

Abstract
Descending input from the rostral ventromedial medulla (RVM) provides positive and negative modulation of spinal nociceptive transmission and has been proposed to be critical for maintaining neuropathic pain. This study tests the hypothesis that neuropathic pain requires the activity of a subset of RVM neurons that are distinguished by co-expression of mu opioid receptor (MOR) and cholecystokinin type 2 receptor (CCK2). Using male Sprague–Dawley rats, we demonstrate that discrete RVM neurons express MOR and CCK2; over 80% of these cells co-express both receptors. Agonist-directed cell lesion in the RVM with the cytotoxin, saporin, using either CCK-saporin to target CCK receptor expressing cells, or dermorphin-saporin to target MOR expressing cells, resulted in concomitant loss of CCK2 and MOR expressing cells, did not alter the basal sensory thresholds but abolished the hyperalgesia induced by microinjection of CCK into the RVM. The findings suggest that these CCK2-MOR co-expressing RVM neurons facilitate pain and can be directly activated by CCK input to the RVM. Furthermore, lesion of these RVM neurons did not affect the initial development of neuropathic pain in the hind paw upon injury to the sciatic nerve, but the abnormal pain states were short lived such that by about day 9 the sensory thresholds had reverted to pre-injury baselines despite the existing neuropathy. These data support our hypothesis and identify CCK2-MOR co-expressing neurons in the RVM as potential therapeutic targets for neuropathic pain.
Keywords: opioid receptor, cholecystokinin receptor, neuropathy, rostral ventromedial medulla, nociception
Introduction
Anatomical, electrophysiological and pharmacological evidence have established the rostral ventromedial medulla (RVM) as an integral relay in the descending modulation of pain (Mason, 2001; Suzuki and Dickenson, 2005; Ren and Dubner, 2007). While descending inhibition from the RVM is well established (Fields et al., 1991), recent findings have pointed to a role of descending facilitation from this region in chronic inflammatory and neuropathic pain states (Urban and Gebhart, 1999; Wei and Pertovaara, 1999; Porreca et al., 2002; Guo et al., 2006). A subset of RVM neurons termed ON-cells accelerates firing immediately before the nocifensive reflex and the response characteristics are consistent with a role in pain facilitation (Fields, 1985). These descending pain facilitation cells are the only cells in the RVM that are inhibited by opioid mu receptor agonists, suggesting they may express mu opioid receptors (MOR) (Heinricher et al., 1994; Marinelli et al., 2002).We have previously lesioned these MOR expressing neurons by agonist-induced internalization of the cytotoxin, saporin, using saporin that was conjugated to the MOR-selective agonist, dermorphin. Microinjection of a low dose of dermorphin-saporin into the RVM of rats with unilateral L5/L6 spinal nerve ligation (SNL) injury reversed the established abnormal pain states in the injured hind paw (Porreca et al., 2001). The findings suggest that these RVM neurons are necessary for the maintenance of experimental neuropathic pain and thus are likely pain facilitatory in nature. Furthermore, if these MOR expressing cells in the RVM were lesioned with dermorphin-saporin prior to SNL injury, the animals exhibited abnormal pain initially but the sensory thresholds reverted to that of control rats by about day 7 (Burgess et al., 2002), indicating that these pain facilitatory RVM neurons act to maintain neuropathic pain but not the initiation of the pain states due to peripheral nerve injury.
Other evidence suggests that cholecystokinin (CCK) may be an important endogenous excitatory input to the RVM under pathophysiological conditions, as a selective antagonist for the CCK type 2 receptor (CCK2), L365,260, acutely reverses both tactile and thermal hypersensitivity when microinjected into the RVM of rats with SNL injury (Kovelowski et al., 2000). In normal rats, the active octapeptide fragment of CCK, sulfated CCK(26–33)(CCK-8(SO3)), selectively activates ON-cells when microinjected into the RVM (Heinricher and Neubert, 2004). It also elicits acute tactile and thermal hypersensitivity that can be abolished by (i) prior lesion of the dorsolateral funiculus, which constitutes the descending projections from the RVM to the spinal cord or (ii) microinjection of a CCK2 antagonist, but not CCK1 antagonist, in the RVM (Xie et al., 2005). Thus, under physiological and pathological conditions, CCK may directly or indirectly activate pain facilitatory RVM neurons via CCK2 receptors.
The present study is the first attempt to define the neuroanatomical features of these pain facilitatory RVM neurons and their role in neuropathic pain. Based on the pharmacological and electrophysiological evidence to date, we hypothesized that RVM neurons that facilitate pain express both CCK2 and MOR. Here we found that in naïve rats the majority of RVM cells that express MOR also express CCK2, and they mediate the hyperalgesia elicited by microinjection of CCK in the RVM and antinociception by RVM morphine. The RVM neurons that co-express CCK2 and MOR are essential for the chronic, but not the initial phase of peripheral nerve injury induced pain states.
Methods
Animals
Male Sprague–Dawley rats (225–300 gm; Harlan; Indianapolis, IN) were maintained on a 12/12 h light/dark cycle and provided food and water ad libitum except during the experimental procedures. All experiments involving animals were performed under protocols approved by the Institutional Animal Care and Use Committee in compliance with policies set forth by the National Institutes of Health.
Tissue preparation
Rats were deeply anesthetized with ketamine/xylazine and tissues perfused transcardially with 4% paraformaldehyde-phosphate-buffered saline (PBS) and post-fixation in the same fixative for 1 h. Brains were cryoprotected in 30% sucrose in PBS and stored at 4°C. Frozen sections (20 μm) were prepared from the brainstem and mounted serially on Superfrost/Plus slides.
Riboprobe synthesis
A 187 bp fragment of rat CCK2 cDNA (nucleotides 73–260, GenBank gi 6978616), or a 338 bp fragment of rat MOR cDNA (nucleotides 628–965, Genebank gi 6981309) was cloned into pCRII vector (Invitrogen, San Diego, CA) and used for probe synthesis. The cRNA probes were synthesized either in the presence of digoxygenin (DIG) labeled UTP, or in the presence of 166 pmol [35S]UTP (PerkinElmer, Boston, MA).
In situ hybridization
Incubations using DIG labeled probes were performed as described in the Nonradioactive In Situ Hybridization Application Manual (Roche Diagnostics Corp., Indianapolis, IN) with minor modifications. Briefly, sections were de-proteinated with 1 μg/ml proteinase K, post-fixed with 4% formaldehyde and acetylated with 0.25% acetic anhydride, and incubated overnight with the DIG-CCK2 cRNA probe (1:50, 50°C) or DIG-MOR cRNA probe (1:50, 42°C) in hybridization buffer (0.1 m Tris-HCl, pH 7.6, 4× SSC, 50% formamide, 1× Denhardt, 10% dextran sulphate, 150 μg/ml yeast tRNA and 150 μg/ml sheared salmon sperm DNA). Sections were treated with RNase A (10 μg/ml, 30 min at 37°C) and rinsed at room temperature (RT) in decreasing concentrations of SSC to a final stringency of 0.1× SSC. The slides were incubated with alkaline phosphatase conjugated anti-DIG Fab(1:500) for 5 h, followed with Fast Red overnight, counterstained with hematoxylin, and mounted with SuperMount. The [35S]-labeled CCK2 cRNA probe was diluted in the hybridization buffer (50% formamide, 0.2 m NaCl, 10 mM Tris, pH 8.0, 10 mm EDTA, and 2× Denhardt's solution) to 4 × 107 cpm/ml. Hybridization was carried out overnight at 50°C. The slides were washed four times in 4× SSC, digested with RNase A (20 μg/ml, 30 min at 37°C), rinsed at RT in decreasing concentration of SSC/1 mm dithiothreitol (DTT) to a final stringency of 0.5× at 50°C for 30 min, dehydrated in graded alcohols/1 mm DTT/0.5× SSC, defatted and dipped in ILFORD K-5D emulsion (Polysciences, Inc., Warrington, PA) and incubated at 4°C for 2 weeks before development. The sections were counterstained with hematoxylin. The protocol for double in situ hybridization was as previously described (Berg-von der Emde et al., 1995). DIG-labeled MOR cRNA was mixed in 1:50 dilution with a hybridization buffer containing 4 × 107 cpm/ml of [35S]-labeled CCK2 cRNA probe. Following hybridization (46°C overnight) and post-hybridization washes as described above, the slides were incubated overnight in 2× SSC containing 0.05% Triton X-100 and 2% normal goat serum at RT. The slides were then incubated with the anti-DIG Fab as above and developed overnight at RT with substrate solution (NTB/BCIP/levamisole). The sections were dehydrated in ethanol/1 mm DTT/2× SSC, dried under vacuum and dipped in ILFORD K5D emulsion without de-fatting to avoid chemo-autoradiographic signals. The slides were developed without nuclear counterstain.
Combined immunohistochemistry and in situ hybridization to determine if CCK2 is expressed exclusively in neurons
Brainstem sections prepared as described above were washed twice in potassium phosphate buffered saline (KPBS), incubated for 1 h in blocking buffer [KPBS, 2% BSA, 4% goat serum, 5 mm DTT, 0.3% Triton X-100, 100 units/ml SUPERaseIn (Ambion, TX)], and overnight with an anti-NeuN antibody (1:200, Chemicon, CA). Sections were rinsed with KPBS and incubated (45 min, RT) with biotin-alkaline phosphatase goat anti-mouse IgG (1:250, Jackson Laboratories), AB complex (Elite Vectastain, Vector Laboratories) and developed with 3,3-diaminobenzidine tetrahydrochloride (DAB). The slides were rinsed and dried under vacuum at RT, hybridized with the [35S] UTP-labeled CCK2 cRNA probe and processed for autoradiography as above. The sections were counterstained with 1% methyl green.
Microscopy
An image-combining computer microscope equipped with a Merzhauser motorized stage and Neurolucida software (Microbrightfield Inc., Baltimore, MD) was used to map the boundaries of the facial nuclei and pyramidal tracts manually using a Nikon 4× objective. The sections were then systematically scored for labeled neurons using a Nikon 40× objective.
RVM cannulation and microinjection
All rats were prepared for bilateral RVM drug administration as described previously (Porreca et al., 2001). Rats were anesthetized with ketamine-xylazine (100 mg/kg, i.p.). The skull was exposed, and two 26-gauge guide cannulae separated by 1.2 mm (Plastics One, Roanoke, VA) were stereotaxically directed toward the lateral portion of the RVM using the atlas of Paxinos and Watson (1986) (anteroposterior, −11.0 mm from bregma; lateral, ±0.6 mm; dorsoventral, −7.5 mm from the dura mata). The guide cannulae were secured to the skull and the animals were allowed to recover for 5 days after surgery before any drug administration. Bilateral injection into the RVM was performed by slowly expelling 0.5 μl of drug solution through a 33-gauge injection cannula inserted through the guide cannula and protruding an additional 1 mm into fresh brain tissue to prevent backflow of drug into the guide cannula. At the termination of the experiments, pontamine blue was injected into the site of RVM injections, and cannula placement was verified histologically. For pretreatment, saporin (50 ng/0.5 μl), dermorphin (1.5 ng/0.5 μl), CCK-8(SO3) (1.9 ng/0.5 μl), dermorphin-saporin (50 ng/0.5 μl) or CCK-8(SO3)-saporin (50 ng/0.5 μl) was administered as a single dose into the RVM bilaterally (0.5 μl/side). In some experiments, rats were given CCK-8(SO3) (30 ng/0.5 μl) or morphine sulfate (10 μg/0.5 μl) bilaterally into the RVM 28 days after pretreatment. Saporin and saporin conjugates were from Advanced Targeting Systems (San Diego, CA).
Spinal nerve injury and sham surgery
SNL injury was performed according to the procedure of Kim and Chung (1992). Sham-operated control rats were prepared in an identical manner without L5/L6 nerve ligation. Rats that exhibited motor deficiency (such as paw dragging or dropping) or showed no tactile allodynia were excluded from further testing.
Tactile and thermal threshold testing
The experimenter was blinded to the drug pretreatment. Response thresholds to innocuous mechanical stimuli were evaluated by determining paw withdrawal after probing the paw with a series of calibrated von Frey filaments (Chaplan et al., 1994). The withdrawal threshold was determined by sequentially increasing and decreasing the stimulus strength (‘up and down’ method), analyzed using a Dixon nonparametric test. Data are expressed as the mean withdrawal threshold. Response thresholds to noxious thermal stimuli were determined by paw withdrawal from a focused beam of radiant heat onto the surface of the hind paw (Hargreaves et al., 1988). Paw-withdrawal latency was determined by a motion detector. The data were expressed as mean ± S.E.M., and analyzed with non-parametric one-way or two-way analyses of variance (ANOVA) where appropriate. Significant differences (baseline value versus that in the following time-points) were calculated for each experimental group using one-way ANOVA. Two-way ANOVA was applied to multiple comparisons between three groups with different RVM pretreatments on either SNL or sham surgery treated rats. Statistical significance is set at 95% confidence level (P < 0.05).
Warm water tail-flick test
Nociceptive testing was performed by placing the distal third of the tail in a water bath maintained at 52°C. The latency to withdrawal was measured to 0.1 s once before and at selected time intervals after morphine injection. A cut-off latency of 10 s was employed to prevent tissue injury. Statistical analysis between treatment groups over time was performed by 2 factor ANOVA and differences within each group were performed by ANOVA followed by Fisher's least significant difference post hoc test.
Results
Neurons in the RVM express CCK2 receptors
Both digoxigenin (DIG)-labeled (Fig. 1A) and [35S]-labeled (Fig. 1B) cRNA probes for CCK2 identified many neurons to be CCK2 positive on serial sections from the brainstem region of naive rats (Fig. 1C). Confining our analysis to the ventral medial aspect of these sections, many labeled cells were found in the RVM region including the nucleus raphe magnus and the nucleus reticularis gigantocellularis pars alpha (GiA). A few cells were seen lateral to the RVM in the nucleus paragigantocellularis-lateral part (LPGi), and many were distributed in the nucleus reticularis gigantocellularis (Gi) dorsal to the RVM. In caudal sections, some labeled cells were also seen in the raphe obscurus which lies in the midline dorsal to the raphe magnus. Many labeled, medium-sized and large pyramidal cell bodies ranging from 25 μm to >50 μm diameter were found throughout the RVM region. The [35S]-labeled cRNA for CCK2 (silver grains) was associated exclusively with cells that were immunoreactive for the neuronal marker NeuN (Fig. 1D). On cells that were not labeled by the anti-NeuN antibody, i.e. cell bodies depicted only by the green nuclear stain, the silver grain density was not different from background. Some CCK2 labeled neurons appeared to cluster. CCK2 labeled (79 ± 26 per section, n = 10) between 10 and 15% of RVM neurons labeled by NeuN (508 ± 109 per section, n = 10).
Figure 1.

Localization of CCK2 receptor mRNA in the RVM. (A) Representative photomicrograph of neurons containing CCK2 mRNA labeled by digoxigenin-labeled cRNA probe of the rat CCK2 and detected by an anti-digoxigenin antibody and Fast Red stain, counterstained with Mayer's hematoxylin (blue nuclei). Scale bar = 25 μm; (B) Neurons containing CCK2 mRNA detected by a [35S]-labeled cRNA probe for rat CCK2 and counterstained with hematoxylin. High-power micrograph shows two large diameter cells labeled with silver grains surrounding the nuclei; (C) Coronal maps of labeled neurons containing CCK2 mRNA (approximately between −11.3 mm and −10.8 mm from bregma). The tracings are arranged at successive caudal (1) to rostral (8) intervals (120 μm). Each dot denotes one labeled neuron. Red line delineates the approximate location of the RVM including GiA and raphe magnus. Thin black lines delineate the approximate location of facial nuclei (VII), pyramids (py) and LPGi lying between VII and RVM. Gi, nucleus reticularis gigantocellularis; (D) Combined NeuN immunoreactivity/[35S]-labeled cRNA probe for rat CCK2 on the same sections. Silver grains produced by [35S] overlaps with NeuN labelling (brown). NeuN-negative cells are visible as clear somata with green nuclear stain.
To show that the cRNA probe labeled CCK2 mRNA specifically, the hybridization reaction was carried out in the presence or absence of unlabeled, sense RNA transcribed from the CCK2 cDNA fragment. The sense RNA blocked the labelling of cell bodies by the DIG-cRNA (data not shown). The DIG-cRNA labeled CCK2 transfected cells but not CCK1 transfected cells (data not shown). A control oligo-dT DNA probe that labels all cell bodies served as positive control for the in situ hybridization throughout.
MOR labeled cells in the RVM
In situ hybridization analysis using a DIG-labeled cRNA for MOR (Fig. 2A) showed similar distribution of MOR positive cells in the RVM as that described previously using a fluorescein-labeled partial cDNA probe (Porreca et al., 2001). These labeled cells were mostly confined to the nucleus raphe magnus and the adjacent reticular formation. In the more lateral aspect of the RVM, the LPGi consisted of fewer MOR positive cells (Fig. 2B). A low density of discrete cell bodies was also evident immediately dorsal to the RVM. Comparing with the number and distribution of cells that were labeled with CCK2 (Fig. 1), MOR positive neurons were relatively concentrated medial ventrally, and fewer in number in the RVM (54 ± 21 per section, n = 20). Consistent with the size of neurons labeled with CCK2, the cell bodies of most MOR labeled neurons were medium- to large diameter.
Figure 2.

Localization of MOR mRNA in the RVM. (A) A representative photomicrograph of neurons containing MOR mRNA in the RVM region using a digoxigenin-labeled cRNA probe of the rat MOR and detected by an anti-digoxigenin antibody and NBT/BCIP (dark blue), counterstained with Mayer's hematoxylin (blue nuclei). Scale bar = 50 μm. (B) Coronal maps of neurons containing MOR mRNA. Tracings are arranged at successive caudal (1) to rostral (8) intervals (120 μm) as for Fig. 1C. Each dot denotes one labeled neuron. Red line delineates the approximate location of the RVM including GiA and raphe magnus. Thin black lines delineate the approximate location of facial nuclei (VII), pyramids (py) and LPGi lying between VII and RVM. Gi, nucleus reticularis gigantocellularis.
Many cells co-express CCK2 and MOR transcripts in the RVM
Double in situ hybridization combining [35S]-labeled CCK2 cRNA and colorimetric DIG-labeled MOR cRNA shows many neurons labeled with both silver grains surrounding the unstained nucleus (CCK2) and dark blue stain (MOR) (Fig. 3). Mapping analysis using nine sections from five rats showed that most of the labeled RVM cells co-expressed CCK2 and MOR. The average cell count per section was 61 ± 13. A small number of RVM cells was labeled with only CCK2; the average cell count per section was 11 ± 7. Very few RVM cells were labeled with only MOR; the average number per section was 2 ± 2.
Figure 3.
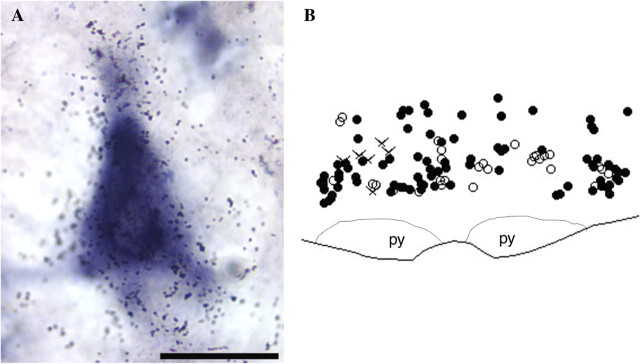
Double hybridization histochemistry for CCK2 and MOR transcripts in the RVM. (A) A high magnification image (100× objective) of a pyramidal neuron in the RVM labeled by both [35S]-labeled CCK2 cRNA probe (silver grains) and DIG-labeled MOR cRNA probe (dark blue). Scale bar = 25 μm; (B) A representative map of cells co-labeled for CCK2 and MOR mRNA (closed circles), CCK2 mRNA only (open circles), and MOR mRNA only (crosses) in the RVM. Based on the analysis of nine sections from five rats, over 80% of labeled RVM neurons co-express both MOR and CCK2, about 15% express only CCK2, and very few cells express only MOR. py, pyramid.
Effects of RVM administration with the cytotoxin saporin covalently linked to CCK-8(SO3)
CCK-8(SO3) was conjugated to the cytotoxin, saporin (Supplementary methods). Initial analysis by competitive binding of CCK-8(SO3)-saporin against [125I]CCK-8(SO3) binding to CCK2 showed that CCK-8(SO3)-saporin had a high affinity of 3.2 nM (Log Ki = −8.5 ± 0.02, n = 2) for CCK2, comparable to 3.6 nM for CCK-8(SO3) (Log Ki = −8.4 ± 0.09, n = 6) (Supplementary Fig. 1). The high affinity binding of the toxin conjugate to CCK receptors is essential for producing the subsequent behavioral effects described below.
A single microinjection of CCK-8(SO3)-saporin was given into the RVM of naïve rats bilaterally at a dose of 1.5 pmol per side. Paw withdrawal thresholds to probing with von Frey filaments was 15 ± 0 g, 28 days after injection (Fig. 4A) and the paw withdrawal latency to radiant heat applied to the plantar aspect of the hind paw was 18.7 ± 0.5 s (Fig. 4B). These values were not significantly different from baseline thresholds prior to RVM injection (P > 0.05). Pretreatment with CCK-8(SO3) or saporin at equivalent dose into the RVM also had no effect on baseline sensory thresholds measured 28 days later (Fig. 4A and B).
Figure 4.

Rats received a bilateral microinjection of saporin (SAP), CCK-8(SO3) (CCK8), CCK-8(SO3)-saporin (CCK8-SAP), dermorphin (DERM), or dermorphin-saporin (DERM-SAP) into the RVM. After 28 days, rats were injected with CCK8 (30 ng/0.5 μl, each side) bilaterally into the RVM and were tested for (A and D) paw withdrawal threshold to von Frey probing or (B and E) paw withdrawal latency to radiant heat. (C and F) Effect of RVM pretreatment with SAP or CCK8-SAP (C) and that of RVM pretreatment with SAP or DERM-SAP (F) on the number of CCK2 and MOR expressing neurons in the RVM 28 days after treatment. Positive cells expressing either CCK2 or MOR transcripts were detected by in situ hybridization using digoxigenin-labeled CCK2 or MOR cRNA probe. The cells were mapped and counted using Neurolucida software. Data are expressed as mean ± S.E.M.; *P < 0.05; ***P < 0.001.
On day 28, these rats were given a higher dose (47 pmol) of CCK-8(SO3) bilaterally into the RVM. Paw withdrawal threshold to von Frey probing and thermal latency to radiant heat in the left hind paw was monitored over a time-course of 120 min. This dose of CCK-8(SO3) has been shown previously to produce transient tactile hypersensitivity and thermal hyperalgesia (Xie et al., 2005). Rats that were pretreated either with saporin or CCK-8(SO3) developed tactile (Fig. 4A) and thermal hypersensitivity (Fig. 4B) within 30–45 min after CCK-8(SO3) microinjection. Both paw withdrawal thresholds in response to von Frey testing and paw withdrawal latency were significantly decreased from baseline values (P < 0.05). However, rats that were pretreated with CCK-8(SO3)-saporin did not exhibit acute tactile hypersensitivity (Fig. 4A) or thermal hyperalgesia (Fig. 4B) upon CCK-8(SO3) microinjection. Neither response differed significantly from the respective pre-injection baseline (P > 0.05).
The RVM from these rats were examined to identify the placement of the cannulae and serial sections prepared for in situ hybridization analysis. There was no evidence of tissue necrosis in the RVM region due to microinjection of CCK-8(SO3) or CCK-8(SO3)-saporin. NeuN labelling of neurons in the RVM indicated a small (about 10–15%) but significant (P < 0.05) reduction in the number of NeuN immunoreactive cells (Supplementary Fig. 2). The sections taken from rats that were pretreated with saporin or CCK-8(SO3) showed similar densities of CCK2 labeled cells in the RVM when compared with that observed in naïve rats above. The RVM of the CCK-8(SO3)-saporin-pretreated rats consisted of markedly fewer CCK2 labeled cells when compared with that of CCK-8(SO3)-, or saporin-pretreated rats. This regional loss of CCK2 labeled cells in CCK-8(SO3)-saporin-pretreated tissues was greatest at the site of the guide cannulae and the effect spread approximately 0.5 mm in radius on the lateral and vertical plane as well as in the caudal-rostral extent. It was noted that the ablation of CCK2 labeled neurons was usually incomplete; some labelling in the RVM may be found in sections distal to the cannula placement in the caudal–rostral extent. The cytotoxin could also extend outside the boundaries of the RVM; there were fewer CCK2 labeled cells in the Gi immediately dorsal to the RVM particularly in sections close to the site of the guide cannulae. Despite the limitations, the cytotoxin treatments produced consistent results within treatment groups.
For quantitative analysis, one section adjacent to the cannula site from each of 5–6 rats from each treatment group was analyzed for significant difference (one-way ANOVA and Fisher's least significant difference test) (Fig. 4C). Statistical analysis showed that CCK-8(SO3)-saporin pretreatment significantly reduced the number of CCK2 expressing neurons in RVM when compared with that of saporin-pretreated rats (P < 0.001). There was a concomitant reduction of MOR labeled cells in the RVM in adjacent sections from the same rats given CCK-8(SO3)-saporin pretreatment when compared with that of saporin-pretreated rats (P < 0.001) (Fig. 4C).
Effects of RVM administration with the cytotoxin saporin covalently linked to dermorphin
Bilateral microinjection of dermorphin-saporin (1.5 pmol each side) or an equivalent dose of saporin or dermorphin into the RVM of naive rats produced no change (P > 0.05) in paw withdrawal thresholds to probing with von Frey filaments or to radiant heat over a period of 28 days (Fig. 4D and E). A dose of 47 pmol of CCK-8(SO3) given bilaterally into the RVM produced significant (P < 0.05) hypersensitivity to von Frey probing (Fig. 4D) and radiant heat (Fig. 4E) in the left hind paw 30–45 min after microinjection in rats that were pretreated with saporin or dermorphin. Rats that were pretreated with dermorphin-saporin, however, did not exhibit acute tactile hypersensitivity or thermal hyperalgesia upon CCK-8(SO3) microinjection (Fig. 4D and E). Their response thresholds to von Frey probing or radiant heat did not differ significantly (P > 0.05) from that prior to CCK-8(SO3) administration into the RVM.
The pretreatment with saporin or dermorphin did not change the density of cells that were labeled with MOR cRNA in the RVM. Dermorphin-saporin pretreatment resulted in a significant reduction in the number of RVM neurons labeled with the MOR cRNA; the greatest loss occurred within a 0.5 mm radius of the cannula site. Quantitative analysis of sections adjacent to the cannula site from multiple rats showed that dermorphin-saporin pretreatment significantly reduced the number of MOR labeled neurons in RVM when compared with that of saporin-pretreated rats (P < 0.001) (Fig. 4F). In situ hybridization using DIG-labeled CCK2 cRNA showed that there was a concomitant reduction in the number of CCK2 labeled cells in the RVM after dermorphin-saporin pretreatment when compared with that of saporin-pretreated rats (P < 0.001) (Fig. 4F).
CCK-8(SO3)-saporin pretreatment attenuates RVM morphine induced antinociception
Rats pretreated with vehicle (water), saporin, CCK-8(SO3), or CCK-8(SO3)-saporin were tested by a bilateral injection of morphine (20 μg total) into the RVM on day 28 after the pretreatment. The tail-flick latency to a 52°C stimulus was measured over 90 min to determine the antinociceptive effect of RVM morphine (Fig. 5). RVM morphine induced antinociception was evident by 30 min after injection in rats pretreated with vehicle, saporin or CCK-8(SO3), but was significantly attenuated in rats that had been pretreated with CCK-8(SO3)-saporin.
Figure 5.

Rats (n = 7–12 per group) received bilateral microinjection of water, saporin (SAP), CCK-8(SO3) (CCK8) or CCK-8(SO3)-saporin (CCK8-SAP) into the RVM. After 28 days, rats were injected with morphine (20 μg/1 µl total) bilaterally into the RVM and tested for tail-flick latency at 52°C before and over 90 min after morphine administration. Significant differences in tail flick latencies among treatment groups over time were determined by 2-factor ANOVA. Antinociceptive responses to morphine obtained in rats treated with CCK-SAP in the RVM were significantly [F(5,23) = 7.40; P = 0.0028] lower than those obtained from the vehicle-treated control group.
Nerve injury-induced tactile and thermal hypersensitivity were not maintained in RVM CCK-8(SO3)-saporin pretreated rats
Bilateral microinjection of saporin, CCK-8(SO3), or CCK-8(SO3)-saporin into the RVM of naive rats did not alter the baseline sensory thresholds acutely upon microinjection or over a period of 28 days (Fig. 6). On the 28th day after RVM microinjections, rats in each treatment group were subdivided into two groups, one underwent sham operation and one subjected to L5/L6 SNL surgery. Behavioral testing began on day 2 after sham or SNL surgery. Figure 6 shows that over the 14-day post surgery period, all rats that received sham surgery maintained sensory thresholds to both tactile and thermal stimuli that were not different from pre-surgery baselines (P > 0.05, One way ANOVA). For rats that received unilateral SNL injury in the left hind paw, all three RVM pretreatment groups developed rapid onset of tactile (Fig. 6A) and thermal (Fig. 6B) hypersensitivity by day 2 after SNL. The paw withdrawal thresholds to tactile stimuli for the RVM saporin, CCK-8(SO3), or CCK-8(SO3)-saporin pretreated group were, respectively, 15 ± 0 g, 14 ± 0.81 g and 15 ± 0 g prior to SNL. On day 2 after SNL, the withdrawal thresholds to tactile stimuli were 2.7 ± 0.49 g, 2.7 ± 0.17 g and 2.6 ± 0.62 g, respectively, which were significantly different from that prior to SNL (P < 0.05). Similarly, the paw withdrawal thresholds to noxious heat for the RVM saporin, CCK-8(SO3), or CCK-8(SO3)-saporin pretreated group were, respectively, 22 ± 2.77 s, 19 ± 0.79 s and 18 ± 1.0 s before SNL. On day 2 after SNL, the sensory thresholds were significantly reduced to 12 ± 0.75 s, 12 ± 0.83 s and 10 ± 1.14 s (P < 0.05).
Figure 6.
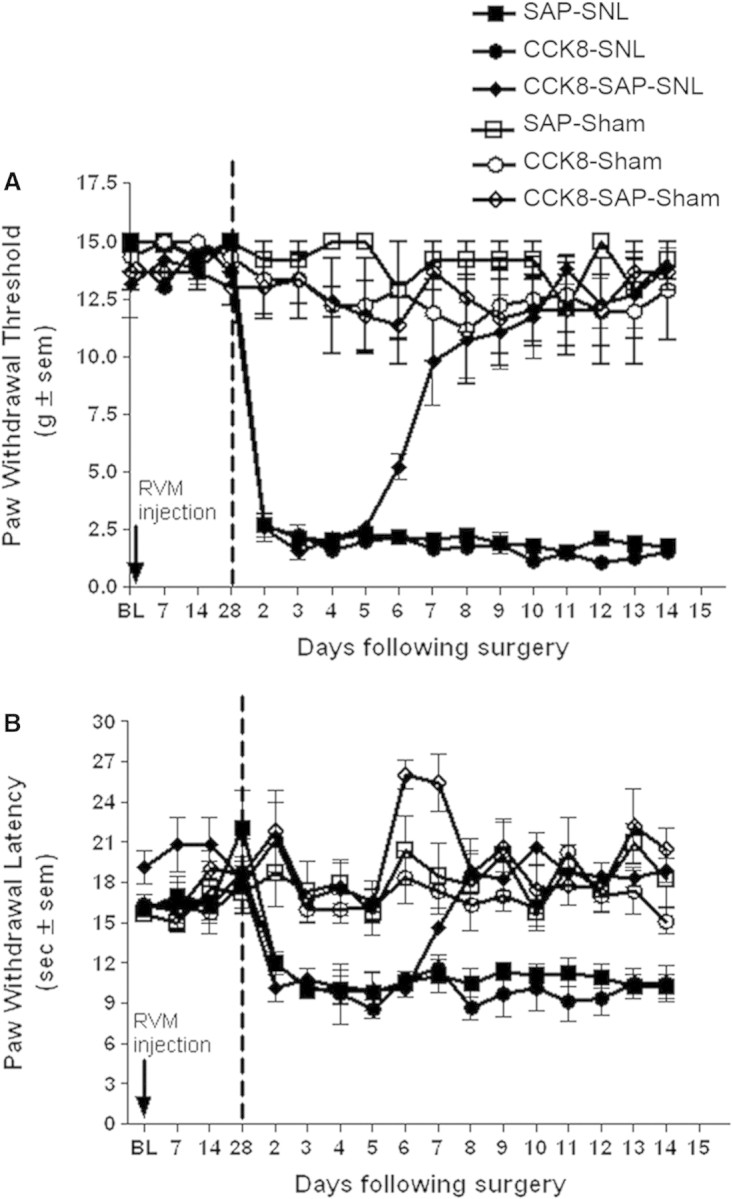
Male Sprague-Dawley rats received a bilateral microinjection of saporin (SAP) (50 ng/0.5 μl, each side), CCK-8(SO3)(CCK8) (1.5 ng/0.5 μl, each side), or CCK-8(SO3)-saporin (CCK8-SAP)(50 ng/0.5 μl, each side) into the RVM. Paw withdrawal threshold to von Frey probing (A) and paw withdrawal latency to radiant heat (B) were determined prior to RVM injection, and on day 7, 14 and 28 after treatment. After 28 days, the rats were subjected to either L5/L6 SNL or sham surgery. Vertical dashed lines represent time of surgery. Behavioral testing was performed once daily from day 2 to day 14 after SNL or sham surgery. Each group consisted of 4–6 rats.
The tactile and thermal hypersensitivity in the ipsilateral hind paw were maintained throughout the 14-day period in SNL rats that were pretreated with RVM saporin or CCK-8(SO3) (P < 0.05). In contrast, rats that were pretreated with RVM CCK-8(SO3)-saporin exhibited a gradual recovery from the SNL-induced tactile hypersensitivity (Fig. 6A) and thermal hyperalgesia (Fig. 6B). Paw-withdrawal thresholds of the CCK-8(SO3)-saporin pretreated rats to tactile stimuli were 5.2 ± 0.57 g, 9.8 ± 1.94 g and 11 ± 1.63 g on day 6, 7 and 8 after SNL, which were significantly different from that on day 2 after SNL (P < 0.05). The tactile sensitivity between day 9 and day 14 ranged from 11 ± 1.62 g to 14 ± 0.82 g, which were not significantly different from the pre-SNL baseline (P > 0.05). Similarly, the paw withdrawal latencies were also reversed in a time course similar to that seen in the tactile hypersensitivity. The thresholds gradually reversed beginning on day 6 after SNL and by day 8, the paw withdrawal latencies (ranging from 18 ± 1.54 s to 21 ± 1.58 s) were not significantly different (P > 0.05) from pre-SNL baseline values.
Discussion
The experimental evidence presented here supports our hypothesis that a discrete subset of neurons in the RVM expresses both MOR and CCK2. Anatomical analysis using riboprobes for MOR (Fig. 2A) or CCK2 (Fig. 1A) transcripts independently label cell bodies that are similar in density, size and distribution in the RVM and adjacent regions; and concurrently, the two probes label predominantly the same cells. The labeled cells are neuronal in nature based on their immunoreactivity for NeuN. The majority of CCK2 and/or MOR labeled cells are located in the raphe magnus and the GiA, regions in which activation of neurons modulates dorsal horn nociceptive sensory processing. Labeled cells are also found in the raphe pallidus, the LPGi and the raphe obscurus, regions that have been implicated in efferent autonomic control (Mason, 2001).
RVM directed microinjection of either CCK-8(SO3)-saporin or dermorphin-saporin resulted in a significant reduction of both CCK2 and MOR expressing cells, which is consistent with the anatomical evidence and further substantiates the cellular co-localization of the two receptors. The cell loss accounts for a very small fraction (<10%, see Supplementary Fig. 2) of RVM neurons suggesting the cytotoxicity incurred by CCK-8(SO3)-saporin is highly selective and argues that the behavioral effects of the pretreatment with either toxin conjugate is likely due to the loss of this discrete cell population. However, as with any manipulation at the systems level, we acknowledge that unintended, off-target or compensatory effects of the toxin pretreatment cannot be precluded.
The loss of CCK2 and MOR expressing cells incurred by CCK-8(SO3)-saporin or dermorphin-saporin treatment did not alter the baseline hind paw sensitivity to tactile or thermal stimuli over a period of 28 days, suggesting that these cells are not essential for the baseline sensory thresholds at least in the endpoints studied here. However, loss of the CCK2-MOR expressing cells resulted in a loss of RVM CCK-induced transient cutaneous sensory hyper-responsiveness, suggesting that this pain facilitating effect of CCK in the RVM is primarily mediated by these CCK2-MOR co-expressing neurons. Furthermore, ablation of these neurons by CCK-8(SO3)-saporin pretreatment attenuated the antinociception of RVM morphine; thus, these CCK2-MOR co-expressing cells are a major site of antinociceptive action of RVM morphine. In naïve rats, this phenotype defines these cells functionally as ON cells.
Pharmacologically, RVM CCK significantly attenuates morphine activation of OFF cell firing and morphine inhibition of tail flick reflex (Heinricher et al., 2001). Higher dose of CCK activates ON cell firing and reduces the tail flick latency (Heinricher and Neubert, 2004), which is consistent with the hyperalgesic effect of RVM CCK shown here. An earlier study found that the distribution of CCK- and enkephalin-immunoreactive terminal fields in the RVM overlap significantly (Skinner et al., 1997). Together, the evidence supports the likelihood that a discrete population of RVM neurons, characterized by their co-expression of CCK2 and MOR, are regulated by endogenous enkephalinergic (pain inhibiting) and CCK (pain facilitating) inputs to the RVM. It is also possible that neurons with the CCK2-MOR phenotype may contribute to other brainstem function such as efferent autonomic control based on their anatomical localization mentioned above (Mason, 2005); further analysis would be necessary to validate such function.
The effects of CCK-8(SO3)-saporin pretreatment on peripheral nerve injury induced abnormal pain states further validate the critical role that this population of RVM neurons has in the descending regulation of spinal nociceptive processing. Nerve injury induced by the ligation of L5/L6 spinal nerve produced a rapid onset of hypersensitivity to touch and noxious heat irrespective of RVM pretreatment with the toxin conjugate. However, only the CCK-8(SO3)-saporin pretreated rats showed a spontaneous, progressive reversal of the sensory thresholds back to control level, i.e. sham-operated or pre-injury baseline, over time, such that by about 9 days after SNL, these injured animals exhibited normal thresholds to both tactile and thermal stimuli despite the sustained injury to the sciatic nerve. This spontaneous reversal of tactile hypersensitivity and thermal hyperalgesia was similarly observed in SNL rats that were pretreated with a single RVM injection of dermorphin-saporin (Burgess et al., 2002). Both lines of evidence support the notion that the RVM neurons that are the target of CCK-8(SO3)-saporin and dermorphin-saporin, i.e. the MOR and CCK2 co-expressing cells, do not participate in the initiation of neuropathic pain but become critical in the maintenance of the abnormal pain states. Previous studies have shown that an excitatory output from the RVM promotes neuropathic pain behavior that can be blocked by RVM lidocaine or a CCK2 selective antagonist. The present data substantiate these findings by demonstrating that the RVM neurons that facilitate neuropathic pain have a CCK2-MOR phenotype. The functional implication of this subset of RVM neurons warrants further evaluation as potential therapeutic targets for neuropathic pain.
Supplementary material
Supplementary material is available at Brain online.
Funding
National Institute of Dental and Craniofacial Research (R01DE 016458 to J.L.); National Institute on Drug Abuse (P01 DA06248 to V.J.H.) and (R01 DA12394 to F.P.).
Supplementary Material
Acknowledgements
The authors thank Dr Sergio Ojeda (Oregon Health Science University) for kindly providing the working protocol for double in situ hybridization and expert consultation. We also thank Sally J. Krajewski and Lynne C. Reichter for technical assistance.
Glossary
Abbreviations:
- CCK2
cholecystokinin type 2 receptor
- cDNA
complementary DNA
- cRNA
complementary RNA
- DIG
digoxigenin
- Gi
nucleus reticularis gigantocellularis
- GiA
nucleus reticularis gigantocellularis pars alpha
- LPGi
nucleus paragigantocellularis-lateral part
- MOR
mu opioid receptor
- RVM
rostral ventromedial medulla
- SNL
spinal nerve ligation
- SSC
sodium citrate buffer
- UTP
uridine trisphosphate
References
- Berg-von der Emde K, Dees WL, Hiney JK, Hill DF, Dissen GA, Costa ME, et al. Neurotrophins and the neuroendocrine brain: different neurotrophins sustain anatomically and functionally segregated subsets of hypothalamic dopaminergic neurons. J Neurosci. 1995;15:4223–37. doi: 10.1523/JNEUROSCI.15-06-04223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SE, Gardell LR, Ossipov MH, Malan TP Jr, Vanderah TW, Lai J, et al. Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci. 2002;22:5129–36. doi: 10.1523/JNEUROSCI.22-12-05129.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci meth. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Fields HL. Nociceptive transmission: the pain system. Science. 1985;228:1522. doi: 10.1126/science.228.4707.1522. [DOI] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219–45. doi: 10.1146/annurev.ne.14.030191.001251. [DOI] [PubMed] [Google Scholar]
- Guo W, Robbins MT, Wei F, Zou S, Dubner R, Ren K. Supraspinal brain-derived neurotrophic factor signaling: a novel mechanism for descending pain facilitation. J Neurosci. 2006;26:126–37. doi: 10.1523/JNEUROSCI.3686-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, McGaraughty S, Tortorici V. Circuitry underlying antiopioid actions of cholecystokinin within the rostral ventromedial medulla. J Neurophysiol. 2001;85:280–6. doi: 10.1152/jn.2001.85.1.280. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Morgan MM, Tortorici V, Fields HL. Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience. 1994;63:279–88. doi: 10.1016/0306-4522(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Neubert MJ. Neural basis for the hyperalgesic action of cholecystokinin in the rostral ventromedial medulla. J Neurophysiol. 2004;92:1982–9. doi: 10.1152/jn.00411.2004. [DOI] [PubMed] [Google Scholar]
- Kovelowski CJ, Ossipov MH, Sun H, Lai J, Malan TP, Porreca F. Supraspinal cholecystokinin may drive tonic descending facilitation mechanisms to maintain neuropathic pain in the rat. Pain. 2000;87:265–73. doi: 10.1016/S0304-3959(00)00290-6. [DOI] [PubMed] [Google Scholar]
- Marinelli S, Vaughan CW, Schnell SA, Wessendorf MW, Christie MJ. Rostral ventromedial medulla neurons that project to the spinal cord express multiple opioid receptor phenotypes. J Neurosci. 2002;22:10847–55. doi: 10.1523/JNEUROSCI.22-24-10847.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason P. Contributions of the medullary raphe and ventromedial reticular region to pain modulation and other homeostatic functions. Annu Rev Neurosci. 2001;24:737–77. doi: 10.1146/annurev.neuro.24.1.737. [DOI] [PubMed] [Google Scholar]
- Mason P. Deconstructing endogenous pain modulations. J Neurophysiol. 2005;94:1659–63. doi: 10.1152/jn.00249.2005. [DOI] [PubMed] [Google Scholar]
- Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan TP, Jr, Ossipov MH, et al. Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the mu-opioid receptor. J Neurosci. 2001;21:5281–8. doi: 10.1523/JNEUROSCI.21-14-05281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–25. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Pain facilitation and activity-dependent plasticity in pain modulatory circuitry: role of BDNF-TrkB signaling and NMDA receptors. Mol Neurobiol. 2007;35:224–35. doi: 10.1007/s12035-007-0028-8. [DOI] [PubMed] [Google Scholar]
- Skinner K, Basbaum AI, Fields HL. Cholecystokinin and enkephalin in brain stem pain modulating circuits. Neuroreport. 1997;8:2995–8. doi: 10.1097/00001756-199709290-00001. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Dickenson A. Spinal and supraspinal contributions to central sensitization in peripheral neuropathy. Neurosignals. 2005;14:175–81. doi: 10.1159/000087656. [DOI] [PubMed] [Google Scholar]
- Urban MO, Gebhart GF. Supraspinal contributions to hyperalgesia. Proc Natl Acad Sci USA. 1999;96:7687–92. doi: 10.1073/pnas.96.14.7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Pertovaara A. MK-801, an NMDA receptor antagonist, in the rostroventromedial medulla attenuates development of neuropathic symptoms in the rat. Neuroreport. 1999;10:2933–7. doi: 10.1097/00001756-199909290-00011. [DOI] [PubMed] [Google Scholar]
- Xie JY, Herman DS, Stiller CO, Gardell LR, Ossipov MH, Lai J, et al. Cholecystokinin in the rostral ventromedial medulla mediates opioid-induced hyperalgesia and antinociceptive tolerance. J Neurosci. 2005;25:409–16. doi: 10.1523/JNEUROSCI.4054-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.