

Abstract
Group II introns are self-splicing ribozymes believed to be the ancestors of spliceosomal introns. Many group II introns encode reverse transcriptases that promote both RNA splicing and intron mobility to new genomic sites. Here we used a circular permutation and cross-linking method to establish sixteen intramolecular distance relationships within the mobile Lactococcus lactis Ll.LtrB-ΔORF intron. Using these new constraints together with thirteen established tertiary interactions and eight published cross-links, we modeled a complete three-dimensional structure of the intron. We also used the circular permutation strategy to map RNA-protein interaction sites through fluorescence quenching and cross-linking assays. Our model provides a comprehensive structural framework for understanding the function of group II ribozymes, their natural structural variations, and the mechanisms by which the intron-encoded protein promotes RNA splicing and intron mobility. The model also suggests an arrangement of active site elements that may be conserved in the spliceosome.
Introduction
Group II introns are ribozyme-based retroelements found in bacteria, mitochondria and chloroplasts (Lambowitz and Zimmerly, 2004). They are the putative ancestors of both nuclear spliceosomal introns and non-LTR retroelements, and thus may have played a major role in the evolution of eukaryotic genomes (Eickbush, 1999). A large number of mobile group II introns have been identified, each consisting of a structurally complex RNA and a multifunctional intron-encoded protein (IEP) having reverse transcriptase (RT) activity. The intron RNA catalyzes its own splicing from precursor RNA, and also its integration into DNA during intron mobility. The IEP assists these reactions by stabilizing the catalytic RNA structure, by providing access to DNA targets, and by reverse transcribing the integrated intron RNA (Lambowitz and Zimmerly, 2004). In addition to their evolutionary significance, group II introns have been harnessed to function as gene targeting vectors (“targetrons”) that can be programmed to insert into virtually any DNA sequence (Perutka et al., 2004). However, in the absence of a complete three-dimensional structure for a mobile group II intron, the mechanisms of group II intron splicing and mobility remain incompletely understood.
Group II intron RNAs have a conserved secondary structure consisting of six helical domains (DI-DVI) arranged around a central wheel (Fig. 1A). There are three major subclasses (IIA, IIB and IIC), each distinguished by specific structural variations (Lambowitz and Zimmerly, 2004; Pyle and Lambowitz, 2006). The six domains fold into a catalytically active tertiary structure aided by fourteen known long-range interactions, which were identified by phylogenetic covariation and biochemical analyses (Fig. 1A). Eight of these interactions involve Watson-Crick base pairs (α-α′, β-β′, γ-γ′, δ-δ′, ε-ε′, IBS1-EBS1, IBS2-EBS2, IBS3-EBS3), three are tetraloop-receptor interactions of known geometries (ζ-ζ′, η-η′ and θ-θ′) and the other three are less defined (λ-λ′, κ-κ′ and μ-μ′).
Figure 1.
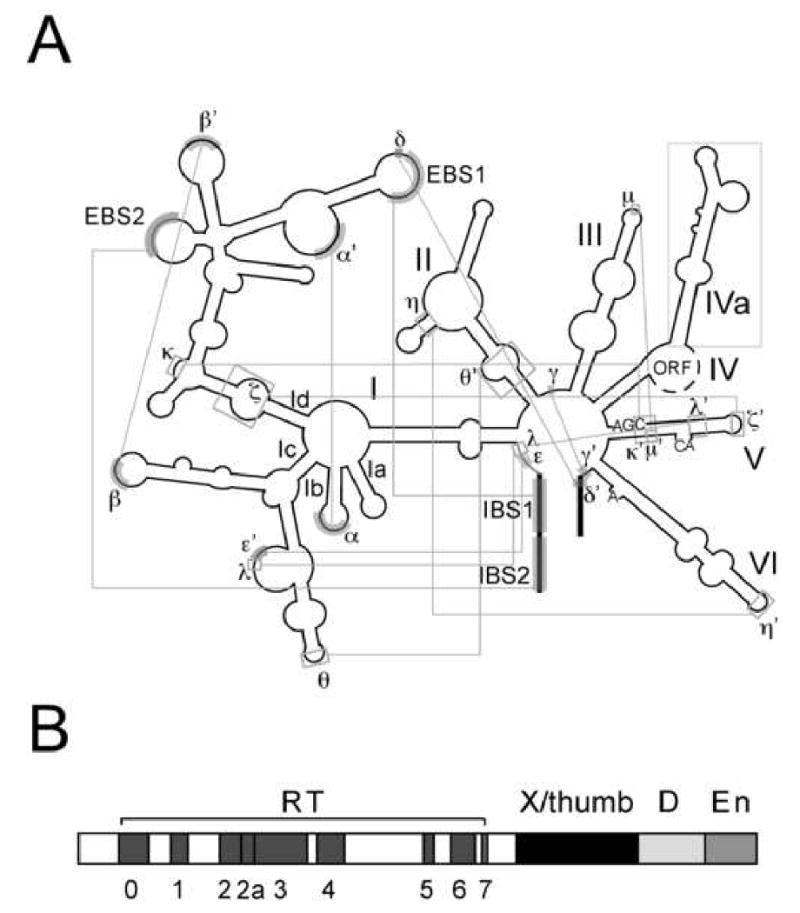
Structure of the IIA Ll.LtrB group II intron and its intron-encoded protein.
A. RNA secondary structure. The intron is indicated by thin black lines and exons by thick black lines. Sequence elements involved in long-range Watson-Crick and non-Watson-Crick interactions are indicated by gray shading and boxes, respectively. The ORF is encoded within a “loop” of DIV (dashed lines).
B. The group II IEP has four domains: reverse transcriptase (RT) with conserved sequence blocks 0-7; X/thumb domain; D, DNA-binding; and En, DNA endonuclease.
Group II ribozymes catalyze splicing via two transesterification reactions that yield spliced exons and an intron lariat, analogous to the splicing mechanism of nuclear spliceosomal introns. During these reactions, the 5′-splice site is positioned at the active site by the IBS1-EBS1 and IBS2-EBS2 pairings (Fig. 1A), while the 3′-splice site is positioned by the interaction δ-δ′ (IIA introns) or IBS3-EBS3 (IIB, IIC introns), as well as by the γ-γ′ pairing of the last nucleotide of the intron (Costa et al., 2000; Pyle and Lambowitz, 2006). In IIB and probably IIC introns, the branch-point A is positioned by a conserved region of DId dubbed the coordination loop, which brings together the bulged A and both exons; however, a conserved motif corresponding to the coordination loop is not present in IIA introns (Pyle and Lambowitz, 2006).
The stem-loop DV is both the catalytic and structural core of group II ribozymes (Fig. 1A). The catalytic face of DV interacts with the 5′ and 3′ splice sites, the J2/3 linker and ε-ε′, with catalytic metal ions proposed in both the AGC triad and AC bulge of DV (Fig. 1A) (Pyle and Lambowitz, 2006, and references therein). The opposite (“binding”) face of DV interacts with DI through the ζ-ζ′ and κ-κ′ interactions. A minimal ribozyme consisting of DV and DI can catalyze 5′ splice site hydrolysis (Koch et al., 1992); however, the inclusion of DIII increases catalytic efficiency (Fedorova and Pyle, 2005). DII is less critical for catalysis, but interacts with DVI and DIc1, presumably anchoring these regions (Costa et al., 1997). Notably, the interaction η-η′ between DII and DVI (Fig. 1A) has been proposed to be dynamic, being formed between the first and second steps of splicing and corresponding to movement of the branch site A between the steps (Chanfreau and Jacquier, 1996).
Virtually all group II introns in bacteria and about half in organelles encode IEPs that promote RNA splicing and/or intron mobility. The IEP is encoded in a “loop” of DIV that extends outside the intron's catalytic core (Fig. 1A). Canonical group II IEPs, exemplified by the LtrA protein encoded by the IIA intron Lactococcus lactis Ll.LtrB, have four conserved domains: reverse transcriptase (RT), which corresponds to the finger and palm regions of retroviral RTs; X/thumb, which corresponds to the RT thumb region and contributes to RNA splicing (“maturase”) activity; D, involved in DNA binding; and En, a DNA endonuclease (Fig. 1B). Studies with the Ll.LtrB intron showed that the RT and X/thumb domains bind specifically to the ribozyme to stabilize its catalytic structure for RNA splicing and reverse splicing, while the D and En domains are not required for splicing but interact with DNA target sites during intron mobility (Lambowitz and Zimmerly, 2004).
The interactions between the IEP and intron RNA are critical for both RNA splicing and intron mobility. The LtrA protein binds to a high-affinity binding site in DIVa of the Ll.LtrB intron and makes additional contacts with core regions to stabilize the ribozyme structure (Matsuura et al., 2001; Wank et al., 1999). After splicing, the IEP remains tightly bound to the lariat RNA in a ribonucleoprotein (RNP) that promotes intron mobility. The RNP recognizes DNA targets for intron mobility using both its RNA and protein subunits. For Ll.LtrB, the IEP recognizes the distal regions of the ∼35 bp target, while the RNA recognizes the central region via the IBS1-EBS1, IBS2-IBS2, and δ-δ′ base pairings (reviewed in Lambowitz and Zimmerly, 2004). After base pairing, the intron RNA reverse splices into the top DNA strand between IBS1 and δ, while the IEP uses the En domain to cleave the bottom DNA strand and then uses the cleaved 3′ end as a primer for reverse transcription of the inserted intron RNA.
Three-dimensional models have proven useful for understanding ribozymes and guiding experimentation. For both RNase P and group I introns, three-dimensional models with essentially correct topologies were constructed based on constraints of cross-links, pseudoknot pairings, tetraloop-receptor interactions, and phylogenetic covariations (Harris et al., 1997; Lehnert et al., 1996). The Harris and Pace model of RNase P, for example, was based on 14 cross-links and five pseudoknot constraints in a ∼400 nt RNA.
In this study we adapted the circular permutation and cross-linking method of Harris and Pace (Harris et al., 1997; Thomas et al., 2000) to determine 16 intramolecular distances within the IIA L. lactis Ll.LtrB-ΔORF intron. By combining these new constraints with 13 established tertiary interactions and 8 published UV-cross-links, we constructed a complete three-dimensional model of the ribozyme. Further, we probed the RNA-IEP interactions using the same circularly permuted RNAs in fluorescence quenching and cross-linking assays. The model provides a structural framework for understanding the ribozyme activity of group II introns, and how the IEP and RNA interact to promote RNA splicing and intron mobility.
Results
Cross-linking strategy
Our cross-linking strategy was adapted from that used for RNase P (Harris et al., 1997; Thomas et al., 2000), in which a photoactivatable azidophenacyl group (APA; cross-linking radius 9 Å) is attached to the 5′ end of circularly permuted (CP) RNAs. Cross-linking is induced by UV irradiation and intramolecular contacts are mapped by primer extension. To analyze a group II intron, CP sequences were generated from a plasmid (pKS-tandem) containing tandem copies of the Ll.LtrB-ΔORF intron separated by a 423-bp exonic linker (Fig. 2A). PCR amplification with appropriate primers resulted in circularly permuted sequences preceded by a T7 promoter. Transcription produced RNAs with guanosine 5′-monophosphorothioate (GMPS) at their 5′ end, to which APA was conjugated via the thio group. The RNA was self-spliced, irradiated with UV light to induce cross-linking, and the products were separated on a native agarose gel, from which the spliced intron band was isolated and analyzed by primer extension.
Figure 2.
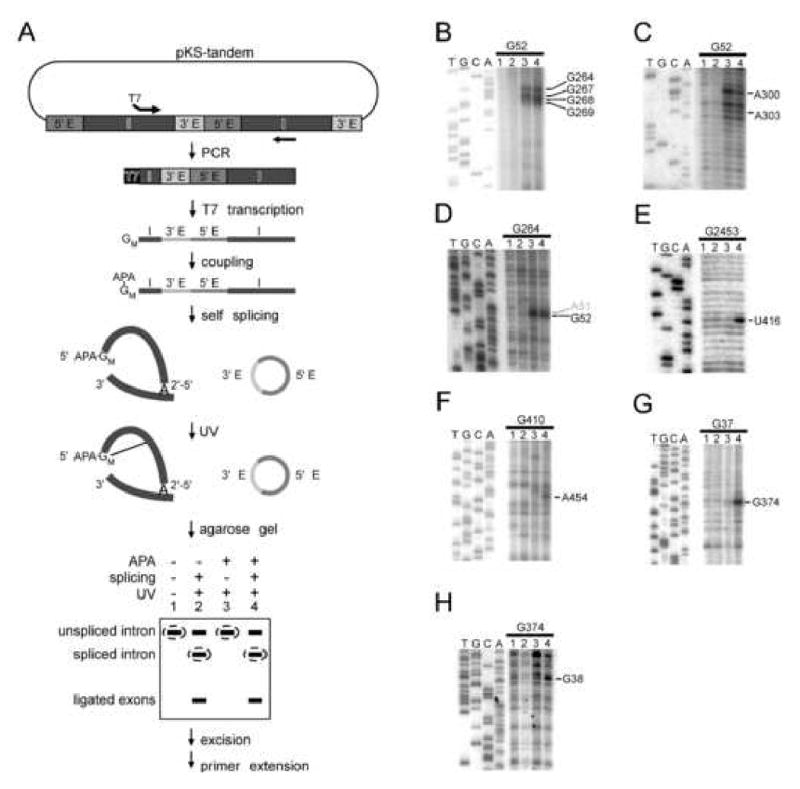
Experimental strategy and representative cross-linking data.
A. Circularly permuted (CP) Ll.LtrB intron was PCR-amplified from plasmid pKS-tandem template, using a 5′ primer that appends a T7 promoter. The PCR product was transcribed by T7 RNA polymerase, with GMPS (GM) incorporated at the 5′ end, to which azidophenacyl (APA) was coupled. The CP RNA was self-spliced, UV-irradiated and electrophoresed in a nondenaturing agarose gel. The indicated bands were excised and cross-links were mapped by primer extension. Four reactions for each CP RNA determined dependence on APA, UV irradiation and splicing.
B-H. Representative primer extension data. Black and gray numbering indicates reproducible and irreproducible stops, respectively. In some cases, the position of the stop was confirmed in a separate gel with greater resolution of the ladder.
B, C. Cross-links demonstrating the interaction α-α′.
D. Reciprocal cross-link for α-α′.
E. The splicing-dependent cross-link G2453xU416, which is in proximity to the η-η′ interaction (see Fig. 3).
F. A splicing-dependent local cross-link in domain II (see Fig. 3).
G,H. Reciprocal splicing-dependent cross-links G37xG374 and G374xG38.
Two key elements of our strategy were the induction of cross-linking after splicing and the separation of products on a nondenaturing agarose gel. Together these steps ensured that cross-links were produced from functional, spliced introns and not misfolded or trans-spliced RNAs. Spliced CP introns are Y-shaped rather than lariats (Fig. 2A), but on a nondenaturing agarose gel all spliced introns comigrate regardless of their cross-linking or permutation state. Thus, all cross-links for a given CP RNA can be mapped in a single primer extension reaction.
Four reactions were done in parallel for each CP RNA. In the first control reaction, CP intron was transcribed without GMPS and the sample was not spliced or UV-irradiated (Fig. 2A lane 1), thus identifying nonspecific stops during primer extension of unspliced intron. In the second control, transcription was also without GMPS, but the RNA was spliced and cross-linked, which identified APA-independent cross-links formed during UV irradiation, and nonspecific primer extension stops from spliced intron template (lane 2). In the third reaction, RNAs were labeled with GMPS and APA, but were incubated in splicing buffer without Mg2+, thereby distinguishing cross-links in unspliced and/or incompletely folded intron (lane 3). In the final reaction, the intron was labeled with GMPS and APA and was spliced and UV-irradiated, which identified cross-links in spliced intron (lane 4). A primer extension stop was considered a putative cross-link if signal was present in lane 4 but not in lanes 1 or 2, as this indicated dependence on both APA and UV. A signal in lane 4, but not in lane 3, could reflect either a conformational change when the RNA assumes a catalytic structure upon Mg2+ addition, or a conformational change during the splicing reaction. Notably, 80% of primer extension stops were the same in lanes 3 and 4, consistent with previous data showing substantial tertiary structure formation in the Ll.LtrB-ΔORF intron in the absence of Mg2+ (Noah and Lambowitz, 2003). The few stops observed in lane 3 but not lane 4 were not used for modeling.
Generation of data
Representative cross-linking data are shown in Fig. 2B-H. The cross-linking method was optimized using a CP RNA with its 5′ end at G52, which is part of the α-α′ interaction (Fig. 1A). As expected, this substrate gave strong cross-links in the α′ region, with reproducible primer extension stops at positions 264-269, 300 and 303 (Fig. 2B,C; Suppl. Table 1). Ninety-two CP substrates were then assayed, some with A to G substitutions at their 5′ ends to allow analysis at positions lacking G's. The method's ability to detect intramolecular contacts was confirmed by seven constructs that yielded cross-links consistent with known tertiary interactions (Suppl. Table 1) and 15 that gave short-range cross-links consistent with the secondary structure (Suppl. Table 2). Fourteen constructs generated sixteen cross-links that were ultimately deemed informative for structure modeling (below) (Table 1). The remaining constructs produced insufficient RNA substrate (e.g., poor PCR or T7 transcription) or no cross-links.
Table 1.
New cross-links for use in structure modeling.
| APA position | Mapped Crosslink | Splicing-dependence |
|---|---|---|
| G5 | C469/G470 | No |
| G14 | A76 | No |
| G14 | A110 | No |
| G37 | G374 | Yes |
| G38 | A110 | Yes |
| A64 | G117 | No |
| G126 | A493/G494/C500 | No |
| G126 | G520/G521/A523 | No |
| G151 | U222 | Yes |
| G347 | G192 | Yes |
| G374 | G38 | No |
| G410 | A454 | Yes |
| G501 | A523 | No |
| G2417 | G279 | Yes |
| G2453 | U416 | Yes |
| G2475 | A446 | Yes |
In confirmation of the G52 cross-links, a reciprocal cross-link occurred from G264 to G52 (Fig. 2D). As expected, neither the G52 nor G264 cross-links were splicing-dependent, indicating that α-α′ is a ground state interaction that persists throughout splicing. Similar cross-linking data were obtained for the β-β′, ε-ε′ and θ-θ′ interactions, with all of these cross-links also being splicing-independent as expected for ground-state interactions (Suppl. Fig. 1; Suppl. Table 1). By contrast, the G2453xU416 cross-link near the η-η′ interaction was observed only in lariat RNA (Fig. 2E), consistent with the formation of η-η′ between the first and second steps of splicing (Chanfreau and Jacquier, 1996). Two other cross-links near η-η′ were also splicing-dependent (G2475xA446, G410xA454; Fig. 2F; Suppl. Fig. 1; Suppl. Table 2), possibly reflecting the same conformational change.
The most valuable constructs were the 14 that provided 16 new cross-linking constraints for modeling (Table 1; Fig. 3). Some of these were corroborated by consistency with other cross-links or published data. For example, G374xG38 was reciprocated by G37xG374 (Fig. 2G,H); G2453xU416 and G2475xA446 are consistent with η-η′. The combination of G14xA76, G14xA110, G38xA110, G37xG374 and G374xG38 suggest the positioning of five sequences along a helical turn of DI(ii). G5xC469/G470 coincides with a prominent cross-link (C6xC469) in a previous study (Noah and Lambowitz, 2003). Three cross-links indicate a sharp bend in DIII (G501xA523, G126xA493/G494/500, G126xG520/G521/A523), consistent with the μ-μ′ interaction (Fedorova and Pyle, 2005). Finally, the G151xU222 cross-link agrees with the previously observed cross-link C(-6)xC153 (Noah and Lambowitz, 2003) in positioning DIc2 next to IBS2-EBS2.
Figure 3.
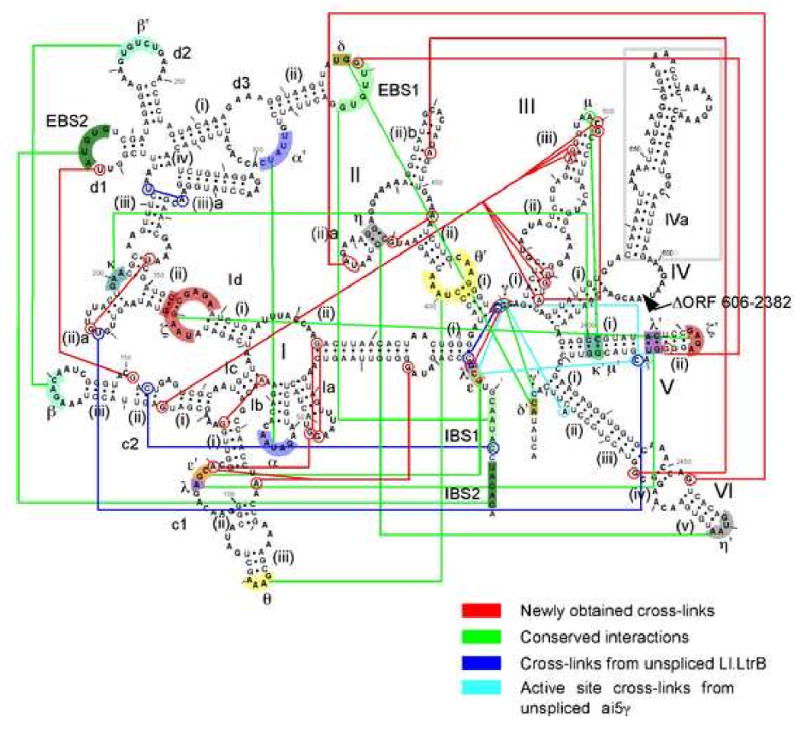
Summary of cross-links and other constraints used in modeling.
Cross-links obtained in this study are indicated by red lines; conserved tertiary interactions are indicated by Greek letters and green lines; cross-links from a previous study with unspliced Ll.LtrB intron are shown by dark blue lines (Noah and Lambowitz, 2003); cross-links mapped to the active site of unspliced aI5γ are indicated by light blue lines (de Lencastre et al., 2005).
A potential artifact is that some cross-links may affect the ability of the RNA to unfold during the primer extension step, or change the propensity for spontaneous RNA cleavage at specific sites (e.g. CA sequences), resulting in primer extension stops that are not cross-links. Indeed, it was found that eight constructs produced stops at 303, a physical impossibility that we attribute to such artifacts. We reasoned that a small number of stops might not correspond to cross-links, and we excluded three putative cross-links because of inconsistency with either known interactions or the combined set of experimental cross-links (Suppl. Table 4).
Constructing the three-dimensional model
The three-dimensional model was constructed based on 37 distance constraints, including the 16 new cross-links, 13 conserved tertiary interactions, four cross-links obtained from unspliced Ll.LtrB-ΔORF intron (Noah and Lambowitz, 2003) and four cross-links from the active site region of unspliced aI5γ intron (de Lencastre et al., 2005) (Fig. 3; Suppl. Table 3). The model was assembled in four successive de novo constructions using the program ERNA-3D with progressive adjustments to optimize satisfaction of distance constraints. Crystal structure data from other RNAs were used to specify the tetraloop-receptor conformations for ζ−ζ′, θ−θ′, and η−η′ (Suppl. Table 3). The remaining helices were arranged manually. The structure was energy-minimized using the program CHARMM to eliminate steric clashes and bond discontinuities, and to optimize distance constraints (see Experimental Procedures). The final model satisfies all constraints with an average distance of 8.4 ± 3.2 Å between constrained atoms (Supplementary Table 3).
Figure 4 presents the model with cylinders indicating helices and black tubes representing the backbone of ligated exons. The core of the molecule consists of DI wrapping around DV, with one face (dark blue) formed by DI(i, ii), Ia, Ib, Ic1 and Ic2, and the other face (light blue) formed by DId(iii,iv), Id1, Id2, and Id3 (Fig. 4A,B). The ζ−ζ′ and κ−κ′ interactions serve as a hinge/turn element to allow the two DI faces to fold toward each other on opposite sides of DV, becoming locked into place by the α-α′ and β-β′ interactions (gray). DIII is mostly internal, just below the core region, with contacts to both DI halves. DII and DVI are on either side of DIII with their distal regions interacting via η-η′ and other contacts. DIV is connected to the bottom of the molecule in a position dictated mainly by steric requirements of DIII and DV. DIVa, the high affinity binding site for the IEP, is arbitrarily angled toward the exon-binding face of DI, where it is near other IEP-binding sites (see below). We note that none of the six CP RNAs tested in DIV gave specific cross-links to other regions (not shown), and it is possible that DIV and/or DIVa are flexible to accommodate changing RNA-protein interactions during RNA splicing and intron mobility (see Discussion).
Figure 4.
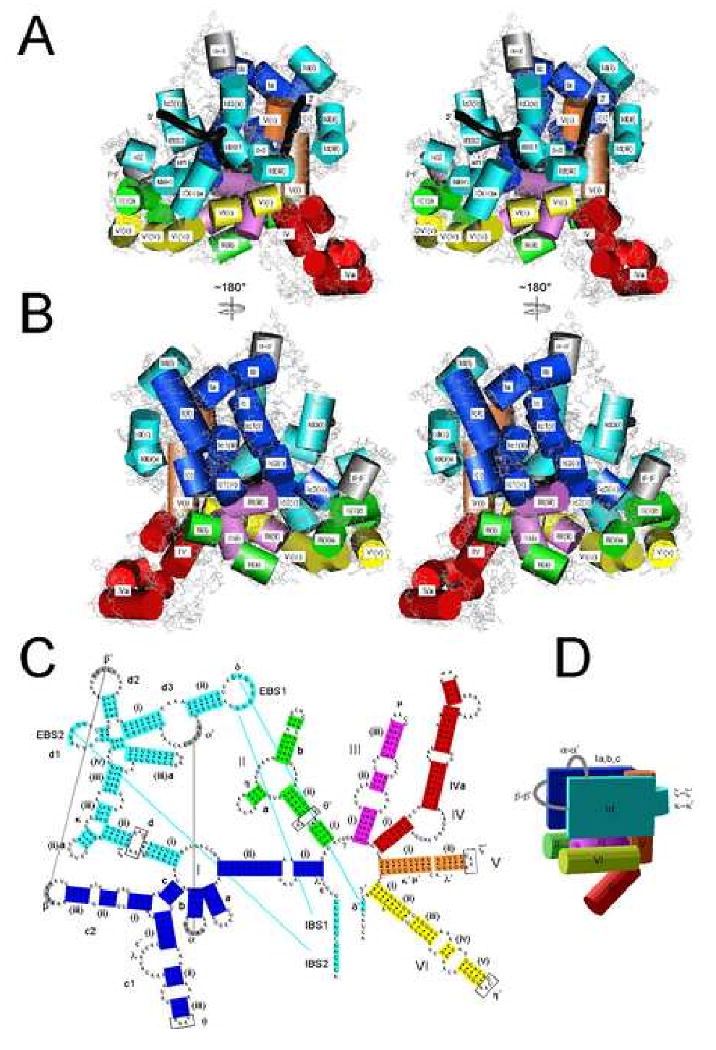
Three-dimensional model of Ll.LtrB-ΔORF lariat RNA and ligated exons.
A, B. Stereoviews of the exon-binding face and opposite face, respectively, with cylinders representing helices and black tubes depicting the phosphate backbone of ligated exons. Domains are colored as in Panel C. Single-stranded regions are shown in stick representation, and in light gray to emphasize the limited experimental data available for modeling these regions.
C. Color coding of RNA domains. Boxed nucleotides indicate single-strands for which specific conformations could be predicted based on crystal structures of tetraloop-receptor interactions. Several base pairs at the ends of helices were unpaired during modeling to allow connection of strands (indicated by the absence of a dot).
D. Schematic showing the arrangement of domains in the ribozyme model.
The 5′ and 3′ exons bind mostly on the surface of the ribozyme via the EBS-IBS and δ-δ′ pairings, but the exon junction is positioned at an internal active site in proximity to ε-ε′, γ-γ′, the branch point A of DVI, and the AGC triad and bulged AC of DV. The overall topology is similar but not identical to two active-site models of IIB introns (Costa et al., 2000; de Lencastre et al., 2005) (Supplementary Fig. 4). Aside from the known differences between IIA and IIB introns, namely the coordination loop and EBS3-IBS3 pairing of IIB introns, the most significant difference in the models is the position of the exons, which are largely internalized in the IIB models, either explicitly or implicitly because of the location of the unmodeled regions. Whether the differences in the models reflect structural differences between the introns or differences in modeling strategies remains to be determined.
Our model is consistent with previously reported protection experiments with the Ll.LtrB-ΔORF ribozyme. Phosphate-backbone and base probes both showed protection of DIII, DIc1(ii,iii), DII(ii)b, DVI(iv,v), as well as DV to a lesser degree (see Figs. 3 and 4 of Matsuura et al. (2001)). In our model, all of these structures are either internal or on the protected sides of surface helices. Exposed regions in the chemical protection experiments were DId(iii,iiia,iv)/Id1, the five-way junction of of DIa, Ib, Ic, Id, and I(ii), the base of DVI, and virtually all of DIVa; all of these regions are on the surface in our model. In addition, the major regions judged to be protected by the LtrA protein (DIVa, DIc2, DId(iii)a, DII(ii)a and DVI(ii,iii)) are on the surface of the model (Matsuura et al., 2001).
Our model is also consistent with hydroxyl radical protection experiments for the minimal D135 construct of aI5γ. In those experiments, the most protected region was DV, followed by the internal loop of DIII, DId(ii), EBS1 and DIb (Fig. 5 of Swisher et al. (2001)). These structures are wholly or partly protected in our model except for DIb. Also buried in both the aI5γ construct and the Ll.LtrB model is the active site region with the catalytically important ε-ε′ and γ-γ′ elements. Further, our model agrees with aI5γ data in having a non-coaxial arrangement of IBS1-EBS1 and IBS2-EBS2 (Qin and Pyle, 1999), and greater exposure of EBS2 compared to EBS1 (Swisher et al., 2001).
Phylogenetic consistency of the model
Group II introns in nature have many variations in their secondary structures, and a valid model should accommodate these differences without perturbing the catalytic core. Major variations among intron classes include: the presence of DId(iii)a and the absence of the EBS3 motif in IIA introns; the absence of a DId1 stem in IIB introns; and the absence of DIc2, Id1 and Id2 in IIC introns. Additional variations among subgroups are summarized in Fig. 5C and Supplementary Table 6. When the sites of these deletions and insertions are mapped onto the Ll.LtrB model (Fig. 5A, B), they are seen to be peripheral, suggesting that their presence or absence does not disrupt the active site core. We conclude that the model is consistent with the natural structural diversity of group II introns and that all group II intron subclasses can be represented in a similar three-dimensional form, with a common core containing the active site.
Figure 5.
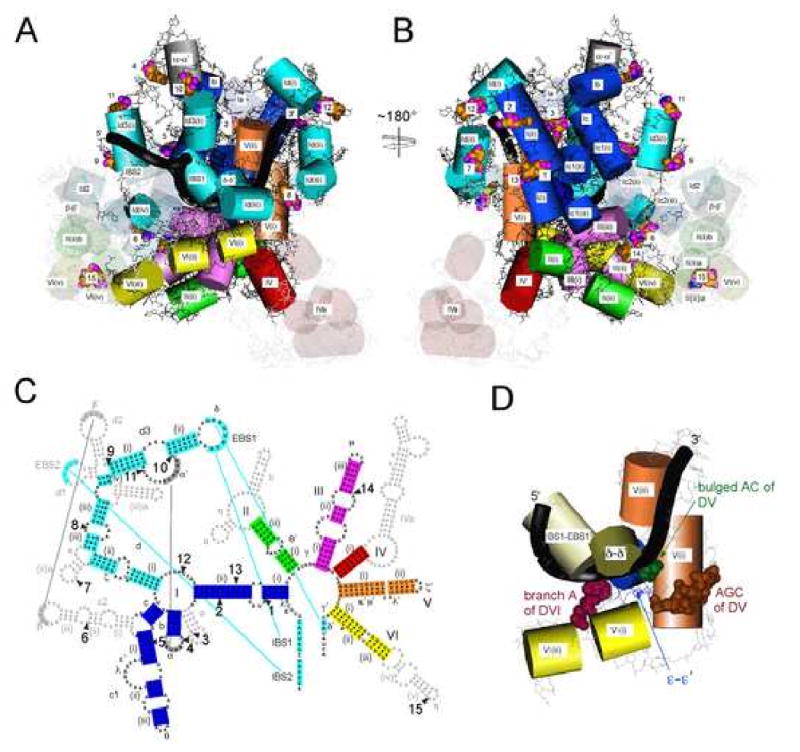
Consistency of the model with known phylogenetic variations of group II introns.
A, B. Semitransparent helices depict substructures in Ll.LtrB that are absent in other intron subclasses, while CPK representations indicate sites of insertions.
C. Secondary structure of Ll.LtrB showing helices absent in other subclasses (gray) or sites of insertions (numbered arrowheads). See Supplementary Table 6 for additional details.
D. Geometry of the group II intron structures having potential analogs in the spliceosome. DV is potentially analogous to U6-ISL, DVI to the U2/UACUAAC box pairing, ε-ε′ to the ACAGAGA box/intron pairing, and IBS1-EBS1 and δ-δ′ to the exon/U5 pairings. CPK atoms depict the branch A in DVI, and the AC bulge and AGC triad in DV.
Sites of protein interaction with the ribozyme
To identify IEP-ribozyme interaction sites, we conjugated a fluorescent probe to the 5′ ends of 77 CP RNAs and assayed fluorescence quenching as a function of LtrA concentration. These experiments were done in a medium containing 50 mM Mg2+ because under these conditions (unlike 5 mM Mg2+) the intron RNA folds into an active conformation that self-splices. Moreover, LtrA still binds to the intron RNA and remains tightly associated with the spliced lariat (Matsuura et al., 2001). Hence, strong quenching in this medium is most likely due to protein binding rather than RNA conformational changes. Figure 6A shows representative data, with fluorescence quenching seen for G561, which lies in the DIVa protein-binding site, and for G2472 in domain VI. In contrast, G155 and G2459 do not show quenching upon LtrA binding, suggesting a lack of proximity to LtrA. Using this assay, we identified 17 positions as giving the highest fluorescence quenching (≥ 40%) upon addition of LtrA protein (Supplementary Table 5, Fig. 7E).
Figure 6.
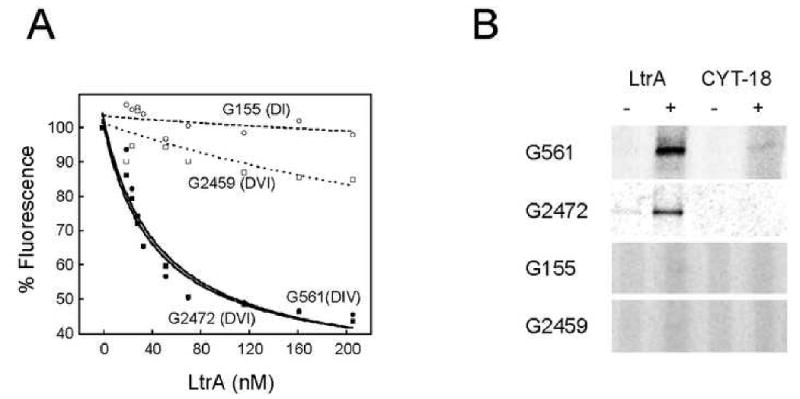
Fluorescence quenching and cross-linking assays for IEP-ribozyme interactions.
A. Representative fluorescence quenching data. CP RNAs with fluorescein conjugated to their 5′ ends were titrated for quenching by 0-200 nM LtrA protein (Experimental Procedures). Data for all constructs tested are summarized in Supplementary Table 5.
B. Representative cross-linking data. CP RNAs with 35S and azidophenacyl at their 5′ ends were incubated with LtrA protein. The complexes were UV-irradiated to induce cross-linking, and digested with RNase T1 to leave a single 35S-labeled nucleotide attached to LtrA (Experimental Procedures). Samples were resolved on a 0.1%SDS/7% polyacrylamide gel, which was dried and analyzed by phosphorimaging. CYT-18 was substituted for LtrA as a specificity control, as was BSA for some experiments (not shown). Data for all positive cross-linking signals are shown in Suppl. Fig. 2, and a summary of all constructs assayed is in Supplementary Table 5.
Figure 7.
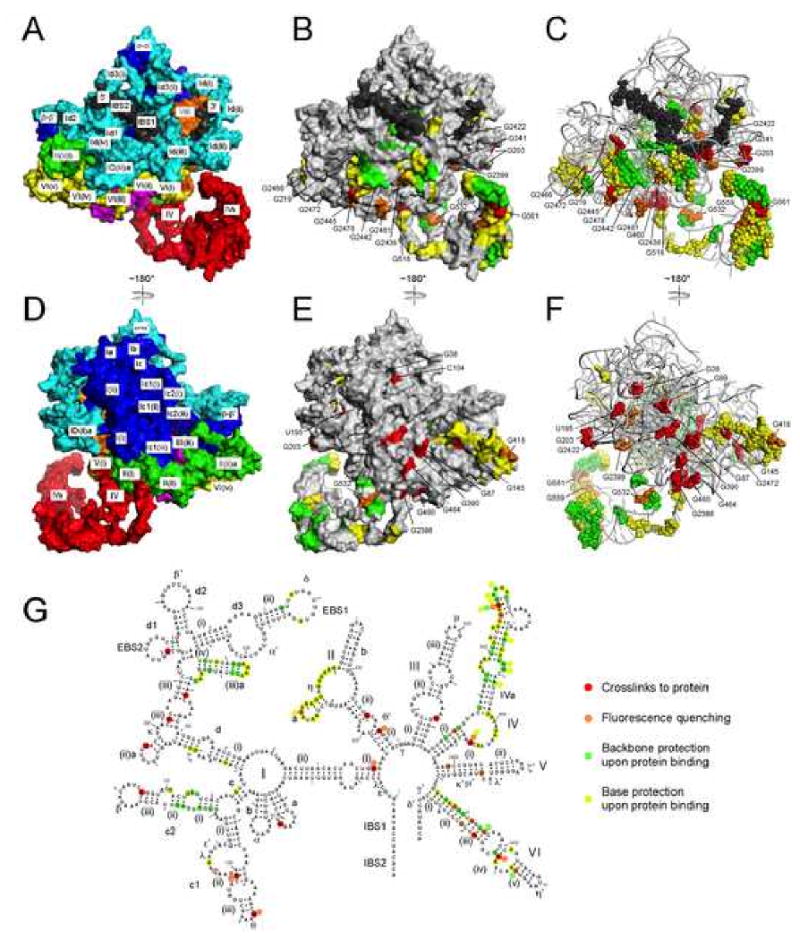
Interaction sites between the IEP and intron RNA.
A, D. Surface representation of the ribozyme model, color-coded as in Figures 4 and 5. Exons are black.
B, E. Gray surface representation of the RNA model, with putative protein contact sites color-coded according to the method of detection. Nucleotide positions identified by cross-linking and fluorescence quenching are specified with residue numbers; a few residues are not visible from the angle shown.
C, F. Alternative view of the model with putative protein contacts shown as spheres.
G. Summary of protein contact data, and definition of the color-coding scheme for panels B, C, E and F. Backbone and base protections are from Matsuura et al. (2001), and include all strong protections in Figures 3 and 4 of that work, as well as moderate protections in regions deemed to be sites of protein contacts as discussed in that manuscript. For residues that gave data by more than one method, the color was determined by the priority of red > orange > green > yellow. The red open circle indicate the nearest exposed residue to G99.
To obtain further evidence for protein contacts, the same 77 CP RNAs were tested for cross-linking to LtrA by constructing CP RNAs with azidophenacyl and 35S attached simultaneously to their 5′ ends (Experimental Procedures). The LtrA-intron complexes were UV-irradiated and digested with RNase T1 to remove all RNA except the 5′-radiolabeled G. Cross-linked proteins were then detected by SDS-PAGE and autoradiography. Cross-linking was carried out in the presence of non-specific competitor RNA, and parallel reactions were performed with the basic protein N. crassa CYT-18 as a control for non-specific cross-links. These assays corroborated the quenching data for the four constructs above (Fig. 6B). In all, 16 positions that cross-link to the IEP were identified (Fig. 7E, Suppl. Fig. 2, Supplementary Table 5).
Figure 7E summarizes protein interaction data from cross-linking and fluorescence quenching assays, as well as published data from phosphate backbone and base protection experiments (Matsuura et al., 2001). Although the four methods identify somewhat different sets of potential contacts, when mapped onto the model they depict a coherent protein-binding surface. The high affinity binding site in DIVa is a major site of interaction identified by all four methods, and the interaction surface extends across the bottom of the RNA, with major contact regions in DIc1(ii,iii), Id(iia, iii, iiia), DII(i, ii) and DVI(ii,iii). In contrast, there is little indication of protein binding on the upper region of the RNA in Fig. 7E,F (e.g., DIb, Id2, Id3). Notably, LtrA interacts near the exon-binding region, thereby positioning the protein to bind DNA exons during intron mobility. The cross-linked residue G99 is not exposed on the surface of the model, but it is sufficiently close that cross-linking may be due to perturbation of the CP RNA's 5′ end, and so the closest exposed residue is indicated (Fig. 7E,F). In addition, we attribute fluorescence quenching at the internal residue G2422 in DV to global compaction of the RNA structure upon protein binding.
Discussion
We have constructed a complete three-dimensional model of a group IIA intron ribozyme, using a cross-linking approach with circularly permuted RNAs. The model depicts lariat intron bound to ligated exons before exon release. In addition, IEP contact sites were mapped onto the ribozyme structure, providing insight into protein facilitation of the RNA splicing and intron mobility reactions. Although the model is of low resolution, it nevertheless provides a comprehensive structural framework for understanding the splicing and mobility reactions of group II introns.
Ribozyme structure and function
In our model, the core of the ribozyme consists of DV bound in a pocket between the two halves of DI, with the κ-κ′ and ζ-ζ′ region being a hinge/turn element on one side, and α-α′ and β-β′ sealing the two halves together on the other side (Fig. 4A, B, D). DIII is largely internalized, with contacts to DV and both halves of DI, potentially stabilizing the bending of DI and its interaction with DV. This organization explains how DIII might function as a catalytic effector of a minimal ribozyme consisting of DI and DV (Pyle and Lambowitz, 2006). The model is also consistent with multiple contacts reported between DIII, DI and DV (Jestin et al., 1997; Su et al., 2005).
Importantly, in the active site region of the model, the catalytic face of DV is oriented inward where it interacts with the catalytically important elements J2/3, γ-γ′, ε-ε′ and the branch A. Thus, the exon junction must be delivered to an internal position for catalysis. This is accomplished in the model by the helical turn of IBS1-EBS1, which allows the 5′ exon to bind mostly on the surface of the ribozyme through the IBS1-EBS1 and IBS2-EBS2 pairings, while the exon junction penetrates internally into the active site. Notably, the δ-δ′ pairing is extended to exon position +2 in the model, consistent with biochemical data for the second base pair (Lambowitz and Zimmerly, 2004). The coordination loop, which positions the splice sites and branch point A residue in IIB and IIC introns (Costa et al., 2000; de Lencastre et al., 2005), is not present in IIA introns, and other interactions presumably fulfill its functions. Nevertheless, the Id(iii) segment that contains the coordination loop in IIB and IIC introns is located near the Ll.LtrB active site, consistent with the expected position of the coordination loop.
In addition to explaining known structural features, the model makes several predictions. Three semi-conserved regions of DI are predicted to help dock DV: the internal loop of DI(i)/(ii) (also suggested by data in Jestin et al., 1997), the DId(ii)a loop near κ, and the loop of DIa. The model also predicts that the single-stranded “wheel” sequences converge at the active site to form a precise network of interactions that may be critical for catalytic function. During modeling, we found the configuration of these strands to be highly constrained by the positioning of helical domains determined by cross-linking data (Suppl. Fig. 5). Notably, the wheel sequences are conserved among intron subclasses, to a degree similar to the ε-ε′ motif, a critical active site component (de Lencastre et al., 2005; Toor et al., 2001). An analogous complex RNA structure formed by single-stranded joining nucleotides has been proposed for the active site of RNase P (Kazantsev et al., 2005).
RNA folding and conformational changes
The Ll.LtrB-ΔORF model explains key aspects of folding of group II ribozymes. Folding of the aI5γ ribozyme was initially described as a slow, apparent two-state process, with a sudden transition from secondary structure to catalytic tertiary structure. Subsequently, the slow step was proposed to be the independent folding of DI to form a scaffold for the docking of DV and DIII (Pyle et al., 2007 and references therein). The rate-limiting step governing assembly of DI is thought to be the introduction of a sharp bend in the ζ and κ region, dubbed a “folding control element.” Folding of DI is rapidly followed by the binding of DV and DIII, and then the remaining group II intron domains. However, both in our model and in the active-site model of aI5γ (de Lencastre et al., 2005), the docking of DV into a fully folded, rigid DI structure would be difficult, particularly after formation of the α-α′ and β-β′ interactions. One possibility suggested by the models is that partial docking of DV occurs before the α-α′ and β-β′ pairings, potentially facilitating the bending and final folding of DI. Once the DI-DV structure is formed, our model readily shows how it could serve as a scaffold for rapid docking of DIII, DII, DIV, and DVI, as proposed (Pyle et al., 2007). While this same folding pathway may apply to all group II ribozymes due to their shared core structure, the rate-limiting steps and stabilizing interactions may vary. In the Ll.LtrB-ΔORF intron, for example, the bend in DI occurs readily, as shown by the formation of α-α′ and β-β′ pairings even in the absence of Mg2+ (this work, Matsuura et al., 2001; Noah and Lambowitz, 2003), while in aI5γ, the bend occurs very slowly unless stabilized by high Mg2+.
The model also explains the exon binding properties reported for the Pylaiella LSU I2 intron, namely that tight 5′ exon binding is dependent on formation of the catalytic core. Indeed, our model suggests that the EBS1, EBS2 and δ′ elements are surrounded by other ribozyme structures. EBS1 in the model interacts with DV, while EBS2 forms contacts with DIc2. The latter contacts are directly supported by the G151xU222 and C(-6)xC153 cross-links and are consistent with exon-dependent protections reported for DIc2 (Costa and Michel, 1999). While 5′ exon binding was inferred to be the final step of ribozyme folding, it is also possible that IBS-EBS pairings occur during secondary structure formation, but that docking into the core occurs late in folding.
Lastly, our detection of three splicing-dependent cross-links near η-η′ is consistent with the proposed conformational change that repositions the branch site between the two splicing steps (Chanfreau and Jacquier, 1996). Other data have indicated that all reactive groups are in proximity prior to splicing, with no large conformational changes being necessary (de Lencastre et al., 2005). It remains possible, however, that the observed conformational changes in the distal region of DVI correspond to fine movements of the branch point at the active site. Another conformational change would appear to be required to release the tightly bound exons after splicing, perhaps the reverse of the conformational change proposed for exon binding (Costa and Michel, 1999).
Facilitation of splicing by the intron-encoded protein
Splicing of Ll.LtrB requires the LtrA protein to stabilize the catalytic RNA structure both in vivo, and at low Mg2+ concentrations in vitro. Previous studies identified a high-affinity LtrA binding site in DIVa, with additional contacts in DI, DII and DVI (Matsuura et al., 2001; Wank et al., 1999). In this study, we used fluorescence quenching and protein cross-linking assays to delineate more precisely the protein-interaction region. Cumulatively, the data suggest that LtrA binding nucleates on DIVa and extends around the bottom half of the molecule, interacting on the exon-binding side with DId(ii)a, Id(iii), Id(iii)a and Id1, along the bottom with DII(i,ii) and DVI(ii,iii), and on the opposite side with DIc1(ii,iii) and to a lesser extent DIc2 (Fig. 7). Consistent with a relatively large RNA binding surface, LtrA binds the intron as a dimer (Saldanha et al., 1999) with predicted dimensions of 190 Å × 90 Å × 90 Å (Blocker et al., 2005), compared to 126 Å × 94 Å × 75Å for the RNA, excluding DIVa (this work).
The protein interaction sites suggest that LtrA could stabilize interactions between the two halves of DI, as well as between domains. Tight binding of LtrA to DIVa would anchor the maturase, while contacts on both the front and back sides may stabilize tertiary interactions thoughout the RNA, particularly in the active site region of DV. Such indirect stabilization may account for the fluorescence quenching observed at G2422 in DV, which is internal and unlikely to interact directly with LtrA (Fig. 7). Unlike the aI5γ intron, the tertiary structure of the Ll.LtrB-ΔORF intron is largely formed at least transiently at low Mg2+ concentrations (this work and Noah and Lambowitz, 2003). Hence LtrA may act late in the folding pathway, either by tertiary structure nucleation, tertiary structure capture, or a combination of the two mechanisms. The large binding surface with contacts to multiple domains may help bring together the two ends of intron (DI-III, DV-VI), which are ordinarily separated by a long ORF sequence. Additionally, it may enable group II intron maturases to compensate for a variety of structural deficiencies acquired during the evolution of different non-self-splicing group II introns.
Mechanism of intron mobility
During intron mobility, the DNA target is recognized by both the IEP and the intron RNA. Initial recognition of distal 5′-exon sequences by the IEP is thought to lead to local DNA melting, which enables the intron RNA to base pair to the adjacent exon sequences via the EBS-IBS and δ-δ′ pairings (Singh and Lambowitz, 2001). Importantly, in our model protein contacts extend up to the exon-binding region of the RNA (Fig. 7), thus enabling the IEP and intron RNA to simultaneously contact the DNA target site. The most critical IEP-RNA interactions involve the RT and X/thumb domains, leaving the D and En domains free to interact with the DNA target site. During the initial stages of the mobility reaction, the IEP must simultaneously maintain some or all of the RT/X domain contacts with the intron RNA in order to stabilize the active ribozyme structure for reverse splicing.
After reverse splicing, additional contacts between the IEP and 3′ exon are required for bottom-strand cleavage by the En domain. The 3′ end of cleaved DNA must then reposition from the En to the RT active site, where it acts as the primer for reverse transcription of the integrated intron RNA. These steps may correspond to the observed bending of the DNA exons into progressively sharper angles (Noah et al., 2006). cDNA synthesis ensues by first copying 9-10 nt of 3′ exon DNA and then extending into the intron. According to our model, reverse transcription of DVI, which is located on the surface of the ribozyme, would not necessarily disrupt other intron structures, but reverse transcription of DV would require gross unfolding of the RNA structure, and thus may be rate limiting. Indeed, in vitro mobility reactions show strong reverse transcription stops between DVI and DV, which are exacerbated by high Mg2+ concentrations (Matsuura et al., 1997).
Parallels with the spliceosome
It is generally believed that nuclear pre-mRNA introns and the spliceosome are evolutionary descendents of group II introns, with snRNAs corresponding to ribozyme fragments that assemble onto intron and reconstitute an RNA-based catalytic center. If so, the geometry of key active site components may be conserved between the two types of introns. Figure 5D shows the segments of the group II model that have putative analogs in the spliceosome, namely DV (analogous to U6-ISL (intramolecular stem-loop of U6)), DVI (U2/UACUAAC box pairing), ε-ε′ (ACAGAGA/intron pairing) and IBS1-EBS1/δ-δ′ (exon/U5 pairings). An equivalent geometry in the spliceosome would place the U6-ISL helix perpendicular to the U2/intron helix, with the ACAGAGA box and exon junctions between them. Consistent with such an arrangement, a study of the B* complex of the spliceosome showed proximity of four RNA elements in activated spliceosomes: the 5′ end of intron (position +10), the ACAGAGA box, the U6-ISL and the U2 sequence that pairs with the branch A motif (Rhode et al., 2006). Although tantalizing, further work is needed to evaluate more precisely the structural relationships between the two types of introns.
Finally, we note that the organization of group II intron domains in our model provides a rationale for why these introns readily evolved into trans-splicing forms, a phenomenon that has occurred multiple times in evolution (Bonen, 1993). The extensive interactions between DI and DV would provide a powerful source of specificity for the reassociation of trans-splicing RNAs, and the assembled core structure of DI wrapped around DV would provide a scaffold for the assembly of remaining domains. Proteins such as group II IEPs or host-encoded splicing factors could assist trans-splicing by bridging the separated RNA domains and stabilizing their interactions. Indeed, the modular organization of group II intron RNAs, with interdomain RNA contacts stabilized by proteins, may be what enabled the evolution of group II introns into spliceosomal introns and snRNAs.
Experimental Procedures
Generation of cross-linking data
The plasmid pKS-tandem contains two tandem copies of Ll.LtrB-ΔORF intron (962 bp) separated by a 423 bp spacer (see Supplementary Methods). To make CP templates, BamHI/ClaI-digested pKS-tandem was PCR amplified with a 5′ primer consisting of 18 nts of intron sequence preceded by a T7 promoter sequence (5′CTAATACGACTCACTATA), and a 3′ primer consisting of 27 nt of intron sequence. Products were either used directly as templates or gel purified using a Qiagen Minielute kit (Qiagen, Valencia CA).
CP RNAs were transcribed with wild-type or R9C mutant T7 RNA polymerase (expression constructs provided by Dr. William McAllister, UMDNJ) at 37°C for 1.5 h in 30 μl of 40 mM Tris-HCl, pH 7.5, 50 mM NaCl, ∼400 ng DNA template, 1 mM ATP, 1 mM UTP, 1 mM CTP, 1 mM GMPS (guanosine-5′-monophosphorothioate; Biolog, Bremen, Germany), 0.3 mM GTP, 0.1% Triton X-100, 5 mM spermidine, 5 mM DTT, 12 mM MgCl2, and 1 μl RNA polymerase (∼0.1 μg). The R9C polymerase was necessary because the presence of a class II terminator within DI of Ll.LtrB prevented clean gel purifications of some CP RNAs. Samples were digested with 10 U DNase I (Amersham, Piscataway, NJ), extracted by phenol-CIA (25:24:1 phenol:chloroform:isoamyl alcohol), ethanol precipitated, and incubated in 100 μl of 20 mM NaHCO3, pH 9.0, 1% SDS, 5 mM azidophenacyl bromide (Sigma-Aldrich, St. Louis, MO) and 40% methanol at room temperature for 1 h in the dark, followed by two phenol-CIA extractions and ethanol precipitation.
End-modified CP RNAs (25 μl in TE; 10 mM Tris-HCl, pH 7.5, 1 mM EDTA) were renatured in a PCR machine as follows: 90°C, 1 min; 75°C, 5 min; and gradual cooling to 43°C over 10 min. After addition of 25 μl of splicing buffer (final concentration of 40 mM Tris-HCl, pH 7.5, 1 M NH4Cl and 100 mM MgCl2), the RNA was incubated for a 25 min at 43°C, then irradiated for 45 min at 4°C with 302 nm UV light. Irradiation was in microfuge tubes with open lids at a distance of 2 cm, screened by a polystyrene Petri dish. Cross-linked RNAs were ethanol precipitated and resolved in a 1.5% agarose gel in TBE. Bands were extracted using a Qiagen Minelute kit, resuspended in 10 mM Tris-HCl, pH 7.5. Cross-links were mapped by primer extension (Supplementary Methods). All putative cross-links were repeated, and nonrepeatable data were discarded.
Modeling of the intron
The three-dimensional model was assembled by four iterative de novo constructions using the program ERNA-3D (http://www.erna-3d.de/), with progressive satisfaction of distance constraints in each cycle. Geometries for ζ-ζ′, η-η′, and θ-θ′ were specified by copying motifs from crystal structures (Supplementary Table 3). The structure was refined by energy minimization using CHARMM (Brooks et al., 1983), interspersed with manual corrections (see Supplementary Methods). In the final construction, helices generated by MC-SYM (Major et al., 1991) were substituted into the model, as well as the crystal structure geometries, to give ideal conformations for these elements. The final model contains no steric clashes or bond discontinuities as monitored by NUCheck of the PDB Validation Suite (http://sw-tools.pdb.org/apps/VAL/index.html). Agreement with data is shown in Supplementary Table 3. We were unable to produce a model with significantly different topology that satisfies the cross-linking data and other constraints. The pdb file is available as a Supplementary Materials File, and also on our web site (http://www.fp.ucalgary.ca/group2introns/).
Fluorescence quenching and RNA-protein cross-linking
For fluorescence quenching experiments, CP RNAs were generated as described above, with GMPS at the 5′ end, and conjugated with 2 mM 5′-IAF (5-iodacetamidofluorescein; Invitrogen, Carlsbad CA) for 2 h at room temperature in dim light. Unincorporated 5′-IAF was removed by addition of 5 mM DTT and gel filtration through a Bio-Spin P-30 column (BioRad; Hercules CA), followed by phenol-CIA extraction and ethanol-precipitation. The RNA was dissolved in 40 mM Tris-HCl, pH 8.0. For fluorescence quenching measurements, 50 nM RNA was incubated with increasing concentrations of LtrA protein (0 to 200 nM) in 200 μl of reaction medium containing 500 mM NH4Cl, 50 mM MgCl2, and 40 mM Tris-HCl, pH 8.0. The high salt concentration is required for optimal LtrA function and also reduces non-specific binding (Saldanha et al., 1999). RNA and protein were allowed to equilibrate for at least 10 min before measuring fluorescence in a fluorimeter (Photon Technology International, Birmingham NJ), with excitation and emission wavelengths of 492 and 515 nm, respectively, using a high-intensity xenon light source. Titration curves of LtrA binding to Ll.LtrB RNA were fit to the hyper decay equation with KaleidaGraph software (Synergy Software, Reading PA).
For RNA-protein cross-linking assays, CP RNAs (500 ng) with 5′ triphosphate ends were dephosphorylated by 25 U Antarctic phosphatase (New England Biolabs, Beverly, MA) according to the manufacturer's protocol. After phenol-CIA-extraction and ethanol precipitation, the RNAs were 5′-labeled with [γ-35S]-ATP (25 μCi; 1,250 Ci/mmol; Perkin Elmer, Boston MA) and T4 polynucleotide kinase (50 U; New England Biolabs) in 50 μl of 70 mM Tris-HCl, pH 7.6, 10 mM MgCl2, and 5 mM DTT for 3 h at 37°C. Samples were phenol-CIA-extracted, filtered through a Biorad spin column, ethanol precipitated and dissolved in 20 μl water. Finally, the RNAs were incubated with 5 mM azidophenacyl bromide in 50 μl of 20 mM Na2CO3, pH 9.0, 50% methanol for 2 h at room temperature in dim light to couple the cross-linker to the 5′ end via the thio group, and then phenol-CIA-extracted, filtered through a spin column, ethanol precipitated and dissolved in distilled water. The coupling efficiency was determined by scintillation counting.
For cross-linking assays, 250,000 cpm of 5′-labeled RNA was renatured in a PCR machine as described above, and incubated for 30 min at 30°C with 100 nM LtrA protein in 8 μl of 500 mM NH4Cl, 50 mM MgCl2, 40 mM Tris-HCl, pH 7.5, and 0.1 mg/ml yeast RNA (Sigma) as a non-specific competitor. As a negative control, 100 nM of N. crassa CYT-18 protein was substituted for LtrA. The presence of the yeast RNA competitor was essential to eliminate nonspecific cross-linking to CYT-18. Complexes were irradiated for 10 min by two 8 W UV lamps (Spectroline, Westbury NY) with a 305-nm filter (Ocean Optics, Dunedin, FL). Samples were boiled for 2 min, cooled to room temperature, and digested with RNase T1 (100 units; Ambion, Austin TX) for 45 min at 37°C, to leave only the 5′-35S-labeled nucleotide attached to the protein. Cross-linked samples were analyzed in a 0.1% SDS/7% polyacrylamide gel, dried and scanned by phosphorimaging.
Supplementary Material
Acknowledgments
This work was supported by CIHR grant MOP-49457 and NSERC grant RGP 203717 (S.Z.) and NIH grant RO1 GM37951 (A.M.L.). Salary support for S.Z. was from the Alberta Heritage Foundation for Medical Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blocker FJ, Mohr G, Conlan LH, Qi L, Belfort M, Lambowitz AM. Domain structure and three-dimensional model of a group II intron-encoded reverse transcriptase. RNA. 2005;11:14–28. doi: 10.1261/rna.7181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonen L. Trans-splicing of pre-mRNA in plants, animals, and protists. FASEB J. 1993;7:40–46. doi: 10.1096/fasebj.7.1.8422973. [DOI] [PubMed] [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- Chanfreau G, Jacquier A. An RNA conformational change between the two chemical steps of group II self-splicing. EMBO J. 1996;15:3466–3476. [PMC free article] [PubMed] [Google Scholar]
- Costa M, Dème E, Jacquier A, Michel F. Multiple tertiary interactions involving domain II of group II self-splicing introns. J Mol Biol. 1997;267:520–536. doi: 10.1006/jmbi.1996.0882. [DOI] [PubMed] [Google Scholar]
- Costa M, Michel F. Tight binding of the 5′ exon to domain I of a group II self-splicing intron requires completion of the intron active site. EMBO J. 1999;18:1025–1037. doi: 10.1093/emboj/18.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Michel F, Westhof E. A three-dimensional perspective on exon binding by a group II self-splicing intron. EMBO J. 2000;19:5007–5018. doi: 10.1093/emboj/19.18.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lencastre A, Hamill S, Pyle AM. A single active-site region for a group II intron. Nat Struct Mol Biol. 2005;12:626–627. doi: 10.1038/nsmb957. [DOI] [PubMed] [Google Scholar]
- Eickbush TH. Mobile introns: retrohoming by complete reverse splicing. Curr Biol. 1999;9:R11–14. doi: 10.1016/s0960-9822(99)80034-7. [DOI] [PubMed] [Google Scholar]
- Fedorova O, Mitros T, Pyle AM. Domains 2 and 3 interact to form critical elements of the group II intron active site. J Mol Biol. 2003;330:197–209. doi: 10.1016/s0022-2836(03)00594-1. [DOI] [PubMed] [Google Scholar]
- Fedorova O, Pyle AM. Linking the group II intron catalytic domains: tertiary contacts and structural features of domain 3. EMBO J. 2005;24:3906–3916. doi: 10.1038/sj.emboj.7600852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris ME, Kazantsev AV, Chen JL, Pace NR. Analysis of the tertiary structure of the ribonuclease P ribozyme-substrate complex by site-specific photoaffinity crosslinking. RNA. 1997;3:561–576. [PMC free article] [PubMed] [Google Scholar]
- Jestin JL, Deme E, Jacquier A. Identification of structural elements critical for inter-domain interactions in a group II self-splicing intron. EMBO J. 1997;16:2945–2954. doi: 10.1093/emboj/16.10.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev AV, Krivenko AA, Harrington DJ, Holbrook SR, Adams PD, Pace NR. Crystal structure of a bacterial ribonuclease P RNA. Proc Natl Acad Sci USA. 2005;102:13392–13397. doi: 10.1073/pnas.0506662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch JL, Boulanger SC, Dib-Hajj SD, Hebbar SK, Perlman PS. Group II introns deleted for multiple substructures retain self-splicing activity. Mol Cell Biol. 1992;12:1950–1958. doi: 10.1128/mcb.12.5.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambowitz AM, Zimmerly S. Mobile group II introns. Ann Rev Genet. 2004;38:1–35. doi: 10.1146/annurev.genet.38.072902.091600. [DOI] [PubMed] [Google Scholar]
- Lehnert V, Jaeger L, Michel F, Westhof E. New loop-loop tertiary interactions in self-splicing introns of subgroup IC and ID: a complete 3D model of the Tetrahymena thermophila ribozyme. Chem Biol. 1996;3:993–1009. doi: 10.1016/s1074-5521(96)90166-0. [DOI] [PubMed] [Google Scholar]
- Major F, Turcotte M, Gautheret D, Lapalme G, Fillion E, Cedergren R. The combination of symbolic and numerical computation for three-dimensional modeling of RNA. Science. 1991;253:1255–1260. doi: 10.1126/science.1716375. [DOI] [PubMed] [Google Scholar]
- Matsuura M, Noah JW, Lambowitz AM. Mechanism of maturase-promoted group II intron splicing. EMBO J. 2001;20:7259–7270. doi: 10.1093/emboj/20.24.7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura M, Saldanha R, Ma H, Wank H, Yang J, Mohr G, Cavanagh S, Dunny GM, Belfort M, Lambowitz AM. A bacterial group II intron encoding reverse transcriptase, maturase, and DNA endonuclease activities: biochemical demonstration of maturase activity and insertion of new genetic information within the intron. Genes Dev. 1997;11:2910–2924. doi: 10.1101/gad.11.21.2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noah JW, Lambowitz AM. Effects of maturase binding and Mg2+ concentration on group II intron RNA folding investigated by UV cross-linking. Biochemistry. 2003;42:12466–12480. doi: 10.1021/bi035339n. [DOI] [PubMed] [Google Scholar]
- Noah JW, Park S, Whitt JT, Perutka J, Frey W, Lambowitz AM. Atomic force microscopy reveals DNA bending during group II intron ribonucleoprotein particle integration into double-stranded DNA. Biochemistry. 2006;45:12424–12435. doi: 10.1021/bi060612h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutka J, Wang W, Goerlitz D, Lambowitz AM. Use of computer-designed group II introns to disrupt Escherichia coli DExH/D-box protein and DNA helicase genes. J Mol Biol. 2004;336:421–439. doi: 10.1016/j.jmb.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Pyle AM, Fedorova O, Waldsich C. Folding of group II introns: a model system for large, multidomain RNAs? Trends Biochem Sci. 2007;32:138–145. doi: 10.1016/j.tibs.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Pyle AM, Lambowitz AM. Group II introns: ribozymes that splice RNA and invade DNA. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA World. Third Edition. Cold Spring Harbor, New York: Cold Spring Harbor Press; 2006. pp. 469–506. [Google Scholar]
- Qin PZ, Pyle AM. Antagonistic substrate binding by a group II intron ribozyme. J Mol Biol. 1999;291:15–27. doi: 10.1006/jmbi.1999.2922. [DOI] [PubMed] [Google Scholar]
- Rhode BM, Hartmuth K, Westhof E, Luhrmann R. Proximity of conserved U6 and U2 snRNA elements to the 5′ splice site region in activated spliceosomes. EMBO J. 2006;25:2475–2486. doi: 10.1038/sj.emboj.7601134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha R, Chen B, Wank H, Matsuura M, Edwards J, Lambowitz AM. RNA and protein catalysis in group II intron splicing and mobility reactions using purified components. Biochemistry. 1999;38:9069–9083. doi: 10.1021/bi982799l. [DOI] [PubMed] [Google Scholar]
- Singh NN, Lambowitz AM. Interaction of a group II intron ribonucleoprotein endonuclease with its DNA target site investigated by DNA footprinting and modification interference. J Mol Biol. 2001;309:361–386. doi: 10.1006/jmbi.2001.4658. [DOI] [PubMed] [Google Scholar]
- Su LJ, Waldsich C, Pyle AM. An obligate intermediate along the slow folding pathway of a group II intron ribozyme. Nucleic Acids Res. 2005;33:6674–6687. doi: 10.1093/nar/gki973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher J, Duarte CM, Su LJ, Pyle AM. Visualizing the solvent-inaccessible core of a group II intron ribozyme. EMBO J. 2001;20:2051–2061. doi: 10.1093/emboj/20.8.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BC, Kazantsev AV, Chen JL, Pace NR. Photoaffinity cross-linking and RNA structure analysis. Methods Enzymol. 2000;318:136–147. doi: 10.1016/s0076-6879(00)18049-8. [DOI] [PubMed] [Google Scholar]
- Toor N, Hausner G, Zimmerly S. Coevolution of group II intron RNA structures with their intron-encoded reverse transcriptases. RNA. 2001;7:1142–1152. doi: 10.1017/s1355838201010251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wank H, San Filippo J, Singh RN, Matsuura M, Lambowitz AM. A reverse transcriptase/maturase promotes splicing by binding at its own coding segment in a group II intron RNA. Mol Cell. 1999;4:239–250. doi: 10.1016/s1097-2765(00)80371-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.