: morphological and biochemical findings. Acta Neuropathol (Berl) 1994;87:106–12. doi: 10.1007/BF00386261. [DOI] [PubMed] [Google Scholar]
- Brooke MH, Neville HE. Reducing body myopathy. Neurology. 1972;22:829–40. doi: 10.1212/wnl.22.8.829. [DOI] [PubMed] [Google Scholar]
- Brown S, McGrath MJ, Ooms LM, Gurung R, Maimone MM, Mitchell CA. Characterization of two isoforms of the skeletal muscle LIM protein 1, SLIM1. Localization of SLIM1 at focal adhesions and the isoform slimmer in the nucleus of myoblasts and cytoplasm of myotubes suggests distinct roles in the cytoskeleton and in nuclear-cytoplasmic communication. J Biol Chem. 1999;274:27083–91. doi: 10.1074/jbc.274.38.27083. [DOI] [PubMed] [Google Scholar]
- Carpenter S, Karpati G, Holland P. New observations in reducing body myopathy. Neurology. 1985;35:818–27. doi: 10.1212/wnl.35.6.818. [DOI] [PubMed] [Google Scholar]
- Curtiss J, Heilig JS. DeLIMiting development. Bioessays. 1998;20:58–69. doi: 10.1002/(SICI)1521-1878(199801)20:1<58::AID-BIES9>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Dawid IB, Toyama R, Taira M. LIM domain proteins. C R Acad Sci III. 1995;318:295–306. [PubMed] [Google Scholar]
- Doxsey SJ, Stein P, Evans L, Calarco PD, Kirschner M. Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell. 1994;76:639–50. doi: 10.1016/0092-8674(94)90504-5. [DOI] [PubMed] [Google Scholar]
- Dubowitz V. Muscle Disorders in Childhood. London: W.B. Saunders; 1978. [Google Scholar]
- Figarella-Branger D, Putzu GA, Bouvier-Labit C, Pouget J, Chateau D, Fardeau M, et al. Adult onset reducing body myopathy. Neuromuscul Disord. 1999;9:580–6. doi: 10.1016/s0960-8966(99)00058-9. [DOI] [PubMed] [Google Scholar]
- Freyd G, Kim SK, Horvitz HR. Novel cysteine-rich motif and homeodomain in the product of the Caenorhabditis elegans cell lineage gene lin-11. Nature. 1990;344:876–9. doi: 10.1038/344876a0. [DOI] [PubMed] [Google Scholar]
- Goebel HH, Halbig LE, Goldfarb L, Schober R, Albani M, Neuen-Jacob E, et al. Reducing body myopathy with cytoplasmic bodies and rigid spine syndrome: a mixed congenital myopathy. Neuropediatrics. 2001;32:196–205. doi: 10.1055/s-2001-17374. [DOI] [PubMed] [Google Scholar]
- Hubner G, Pongratz D. Reducing body myopathy—ultrastructure and classification (author's transl) Virchows Arch A Pathol Anat Histol. 1981;392:97–104. doi: 10.1007/BF00430552. [DOI] [PubMed] [Google Scholar]
- Hubner G, Pongratz D. Granular body myopathy (so-called reducing body myopathy) Pathologe. 1982;3:111–3. [PubMed] [Google Scholar]
- Ikezoe K, Nakagawa M, Osoegawa M, Kira J, Nonaka I. Ultrastructural detection of DNA fragmentation in myonuclei of fatal reducing body myopathy. Acta Neuropathol (Berl) 2004;107:439–42. doi: 10.1007/s00401-004-0831-5. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Illing ME, Kopito RR. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil Cytoskeleton. 2002;53:26–38. doi: 10.1002/cm.10057. [DOI] [PubMed] [Google Scholar]
- Joshi HC, Palacios MJ, McNamara L, Cleveland DW. Gamma-tubulin is a centrosomal protein required for cell cycle-dependent microtubule nucleation. Nature. 1992;356:80–3. doi: 10.1038/356080a0. [DOI] [PubMed] [Google Scholar]
- Jurata LW, Gill GN. Structure and function of LIM domains. Curr Top Microbiol Immunol. 1998;228:75–113. doi: 10.1007/978-3-642-80481-6_4. [DOI] [PubMed] [Google Scholar]
- Kadrmas JL, Beckerle MC. The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol. 2004;5:920–31. doi: 10.1038/nrm1499. [DOI] [PubMed] [Google Scholar]
- Keith A, Brownell W. Reducing body myopathy in an adult. J Neurol Sci. 1990;98:336. [Google Scholar]
- Kiyomoto BH, Murakami N, Kishibayashi J, Sunohara N, Nonaka I. Reducing bodies in distal myopathy with rimmed vacuole formation. Acta Neuropathol (Berl) 1995a;89:109–11. doi: 10.1007/BF00294268. [DOI] [PubMed] [Google Scholar]
- Kiyomoto BH, Murakami N, Kobayashi Y, Nihei K, Tanaka T, Takeshita K, et al. Fatal reducing body myopathy. Ultrastructural and immunohistochemical observations. J Neurol Sci. 1995b;128:58–65. doi: 10.1016/0022-510x(94)00204-2. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Nihei K, Kuwajima K, Nonaka I. Reducing body myopathy—a case report. Rinsho Shinkeigaku. 1992;32:62–7. [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–30. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Liewluck T, Hayashi YK, Ohsawa M, Kurokawa R, Fujita M, Noguchi S, et al. Unfolded protein response and aggresome formation in hereditary reducing-body myopathy. Muscle Nerve. 2007;35:322–6. doi: 10.1002/mus.20691. [DOI] [PubMed] [Google Scholar]
- McGrath MJ, Cottle DL, Nguyen MA, Dyson JM, Coghill ID, Robinson PA, et al. Four and a half LIM protein 1 binds myosin-binding protein C and regulates myosin filament formation and sarcomere assembly. J Biol Chem. 2006;281:7666–83. doi: 10.1074/jbc.M512552200. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Shashidharan P, Perl DP, Jenner P, Olanow CW. Aggresome-related biogenesis of Lewy bodies. Eur J Neurosci. 2002;16:2136–48. doi: 10.1046/j.1460-9568.2002.02301.x. [DOI] [PubMed] [Google Scholar]
- Ng EK, Lee SM, Li HY, Ngai SM, Tsui SK, Waye MM, et al. Characterization of tissue-specific LIM domain protein (FHL1C) which is an alternatively spliced isoform of a human LIM-only protein (FHL1) J Cell Biochem. 2001;82:1–10. doi: 10.1002/jcb.1110. [DOI] [PubMed] [Google Scholar]
- Nomizu S, Person DA, Saito C, Lockett LJ. A unique case of reducing body myopathy. Muscle Nerve. 1992;15:463–6. doi: 10.1002/mus.880150408. [DOI] [PubMed] [Google Scholar]
- Oakley CE, Oakley BR. Identification of gamma-tubulin, a new member of the tubulin superfamily encoded by mipA gene of Aspergillus nidulans. Nature. 1989;338:662–4. doi: 10.1038/338662a0. [DOI] [PubMed] [Google Scholar]
- Oh SJ, Meyers GJ, Wilson ER, Jr, Alexander CB. A benign form of reducing body myopathy. Muscle Nerve. 1983;6:278–82. doi: 10.1002/mus.880060406. [DOI] [PubMed] [Google Scholar]
- Ohsawa M, Liewluck T, Ogata K, Iizuka T, Hayashi Y, Nonaka I, et al. Familial reducing body myopathy. Brain Dev. 2006;29:112–116. doi: 10.1016/j.braindev.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Quinzii CM, Vu TH, Min KC, Tanji K, Barral S, Grewal RP, et al. X-Linked Dominant Scapuloperoneal Myopathy Is Due to a Mutation in the Gene Encoding Four-and-a-Half-LIM Protein 1. Am J Hum Genet. 2008;82:208–13. doi: 10.1016/j.ajhg.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmann H, Goebel HH, Schneider C, Toyka KV. Familial mixed congenital myopathy with rigid spine phenotype. Muscle Nerve. 1997;20:411–7. doi: 10.1002/(sici)1097-4598(199704)20:4<411::aid-mus2>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Schessl J, Zou Y, McGrath MJ, Cowling BS, Maiti B, Chin SS, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 2008;118:904–12. doi: 10.1172/JCI34450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcen D. Myofibrillar myopathies. Curr Opin Neurol. 2008;21:585–9. doi: 10.1097/WCO.0b013e32830a752b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde A, Nakano S, Kusaka H, Nakaya Y, Sawada H, Kohara N, et al. Nucleolar characteristics of reducing bodies in reducing body myopathy. Acta Neuropathol (Berl) 2004;107:265–71. doi: 10.1007/s00401-003-0806-y. [DOI] [PubMed] [Google Scholar]
- Tome FM, Fardeau M. Congenital myopathy with "reducing bodies" in muscle fibres. Acta Neuropathol (Berl) 1975;31:207–17. doi: 10.1007/BF00684560. [DOI] [PubMed] [Google Scholar]
- Windpassinger C, Schoser B, Straub V, Hochmeister S, Noor A, Lohberger B, et al. An X-Linked Myopathy with Postural Muscle Atrophy and Generalized Hypertrophy, Termed XMPMA, Is Caused by Mutations in FHL1. Am J Hum Genet. 2008;82:88–99. doi: 10.1016/j.ajhg.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.