

Abstract
Oncogenic EGFRvIII is a naturally occurring oncoprotein and is expressed in about 40–50% of human glioblastomas, particular those that arise de novo. To understand the molecular mechanisms by which this oncoprotein alters transforming phenotypes, and since our previous work indicated that SHP-2 protein tyrosine phosphatase activity modulated EGFRvIII activation and downstream signaling, we examined whether SHP-2 plays a role in EGFRvIII-induced oncogenesis by using both PTEN-deficient U87MG.EGFRvIII and PTEN-intact LN229.EGFRvIII cells. Inhibition of SHP-2 expression by Shp-2 siRNA inhibited cell growth, transformation and altered morphology of these EGFRvIII transformed GBM cells. Ectopic expression of a PTPase-inactive form of SHP-2, SHP-2 C459S, but not its wild-type SHP-2 or either of two SH2 domain mutants, abrogated transformation of EGFRvIII-expressing glioblastomas in soft agar and in nude mice. SHP-2 C459S cells grew slower and exhibited a more flattened morphology with more organized actin stress fibers under both full growth and low serum conditions. Furthermore, shp-2 +/− and −/− mouse embryonic fibroblasts (MEFs) could not be transformed by EGFRvIII while shp-2 +/+ MEFs displayed a fully transformed phenotype upon introduction of EGFRvIII, again indicating a requirement for functional SHP-2 in EGFRvIII transformation. Moreover, the SHP-2 PTPase activity inhibitor NSC-87877 inhibited endogenous SHP-2 activity, Erk phosphorylation and transformation in both GBM cell lines. EGFRvIII expression recruited SHP-2 to the receptor complex to transduce signals and also increased SHP-2 phosphorylation at Tyr542. Inhibition of EGFRvIII-induced cell growth and transformation by SHP-2 C459S or shp-2 siRNA was mediated by its ability to block cell cycle progression at different phases in these GBM cells. These data indicate that differential activation of SHP-2 phosphorylation at Tyr542 in these two GBM cell lines likely results in increased different PTPase activity and distinct mechanisms of cell cycle progression andSHP-2, in particular its PTPase activity, plays a critical role in EGFRvIII-mediated transformation.
Keywords: SHP-2, EGFRvIII, transformation, cell cycle, human glioblastoma cells
Introduction
The epidermal growth factor receptor (EGFR) gene is often amplified and mutated in human cancers [1, 2]. EGFRvIII is the most common mutation of EGFR and frequently correlates with advanced diseases including malignant gliomas, breast carcinomas, non-small cell lung carcinomas, prostate and ovarian cancers [3–5]. This mutated oncoprotein is expressed on the cell surface but is not present in any normal adult tissues, making it an attractive target for anticancer therapy [4, 6]. EGFRvIII is constitutively phosphorylated in the absence of EGF [7]. This constitutive kinase activity has been shown to dysregulate signals leading to increased tumorigenicity and resistance to therapy [7, 8]. Targeting EGFRvIII for cancer therapy by using monoclonal antibodies or kinase inhibitors is under investigation [5, 9]. However, the intracellular signals activated by oncogenic EGFRvIII are poorly understood. Therefore, understanding the signaling pathways of EGFRvIII may provide new strategies for cancer treatment.
Activated EGFR initiates a cascade of phosphorylation events that transduce signals to change a variety of biological processes [10–12]. These protein phosphorylation events are regulated by a set of protein kinases and protein phosphatases. One of the protein tyrosine phosphatases (PTPase), SHP-2 (also termed PTPN11, PTP1D, PTP2C, PTPL1, SHPTP3 and Syp) [13–15], has been shown to be involved in positively regulating signals initiated by epithelial growth factor (EGF) and also other growth factors such as platelet-derived growth factor (PDGF) and insulin to change biological phenotypes [16, 17]. Upon RTK activation, SHP-2 interacts with phosphotyrosine residues of receptors or adaptor molecules to transduce signals and regulates downstream Ras/ERK or PI3-K/Akt pathways. SHP-2 plays an important role in cell proliferation and differentiation [18]. A catalytically inactive form of SHP-2 (C459S) reduced mitogenesis stimulated by insulin and EGF [19, 20]. A N-terminal deletion of SHP-2 not only reduced ERK activity induced by IGF-I, EGF and PDGF, but also inhibited the expression of the beta subunit of platelet-derived growth factor receptor in mouse embryonic fibroblast cells [21, 22]. Additional evidence indicates that SHP-2 plays a critical role in the regulation of cell spreading, migration, and cytoskeletal organization of fibroblasts and epithelial cells [23, 24]. Importantly, the catalytic activity of SHP-2 is required for most of these biological processes [19, 20].
Recent studies showed that SHP-2 plays an important role in cell transformation and in human diseases. SHP-2 mediates v-Src-induced morphological transformation in fibroblasts and is required for hematopoietic cell transformation [25, 26]. Gain-of-function of SHP-2 was found in Noonan syndrome-associated leukemias [27]. SHP-2 is also mutated in human cancers and, interestingly, these mutations resulted in an activated form of SHP-2 to enhance transformation [28–30]. SHP-2 was overexpressed in breast tumors [31]. However, the role and mechanism of adaptor recruitment, including SHP-2, following activation of EGFR by oncogenic mutation, have not been defined. Previously we showed that overexpression of SHP-2 C459S in EGFRvIII transformed glioma cells inhibited MAPK activity and downregulated EGFRvIII tyrosine phosphorylation (pTyr), suggesting a SHP-2-mediated downstream signaling loop can feedback to regulate EGFRvIII pTyr [32]. Therefore, we examined whether SHP-2 couples EGFRvIII signals to downstream events to regulate EGFRvIII-mediated biological responses, specially cell growth and transformation
In this study, we demonstrate that the PTPase activity of SHP-2 is required for the naturally occurring EGFRvIII oncoprotein to transform glial cells and mouse embryonic fibroblasts. Cells expressing PTPase-inactive SHP-2 C459S or shp-2 siRNA demonstrated reduced transforming efficiency and this inhibited phenotype correlated to a block in cell cycle progression. These results suggest that the SHP-2 PTPase is functionally coupled to activated EGFRvIII and that EGFRvIII requires SHP-2 to transform cells. Importantly, SHP-2 may represent a good therapeutic target in EGFRvIII transformed human cancer cells such as GBMs.
Materials and methods
Materials
Fetal bovine serum was purchased from HyClone (Logan, UT). Dulbecco’s Modified Eagle’s Medium (DMEM) was obtained from Mediatech Cellgro (Kansas City, MO). Penicillin, streptomycin, lipofectamine and lipofectamine 2000 were from Invitrogen Life Technologies (Carlsbad, CA). Anti-SHP-2 and anti-actin antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and Sigma separately. The anti-SHP-2 Tyr542 antibody was obtained from Epitomics (Burlingame, CA). Anti-phospho-Cdc2(Tyr15), anti-cdc2, anti-phospho-Akt (Ser473) and anti-Akt antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-phospho-Erk and anti-Erk1/2 antibodies were from Promega Corporation (Madison, WI). SHP-2 PTPase substrate DiFMUP (6,8-difluoro-4-methylumbelliferyl phosphate) was obtained from Invitrogen (Carlsbad, CA) and its inhibitor NSC-87877 (8-Hydroxy-7-(6-sulfonaphthalen-2-yl)diazenyl-quinoline-5-sulfonic acid, Disodium Salt) from Calbiochem (San Diego, CA). All other chemicals were obtained from commercial sources.
Cell Culture and Cell Transfection
All of the cell lines were cultured in DMEM with 10 % fetal bovine serum supplemented with 100U/ml streptomycin/penicillin and plus different selection reagents as indicated below at 37°C in a humidified atmosphere of 5% CO2 incubator. The human GBM cell line U87MG.EGFRvIII was obtained from Dr. Webster Cavenee (Ludwig Cancer Institute, San Diego, CA) and previously described [32]. U87MG.EGFRvIII cells were grown with 400μg/ml G418. The EGFRvIII stable transfectants expressing different Shp-2 mutants (shp-2 wild-type; the PTPase-inactive shp2, shp-2 C459S; the two SH2 domain mutants that are incapable of binding Tyr(P) residues, shp-2 R32E and shp-2 R138) were cultured in 40 μg/ml hygromycin [32]. LN229.EGFRvIII cells were obtained from Dr. Hui-Kuo G. Shu (Department of Radiation Oncology, Emory University, Atlanta, Georgia) and cultured with 1.2 μg/ml puromycin. Wild-type (Shp-2+/+), heterozygous and homozygous Shp-2 mutant (Shp-2+/− and Shp-2−/−) mouse embryonic fibroblast cell lines (MEF) were obtained from Dr. Gen-Sheng Feng (The Burnham Institute, La Jolla, CA) and were previously described [21]. The MEF cell lines were transfected with either empty vector or a vector containing the EGFRvIII oncogene (1726/zeoGW-EGFRvIII) by lipofectamine as described previously [33]. The shp2+/+ and shp2+/− stable clones were selected with 50 μg/ml zeocin and the shp2−/− clones were selected with 10 μg/ml zeocin.
RNA Interference Studies
Small interfering RNA (siRNA) specific for shp-2 (5′-GAAUAUGGCGUCAUGCGUGTT-3′ and 5′-CACGCAUGACGCCAUAUUCTT-3′) [34] and scrambled shp-2 siRNA 5′-AGUUAUAAGGCGGUCGUGCTT-3′ and 5′-GCACGACCGCCUUAUAACUTT-3′ were synthesized by Dharmacon Research, Inc. (Lafayette, CO). SiRNAs were transfected into U87MG.EGFRvIII and LN229.EGFRvIII cells by using 3 μl of Lipofectamine 2000 reagent from Invitrogen Life Technologies (Carlsbad, CA) [34] for 6 hour and then recovered in DMEM plus 10% FBS.
MTT Assay
For cell proliferation, 2000–4000 cells were seeded in 96 well plates. At each time point as indicated in the figure, 20μl of MTT solution (5 mg/ml in PBS) was added to each well. After 3to 4 hour incubation at 37 °C, 100 μl of the extraction buffer (20% w/v of SDS, 50% N,N-dimethylformamide, pH 4.7) was added and incubated at 37°C for overnight. The optical density at 570nm was measured using an enzyme-linked immunosorbent assay reader at specific time points as designated.
Soft Agar Assay and Tumorigenicity Studies
Cells were trypsinized and suspended in 2ml top agar containing 10% serum and 0.3% agarose. The mixtures were then plated in 6-well plates containing 2ml bottom agar with 10% serum and 0.6% agarose. The plates were incubated at 37 °C at 5% CO2 for 2to 4 weeks. The colonies were stained using a 0.25-mg/ml solution of [2-(p-iodophenyl)-3-(p-nitropheyl)-5-phenyl tetrazolium chloride and incubated at 37 °C for overnight and photographed. 1×105 U87MG.EGFRvIII expressing Shp-2 mutants and 3000 EGFRvIII expressing mouse embryonic fibroblast (MEF) were used for this assay. For shp-2 siRNA (see below) study, U87MG.EGFRvIII and LN229.EGFRvIII cells were transiently transfected with 20 picomoles scrambled shp-2 siRNA and shp-2 siRNA for 6 hour by lipofectamine 2000 and recovered in DMEM plus 10% serum for two days. Then cells were split and 1–2×105 cells were used for soft agar assay. For SHP-2 inhibitor study, NSC-87877 was added to mix with the cells in the top agar. For tumor formation in vivo, cells were trypsinized and suspended in DMEM. 5×105 cells per 0.1 ml were injected into each flank of the nude mice. Tumors were monitored weekly after inoculation. Three to four weeks later, mice were sacrificed and tumors were dissected and photographed.
Western Blot, Immunoprecipitation and Immunofluorescence Analysis
For Western blot analysis, cells were washed with PBS twice before harvest and then lysed in lysis buffer (20 mM Tris-HCl. PH 7.5, 0.15 M NaCl, 1% Triton x-100, 1 mM PMSF, 1 mM Na3VO4, 1 mM EGTA, 1μg/ml leupeptin, 2μg/ml aprotinin, 10μg/ml pepstatin). Protein concentration was measured and equal amount of protein (25 μg) was loaded for electrophoresis through SDS/8.0% polyacrylamide gels. An enhanced chemiluminescence detection kit was used to detect the proteins (Amersham, Buckinghamshire, UK). For immunoprecipitation, cells were lysed in lysis buffer containing 50mM Tris, pH 7.5, 150mM NaCl, 2mM EGTA, 0.1% Triton-X-100, 1mM PMFS, 10μg/ml aprotinin and leupeptin, and 1mM orthovanadate. After incubating for 30 minutes on ice, soluble fraction protein concentration was measured and equal amount of protein was incubated with the appropriate antibody for overnight at 4°C. The immune complexes were collected with protein A-Sepharose (Sigma) for 1 h, washed three times with lysis buffer and boiled for 5 min in SDS-polyacrylamide gel electrophoresis sample buffer (250mM Tris, pH 6.8, 10% SDS, 10% β-mercaptoethanol, and 40% glycerol). Proteins were resolved by SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose, incubated with indicated antibodies, and visualized by ECL detection system (Amersham, Buckinghamshire, UK). For immunofluorescent analysis, cells were plated in 12-well plates on glass cover slips and cultured as indicated in figure legend. Cells were washed twice with PBS, and fixed in 3.7% paraformaldehyde for 20min. Fixed cells were washed twice with PBS and permeabilized with 0.5% Triton X-100 in PBS for 5min, and then blocked in PBS containing 10% bovine albumin. Cells were stained with rhodamine-conjugated phalloidin (Sigma) in PBS containing 2% bovine albumin for 2 hour at 37°C in a humidified chamber. Images were captured and analyzed using a fluorescent Nikon Eclipse E 600 light microscope (Nikon Co., Natick, MA) and a Magnafire CCD camera (Optronics, Goleta, California).
Flow Cytometry Analysis
The cells were trypsinized and harvested. After centrifugation, the cell pellet was washed with PBS and fixed with 70% ice cold ethanol for 15 minutes, and resuspended in phosphate-buffered saline containing RNase A for 30 min at room temperature. Cells were washed with PBS and propidium iodide (75μg/ml) was then added to evaluate nuclear staining. DNA content analysis was performed by a fluorescence-activated cell sorter with CellQuest software (FACS Caliber, Becton Dickinson, San Jose, CA). Data were analyzed by ModiFitLT 3.0 software (Immunocytometry System, BD Bioscience) for cell cycle distribution.
Immune Complex PTP Assay
Cells were lysed in ice-cold PTP lysis buffer, 25 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, with protease inhibitor cocktail [35]. 500 μg protein of the cell lysate was immunoprecipitated with anti-SHP-2 antibody plus protein A-Sepharose for 2 to 4 hours at 4°C. The SHP-2 immune complex was resuspended in 100 μl of reaction buffer containing 20 μM DiFMUP and then incubated at 37 °C for 30 min in a water bath. During the incubation period, shake the tube to mix the beads every 5 to 10 minutes. After a brief centrifugation, 100 μl supernatants were transferred into a black 96-well plate and the DiFMU fluorescence signal was measured at an excitation of 360 nm and an emission of 460 nm. The remaining immune complexes were boiled and used for SHP-2 immunoblot. To determine NSC-87877 IC50 for SHP-2 from these GBM cells, the assay was performed in the presence of this inhibitor.
Results
Catalytically inactive SHP-2 inhibits growth of human GBM cells
We have previously shown that oncogenic EGFRvIII human GBM cells are more tumorigenic and demonstrate resistance to treatment [32, 36]. Constitutive phosphorylation of EGFRvIII and its enhanced downstream signaling pathways, including MAPK and PI3K/Akt activation, likely contribute to these biological responses [32, 37, 38]. SHP-2 participates in RTK pathways and regulates both MAPK and Akt activities [18, 32, 39]. Previously, we reported that PTPase-inactive SHP-2 C459S inhibited basal MAPK activity in EGFRvIII transfected glioma cells, indicating that SHP-2 modulates signaling downstream of EGFRvIII [32]. In this study, we analyzed whether SHP-2 is required for EGFRvIII cell proliferation and transformation in GBMs. We addressed these questions in PTEN-negative U87MG.EGFRvIII and the PTEN-positive LN229.EGFRvIII GBM cells. Shp-2 siRNA was introduced into these cells to inhibit SHP-2 expression and its effects on these biological responses were monitored. Introduction of 20 picomoles shp-2 siRNA [34] resulted in a dramatic reduction of SHP-2 protein expression compared to control scrambled shp-2 siRNA on day three after transient transfection in both GBM cells (Fig. 1A). We thus used this same strategy to evaluate whether inhibition of SHP-2 expression affected cell proliferation. Cells transfected with control or shp-2 siRNA were counted at indicated time points and we found that shp-2 siRNA suppressed GBM cell growth compared to control cells (Fig. 1B). To further evaluate the influence of distinct SHP-2 mutants on the growth of EGFRvIII subclones, we employed clones expressing wild-type SHP-2, PTPase-inactive SHP-2 (SHP-2 C459S), and two distinct SH2 domain point mutants of SHP-2 (SHP-2 R32E, SHP-2 R138E) to evaluate cell growth in 10% serum by the MTT assay which was performed daily after cell seeding. PTPase-inactive SHP-2 C459S expressing EGFRvIII subclones exhibited a slower growth rate compared to other SHP-2 subclones and empty vector controls (Fig. 1C). Similar results were obtained with LN229.EGFRvIII cells expressing SHP-2 C459S (data not shown). Taken together, these data suggest that SHP-2, in particular the PTPase activity of SHP-2, contributes to GBM cell growth.
Fig. 1. PTPase-defective SHP-2 inhibits growth of EGFRvIII GBM cells.
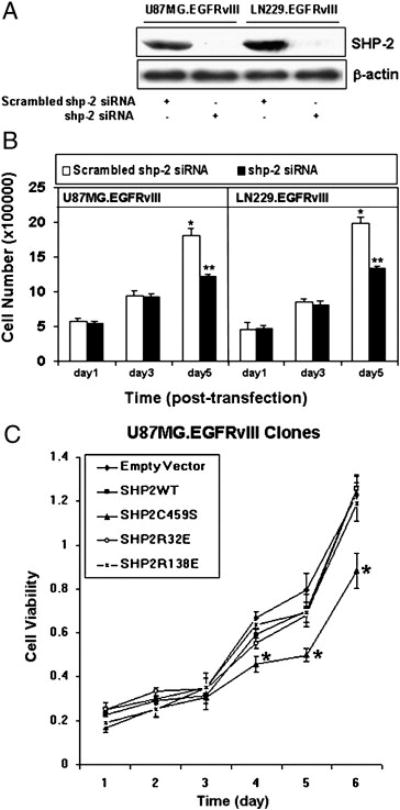
(A) U87MG.EGFRvIII and LN229.EGFRvIII cells were transiently transfected with 20 picomoles of scrambled or shp-2 siRNA for 6 hour and cultured in DMEM plus 10% FBS for 72 h. Equal amounts of protein were loaded for SDS-PAGE gel electrophoresis and western blots were performed with anti-SHP-2 and anti-β-actin antibodies. (B) 20 picomoles of scrambled shp-2 siRNA and shp-2 siRNA were transfected to U87MG.EGFRvIII and LN229.EGFRvIII cells for 6 h and cell number was then counted at the indicated time points. Each column represents the mean ± S.D., n = 4. Data are summarized from four independent experiments giving similar growth profile. A significant difference is presented (* vs **, P<0.05). (C) 4000 cells of distinct EGFRvIII clones expressing different Shp-2 mutants were seeded in 96 well plates and cultured in 10% FBS. The MTT assay was performed at different experimental time points as noted. The data are shown as means ± S.D. of triplicate samples. This is one representative of three separate experiments. Significant differences among the same time points are presented (*, P<0.05).
PTPase-inactive SHP-2 inhibits GBM transformation and tumorgenicity in vivo and alters F-actin stress fiber organization
Since SHP-2 is required for GBM cell growth, we examined whether SHP-2 participates in GBM cell transformation. We used siRNA to inhibit SHP-2 expression in transient transfection assays to examine whether inhibition of SHP-2 affected cell morphology and transformation of the GBM cells. We found that cells transfected with shp-2 siRNA (Fig. 2A, b, d) formed fewer colonies after 3 to 4 weeks than cells transfected with scrambled shp-2 siRNA (Fig. 2A, a, c). Consistent with this, cells with shp-2 siRNA showed a more flattened morphology (Fig. 2A, f, h). Thus, these data suggest that SHP-2 regulates EGFRvIII cell transformation in GBM cells. Interestingly the expression of PTPase-inactive SHP-2 C459S, but not its SH2 domain point mutants, SHP-2 R32E and SHP-2 R138E, also caused morphological changes in GBM subclones (Fig. 2B, upper panel). Clones expressing SHP-2 C459S showed a more flattened cell shape in culture in the presence of 10% serum. Conversely, vector control cells grew in foci typical of highly transformed GBM cells. A similarly transformed morphology was also observed in cells expressing wild-type SHP-2, and the SHP-2 R32E and SHP-2 R138E mutants. Therefore, only expression of PTPase-inactive SHP-2 C459S profoundly altered GBM cell morphology.
Fig. 2. Catalytically inactive SHP-2 PTPase alters EGFRvIII GBM cell morphology, inhibits cell transformation and tumorgenicity and results in more organized actin stress fibers in EGFRvIII cells.



(A) U87MG.EGFRvIII (a, b, e and f) and LN229.EGFRvIII cells (c, d, g and h) were transiently transfected with scrambled or shp-2 siRNA for 6 h and cultured in DMEM plus 10% FBS for 48 h. Then cells were split for soft agar transformation assays (n = 3–4). The same transfected cells were cultured for 5 days in the presence of 10%FBS and photos were taken using phase contrast microscopy. The data were accrued over three separate experiments. (B) Clones expressing Shp-2 mutants were cultured in the presence of 10% FBS and photographed using phase contrast microscopy (upper panel). The middle panel shows that PTPase-inactive SHP-2 C459S inhibited transformation in soft agar. 0.5–1×105 cells expressing Shp-2 mutants were used for this analysis. Photographs were taken 2–3 weeks after seeding (n=3). The lower panel shows that expression of SHP-2 C459S inhibited tumor formation in vivo. U87MG.EGFRvIII cells expressing shp-2 cDNA constructs were injected into flanks of nude mice (0.5 million cells per injection). Three to four weeks later, tumors were removed upon sacrifice of the animals. Representative tumors from a total of 10 injections are shown. The average size of the tumors was calculated as (mm3): Empty Vector, 1057.6±348.3; SHP-2 wild-type, 1982±811.2 and SHP-2 C459S, 0.021±0.04. A ruler for size reference is shown at the bottom. (C) U87MG.EGFRvIII cells expressing vector (a, d), shp-2 wild-type (b, e) and shp-2 C459S (c, f) were cultured on coverslips in 10% serum. After cells were attached, cells were either kept in 10% FBS medium (a, b, c) or in 0.5% FBS medium (d, e, f) for another 72 h. Then cells were fixed and stained with rhodamine-conjugated phalloidin and demonstrate that PTPase defective Shp2 C459S results in a more organized pattern of actin stress fibers in EGFRvIII cells. These data are representative of three different experiments (n=3).
Changes in cell morphology have been shown to be associated with malignant transformation in many tumor cell types. To investigate whether the morphological changes induced by PTPase-inactive SHP-2 C459S expression correlated with a reduction in the transforming properties of these cells, we compared the ability of U87MG.EGFRvIII cells expressing empty vector, wild type SHP-2, PTPase-inactive SHP-2 C459S, or either of two SH2 mutants, SHP-2 R32E and SHP-2 R138E, to grow in soft agar. We found that vector control and the distinct SH2 mutant-expressing cells readily grew in soft agar at similar cell densities, but the PTPase-inactive SHP-2 C459S-expressing cells lost their ability to grow under the same conditions (Fig. 2B, middle panel). Thus, the PTPase activity of SHP-2 was shown to be required for EGFRvIII transformation. Moreover, ectopic expression of PTPase-inactive SHP-2 reverted EGFRvIII-mediated transformation.
We then assayed GBM cells for their ability to form tumors in nude mice. We focused on U87MG.EGFRvIII cells expressing empty vector control, SHP-2 wild-type and the PTPase-inactive SHP-2 C459S form, since the SH2 domain mutants, SHP-2 R32E and SHP-2 R138E, did not affect cell growth or transformation in soft agar. In mice injected with vector control and SHP-2 wild type cells, tumors grew 3 to 4weeks after inoculation (Fig. 2B, lower panel). But in mice injected with SHP-2 C459S-expressing cells, only very small tumors formed when examined by dissection within the same time period after inoculation (Fig. 2A, lower panel). Notably, SHP-2 wild type cells grew larger tumors than vector control cells. The illustrated tumors are one representative example from ten separate experimental injections. These studies indicate that expression of the PTPase-inactive SHP-2 C459S alters cell morphology and suppresses transformation mediated by the EGFRvIII oncoprotein.
The phenotypes of U87MG.EGFRvIII subclones stably transfected with either vector, a SHP-2 wild-type allele, or either of the two SHP-2 SH2 mutants, SHP-2 R32E and SHP-2 R138E, were similar to that of the U87MG.EGFRvIII cells (Fig. 2B). However, U87MG.EGFRvIII cells transfected with PTPase-inactive SHP-2 C459S showed a markedly flattened morphology (Fig. 2B). To investigate whether changes induced by SHP-2 C459S expression were associated with alterations of F-actin structure, actin filaments were visualized with rhodamine-conjugated phalloidin, as we previously showed in EGFRvIII cells in our laboratory [33]. Immunofluorescence analysis showed that EGFRvIII cells expressing either vector or wild-type SHP-2 did not exhibit well-defined stress fibers under full growth serum conditions, which correlated with a transformed phenotype (Fig. 2C, a, b). However, SHP-2 C459S cells showed a markedly increased and organized actin stress fiber pattern, which correlated with an untransformed phenotype (Fig. 2C, c). When cells were cultured in low serum, they exhibited a more elongated cell morphology (Fig. 2C, d, e, f). Notably, SHP-2 C459S cells also demonstrated a clear and organized pattern of actin filaments (Fig. 2C, f), consistent with an untransformed phenotype.
SHP-2 is required for EGFRvIII transformation of mouse embryonic fibroblasts (MEFs)
We have shown that the SHP-2 PTPase domain is important for EGFRvIII-mediated transformation of human GBM cells. To further investigate the general importance of SHP-2 in EGFRvIII-mediated cell transformation, we employed MEFs that express wild-type Shp-2 (Shp-2 +/+) or allelic deletion of Shp-2 (Shp-2 +/− and Shp-2 −/−) [21]. Shp-2 MEFs were transfected with the EGFRvIII oncogene and stable clones were selected. Subclones were examined by western blot analysis and those expressing similar amounts of EGFRvIII were used for transformation studies (Fig. 3A). Soft agar assays indicated that cells with wild-type SHP-2 and EGFRvIII expression grew colonies efficiently (Fig. 3B, d), but cells containing only Shp-2 +/− or Shp-2 −/− were not transformed by EGFRvIII (Fig. 3B, e, f). Therefore, EGFRvIII transformed only Shp-2 +/+ cells and was unable to transform Shp-2 +/− or Shp-2 −/− cells. The same concentration of zeocin was used to select Shp-2 +/+ and Shp-2 +/− clones transfected with EGFRvIII, and EGFRvIII only transformed shp-2 +/+ cells, indicating that the transformation of shp-2 MEFs was due to EGFRvIII. Zeocin did not influence transformation of shp-2 MEFs in these studies [21]. These results further support that SHP-2 plays a critical and general role in EGFRvIII-induced cell transformation in both glial cells and fibroblasts, indicating a positive coupling between EGFRvIII signaling and SHP-2 PTPase activity that is required to maintain oncogenic transformation.
Fig. 3. Soft agar colony formation in shp-2 mouse embryonic fibroblasts.


(A) Shp-2 +/+. Shp-2 +/− and Shp-2 −/− MEF cells were transfected with EGFRvIII and clones were selected. Equal amounts of protein were loaded for western blot analysis. The blots were probed with anti-EGFR (upper panel) and anti-SHP-2 antibodies (lower panel). The upper panel shows the introduced EGFRvIII and the lower panel shows the intact SHP-2 (upper band) and the deleted SHP-2 (lower band) [21]. (B) 2500 MEF shp-2+/+ (a, d), shp-2+/− (b, e) and shp-2−/− (c, f) cells with vector (a, b, c) or with EGFRvIII (d, e, f) expression were suspended in 2 ml of medium containing 0.3% agarose and 10% serum and seeded on top of 2 ml 0.6% agarose layer in 6-well plate. After 3 weeks, the cells were stained and photographed. The data is shown from three separate experiments (n=3).
Pharmacologic inhibition of endogenous SHP-2 PTPase activity reduced EGFRvIII-mediated GBM transformation
Based on our observations, SHP-2 phosphatase activity is required for GBM transformation. We next employed a pharmacologic SHP-2 inhibitor, NSC-87877, that has been shown to inhibit SHP-2 PTPase activity in cells [35] to test the role of endogenous SHP-2 in EGFRvIII-mediated GBM transformation. Although this inhibitor also inhibits the protein tyrosine phosphatase, SHP-1 (Calbiochem, San Diego, CA), these GBM cells have no detectable SHP-1 (data not shown). This inhibitor’s IC50 values for SHP-2 from U87MG.EGFRvIII and LN229.EGFRvIII cells are 5.5 μM and 1.8 μM, respectively, in an in vitro SHP-2 PTPase activity assay (Fig. 4A), indicating the SHP-2 PTPase activity may be differentially regulated in these cells. Incubation of NSC-87877 with U87MG.EGFRvIII and LN229.EGFRvIII cells inhibited SHP-2 PTPase activity in a dose-dependent manner (Fig. 4B). It is well known that SHP-2 is required for Erk activation. To evaluate whether NSC-87877 can inhibit Erk activation in these GBM cells, the cells were treated with this inhibitor and Erk phosphorylation was monitored. Fig. 4C shows that NSC-87877 inhibited Erk phosphorylation at 50 μM and this inhibition was in a dose-dependant manner. The inhibitor also inhibited colony formation of U87MG.EGFRvIII and LN229.EGFRvIII cells in soft agar (Fig. 4D). NSC-87877 more potently inhibited LN229.EGFRvIII cells in anchorage-independent growth than U87MG.EGFRvIII cells at the same dose. This result is consistent with the inhibitory effect of this inhibitor on SHP-2 PTPase activity from these two cell lines in the in vitro PTPase assay. Thus, it further suggests that the functional status of SHP-2 may be different in these two cells. These data collectively indicate that SHP-2 PTPase activity is required for oncogenic EGFRvIII-induced GBM transformation regardless of the PTEN status in these cells.
Fig. 4. The SHP-2 inhibitor NSC-87877 inhibited endogenous SHP-2 PTPase activity and inhibited EGFRvIII-induced transformation in human GBM cells.


(A) To determine IC50 of NSC-87877 against SHP-2, SHP-2 was immunoprecipitated from U87MG.EGFRvIII and LN229.EGFRvIII cell lysates and incubated with different concentrations of NSC-87877 in the SHP-2 PTPase assay. The data shows the percentage of inhibition for SHP-2 PTPase activity and present one of three separate experiments. (B) 0.5–1.0×106 cells were plated in 10-cm dish and cultured for overnight. NSC-87877 (μM) was added in DMEM/10%FBS for 24 to 48 hours. The cells were harvested and SHP-2 PTPase activity was determined. The data is presented as percentage of SHP-2 PTPase activity and it is summarized from two to three different experiments (n=2–3). The immunoblots show the amount of immunoprecipitated SHP-2 in SHP-2 PTPase activity assay. (C) Cells were serum starved for 3 hour and then treated with NSC-87877 in the absence of serum for another 3 hour. Equal amounts of protein were loaded for gel electrophoresis. The blots were probed with anti-phospho-Erk and total Erk antibodies (n=2). (D) Cells were trypsinized and 0.5–1×105 cells were used for soft agar assay along with 100μM NSC-87877. After 3 to 4 weeks of incubastion, the cells were stained and photographed. The data are from three individual experiments performed in duplicates and the representative images are shown (n=3)
EGFRvIII increased EGFRvIII/SHP-2 complex formation and SHP-2 phosphorylation at Tyr542
After studying the importance of SHP-2 PTPase activity in EGFRvIII-transformed GBM cells, we explored how EGFRvIII expression activated endogenous SHP-2. First, we performed an immunoprecipitation experiment and found that SHP-2 can be coprecipitated with EGFRvIII in U87MG.EGFRvIII cells (Fig. 5A). This interaction between EGFRvIII and SHP-2 was sensitive to the kinase inhibitor AG1478 and suggests that the recruitment of SHP-2 to the receptor required phosphorylated EGFRvIII. Interestingly, NSC-87877 also inhibited EGFRvIII/SHP-2 complex formation. We also noticed that NSC-87877 inhibited EGFRvIII phosphorylation (Fig. 5A) and Erk phosphorylation (Fig. 4C). This result is consistent with our previous observation that downstream signaling such as Erk activation regulates upstream EGFRvIII receptor activity [32]. Since phosphorylation of SHP-2 at its major site Tyr542 increased SHP-2 PTPase activity [40], we monitored SHP-2 phosphorylation status in these GBMs. We found that EGFRvIII expression induced SHP-2 phosphorylation at Tyr542 and its PTPase dramatically in PTEN-deficient U87MG cells, but only modestly in PTEN-intact LN229 cells (Fig. 5B and data not shown). These results suggest that EGFRvIII expression recruits SHP-2 to an activated complex and induces SHP-2 Tyr542 and its PTPase activity in GBM cells. But the regulation of SHP-2 PTPase activity may be different in these two distinct cell lines.
Fig. 5. EGFRvIII expression recruits SHP-2 and enhances SHP-2 phosphorylation at Tyr542.


(A) 1×106 cells were seeded in 10 cm dishes and then serum starved for 18 to 24 h. 100μM NSC-87877 was added for 18 to 24 h and 5μM AG1478 was added for 30 min. The cells were harvested and the cell lysates used for western blot analysis and immunoprecipitation with anti-SHP-2 antibody followed by SDS-PAGE and immunoblotting with different antibodies (n=3). (B) After serum starvation for 24 h, the cells were collected and equal amounts of protein were used for western blot analysis. The blot was probed with anti-SHP-2 p542 and anti-SHP-2 antibodies (n=3–5).
SHP-2 is required for cell cycle progression in PTEN-deficient and PTEN–intact EGFRvIII-containing GBM cells
To examine mechanisms by which PTPase-inactive SHP-2 C459S mediates suppression of cell growth and transformation, we examined cell cycle populations in SHP-2 mutant expressing EGFRvIII cells under different serum concentrations. In 10% serum, PTPase inactive SHP-2 C459S increased the G2/M population and induced a concomitant decrease in the G0/G1 population (Fig. 6A, c), as compared to empty vector control and SHP-2 wild type cells (Fig. 6A, a, b). The increase in G2/M by SHP-2 C459S was partially abrogated by 0.5% serum (Fig. 6A, f). Thus, the delay in G2/M cell cycle progression by SHP-2 C459S may contribute to the inhibition of EGFRvIII mediated cell growth and transformation.
Fig. 6. PTPase defective Shp-2 expression results in G2/M cell cycle arrest and decreases Cdc2(Tyr15) phosphorylation in U87MG.EGFRvIII cells.



(A) 5×105 U87MG.EGFRvIII cells expressing empty vector (a, d), Shp-2 wild-type (b, e) and PTPase-defective Shp-2 C459S (c, f) were seeded in 10-cm dishes in the presence of 10% serum. After cells attached to the dish, one group of cells was maintained in 10% FBS (a, b, c) and in an additional group, 10% FBS medium was replaced with 0.5% FBS (d, e, f). All cells were then cultured for 72 h. Cells were then trypsinized and the DNA content was analysized by a fluorescence-activated cell sorter. The cell distribution in G0/G1, S, and G2/M phase and the percentage of cells are indicated in the histogram. Similar results were obtained in four independent experiments (n=4). (B) Cells were split and allowed to attach to plates in 10% FBS. For one group, 10% FBS medium was replaced with 0.5% FBS and all cells were cultured for an additional 72 h. Cells were then harvested for western blot analysis. Equal amounts of protein were loaded and examined with anti-phospho-Cdc2(Tyr15), anti-Cdc2 and anti-actin antibodies. Three separate experiments were performed (n=3). (C) Cells were transfected with different amounts of scrambled shp-2 siRNA (a, c) and shp-2 siRNA (b, d) for 6 h and cultured in 10% serum for an additional 5 days. Cells were then trypsinized and the DNA content was analysized. The cell distribution in G0/G1, S, and G2/M phase is shown (a and b are U87MG.EGFRvIII cells; c and d are LN229.EGFRvIII cells). Similar results were obtained in two to three independent experiments (n=2–3).
Cell cycle progression is regulated by a set of cyclins and cyclin dependent kinases. Perturbation of these molecules may result in growth arrest at certain growth phases or checkpoints. For integrity of the G2/M checkpoint, the phosphorylation status of Cdc2(Tyr15) is of critical importance. Therefore, we examined Cdc2(Tyr15) phosphorylation status in cells expressing SHP-2 mutants under both full serum (10%) and low serum (0.5%) culture conditions. We observed a dramatic decrease in Cdc2(Tyr15) phosphorylation in PTPase-inactive SHP-2 C459S clones compared to that of vector and SHP-2 wild-type clones in 10% serum (Fig. 6B). The decrease in Cdc2(Tyr15) phosphorylation suggests that the cyclin dependent kinase, Cdc2, is activated and results in growth arrest at M phase [41]. We also noticed that the inhibition of Cdc2(Tyr15) phosphorylation could be partially overridden by low serum conditions, which correlated to the reduction in the G2/M population under these conditions (Fig. 6B, lane 6, Fig. 6A, f). These results suggest that the PTPase activity of SHP-2 is required for cell cycle progression in cells expressing EGFRvIII and that PTPase-inactive SHP-2 C459S growth-arrested EGFRvIII cells at G2/M by dephosphorylating Cdc2(Tyr15).
Since we have observed that shp-2 siRNA expression inhibited both PTEN-deficient U87MG.EGFRvIII and PTEN-intact LN229.EGFRvIII cell proliferation and transformation (Fig. 1B and 2A). We next also assayed cell cycle distribution in these GBM cell lines. Shp-2 siRNA was used for transient transfections and cell cycle analysis was done on day 5, when SHP-2 expression was still suppressed after transfection. We found that shp-2 siRNA expression increased the G2/M population in U87MG.EGFRvIII cells and the G0/G1 fraction of LN229.EGFRvIII cells (Fig. 6C). These results further suggest that SHP-2 regulates cell cycle progression of these GBMs.
SHP-2 inhibition by siRNA alters downstream signaling pathways in GBM cells
Many studies have shown that SHP-2 is required to transduce signals to downstream MEK/ERK and PI-3K/AKT pathways to transform cells [42, 43]. Therefore, we examined the activation of EGFRvIII, MEK/ERK and PI-3K/AKT and the cdc2 cyclin dependent kinase following SHP-2 inhibition. Immunoblot analysis was performed against phosphorylated EGFRvIII, Erk, AKT and Cdc2 along with their counterparts, respectively. Consistent with our previous observation with SHP-2 C459S expression in U87MG.EGFRvIII cells [32], shp-2 siRNA expression dramatically inhibited EGFRvIII and Erk phosphorylation, while it had a modest or no effect on their total expression level in U87MG.EGFRvIII cells (Fig. 7). Shp-2 siRNA expression had no effect on Akt phosphorylation in these cells (Fig. 7), but it inhibited cdc2 phosphorylation at Tyr15 at at the same concentration (20 picomoles) used for transformation assay in Fig. 2A.
Fig. 7. The effects of Shp-2 siRNA on the activation of EGFRvIII, Erk, Akt and Cdc2 in EGFRvIII GBM cells.

Cells were transfected with different amounts of scrambled shp-2 siRNA and shp-2 siRNA for 6 h and cultured in 10% serum for an additional 5 days. Cells were collected for western blot analysis. The transfected cells were lysed and equal amounts of protein were used for electrophoresis and the blots were probed with different antibodies as indicated. The data are one representative of two to three separate experiments (n=2–3).
Inhibition of cdc2 phosphorylation at Tyr15 may contribute to subsequent G2/M arrest of U87MG.EGFRvIII cells. On the other hand, in PTEN-functional LN229.EGFRvIII cells, shp-2 siRNA expression resulted in a dramatic decrease of total EGFRvIII protein and a modest decrease of other signaling molecules including Erk, Akt and cdc2 in a dose-dependent manner (Fig. 7) (see discussion section). Taken together, these data suggest that SHP-2 regulates its downstream MEK/ERK and PI-3K/Akt signaling pathways in a distinct manner in these two cell lines and therefore may perturb cell cycle progression at different checkpoints.
Discussion
Oncogenic EGFRvIII is expressed in approximately 50% of human GBMs, particularly de novo GBMs [6, 44–46]. EGFRvIII pTyr and kinase activity are important for its transforming functions both in vivo and in vitro [7, 47, 48]. However, little is known about the signaling pathways that EGFRvIII utilizes to enhance transformation. In this study, we continued to explore the relationship between the distinct domains of SHP-2 and EGFRvIII, since our previous work indicated that SHP-2 PTPase activity modulated EGFRvIII pTyr and downstream signaling [32]. We showed that inhibition of SHP-2 expression by its siRNA and expression of PTPase-inactive SHP-2 potently suppressed growth and tumorigenicity of human GBM cells (Figs. 1 and 2). Consistent with this, actin filaments expressed in EGFRvIII cells expressing PTPase-inactive SHP-2 C459S were more organized than those in parental EGFRvIII cells under both full serum and low serum conditions (Fig. 2C). Additional experiments with shp-2 MEFs indicated that EGFRvIII required functional SHP-2 to maintain oncogenic transformation (Fig. 3). The SHP-2 PTPase activity inhibitor NSC-87877 inhibited endogenous SHP-2 PTPase activity and transformation of PTEN-deficient U87MG.EGFRvIII and PTEN-intact LN229.EGFRvIII cells (Fig. 4). EGFRvIII expression increased the engagement of SHP-2 to the receptor and increased its PTPase activity and phosphorylation at Tyr542 (Fig. 5). SHP-2-mediated GBM proliferation and transformation occurred through a regulation of cell cycle progression in these cells. Our data suggest that SHP-2 may mediate cell cycle progression in part at the G2/M phase in U87MG.EGFRvIII cells and G0/G1 phase in LN229.EGFRvIII cells (Fig. 6). Coordinately, Cdc2(Tyr15) phosphorylation was decreased in SHP-2 C459S U87MG.EGFRvIII cells and after shp-2 siRNA expression in U87MG.EGFRvIII cells (Fig. 6B and 7).
EGFRvIII C-terminal tyrosine residues are constitutively phosphorylated in U87MG.EGFRvIII cells and LN229.EGFRvIII cells and these phosphorylation sites are likely to be important for activating downstream signaling pathways. Many studies have shown that SHP-2 is required to activate wild-type EGFR signaling pathways. EGFRvIII shares the same phosphorylation sites as wild-type EGFR and, thus, EGFRvIII may use similar signaling molecules, including SHP-2, to transduce signals and affect biological responses. Here our results indicated that expression of PTPase-inactive SHP-2 C459S, but not its SH2 domain point mutants, dramatically inhibited cell growth and tumorigenicity in EGFRvIII-expressing U87MG.EGFRvIII cells. And the SHP-2 PTPase activity inhibitor NSC-87877 inhibited both U87MG.EGFRvIII and LN229.EGFRvIII transformation. These data suggest that this suppression of transformation was dependent on the PTPase activity of SHP-2. In addition, our data also suggest that inhibition of SHP-2 PTPase activity more specifically inhibited transformation than proliferation. Because SHP-2 C459S clone only showed slower growth rate compared to other clones. But SHP-2 C459S clone completely lost its ability to grow in soft agar. Therefore, inhibition of EGFRvIII transformation by SHP-2 C459S was not only due to inhibition of its cell proliferation. Future work will focus on looking for SHP-2 interacting proteins and/or substrates that may play a role in the growth, differentiation and transformation of astrocytes.
The pharamacologic inhibitor NSC-87877 also inhibited SHP-2 PTPase activity [35] and inhibited GBM cell transformation (Fig. 4), indicating the PTPase activity of SHP-2 is required for EGFRvIII oncogenesis in GBMs. With 24–48 hours of incubation with 50–100μM NSC-87877, it was observed that inhibition of SHP-2 PTPase activity was 30–40% in these GBM cells (Fig. 4B). NSC-87877 also inhibited GBM anchorage-independent growth of these two GBM cells in a dose-dependent manner (Fig. 4D and data not shown). Complete inhibition of colonies of GBM cell lines in soft agar was observed with higher concentrations (200–400μM) of NSC-87877 (data not shown), and suggest that more potent SHP-2 inhibitors may have a more profound inhibitory effects on GBM phenotype. The requirement of higher NSC-87877 for inhibition of U87MG.EGFRvIII transformation may be due to the enhanced PTPase activity from higher basal SHP-2 Tyr542 phosphorylation induced by EGFRvIII (Fig. 5B). Regardless, these data indicate that NSC-87877 specifically inhibits EGFRvIII GBM transformation even in the setting of nonfunctional PTEN. Therefore, SHP-2 may be a more general therapeutic target than EGFRvIII in GBM cells since EGFR tyrosine kinase inhibitors seem to be efficacious only in the setting of functional PTEN [49]. Thus, SHP-2 inhibition provides a valuable tool to further explore GBM transformation and tumorgenicity, particularly in those tumors that express oncogenic EGFRvIII.
PTPase-inactive SHP-2 C459S and shp-2 siRNA inhibited cell proliferation and transformation of EGFRvIII cells by perturbing cell cycle progression. PTPase-inactive SHP-2 and shp-2 siRNA caused significant cell growth arrest at G2/M in U87MG.EGFRvIII cells (Fig. 6). Concomitantly, SHP-2 C459S and shp-2 siRNA inhibited MAPK activity in U87MG.EGFRvIII cells [32] (Fig. 7) and the MAPK pathway inhibitor PD98059 inhibited U87MG.EGFRvIII proliferation [50]. Since catalytically inactive SHP-2 reduces MAPK activity in EGFRvIII cells [32], it is likely that SHP-2 PTPase-regulated MAPK contributes to cell cycle regulation in U87MG.EGFRvIII cells. Similarly, it has been reported that SHP-2 is required to arrest MEFs at the G2/M phase in response to DNA damage reagent treatment [51]. Other studies have demonstrated that MAPK activation is important for the G2/M transition [52]. Interestingly, a recent study shows that siRNA against EGFRvIII in both U87MG.EGFRvIII and U373.EGFRvIII cells also caused growth arrest at G2/M [53]. The key regulatory element in the control of the G2/M phase, Cdc2, also exhibited a decrease in phosphorylation at tyrosine residue 15 (Figs. 6B). Cdc2(Tyr15) phosphorylation was also decreased by PD98059 treatment in our studies (data not shown), indicating the activation of Cdc2 kinase activity and the transition of the cells from G2 to M phase required MAPK activation [41, 54]. Interestingly, nonfunctional SHP-2 also has been shown to increase the mitotic cell population in response to DNA damage [51]. Phosphorylation of Cdc2(Tyr15) was increased in U87MG.EGFRvIII cells expressing SHP-2 C459S under low serum (0.5% FBS) conditions when compared to growth in full serum (Fig. 6B). Since low serum results in an accumulation of the G1 population, which is consistent with the decrease of Cdc activity and the increased phosphorylation of Cdc2(Tyr15), these data suggest that low serum tipped the balance from G2/M to G1 and, coordinately, phosphorylation of Cdc2(Tyr15) was increased in these cells. Taken together, these data indicate that PTPase-inactive SHP-2 and shp-2 siRNA expression arrest U87MG.EGFRvIII cells at G2/M and this association correlates with a decrease in Cdc2(Tyr15) phosphorylation, but an increase in Cdc2(Tyr15) activity.
Inhibition of SHP-2 by shp-2 siRNA arrested LN229.EGFRvIII cells at the G0/G1 phase rather than at G2/M which was observed in U87MG.EGFRvIII cells (Fig. 6C). When comparing the activation status of the EGFRvIII oncoproteins in these two cell lines following shp-2 siRNA expression, we found that introduction of shp-2 siRNA resulted in a dramatic decrease of EGFRvIII expression in LN229.EGFRvIII, but not U87MG.EGFRvIII, cells (Fig. 7). At the same time, expression of other signaling molecules including Erk, Akt and Cdc2 in LN229.EGFRvIII cells was also decreased to different levels (Fig. 7). The crystal structure of SHP-2 showed that the SHP-2 N-SH2 domain interacts with its PTP domain to keep SHP-2 PTPase inactive under unstimulated conditions [55] When the N-SH2 domain of SHP-2 interacts with phosphotyrosine residue of activated RTK or adaptor molecules, its PTP domain is released and its PTPase is activated. Our results showed that EGFRvIII expression resulted in its autophosphorylation which further recruited SHP-2 to U87MG.EGFRvIII, but not LN229.EGFRvIII, cells. This recruitment might result in the release of the SHP-2 PTP domain to increase its PTPase activity. Furthermore, we propose that the increased tyrosine phosphorylation of SHP-2 at Tyr542 we observed in U87MG.EGFRvIII cells further enhanced the SHP-2 PTPase activity, as has been observed in other systems [40]. On the other hand, there was no detectable recruited SHP-2 to EGFRvIII in LN229.EGFRvIII cells and there was only a modest increase of SHP-2 Tyr542 and its PTPase activity in this cell line. The different functional status of SHP-2 in these two cell lines likely alters downstream biochemical changes and cell cycle progression differently in distinct cell types. Importantly, our data suggest that SHP-2 regulates GBM proliferation and transformation independent of PTEN status. In addition, SHP-2 may function downstream of PTEN in these GBM cells. Further experiments are now ongoing to specifically explore the relationship between PTEN status and SHP-2.
In conclusion, our data indicate that SHP-2 is required for EGFRvIII-induced transformation in human GBMs and mouse fibroblasts and also demonstrate that SHP-2 PTPase activity couples EGFRvIII signals to transformation. To our knowledge, this is the first study showing that the protein tyrosine phosphatase, SHP-2 mediates oncogenic EGFRvIII signals. These effects may occur by perturbing cell cycle progression through G2/M and G0/G1 in distinct cell lines. In refractory human cancers expressing the activated EGFRvIII oncoprotein, therapies that target the SHP-2 PTPase may represent a novel and more broadly applicable therapeutic strategy.
Acknowledgments
This study was supported by grants from The National Institutes of Health (RO1 CA-90586 and R56-CA-90586-06A1), The Department of Veterans Affairs (Merit Review Program) and The Brain Tumor Society to D.M.O.
Abbreviations
- EGFR
epithelial growth factor receptor
- EGFRvIII
an in-frame deletion of an 801bp sequence of epithelial growth factor receptor
- Erk
extracellular signal-related kinases
- MAPK
Mitogen activated protein kinase
- PI3K
phosphatidylinositol 3′-kinase
- cdc2
cyclin-dependent kinase 1
- MEF
mouse embryonic fibroblast
- pTyr
tyrosine phosphorylation
- PTEN
tensin homologue deleted on chromosome 10
- PTP
protein tyrosine phosphatase
- GBM
glioblastoma multiforme
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for spublication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Laskin JJ, Sandler AB. Epidermal growth factor receptor: a promising target in solid tumours. Cancer Treat Rev. 2004;30:1–17. doi: 10.1016/j.ctrv.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Nagane M, Lin H, Cavenee WK, Huang HJ. Aberrant receptor signaling in human malignant gliomas: mechanisms and therapeutic implications. Cancer Lett. 2001;162(Suppl):S17–S21. doi: 10.1016/s0304-3835(00)00648-0. [DOI] [PubMed] [Google Scholar]
- 3.Jungbluth AA, Stockert E, Huang HJ, Collins VP, Coplan K, Iversen K, Kolb D, Johns TJ, Scott AM, Gullick WJ, Ritter G, Cohen L, Scanlan MJ, Cavenee WK, Old LJ. A monoclonal antibody recognizing human cancers with amplification/overexpression of the human epidermal growth factor receptor. Proc Natl Acad Sci U S A. 2003;100:639–644. doi: 10.1073/pnas.232686499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997;57:4130–4140. [PubMed] [Google Scholar]
- 5.Wikstrand CJ, Hale LP, Batra SK, Hill ML, Humphrey PA, Kurpad SN, McLendon RE, Moscatello D, Pegram CN, Reist CJ. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 1995;55:3140–3148. [PubMed] [Google Scholar]
- 6.Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, Biegel JA, Hayes RL, Wong AJ. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 55:5536–5539. [PubMed] [Google Scholar]
- 7.Moscatello DK, Montgomery RB, Sundareshan P, McDanel H, Wong MY, Wong AJ. Transformational and altered signal transduction by a naturally occurring mutant EGF receptor. Oncogene. 1996;13:85–96. [PubMed] [Google Scholar]
- 8.Nagane M, Levitzki A, Gazit A, Cavenee WK, Huang HJ. Drug resistance of human glioblastoma cells conferred by a tumor-specific mutant epidermal growth factor receptor through modulation of Bcl-XL and caspase-3-like proteases. Proc Natl Acad Sci U S A. 1998;95:5724–5729. doi: 10.1073/pnas.95.10.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han Y, Caday CG, Nanda A, Cavenee WK, Huang HJ. Tyrphostin AG 1478 preferentially inhibits human glioma cells expressing truncated rather than wild-type epidermal growth factor receptors. Cancer Res. 1996;56:3859–3861. [PubMed] [Google Scholar]
- 10.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 11.Hunter T. Signaling--2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 12.Pawson T, Gish GD, Nash P. SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 2001;11:504–511. doi: 10.1016/s0962-8924(01)02154-7. [DOI] [PubMed] [Google Scholar]
- 13.Freeman RM, Jr, Plutzky J, Neel BG. Identification of a human src homology 2-containing protein-tyrosine-phosphatase: a putative homolog of Drosophila corkscrew. Proc Natl Acad Sci U S A. 1992;89:11239–11243. doi: 10.1073/pnas.89.23.11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmad S, Banville D, Zhao Z, Fischer EH, Shen SH. A widely expressed human protein-tyrosine phosphatase containing src homology 2 domains. Proc Natl Acad Sci U S A. 1993;90:2197–2201. doi: 10.1073/pnas.90.6.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng GS, Hui CC, Pawson T. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science. 1993;259:1607–1611. doi: 10.1126/science.8096088. [DOI] [PubMed] [Google Scholar]
- 16.Lechleider RJ, Freeman RM, Jr, Neel BG. Tyrosyl phosphorylation and growth factor receptor association of the human corkscrew homologue, SH-PTP2. J Biol Chem. 1993;268:13434–13438. [PubMed] [Google Scholar]
- 17.Yamauchi K, Milarski KL, Saltiel AR, Pessin JE. Protein-tyrosine-phosphatase SHPTP2 is a required positive effector for insulin downstream signaling. Proc Natl Acad Sci U S A. 1995;92:664–668. doi: 10.1073/pnas.92.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qu CK. The SHP-2 tyrosine phosphatase: signaling mechanisms and biological functions. Cell Res. 2000;10:279–288. doi: 10.1038/sj.cr.7290055. [DOI] [PubMed] [Google Scholar]
- 19.Milarski KL, Saltiel AR. Expression of catalytically inactive Syp phosphatase in 3T3 cells blocks stimulation of mitogen-activated protein kinase by insulin. J Biol Chem. 1994;269:21239–21243. [PubMed] [Google Scholar]
- 20.Bennett AM, Hausdorff SF, O’Reilly AM, Freeman RM, Neel BG. Multiple requirements for SHPTP2 in epidermal growth factor-mediated cell cycle progression. Mol Cell Biol. 1996;16:1189–1202. doi: 10.1128/mcb.16.3.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi ZQ, Lu W, Feng GS. The Shp-2 tyrosine phosphatase has opposite effects in mediating the activation of extracellular signal-regulated and c-Jun NH2-terminal mitogen-activated protein kinases. J Biol Chem. 1998;273:4904–4908. doi: 10.1074/jbc.273.9.4904. [DOI] [PubMed] [Google Scholar]
- 22.Lu X, Qu CK, Shi ZQ, Feng GS. Downregulation of platelet-derived growth factor receptor-beta in Shp-2 mutant fibroblast cell lines. Oncogene. 1998;17:441–448. doi: 10.1038/sj.onc.1201988. [DOI] [PubMed] [Google Scholar]
- 23.Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. J Biol Chem. 1998;273:21125–21131. doi: 10.1074/jbc.273.33.21125. [DOI] [PubMed] [Google Scholar]
- 24.Manes S, Mira E, Gomez-Mouton C, Zhao ZJ, Lacalle RA, Martinez AC. Concerted activity of tyrosine phosphatase SHP-2 and focal adhesion kinase in regulation of cell motility. Mol Cell Biol. 1999;19:3125–3135. doi: 10.1128/mcb.19.4.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hakak Y, Hsu YS, Martin GS. Shp-2 mediates v-Src-induced morphological changes and activation of the anti-apoptotic protein kinase Akt. Oncogene. 2000;19:3164–3171. doi: 10.1038/sj.onc.1203655. [DOI] [PubMed] [Google Scholar]
- 26.Chen J, Yu WM, Daino H, Broxmeyer HE, Druker BJ, Qu CK. SHP-2 phosphatase is required for hematopoietic cell transformation by Bcr-Abl. Blood. 2007;109:778–785. doi: 10.1182/blood-2006-04-019141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, Curtiss NP, Gotlib J, Meshinchi S, Le Beau MM, Emanuel PD, Shannon KM. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103:2325–2331. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 28.Tartaglia M, Martinelli S, Iavarone I, Cazzaniga G, Spinelli M, Giarin E, Petrangeli V, Carta C, Masetti R, Arico M, Locatelli F, Basso G, Sorcini M, Pession A, Biondi A. Somatic PTPN11 mutations in childhood acute myeloid leukaemia. Br J Haematol. 2005;129:333–339. doi: 10.1111/j.1365-2141.2005.05457.x. [DOI] [PubMed] [Google Scholar]
- 29.Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 30.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A, Sugarbaker DJ, Richards WG, Du J, Girard L, Minna JD, Loh ML, Fisher DE, Velculescu VE, Vogelstein B, Meyerson M, Sellers WR, Neel BG. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64:8816–8820. doi: 10.1158/0008-5472.CAN-04-1923. [DOI] [PubMed] [Google Scholar]
- 31.Zhou X, Coad J, Ducatman B, Agazie YM. SHP2 is up-regulated in breast cancer cells and in infiltrating ductal carcinoma of the breast, implying its involvement in breast oncogenesis. Histopathology. 2008;53:389–402. doi: 10.1111/j.1365-2559.2008.03103.x. [DOI] [PubMed] [Google Scholar]
- 32.Zhan Y, O’Rourke DM. SHP-2-dependent mitogen-activated protein kinase activation regulates EGFRvIII but not wild-type epidermal growth factor receptor phosphorylation and glioblastoma cell survival. Cancer Res. 2004;64:8292–8298. doi: 10.1158/0008-5472.CAN-03-3143. [DOI] [PubMed] [Google Scholar]
- 33.Boockvar JA, Kapitonov D, Kapoor G, Schouten J, Counelis GJ, Bogler O, Snyder EY, McIntosh TK, O’Rourke DM. Constitutive EGFR signaling confers a motile phenotype to neural stem cells. Mol Cell Neurosci. 2003;24:1116–1130. doi: 10.1016/j.mcn.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 34.Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, Higuchi M, Takahashi A, Kurashima Y, Teishikata Y, Tanaka S, Azuma T, Hatakeyama M. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem. 2004;279:17205–17216. doi: 10.1074/jbc.M309964200. [DOI] [PubMed] [Google Scholar]
- 35.Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, Sebti SM, Lawrence NJ, Wu J. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol. 2006;70:562–570. doi: 10.1124/mol.106.025536. [DOI] [PubMed] [Google Scholar]
- 36.O’Rourke DM, Nute EJ, Davis JG, Wu C, Lee A, Murali R, Zhang HT, Qian X, Kao CC, Greene MI. Inhibition of a naturally occurring EGFR oncoprotein by the p185neu ectodomain: implications for subdomain contributions to receptor assembly. Oncogene. 1998;16:1197–1207. doi: 10.1038/sj.onc.1201635. [DOI] [PubMed] [Google Scholar]
- 37.Kapoor GS, O’Rourke DM. Mitogenic signaling cascades in glial tumors. Neurosurgery. 2003;52:1425–1434. doi: 10.1227/01.neu.0000065135.28143.39. discussion 1434–1435. [DOI] [PubMed] [Google Scholar]
- 38.Kapoor GS, Zhan Y, Johnson GR, O’Rourke DM. Distinct domains in the SHP-2 phosphatase differentially regulate epidermal growth factor receptor/NF-kappaB activation through Gab1 in glioblastoma cells. Mol Cell Biol. 2004;24:823–836. doi: 10.1128/MCB.24.2.823-836.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu CJ, O’Rourke DM, Feng GS, Johnson GR, Wang Q, Greene MI. The yrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene. 2001;20:6018–6025. doi: 10.1038/sj.onc.1204699. [DOI] [PubMed] [Google Scholar]
- 40.Lu W, Gong D, Bar-Sagi D, Cole PA. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol Cell. 2001;8:759–769. doi: 10.1016/s1097-2765(01)00369-0. [DOI] [PubMed] [Google Scholar]
- 41.Laird AD, Shalloway D. Oncoprotein signalling and mitosis. Cell Signal. 1997;9:249–255. doi: 10.1016/s0898-6568(96)00176-3. [DOI] [PubMed] [Google Scholar]
- 42.Dance M, Montagner A, Salles JP, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal. 2008;20:453–459. doi: 10.1016/j.cellsig.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 43.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008;27:179–192. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 44.Kuan CT, Wikstrand CJ, Bigner DD. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr Relat Cancer. 2001;8:83–96. doi: 10.1677/erc.0.0080083. [DOI] [PubMed] [Google Scholar]
- 45.Pedersen MW, Tkach V, Pedersen N, Berezin V, Poulsen HS. Expression of a naturally occurring constitutively active variant of the epidermal growth factor receptor in mouse fibroblasts increases motility. Int J Cancer. 2004;108:643–653. doi: 10.1002/ijc.11566. [DOI] [PubMed] [Google Scholar]
- 46.Wikstrand CJ, Cole VR, Crotty LE, Sampson JH, Bigner DD. Generation of anti-idiotypic reagents in the EGFRvIII tumor-associated antigen system. Cancer Immunol Immunother. 2002;50:639–652. doi: 10.1007/s00262-001-0243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, Huang HJ. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A. 1994;91:7727–7731. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Batra SK, Castelino-Prabhu S, Wikstrand CJ, Zhu X, Humphrey PA, Friedman HS, Bigner DD. Epidermal growth factor ligand-independent, unregulated, cell-transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ. 1995;6:1251–1259. [PubMed] [Google Scholar]
- 49.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 50.Klingler-Hoffmann M, Fodero-Tavoletti MT, Mishima K, Narita Y, Cavenee WK, Furnari FB, Huang HJ, Tiganis T. The protein tyrosine phosphatase TCPTP suppresses the tumorigenicity of glioblastoma cells expressing a mutant epidermal growth factor receptor. J Biol Chem. 2001;276:46313–46318. doi: 10.1074/jbc.M106571200. [DOI] [PubMed] [Google Scholar]
- 51.Yuan L, Yu WM, Qu CK. DNA damage-induced G2/M checkpoint in SV40 large T antigen-immortalized embryonic fibroblast cells requires SHP-2 tyrosine phosphatase. J Biol Chem. 2003;278:42812–42820. doi: 10.1074/jbc.M305075200. [DOI] [PubMed] [Google Scholar]
- 52.Lorimer IA, Wikstrand CJ, Batra SK, Bigner DD, Pastan I. Immunotoxins that target an oncogenic mutant epidermal growth factor receptor expressed in human tumors. Clin Cancer Res. 1995;1:859–864. [PubMed] [Google Scholar]
- 53.Fan QW, Weiss WA. RNA interference against a glioma-derived allele of EGFR induces blockade at G2M. Oncogene. 2005;24:829–837. doi: 10.1038/sj.onc.1208227. [DOI] [PubMed] [Google Scholar]
- 54.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 55.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]