

Abstract
Due in part to the increasing availability of crystallographic protein structures as well as rapid improvements in computing power, the past few decades have seen an explosion in the field of computer-based rational drug design. Several algorithms have been developed to identify or generate potential ligands in silico by optimizing the ligand-receptor hydrogen bond, electrostatic, and hydrophobic interactions. We here present AutoGrow, a novel computer-aided drug design algorithm that combines the strengths of both fragment-based growing and docking algorithms. To validate AutoGrow, we recreate three crystallographically resolved ligands from their constituent fragments.
Keywords: AutoGrow, Rational Drug Design, Docking, Evolutionary Algorithm
Introduction
Due in part to the increasing availability of crystallographic protein structures as well as rapid improvements in computing power, the past few decades have seen an explosion in the field of computer-based drug discovery. Algorithms developed to identify, generate, and optimize potential ligands in silico by optimizing the ligand-receptor hydrogen bond, electrostatic, and hydrophobic interactions have contributed to the development of a number of FDA-approved drugs (1).
Many computer-aided drug design techniques have been developed in recent years (reviewed in reference 2). Among these, comprehensive physics-based approaches, such as thermodynamic integration (3), single-step perturbation (4), and free energy of perturbation (5), can yield very accurate binding free energies but are limited in the diversity of compounds generated and come at a high computational expense. Even so, physics-based ligand optimization methods based on density functional theory (6) and electron-nuclear attraction potentials (7) are promising new approaches.
Fragment-based growing strategies, which create novel structures by adding interacting moieties to a fixed scaffold, are generally more popular due to the lower computational expense and the higher degree of compound diversity generated (8–17). As these algorithms typically limit moiety addition to a library of fragments, finding good ligands is computationally efficient. Moreover, the scoring functions used to predict the overall binding free energies of new ligands are generally faster, though not as accurate as the comprehensive physics-based approaches described above. Notably, although many fragment-based growing strategies assume that the position or binding mode of the “core” scaffold does not change upon fragment addition, this assumption is not valid in many cases.
Ligand docking is another common drug development technique. Docking programs are commonly employed in virtual screening applications to search through databases of pre-defined compounds, position the ligands correctly within the receptor active site, and identify those ligands predicted to bind with high affinity. Developing and improving docking scoring functions is an area of active interest (see, for example, references 18–39). Like fragment-based growing scoring functions, docking scoring functions are typically faster and less accurate than more rigorous physics-based approaches. Unlike many growing techniques, docking typically accounts for the mobility of all portions of the ligand. However, rather than generate novel compounds, docking techniques are only able to suggest good binders from among those compounds contained in a database of pre-defined ligand candidates.
The current work is motivated by the desire to overcome the inherent weaknesses of both fragment-growing and docking algorithms. In this work, we introduce AutoGrow, a novel drug design algorithm that combines elements of both techniques. AutoGrow uses a growing strategy to build upon an initial “core” scaffold; molecular fragments are added at random to this scaffold, thereby generating a population of novel compounds. Rather than assuming that the common “core” scaffold of the members of this population is static within the active site, AutoGrow dynamically redocks each novel compound into the protein receptor upon addition of varied fragments, producing ab initio poses for every molecule. An evolutionary algorithm then evaluates the docking scores of each population member, and the best binders become founders of the subsequent generation.
As generation after generation is created, each based on the most fit individuals of the previous generation, a larger inhibitor with a higher predicted binding affinity eventually evolves. While not necessarily drug-like, these predicted inhibitors often provide information that is useful early in the lead optimization process, including the identification of potential new protein-ligand interactions as well as novel drug scaffolds. To validate the AutoGrow algorithm, we recreate ATP, the known substrate of Trypanosoma brucei RNA editing ligase 1 (40); oseltamivir, a nanomolar-affinity antiviral inhibitor of the neuraminidase enzyme (41); and aminoimidazole 4-carboxamide ribonucleotide (AMZ), a nanomolar-affinity inhibitor of AICAR transformylase (42).
Methods and Materials
Evolutionary algorithms are ideally suited to complex problems such as those associated with de novo drug discovery (43). These algorithms typically include three operators, modeled on the three natural operators of biological evolution: selection, crossover, and mutation. The evolutionary procedure is divided into generations, where each generation consists of a population of individuals derived from selection of the most fit members of the previous generation. The internal variation of each generation is exploited via crossover, wherein the characteristics of two “parent” individuals are combined to create a new “child” individual. External variation is introduced into each generation via mutation, wherein new individuals are created by making small, usually random changes to individuals already present in the population. As generation after generation is created, each based on the most fit individuals of the previous generation as well as additional individuals derived by exploiting internal and external variation, a solution eventually evolves. In the AutoGrow implementation, each generation consists of multiple potential ligands. The algorithm evolves ligands that are predicted to bind to a given target protein with high affinity.
AutoGrow uses AutoDock as the selection operator (36). For each generation, all ligand files are docked into the target protein, and for each dock, AutoDock returns a predicted binding affinity. Those ligands that bind within the active site and have the most favorable predicted binding affinities are selected for inclusion in the next generation. AutoDock is a good selection operator; the program is free, takes into account full ligand flexibility, and has a well-tested scoring function (44). For these reasons, AutoGrow has been coded to interface with AutoDock by default. With only slight modification to the code, however, AutoGrow could use any number of docking programs as its selection operator. AutoGrow is open source, and we encourage source-code modifications of this type.
In order to exploit the internal variation present in each generation after the first, a crossover operator is used. First, individual pairs (“couples”) are selected from among those ligands that were the most fit of the previous generation. New hybrid “children” ligands are then formed by randomly mixing and matching the attached moieties of the two “parents” (Figure 1A).
Figure 1.

An illustration of the mutation and crossover operators. A) A crossover, or “child” ligand is formed by selecting an individual pair (“couple”) and creating a new hybrid ligand by randomly mixing and matching the moieties of the two “parents.” B) A “mutant” ligand is formed by replacing an appropriate scaffold linker hydrogen atom with a randomly chosen molecular fragment. Compound fragments are color coded to better illustrate crossover and mutation.
In order to introduce external variation into each generation, a mutation operator is used. First, a number of individuals are selected from among those that were the most fit of the previous generation. For each of these individuals, an appropriate hydrogen atom (the “scaffold linker hydrogen”) is randomly selected. A molecular fragment is then chosen randomly from a fragment library, and one of the hydrogen atoms of that fragment is likewise selected at random (the “fragment linker hydrogen”). A new “mutant” ligand is created by linking the scaffold and fragment through their respective linker hydrogen atoms, replacing those hydrogen atoms with a single bond. Thus, “mutants” are similar to, but distinct from, other population members (Figure 1B).
The AutoGrow mutation operator draws upon fragment libraries in order to make small modifications to evolving ligands. By default, AutoGrow comes with large- and small-fragment libraries, although customized, user-generated libraries are straightforward to incorporate. When larger refinements are desired, the mutation operator can draw upon the default library of large molecular fragments. To generate the large-fragment library, 32,091 compounds were downloaded from the ZINC database (45). Those ligands with more than 19 atoms were discarded, thereby preserving only 2,437 small, fragment-like molecules ranging in size from 8 to 19 atoms (average: 16.7 atoms, standard deviation: 2.1, Figure 2a). Collectively, these fragment-like molecules constitute the default large-fragment library. Ten representative large fragments from the library are shown in Figure 3a.
Figure 2.

Default large- and small-fragment libraries used by the mutation operator. A) The large-fragment library contains 2,437 fragments ranging in size from 8 to 19 atoms (average: 16.7 atoms, standard deviation: 2.1). B) The small-fragment library contains 46 fragments ranging in size from three to fifteen atoms (average: 9.6 atoms, standard deviation: 2.8).
Figure 3.
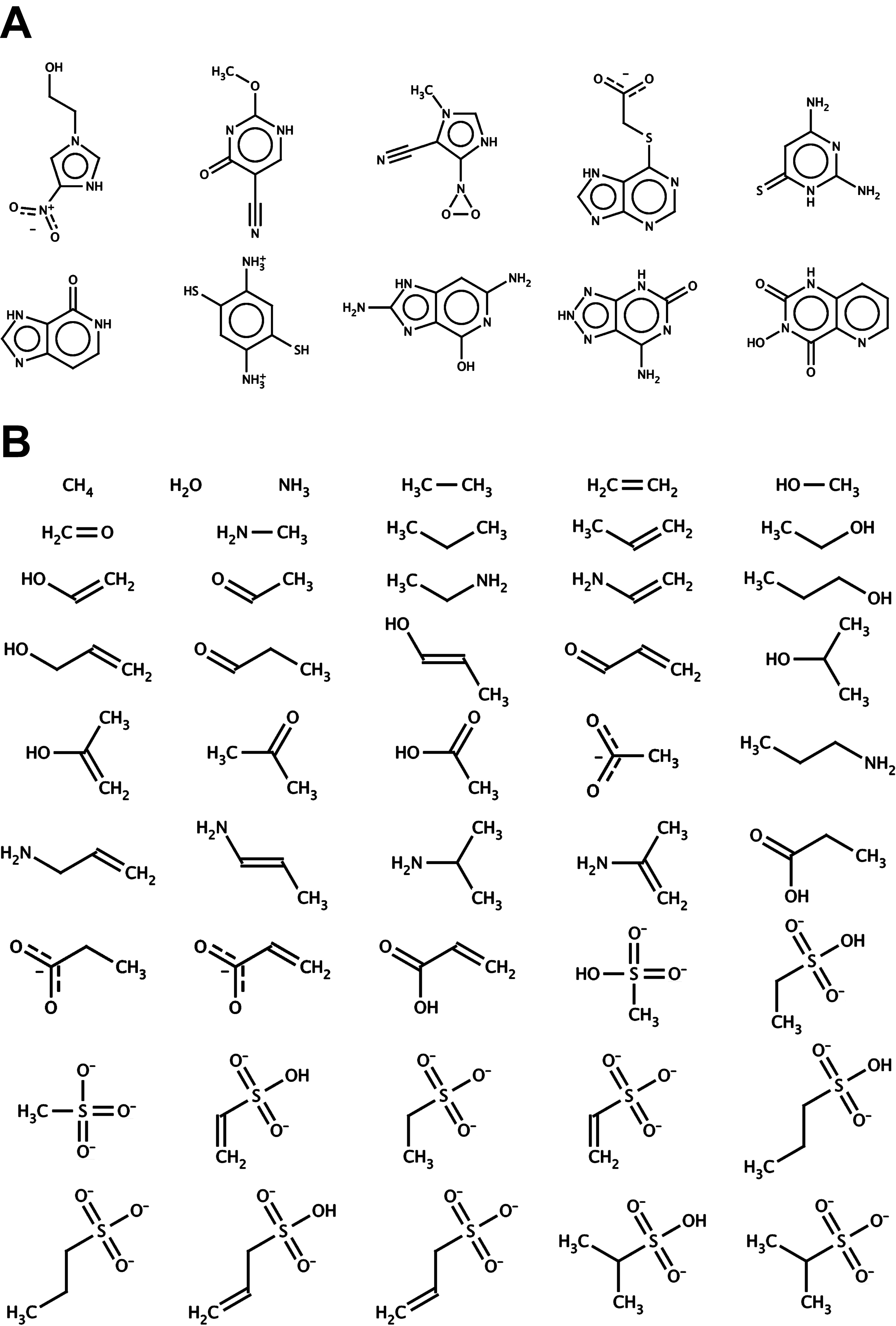
The large- and small-fragment libraries. A) Ten representative fragments were selected from the large-fragment library, which contains 2,437 fragments total. B) All 46 molecular fragments of the small-fragment library.
When small refinements are desired, the mutation operator can draw upon the default library of smaller fragments. To generate the small-fragment library, 46 fragments were created manually using the program molden (46). Most of these small fragments contained one to three carbon atoms, connected by varying combinations of single and double bonds. To these carbon backbones amino, hydroxyl, sulfonate, sulfonic-acid, carboxylate, carboxyl, and ketone moieties were added. Additionally, ammonia and water were included in the library. The size of the small fragments ranged from three to fifteen atoms (average: 9.6 atoms, standard deviation: 2.8, Figure 2b). All 46 fragments of the small-fragment library are shown in Figure 3b.
To control the AutoGrow algorithm, user-specified variables must be included in a user-input file. A number of the AutoGrow variables in the user-input file correspond to AutoGrid and AutoDock parameters defined in a typical AutoGrid grid-parameter file (gpf) or AutoDock docking-parameter file (dpf), respectively. Although AutoGrow provides default AutoGrid and AutoDock parameters, these values are receptor dependent and should be modified as necessary. The AutoGrow “best dock criteria” variable allows the user to specify whether AutoGrow should judge the fitness of a given ligand based on the AutoDock-predicted binding energy of its lowest-energy or most-populated cluster. In addition to specifying the parameters that AutoGrow will pass to the AutoDock and AutoGrid programs, the user must also specify the parameters of the AutoGrow evolutionary algorithm itself. These parameters include the number of best-fit ligands from each generation to become the founding members of the next generation, as well as the number of “children” and “mutant” ligands derived from those founders via the “crossover” and “mutation” operator, respectively.
The user can place a number of constraints on the growing ligands. User-defined variables specifying the maximum number of generations for which AutoGrow will run as well as the maximum number of atoms allowed per ligand can be used to prevent ligands from growing too large. The user can also require that the evolving ligands dock at a specific location within the active site and that fragments be added only to selected “core” scaffold hydrogen atoms. Details regarding AutoGrow installation, configuration, and execution can be found in the supplementary materials. A summary of all AutoGrow user-specified variables is given in Table S1.
Ligand and Receptor Preparation/Docking Parameters for the Validation Studies
For each of the three validation studies, the receptor proteins were processed prior to AutoGrow execution. Optimized AutoGrid and AutoDock parameters for each of the systems were either taken from the literature or determined here.
For the TbREL1-ATP complex, the protein was processed as described by Amaro et. al (47). To use AutoGrow to regenerate the ATP substrate, we first identified AutoDock parameters that could successfully redock the co-crystallized ATP molecule back into the 1XDN structure when three crystallographic active-site water molecules essential for proper ATP binding were retained (Table S2). These AutoDock parameters, tailored specifically for 1XDN, were used in all subsequent AutoGrow-guided AutoDock runs. Decomposing the co-crystallized ATP ligand and adding hydrogen atoms as necessary generated three ATP-derived molecules. Two of these, a triphosphate with a terminal hydroxyl group and an adenine, constituted a fragment library. The third, (3R,4S)-5-(Methyl)tetrahydrofuran-3,4-diol, served as the initial “core” scaffold. Fragments were allowed to grow from four scaffold linker hydrogen atoms: the three equivalent hydrogen atoms of the methyl group and the 5C hydrogen trans to the two hydroxyl groups of the tetrahydrofuran ring (Figure 4a).
Figure 4.

New fragment libraries were created to see if AutoGrow could recreate (A) ATP, (B) oseltamivir, and (C) AMZ. In all cases, the initial “core” scaffold is shown above the two fragments of each library, and yellow circles depict linker hydrogen atoms. A) To regenerate ATP, a library was created containing only two fragments: a triphosphate with a terminal hydroxyl group and an adenine. (3R,4S)-5-(Methyl)tetrahydrofuran-3,4-diol served as the initial scaffold, and fragments were allowed to grow from four scaffold hydrogen atoms. B) To regenerate oseltamivir, a library was created containing only two fragments: pentan-3-ol and acetamide. (5R)-5-aminocyclohex-1-ene-1-carboxylate served as the scaffold, and fragments were allowed to grow from two scaffold linker hydrogen atoms. C) To regenerate AMZ, a library was created containing only two fragments: methoxy(oxo)phosphinate and 5-amino-1H-imidazole- 4-carboxamide. (3R,4S)-oxolane-3,4-diol served as the initial scaffold, and fragments were allowed to grow from two scaffold linker hydrogen atoms.
For the neuraminidase-oseltamivir complex, oseltamivir was extracted from the active site, and hydrogen atoms were added using the PRODRG server (48). AutoDockTools (ADT) version 1.5.2 (49) was then used to remove non-polar ligand hydrogen atoms, to add protein-receptor polar hydrogen atoms, and to assign Gasteiger charges (50) to all atoms of the system. For this system, we identified AutoDock parameters that could successfully redock the co-crystallized oseltamivir molecule back into the 2HU4 structure, similar to parameters reported in the literature (51) (Table S2). These optimized AutoDock parameters were used in all subsequent AutoGrow-guided AutoDock runs. The co-crystallized oseltamivir molecule was decomposed into three oseltamivir-derived molecules. Two of these, pentan-3-ol and acetamide, comprised the fragment library. The third, (5R)-5-aminocyclohex-1-ene-1-carboxylate, served as the initial “core” scaffold. Fragments were allowed to grow from two scaffold linker hydrogen atoms (Figure 4b).
For the AICAR transformylase-AMZ complex, we first removed from the crystal structure the AMZ ligand, all but six crystallographic water molecules judged important for AMZ binding, and all potassium ions (52). Xanthosine-5'-monophosphate (XMP) and N-[(s)-(4-{[(2-amino-4-hydroxyquinazolin- 6-yl)(dihydroxy)-lambda~4~-sulfanyl]amino}phenyl)(hydroxy)methyl]-l-glutamic acid (compound 354), two ligands that were co-crystallized with AMZ, were retained as part of the receptor structure. As ADT 1.5.2 does not assign hydrogen atoms to water molecules, the system was processed with ICM (Molsoft), which added hydrogen atoms to the protein and to the water oxygen atoms, appropriately protonated histidines, flipped certain protein amide groups, and corrected terminal phase angles. Hydrogen atoms were added to the AMZ ligand with Discovery Studio (Accelrys). ADT (49) was then used to remove nonpolar hydrogens and assign Gasteiger charges (50) to both the protein and the ligand structures. We then generated new optimized AutoDock parameters that could successfully redock the co-crystallized AMZ molecule back into the 1P4R structure (see Table S2). These tailored AutoDock parameters were used in all subsequent AutoGrow-guided 1P4R AutoDock runs. The co-crystallized AMZ ligand was decomposed into three AMZ-derived ligands; hydrogens were added to these ligands using the PRODRG server (48). Two of these new molecules, methoxy(oxo)phosphinate and 5-amino-1H-imidazole-4-carboxamide, constituted the fragment library. The third, (3R,4S)-oxolane-3,4-diol, served as the initial “core” scaffold. Fragments were allowed to grow from two scaffold linker hydrogen atoms (Figure 4c).
AutoGrow Parameters
For each of the three test systems, we ran AutoGrow using an initial generation size of fifty compounds. Each subsequent generation also contained fifty compounds. Ten of those compounds were taken from the previous generation, based on both the score of the best AutoDock cluster (either the most-populated or the lowest-energy cluster, depending on the system) and successful active-site binding. An additional twenty “children” and twenty “mutants” were created from these ten primary individuals, subject to the requirement that all compounds contain fewer than seventy atoms. The first generation initially contained only the scaffold and 49 “mutants,” as no previous generation existed from which “parents” could be drawn for crossover production.
AutoGrid version 4.0 was used to create affinity grids centered on the active site of each protein receptor. Grid dimensions and spacing are given in Table S2. Affinity grids were calculated for all of the atom types present in the target ligand (ATP, oseltamivir, and AMZ, respectively). AutoGrow docked all generated ligands into their corresponding protein receptors with AutoDock 4.0.1 (36) using the AutoGrid and AutoDock parameters given in Table S2.
Results/Discussion
In the current work, we introduce AutoGrow, a new computer-aided drug design algorithm that uses a growing strategy to build upon an initial “core” scaffold. Molecular fragments are added at random to this scaffold, thereby generating a population of novel ligands. These ligands are subsequently docked into the target protein receptor. An evolutionary algorithm evaluates the docking scores of each population member, and the best binders become founders of the subsequent generation. As generation after generation is created, each based on the most fit individuals of the previous generation, a larger inhibitor with higher predicted binding affinity eventually evolves.
Method Validation
In order to validate the AutoGrow algorithm, we attempted to recreate several known small-molecule protein ligands, including ATP, the natural substrate of Trypanosoma brucei RNA editing ligase 1 (TbREL1); oseltamivir, a nanomolar-affinity antiviral inhibitor of the influenza neuraminidase enzyme; and aminoimidazole 4-carboxamide ribonucleotide (AMZ), a nanomolar-affinity inhibitor of AICAR transformylase.
AutoGrow limits combinatorial explosion by drawing upon fragment libraries and using an evolutionary algorithm. As a result, AutoGrow often generates compounds with higher predicted binding affinities than the initial scaffold, despite the fact that most modifications to a good scaffold reduce predicted binding affinity. However, the use of even a moderately sized fragment library leads to a computationally intractable number of possible outcomes (ligands); consequently, AutoGrow is nondeterministic. If the goal is to produce a ligand that has a higher predicted binding affinity than an initial scaffold, a solution can be reasonably expected. If the goal is to produce one specific ligand out of the millions of possible compound outcomes, success is unlikely.
In order to have any reasonable chance of recreating a specific compound such as ATP, oseltamivir, or AMZ, the number of possible combinations must be significantly reduced. To achieve this reduction, instead of using the AutoGrow default large- or small-fragment libraries, we cut each of these specific compounds into three pieces, producing two fragments and one initial “core” scaffold for each. Fragments were allowed to grow from appropriate scaffold linker hydrogens, though all fragment hydrogen atoms were allowed to serve as linkers (Figure 4). Despite these limitations imposed on the fragment-library size and the number of scaffold linker hydrogen atoms, this setup can still produce hundreds of distinct compounds after only several generations.
Validation Using ATP
ATP, the natural substrate of TbREL1, is a good candidate for validation, as the ATP-bound complex has been crystallized at 1.2 Å resolution (40). AutoGrow created ATP in the second generation via mutation by adding the adenine fragment to the second best ligand of the first generation; ATP was the highest scoring ligand of the second generation (Table S3). The ATP adenosine moieties of both the co-crystallized and docked ligand sit deep within the binding site and interact with the protein via a hydrogen bond with E86 and via pi-pi interactions with F209. Additionally, the N1 adenine nitrogen accepts a hydrogen bond from a co-crystallized water molecule. Two conserved residues, E159 and N92, interact with the ATP ribose moiety via hydrogen bonds.
The co-crystallized and AutoGrow-generated ligands do not overlap in the region of the ATP polyphosphate tail. A magnesium ion, present in the crystal structure but absent in the protein receptor used for docking, coordinates many of the polyphosphate oxygen atoms; in the absence of magnesium, the position of the polyphosphate tail is naturally altered (Figure 5). The RMSD between the co-crystallized ATP and the docked, AutoGrow-generated ATP with all hydrogen atoms removed is 3.88 Å. When the polyphosphate tail is removed (neglecting all atoms including and beyond the alpha phosphate), the RMSD difference falls to 0.39 Å.
Figure 5.

ATP co-crystallized with TbREL1 (rendered in CPK) superimposed on the AutoGrow-generated ATP docked into the protein receptor (rendered in licorice). Black lines represent hydrogen bonds. The co-crystallized and docked ligands do not overlap in the region of the ATP polyphosphate tail because a magnesium ion, present in the crystal structure but absent in the protein receptor used for docking, coordinates many of the polyphosphate oxygen atoms.
Validation Using Oseltamivir
Oseltamivir is a nanomolar-affinity antiviral compound that inhibits the neuraminidase enzyme; the oseltamivir-neuraminidase (N1) complex has been crystallized at 2.50 Å resolution (41). Although AutoGrow failed to recreate oseltamivir after eight generations, inspection of the final set of compounds revealed that AutoGrow had created a very similar compound (Figure 6). This compound, the third best ligand of the second generation (Table S3), was created as a result of a crossover between the eighth and ninth best ligands of the first generation. Like oseltamivir, the new compound is predicted to make hydrogen bond interactions with the side chains of R292, R121, E119, D151, and R152 (Figure 6). However, the linking amide nitrogen of oseltamivir, a potential hydrogen bond acceptor, is absent in the new compound. Interestingly, the crystal structure of the neuraminidase-oseltamivir complex demonstrates that this nitrogen does not participate in hydrogen bonding (41). In the new compound, the amide nitrogen is further away from the central ring, allowing it to potentially form hydrogen bonds with the backbone carbonyls of W178 and D151. The RMSD between the co-crystallized oseltamivir and the docked, AutoGrow-generated novel compound with all hydrogen atoms removed is 1.95 Å, suggesting that even with the altered acetamide moiety, the binding of the novel compound closely mimics that of oseltamivir (Figure 6).
Figure 6.

On the left, oseltamivir co-crystallized with neuraminidase (rendered in CPK) superimposed on the AutoGrow-generated novel compound docked into the protein receptor using AutoDock 4.0 (rendered in licorice). Black lines represent hydrogen bonds. On the right, a two dimensional representation of oseltamivir and the AutoGrow-generated novel compound demonstrates their similarity.
Validation Using AMZ
AMZ is a nanomolar-affinity inhibitor of AICAR transformylase; the AMZ-protein complex has been crystallized at 2.55 Å (42). AutoGrow was able to recreate AMZ in the third generation via mutation by adding the 5-amino-1H-imidazole-4-carboxamide fragment to the sixth best ligand of the second generation; AMZ was the highest scoring ligand of the third generation (Table S3).
The AutoDock-predicted binding pose of the AutoGrow-generated AMZ ligand was nearly identical to the crystallographic pose, with a heavy-atom RMSD of only 0.48 Å (Figure 7). The crystal structure and predicted docked pose of AMZ bound to AICAR transformylase reveal a number of protein-ligand and water-ligand hydrogen bonds. The hydroxyl group of the Y208 side chain and the guanidine groups of the R588 and R207 side chains all make hydrogen bonds with the non-bridging phosphates of the AMZ phosphinate moiety, as do four co-crystallized water molecules buried within the active site. The carboxylate group of the D339 side chain and the G316 backbone carbonyl accept hydrogen bonds from the two hydroxyl groups of the central (3R,4S)-oxolane-3,4-diol core, and two additional co-crystallized water molecules donate hydrogen bonds to the oxygen atoms of those same groups. Additionally, the carbonyl oxygen of the N431 side chain accepts a hydrogen bond from the amino group attached to the AMZ imidazole ring, and the benzene ring of compound 354 co-crystallized with AMZ stabilizes that imidazole via pi-pi interactions (not shown in Figure 7). Finally, the guanidine group of the R451 side chain donates two hydrogen bonds to the amide carbonyl oxygen of AMZ, and the F541 backbone carbonyl accepts a hydrogen bond from the AMZ amide nitrogen.
Figure 7.

AMZ co-crystallized with AICAR transformylase (rendered in CPK) superimposed on the AutoGrow-generated AMZ docked into the protein receptor (rendered in licorice). Black lines represent hydrogen bonds. For clarity, water molecules present in the active site, which are essential for AMZ binding, are not shown.
Advantages and Limitations of the Current Method
AutoGrow has a number of advantages over other de novo drug design algorithms. AutoGrow is novel in the way it combines an evolutionary algorithm with a fragment-based growing method to explore new chemical space and a fitness function based on a fully flexible ligand docking score. Several other programs also combine fragment-based growing with an evolutionary algorithm, including Chemical Genesis (53), LEA (54), and LigBuilder (55), but these algorithms do not incorporate full flexible-ligand docking into their respective fitness functions. AutoGrow performs a separate docking of the entire molecule (moieties and scaffold) for each ligand it creates, allowing the scaffold position to change in response to the addition of any new fragments. Although the ab initio redockings increase the computational expense, in many cases they allow for a more realistic prediction of the ligand binding pose.
Two programs, ADAPT (56) and SYNOPSIS (57), are similar to AutoGrow in that they are fragment-based evolutionary algorithms that do incorporate docking scores into the fitness function. ADAPT uses DOCK (56), and SYNOPSIS is designed to work with any number of user-generated fitness functions. For the latter, Vinkers et al. gave one example where an in house docking-program score was used to determine fitness (57). Despite these similarities, however, AutoGrow differs from ADAPT and SYNOPSIS in the way its evolutionary algorithm is implemented. ADAPT and AutoGrow have similar crossover operators, but very different mutation operators. While ADAPT generates its initial population of ligands by adding fragments to a core scaffold, in subsequent generations it “mutates” ligands by either changing atom types or altering local connectivity. AutoGrow “mutates” ligands by adding novel fragments at each generation, not just the first. In contrast, SYNOPSIS and AutoGrow have somewhat similar mutation operators. However, SYNOPSIS has no crossover operator whereby two ligands can be combined to produce a third with chemical properties of the two “parents.”
AutoGrow has several limitations. As AutoDock is used as the selection operator, the results are dependent on the accuracy of the AutoDock scoring function, which has a standard error of 2.177 kcal mol−1 (36). The AutoDock scoring function used to predict ligand free energies of binding is much faster and less accurate than comprehensive physics-based approaches like thermodynamic integration (3), single-step perturbation (4), and free energy of perturbation (5). Nevertheless, a recent validation study (58) showed that AutoDock performs well compared to other common docking programs like DOCK (21), FleX (33), and GOLD (34), and AutoDock is the most cited docking software in the literature (44). Additionally, unlike many other docking programs, AutoDock is freely distributed.
AutoDock employs a Lamarckian genetic algorithm that is computationally intensive and time consuming; one study found that AutoDock was slower than DOCK, FleX, GOLD, and ICM (58). AutoGrow generates novel compounds quickly, but its dependence on AutoDock impacts the efficiency of the algorithm. The extent of impact depends on the exact AutoDock parameters used and the computer hardware employed. As an example, an analysis of the AutoGrow data from one of our own ongoing projects shows that AutoDock docked each of our AutoGrow-generated ligands in 4.1 hrs ± 1.9. Running on 51 processors, each generation of 50 ligands finished in 6.5 hrs ± 2.6.
Inaccuracies in the predicted binding energies may arise because AutoDock only approximates ligand flexibility via torsional rotation and molecule translation and rotation. All bond lengths are fixed so ligand flexibility is not truly complete; however, the effects due to changes in bond length are expected to be minor. Additionally, AutoDock uses an implicit desolvation term that is not as accurate as more computationally expensive explicitly solvated molecular dynamics techniques.
Although AutoGrow is not presently coded to interface with alternate docking programs, therefore precluding an explicit comparison, we anticipate that incorporating a faster docking and scoring module will improve the computational performance. Other methods to improve performance, such as incorporating a parallelized version of AutoDock (59), can also be envisioned. As AutoDock is free, performs well compared to many other docking programs (58), and has a well-tested scoring function (44), AutoGrow has been coded to interface with AutoDock by default; with only slight modification to the code, however, AutoGrow could interface with any number of docking programs. We note that AutoGrow is open source, and we encourage source-code modifications of this type.
Other AutoGrow limitations are typical of any computer-aided drug design method. With the number of chemically feasible, drug-like molecules being on the order of 1060 to 10100 (1), the size of chemical space is far too large to be completely scanned by any computer algorithm. While AutoGrow can in many cases generate compounds predicted to bind with high affinity, one cannot expect it to find the single best binder. As evolutionary algorithms are often nondeterministic, we recommend running AutoGrow on the same initial scaffold several times in order to obtain multiple potential inhibitor candidates for subsequent evaluation and optimization.
The quality of the AutoGrow results is also affected by the fragment database used. Medicinal chemists should carefully filter AutoGrow results, eliminating or modifying metabolically reactive groups and other undesirable moieties. AutoGrow is unlikely to produce good de novo drug candidates if the default fragment databases are used. The program does not take into account Lipinski’s Rule of Five (60) or ADME-Tox properties. Consequently, one should not expect AutoGrow-generated compounds to be drug-like. However, the compounds that are generated can help the medicinal chemist identify novel and potentially important receptor-ligand interactions. AutoGrow-suggested compounds many also elucidate new drug scaffolds, thereby providing useful information during the initial stages of drug development. We recommend using AutoGrow to supplement the medicinal chemist’s creativity, rather than to replace it.
AutoGrow can be downloaded for free at http://autogrow.ucsd.edu
Supplementary Material
Acknowledgements
REA is funded by NIH F32-GM077729 and NSF MRAC CHE060073N. Funding by NIH GM31749, NSF MCB-0506593 and MCA93S013 (to JAM) also supported this work. Additional support from the Howard Hughes Medical Institute, the National Center for Supercomputing Applications, the San Diego Supercomputing Center, the W.M. Keck Foundation, Accelrys, Inc., the National Biomedical Computational Resource and the Center for Theoretical Biological Physics is gratefully acknowledged. We would also like to thank Meghan Cain for helpful discussions and code debugging.
Footnotes
Supporting Information
Technical details regarding AutoGrow installation, configuration, and execution can be found in this section. A summary of all AutoGrow user-specified variables is given in Table S1. Optimized AutoDock parameters for all validation systems are found in Table S2. The structures and AutoDock-predicted binding energies of the target ligands are found in Table S3. The AutoGrow workflow is shown in Figure S1.
References
- 1.Schneider G, Fechner U. Computer-based de novo design of drug-like molecules. Nature Reviews Drug Discovery. 2005;4:649. doi: 10.1038/nrd1799. [DOI] [PubMed] [Google Scholar]
- 2.Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 3.Oostenbrink BC, Pitera JW, van Lipzig MM, Meerman JH, van Gunsteren WF. Simulations of the estrogen receptor ligand-binding domain: affinity of natural ligands and xenoestrogens. J Med Chem. 2000;43:4594–4605. doi: 10.1021/jm001045d. [DOI] [PubMed] [Google Scholar]
- 4.Oostenbrink C, van Gunsteren WF. Free energies of binding of polychlorinated biphenyls to the estrogen receptor from a single simulation. Proteins. 2004;54:237–246. doi: 10.1002/prot.10558. [DOI] [PubMed] [Google Scholar]
- 5.Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, et al. FEP-guided selection of bicyclic heterocycles in lead optimization for non-nucleoside inhibitors of HIV-1 reverse transcriptase. J Am Chem Soc. 2006;128:15372–15373. doi: 10.1021/ja066472g. [DOI] [PubMed] [Google Scholar]
- 6.von Lilienfeld OA, Lins RD, Rothlisberger U. Variational particle number approach for rational compound design. Phys Rev Lett. 2005;95:153002. doi: 10.1103/PhysRevLett.95.153002. [DOI] [PubMed] [Google Scholar]
- 7.Wang M, Hu X, Beratan DN, Yang W. Designing molecules by optimizing potentials. J Am Chem Soc. 2006;128:3228–3232. doi: 10.1021/ja0572046. [DOI] [PubMed] [Google Scholar]
- 8.Gillet VA, Johnson AP, Mata P, Sike S. Automated structure design in 3D. Tetrahedron. 1990;3:681. [Google Scholar]
- 9.Moon JB, Howe WJ. Computer design of bioactive molecules: A method for receptor-based de novo ligand design. Proteins: Structure, Function, and Genetics. 1991;11:314. doi: 10.1002/prot.340110409. [DOI] [PubMed] [Google Scholar]
- 10.Bohm H-J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. JComputAided MolDes. 1992;6:61. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- 11.Gillet V, Johnson AP, Mata P, Sike S, Williams P. SPROUT: a program for structure generation. JComputAided MolDes. 1993;7:127. doi: 10.1007/BF00126441. [DOI] [PubMed] [Google Scholar]
- 12.Rotstein SH, Murcko MA. GroupBuild: a fragment-based method for de novo drug design. JMedChem. 1993;36:1700. doi: 10.1021/jm00064a003. [DOI] [PubMed] [Google Scholar]
- 13.Bohacek RS, McMartin C. Multiple Highly Diverse Structures Complementary to Enzyme Binding-Sites - Results of Extensive Application of a de-Novo Design Method Incorporating Combinatorial Growth. Journal of the American Chemical Society. 1994;116:5560. [Google Scholar]
- 14.Clark DE, Frenkel D, Levy SA, Li J, Murray CW, Robson B, et al. PRO_LIGAND: An approach to de novo molecular design. 1. Application to the design of organic molecules. JComputAided MolDes. 1995;9:13. doi: 10.1007/BF00117275. [DOI] [PubMed] [Google Scholar]
- 15.Westhead DR, Clark DE, Frenkel D, Li J, Murray CW, Robson B, et al. PRO_LIGAND: An approach to de novo molecular design. 3. A genetic algorithm for structure refinement. JComputAided MolDes. 1995;9:139. doi: 10.1007/BF00124404. [DOI] [PubMed] [Google Scholar]
- 16.DeWitte RS, Shakhnovich EI. SMoG: de Novo Design Method Based on Simple, Fast, and Accurate Free Energy Estimates. 1. Methodology and Supporting Evidence. Journal of the American Chemical Society. 1996;118:11733. [Google Scholar]
- 17.Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? Journal of Computational Chemistry. 2000;21:1049. [Google Scholar]
- 18.Sheridan RP, Nilakantan R, Rusinko A, Bauman N, Haraki KS, Venkataraghavan R. 3DSEARCH: A System for Three-Dimensional Substructure Searching. J Chem Inf Comp Sci. 1989;29:255. [Google Scholar]
- 19.Van Drie JH, Weininger D, Martin YC. ALADDIN: an integrated tool for computer-assisted molecular design and pharmacophore recognition from geometric, steric, and substructure searching of three-dimensional molecular structures. JComputAided MolDes. 1989;3:225. doi: 10.1007/BF01533070. [DOI] [PubMed] [Google Scholar]
- 20.DesJarlais RL, Sheridan RP, Seibel GL, Dixon JS, Kuntz ID, Venkataraghavan R. Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure. JMedChem. 1988;31:722. doi: 10.1021/jm00399a006. [DOI] [PubMed] [Google Scholar]
- 21.Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. JComputAided MolDes. 2001;15:411. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 22.Ewing TJA, Kuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. Journal of Computational Chemistry. 1997;18:1175. [Google Scholar]
- 23.Kuntz ID. Structure-Based Strategies for Drug Design and Discovery. Science. 1992;257:1078. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 24.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. JMolBiol. 1982;161:269. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 25.Kuntz ID, Meng EC, Shoichet BK. Structure-based molecular design. 1994;27:117. [Google Scholar]
- 26.Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy evaluation. Journal of Computational Chemistry. 1992;13:505. [Google Scholar]
- 27.Lawrence MC, Davis PC. CLIX: a search algorithm for finding novel ligands capable of binding proteins of known three-dimensional structure. Proteins. 1992;12:31. doi: 10.1002/prot.340120105. [DOI] [PubMed] [Google Scholar]
- 28.Hart TN, Ness SR, Read RJ. Critical evaluation of the research docking program for the CASP2 challenge. Proteins. 1997 Suppl 1:205. doi: 10.1002/(sici)1097-0134(1997)1+<205::aid-prot27>3.3.co;2-p. [DOI] [PubMed] [Google Scholar]
- 29.Hart TN, Read RJ. A multiple-start Monte Carlo docking method. Proteins. 1992;13:206. doi: 10.1002/prot.340130304. [DOI] [PubMed] [Google Scholar]
- 30.Lauri G, Bartlett PA. CAVEAT: a program to facilitate the design of organic molecules. JComputAided MolDes. 1994;8:51. doi: 10.1007/BF00124349. [DOI] [PubMed] [Google Scholar]
- 31.Miller MD, Kearsley SK, Underwood DJ, Sheridan RP. FLOG: a system to select 'quasi-flexible' ligands complementary to a receptor of known three-dimensional structure. JComputAided MolDes. 1994;8:153. doi: 10.1007/BF00119865. [DOI] [PubMed] [Google Scholar]
- 32.Rarey M, Kramer B, Lengauer T. The particle concept: placing discrete water molecules during protein-ligand docking predictions. Proteins. 1999;34:17. [PubMed] [Google Scholar]
- 33.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. JMolBiol. 1996;261:470. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 34.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. JMolBiol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 35.Lambert MH. Docking Conformationally Flexible Molecules into Protein Binding Sites. In: Charifson PS, editor. Docking Conformationally Flexible Molecules into Protein Binding Sites. New York: Dekker; 1997. [Google Scholar]
- 36.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19:1639–1662. [Google Scholar]
- 37.Venkatachalam CM, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. Journal of Molecular Graphics and Modelling. 2003;21:289. doi: 10.1016/s1093-3263(02)00164-x. [DOI] [PubMed] [Google Scholar]
- 38.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. JMedChem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 39.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. JMedChem. 2004;47:1750. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 40.Deng J, Schnaufer A, Salavati R, Stuart KD, Hol WG. High resolution crystal structure of a key editosome enzyme from Trypanosoma brucei: RNA editing ligase 1. JMolBiol. 2004;343:601. doi: 10.1016/j.jmb.2004.08.041. [DOI] [PubMed] [Google Scholar]
- 41.Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, Blackburn GM, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- 42.Li C, Xu L, Wolan DW, Wilson IA, Olson AJ. Virtual screening of human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase against the NCI diversity set by use of AutoDock to identify novel nonfolate inhibitors. J Med Chem. 2004;47:6681–6690. doi: 10.1021/jm049504o. [DOI] [PubMed] [Google Scholar]
- 43.Holland JH. Adaptation in Natural and Artificial Systems. Ann Arbor MI: University of Michigan Press; 1975. [Google Scholar]
- 44.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 45.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. JChemInfModel. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaftenaar G, Noordik JH. Molden: a pre- and post-processing program for molecular and electronic structures. J Comput Aided Mol Des. 2000;14:123–134. doi: 10.1023/a:1008193805436. [DOI] [PubMed] [Google Scholar]
- 47.Amaro RE, Schnaufer A, Interthal H, Hol W, Stuart KD, McCammon JA. Discovery of drug-like inhibitors of an essential RNA-editing ligase in Trypanosoma brucei. Proceedings of the National Academy of Sciences. 2008:17278. doi: 10.1073/pnas.0805820105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 49.Sanner MF. A component-based software environment for visualizing large macromolecular assemblies. Structure. 2005;13:447–462. doi: 10.1016/j.str.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 50.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity--a rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 51.Cheng LS, Amaro RE, Xu D, Li WW, Arzberger PW, McCammon JA. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neuraminidase. J Med Chem. 2008;51:3878–3894. doi: 10.1021/jm8001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheong CG, Wolan DW, Greasley SE, Horton PA, Beardsley GP, Wilson IA. Crystal structures of human bifunctional enzyme aminoimidazole-4-carboxamide ribonucleotide transformylase/IMP cyclohydrolase in complex with potent sulfonyl-containing antifolates. J Biol Chem. 2004;279:18034–18045. doi: 10.1074/jbc.M313691200. [DOI] [PubMed] [Google Scholar]
- 53.Glen RC, Payne AW. A genetic algorithm for the automated generation of molecules within constraints. J Comput Aided Mol Des. 1995;9:181–202. doi: 10.1007/BF00124408. [DOI] [PubMed] [Google Scholar]
- 54.Tschinke V, Cohen NC. The NEWLEAD program: a new method for the design of candidate structures from pharmacophoric hypotheses. J Med Chem. 1993;36:3863–3870. doi: 10.1021/jm00076a016. [DOI] [PubMed] [Google Scholar]
- 55.Wang R, Gao Y, Lai L. LigBuilder: A Multi-Purpose Program for Structure-Based Drug Design. Journal of Molecular Modeling. 2000;6:498. [Google Scholar]
- 56.Pegg SC, Haresco JJ, Kuntz ID. A genetic algorithm for structure-based de novo design. J Comput Aided Mol Des. 2001;15:911–933. doi: 10.1023/a:1014389729000. [DOI] [PubMed] [Google Scholar]
- 57.Vinkers HM, de Jonge MR, Daeyaert FF, Heeres J, Koymans LM, van Lenthe JH, et al. SYNOPSIS: SYNthesize and OPtimize System in Silico. J Med Chem. 2003;46:2765–2773. doi: 10.1021/jm030809x. [DOI] [PubMed] [Google Scholar]
- 58.Bursulaya BD, Totrov M, Abagyan R, Brooks CL., 3rd Comparative study of several algorithms for flexible ligand docking. J Comput Aided Mol Des. 2003;17:755–763. doi: 10.1023/b:jcam.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- 59.Khodade P, Prabhu R, Chandra N, Raha S, Govindarajan R. Parallel implementation of AutoDock. Journal of Applied Crystallography. 2007;40:598–599. [Google Scholar]
- 60.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.