

Abstract
Genome-wide expression profiling has aided the understanding of the molecular basis of neuronal diversity, but achieving broad functional insight remains a considerable challenge. Here, we perform the first systems-level analysis of microarray data from single neuronal populations using weighted gene co-expression network analysis to examine how neuronal transcriptome organization relates to neuronal function and diversity. We systematically validate network predictions using published proteomic and genomic data. Several network modules of co-expressed genes correspond to interneuron development programs, in which the hub genes are known to be critical for interneuron specification. Other co-expression modules relate to fundamental cellular functions, such as energy production, firing rate, trafficking, and synapses, suggesting that fundamental aspects of neuronal diversity are produced by quantitative variation in basic metabolic processes. We identify two transcriptionally distinct mitochondrial modules and demonstrate that one corresponds to mitochondria enriched in neuronal processes and synapses, whereas the other represents a population restricted to the soma. Finally, we show that galectin-1 is a new interneuron marker, and we validate network predictions in vivo using Rgs4 and Dlx1/2 knockout mice. These analyses provide a basis for understanding how specific aspects of neuronal phenotypic diversity are organized at the transcriptional level.
Keywords: development, microarray, mitochondria, neuronal diversity, systems biology
Introduction
The mammalian central nervous system comprises a large number of local and regional circuits that are formed by precise connections between neurons, which themselves are quite diverse from the perspective of morphology, physiological activity, and expression of specific neurotransmitters, peptides, calcium-binding proteins, and numerous other molecules (McKay and Hockfield, 1982; McConnell, 1991, 1995; Polleux, 2005). These aspects of neuronal diversity may influence network function or information processing (Yoshimura and Callaway, 2005). Therefore, determining functions of neuronal circuits depends on understanding the differences that are present between specific neurons. Diversity between neurons is often studied from the perspective of genes responsible for various aspects of neuronal specification (Molyneaux et al, 2007). Earlier studies have identified important genes involved in the differentiation of specific subtypes of neurons, such as corticospinal motor neurons (Chen et al, 2005; Molyneaux et al, 2005) and midbrain dopaminergic neurons (Ferri et al, 2007). However, earlier work has not addressed the general issue of diversity in basic cellular or metabolic functions between neurons.
Studies of cellular heterogeneity have used various methods, such as laser capture microdissection (Bonaventure et al, 2002) and fluorescence-activated cell sorting (Arlotta et al, 2005; Lobo et al, 2006) to study neuronal subtypes. Sugino et al (2006) used both genetic and tracer manipulations to label 12 specific neuronal populations in the adult mouse, which were sorted to purity and subsequently analyzed using gene expression microarrays. Their work resulted in a basic molecular taxonomy of neuronal populations, in which glutamatergic and GABAergic neurons were distinguished by characteristic gene expression patterns and divergence in expression patterns reflected known biological differences between populations (Sugino et al, 2006). Several of these studies take the important and arduous step of functional validation at the level of single genes (Arlotta et al, 2005; Lobo et al, 2006). However, it has been challenging to attain a systems-level understanding of the transcriptome with clear relationships underlying biological or molecular function.
The use of co-expression networks can circumvent this problem because it allows for the examination of gene expression from a system's perspective (Stuart et al, 2003; Lee et al, 2004). Weighted gene co-expression network analysis (WGCNA) groups functionally related genes into modules in an unsupervised manner (Zhang and Horvath, 2005; Horvath et al, 2006; Oldham et al, 2006, 2008), based on the self-organizing properties of complex systems (Barabasi and Albert, 1999; Ravasz et al, 2002). The modularity of the system allows us to consider its components independently, and the relationships between genes within modules can be identified, facilitating gene annotation based on network position without any a priori assumptions about gene function.
Here, we provide a systems biology view of gene expression that we relate to cellular diversity and function within the nervous system. We examined the data set generated by Sugino et al (2006) using WGCNA and identified multiple co-expression modules that are related to fundamental phenotypic features of neurons and the proteome, showing a clear correspondence between gene and protein expression on a large scale. For example, we observed several modules related to interneuron development and show that galectin-1, a hub gene in one module, is a marker of a specific interneuron type. We identify modules that correspond to basic cellular functions, such as energy production and synaptic structure. This suggests that quantitative variation in these modules is a major factor in neuronal diversity. Two modules were highly enriched with mitochondrial genes, but had distinct gene expression patterns, leading to the hypothesis that these modules correspond to synaptic and non-synaptic mitochondria. We tested this hypothesis experimentally, providing a novel molecular basis for examining mitochondrial heterogeneity in neurons. Finally, we used Rgs4 and Dlx1/2 knockout mice to validate predictions based on network organization in vivo, showing that module membership and network position have significant power to predict changes in gene expression. These results provide a systems-level framework for understanding neuronal diversity on a molecular level and its relationship to the physiological and structural aspects of the cell, which can be used to generate hypotheses about processes such as development, neuronal function, and disease (Hood et al, 2004).
Results
Network construction and module detection
We analyzed a unique, high-quality microarray data set that measured gene expression within single neuron classes from Sugino et al (2006). We downloaded data from 36 Affymetrix MOE430A microarrays with 22 690 probe sets from 12 neuronal populations, each with three replicates, from GEO DataSets (GSE2882). The individual neuronal populations analyzed represented a diverse set of excitatory and inhibitory subtypes from multiple brain regions (Supplementary Table 1).
To study how these phenotypic differences were reflected in the transcriptional profile of each neuronal subtype, we performed WGCNA (Zhang and Horvath, 2005), which allows unbiased visualization of the higher-order relationships between genes based on expression. The co-expression network is based on topological overlap (TO) between genes, which simultaneously considers not only the correlation of two genes with each other, but also the degree of their shared correlations within the network (Ravasz et al, 2002; Zhang and Horvath, 2005), providing a more robust measure of relatedness than correlations alone (Yip and Horvath, 2007). Genes are clustered based on TO, and those with similar expression patterns are grouped together (Box 1). The result of clustering can be viewed as a dendrogram, in which each branch corresponds to a group of co-expressed genes (a module) that is designated a color and a number and will be referred to by both its color and number for the rest of this manuscript (Figure 1). We identified 13 modules that had characteristic patterns of gene expression and enrichment for specific gene ontology (GO) categories (Supplementary Tables 2 and 3). Using these module definitions, we studied the importance of these functionally related groups of genes to the developmental, physiological, and structural characteristics of neuronal populations. We have also included two resources for further exploring this network data set, including a gene neighborhood explorer tool (MultiTOM; http://www.genetics.ucla.edu/labs/horvath/MTOM/) and a table with calculated kME values for all genes in all modules (see Materials and methods). These resources alone, or in combination, can be used to explore the functions of any of the genes in the network, beyond the analyses presented here.
Figure 1.

Network construction and modular organization. This dendrogram represents a visual summary of the network, emphasizing its modular organization. The network itself contains 4097 genes, of which 2983 are assigned to 13 modules. Each gene is represented by a vertical line on the x-axis, and the genes are grouped into branches based on their TO. The y-axis on the dendrogram represents the dissimilarity in expression (1-TO) between neighboring genes in the dendrogram. Branches are isolated using an automatic module detection algorithm (Langfelder et al, 2008) and assigned a color, which is shown on the horizontal bar below the dendrogram. The gray areas denote locations where no group of co-expressed genes is detected, and the genes within these areas are not assigned to any of the modules.
Gene expression within modules
We condensed the gene expression pattern within a module to a ‘module eigengene' (ME), which is a weighted summary of gene expression in the module (Oldham et al, 2008). The ME also allows us to determine the network position of a gene by calculating the ME-based connectivity (kME), or the absolute value of the correlation between the expression of a gene and a ME. This value can be interpreted as a measure of module membership or intramodular connectivity (Dong and Horvath, 2007; Horvath and Dong, 2008), and genes with high connectivity are hubs or central genes, which are natural targets for testing hypotheses about modular function (Horvath et al, 2006; Oldham et al, 2008).
As multiple genetic strains of mice were used in this analysis (GIN, G42, G30, YFPH, and C57/Bl6), we were concerned that some modules could be artifacts of genetic background. Therefore, we compared each ME to neuronal subtypes or genetic strains of mice using a Pearson correlation. With the exception of the orange (#8) module, each module showed a higher correlation with specific cell types than genetic background (Supplementary Table 4). Importantly, this systematic analysis of the effect of genetic background was not possible using the standard measures of differential expression, since strain and neuronal sub-type are confounded in some cases (Sugino et al, 2006).
Systematic validation of co-expression relationships
We and others have shown earlier the reproducibility and validity of co-expression modules identified by WGCNA using similarly sized data sets (Drake et al, 2006; Horvath et al, 2006; Oldham et al, 2006, 2008; Fuller et al, 2007; Miller et al, 2008). To provide a level of systematic validation in these data, we confirmed network predictions at the level of both the transcriptome and proteome. First, we assessed the module significance at the statistical level by comparing mean TO between the genes within a module to a random group of genes (Oldham et al, 2008), which provided statistical support for every module identified (Table I). As a second systematic validation of the co-expression relationships defined here, we used a large, independent data set to test whether these co-expression relationships might be conserved in other cell types and tissues. We obtained all publicly available expression data collected on the Affymetrix MOE430A platform (n=3739 microarrays) for the top 100 most highly connected genes in each module from the Celsius database (Day et al, 2007). Using these data, we calculated the mean correlation between the most highly connected genes within each module and compared this value to the mean correlation between a randomly selected group of genes. Each module showed strong conservation of gene co-expression in this data set, providing another level of independent validation of the co-expression relationships identified here (Table I). As a third step of systematic validation of network connectivity, we tested the hypothesis that genes within a module would be functionally related and more likely to interact with each other at the protein level. We found that highly connected genes within a module were far more likely to interact with each other than random groups of genes (Table I), consistent with earlier data showing that interacting proteins are likely to be co-expressed (Jansen et al, 2002; Bhardwaj and Lu, 2005; Oldham et al, 2008). These data extend these observations to formally and systematically validate co-expression relationships identified within this current data set.
Table 1.
Systematic validation and characterization of modules
| Module | Network validation | Module validation (P-value) | Module characterization | ||||
|---|---|---|---|---|---|---|---|
| Transcriptome (P-value) | Proteome (P-value) | Top three hub genes | Gene ontology (P-value) | Cell types | |||
| 1 | Black | 2.63E−66 | 8.24E−33 | 0.002 | Diras1, Plk2, Mast3 | Cellular protein metabolism (P=1.2e−3) | Amygdala and hippocampal pyramidal neurons |
| 2 | Blue | 1.04E−114 | 2.28E−19 | <0.001 | Hadhb, Gas6, Ppp1cc | Mitochondria (P=1.9e−6) | Sst- and Pvalb-positive interneurons; Somatosensory layer V pyramidal neurons |
| 3 | Brown | 6.86E−201 | 1.32E−86 | <0.001 | Tmod2, Rab2, Ywhaz | Protein transport (P=2.1e−6) | Basolateral amygdala, hippocampal, and layers V & VI pyramidal neurons |
| 4 | Green | 3.24E−199 | 3.62E−107 | <0.001 | Crym, Dnclc1, Klhl2 | Synaptic transmission (P=3.7e−4) | Glutamatergic neurons |
| 5 | Green yellow | 2.19E−31 | 9.93E−12 | <0.001 | Slc38a1, Fliih, Phyhipl | Nonsignificant | GABAergic interneurons |
| 6 | Light yellow | 3.12E−25 | 5.59E−35 | <0.001 | Arx, Dlx1, Nxph1 | Signaling/signal transduction (P=5.3e−4) | Telencephalic interneurons |
| 7 | Midnight blue | 2.71E−120 | 3.77E−53 | <0.001 | Ppp1ca, Slc25a22, Synj2 | Coated vesicle (P=3.5e−3) | Basolateral amygdala and layers V & VI pyramidal neurons |
| 8 | Orange | 9.99E−7 | 3.38E−31 | 0.001 | Pdxk, Klhl13, Amy1 | Apoptosis (P=9.0e−3) | Cck positive and LGN interneurons |
| 9 | Pink | 3.16E−161 | 8.23E−77 | <0.001 | Baiap2, Skil, Lmo7 | Protein transport (P=9.8e−3) | Glutamatergic neurons |
| 10 | Purple | 1.01E−158 | 3.94E−98 | <0.001 | Uhmk1, Oaz1, Atp6v1b2 | Endoplasmic reticulum (P=9.0e−3) | Basolateral amygdala and layers V & VI pyramidal neurons |
| 11 | Red | 3.30E−4 | 9.36E−82 | <0.001 | Tacr3, Lhx1, Sdsl | Lipid biosynthesis (P=5.4e−3) | LGN interneurons |
| 12 | Turquoise | 8.12E−203 | 8.65E−28 | <0.001 | Uqcrfs1, Atp5b, Idh3a | Mitochondria (P=2.5e−11) | Pvalb-positive interneurons; layer V pyramidal neurons |
| 13 | Yellow | 1.08E−14 | 1.84E−12 | <0.001 | Snca, Gpm6a, Wasf1 | Ion transport (P=5.7e−4) | Sst positive, Pvalb positive, and LGN interneurons |
Using multiple different data types and analyses, we validated the network on the transcriptional and proteomic levels and evaluated the biological functions of each module. To validate the transcriptional level (‘transcriptome' column), we examined the top 100 most highly connected genes within each module in a very large, independent data set of many thousands of microarrays (Day et al, 2007). This showed that co-expression relationships identified here are conserved and highly significant compared with a similarly sized group of randomly selected genes (P<0.004). We validated co-expression relationships on the proteomic level using a database of protein–protein interactions (‘proteomics' column) (Peri et al, 2003). Here, genes that are highly connected within the same module (kME>0.7) were significantly more likely to interact with each other than random groups of genes (P<0.004). The ‘module validation' column shows that the mean TO within each module is significantly greater than what would be expected by chance, showing strong statistical support for every module (P<0.004). The ‘top three hub genes' column displays the top three genes that have the highest kME within each module. The ‘gene ontology' column displays the most characteristic category for each module, and the ‘cell types' column provides a summary of the associated cell populations for each module.
Validation and annotation of modules
We next moved to the level of individual modules to characterize functional groupings and validate module predictions. In some cases, MEs corresponded to known expression patterns within a group of neuronal subtypes. For example, the distinction between glutamatergic and GABAergic neurons observed by hierarchical clustering of gene expression (Sugino et al, 2006) was reflected by the green (#4) module because genes positively correlated with the green ME reflect glutamatergic neuronal identity, whereas the genes inversely correlated with the green ME reflect GABAergic neuronal identity (Figure 2A). The GABA vesicular transporter (Slc32a1) is a hub negatively correlated with the green ME (kME=0.97), which is consistent with its central role in inhibitory neurotransmission (McIntire et al, 1997). These data suggest that negatively correlated hubs are also biologically significant within the network. Another example is the red (#11) module that corresponds to LGN interneurons (Figure 2B), which is consistent with earlier data showing that it has the most distinct expression pattern among these neuronal types (Sugino et al, 2006).
Figure 2.

Network modules correspond to known and novel functional distinctions between neuronal subtypes. Heat maps depicting expression of genes (rows) across all samples (columns) are shown for six selected modules: green (#4), red (#11), turquoise (#12), blue (#2), light yellow (#6), and pink (#9). The remaining modules are depicted in Supplementary Figure 1. Within the heat map, red corresponds to high expression and green corresponds to genes that are expressed at a low level. A weighted summary of gene expression (or the module eigengene) is shown below each heat map as a barplot. The black horizontal bar above the heat map denotes the association between the module and neuronal subtypes, and the significance of this association using the Kruskal–Wallis test is reported as the P-value below the heat map. A map of the different neuronal subtypes across the heat maps is located in the upper left. (A) The green (#4) module contains 419 genes and corresponds to genes highly expressed in either glutamatergic or GABAergic neurons (P=2.9e−7). (B) The red (#11) module contains 263 genes in LGN interneurons (P=0.005). (C) The turquoise (#12) module contains 358 genes that are highly expressed in cingulate parvalbumin-positive interneurons and layer V pyramidal neurons (P=9.1e−6). (D) The blue (#2) module contains 252 genes that are highly expressed in hippocampal somatostatin-positive interneurons, cingulate parvalbumin-positive interneurons, and layer V pyramidal neurons (P=1.6e−6). (E) The light yellow (#6) module contains 121 genes that are regulated specifically in telencephalic interneurons, but not other interneurons (P=4.3e−7). (F) The pink (#9) module contains 249 genes that are highly expressed in glutamatergic neurons and downregulated in somatostatin- and parvalbumin-positive interneurons (P=3.0e−7).
The identification of modules that reiterate earlier identified differences shows that WGCNA can detect known functional distinctions in an unsupervised manner. Moreover, the identification of hub genes within these modules shows the arrangement of gene expression within each module and which genes are most central. However, many modules are not as easily defined because they correspond to subsets of both glutamatergic and GABAergic neurons and are not confined by known anatomical or neurochemical differences (Figure 2; Supplementary Figure 1). An overview of the modules reflects a complex picture of gene expression within multiple neuronal subtypes. The glutamatergic neurons are represented by the green (#4) (Figure 2A), pink (#9)(Figure 2D), black (#1), brown (#3), and midnight blue (#7) modules, whereas the red (#11) (Figure 2B), light yellow (#6) (Figure 2C), green yellow (#5), orange (#8), and yellow (#13) modules correspond to GABAergic neurons. The turquoise (#12) (Figure 2E), blue (#2) (Figure 2F), and purple (#10) modules represent subsets of both glutamatergic and GABAergic neurons.
To further evaluate individual modules in a systematic manner, we used the ME and the Allen Brain Atlas (ABA) to observe module expression in vivo (Lein et al, 2007). We were able to accomplish this for the three modules that corresponded to recognizable brain regions (black (#1), brown (#3), and midnight blue (#7)), as only in these areas could RNA in situ hybridization data be reliably used to verify the expression of genes within a module. The black (#1) module corresponded to pyramidal neurons in the hippocampus and amygdala, and we found that 88% (22/25) were expressed in the hippocampal pyramidal layer in the ABA (Supplementary Table 5). Within the amgydala, 80% (20/25) were expressed in the basolateral amygdala, whereas 64% (16/25) were expressed in the lateral amygdala. We repeated this analysis in the brown (#3) module, which corresponded to pyramidal neurons in layer V and in the basolateral amygdala. Among this group of genes, 84% (21/25) were expressed in layer V and 76% (19/25) were expressed in the basolateral amygdala (Supplementary Table 6). Finally, the midnight blue (#7) module corresponds to cortical pyramidal neurons within layer V and layer VI, as well as pyramidal neurons within the basolateral amygdala. Within the genes in the midnight blue (#7) module, 88% (22/25) were expressed in layer V and layer VI and 84% (21/25) were expressed in the basolateral amygdala (Supplementary Table 7). These results provide further evidence that the MEs are reflective of the actual expression of the genes within those modules. Such co-expression relationships may be useful in disease gene identification because specific cell classes have been associated with neuropsychiatric diseases (Howard et al, 2005).
We next annotated the modules systematically using GO analysis and the identity of the hub genes (Table I) to determine the biological functions of each module. Of the five modules that correspond to glutatmatergic neurons, four (the black (#1), brown (#3), pink (#9), and midnight blue (#7) modules) were enriched in GO trafficking functions. The black (#1), brown (#3), and pink (#9) module were all enriched in genes involved in protein transport (P=0.029, P=2.1e−6 and P=0.0098), but the black (#1) module was more highly enriched in genes involved in cellular protein metabolism (P=0.0012), suggesting that these modules have slightly different functions. The midnight blue (#7) module was enriched in genes localized to coated vesicles (P=0.0034), and one of its major hub genes, Synj2, is known to be involved in vesicle formation (Malecz et al, 2000). The green (#4) module corresponded to the difference between glutamatergic and GABAergic neurons and was involved in synaptic transmission (P=3.7e−4).
Modules that were associated with GABAergic neurons had multiple, diverse functions. For example, the light yellow (#6) and red (#11) modules corresponded to developmentally distinct groups of interneurons, and hubs within both of these modules are known to influence neuronal development, including Arx, Dlx1, and Lhx1 (Shawlot and Behringer, 1995; Anderson et al, 1997a; Kitamura et al, 2002). The yellow (#13) module was likely related to neuronal physiology because it was enriched in genes involved in ion transport (P=5.6e−4). The orange (#8) module was enriched in genes involved in apoptosis (P=0.0089), but this module also showed a greater correlation with the YFPH strain than specific cell types. Therefore, this module might either be related to injury after dissection or strain differences between animals.
Finally, there were three modules that corresponded to subsets of both glutatmatergic and GABAergic neurons, and each of these modules represented basic cellular functions. The blue (#2) and turquoise (#12) modules were both enriched in genes localized to the mitochondria (P=1.8e−6 and P=2.4e−11). The purple (#10) module was related to trafficking because it was enriched in genes localized to the endoplasmic reticulum (ER) (P=0.0091). We used these basic classifications to more thoroughly examine how differences in development and basic cellular functions contribute to neuronal diversity.
Developmental diversity
Classically, neuronal diversity has been studied through specific marker genes that correspond to differences in specification or origin. We hypothesized that gene expression would reflect these relationships and used the light yellow (#6) module to examine interneuron development from the network perspective. The light yellow (#6) module contains genes that are specifically expressed or repressed in the subset of interneurons derived from the subpallium (Figure 2E), such as all of the Distalless transcription factors that are expressed in the brain (Panganiban and Rubenstein, 2002). In addition, GO analysis showed that both cell migration (P=0.03) and neuron differentiation (P=0.03) were overrepresented within the module (Supplementary Table 2). These data suggest that this module relates to interneuron development and the molecular pathways within this cell class that persist into adulthood.
For computational efficiency, the original network was generated using the subset of genes showing the most variance between samples. Therefore, we examined all genes on the array for significant connectivity to the light yellow (#3) module and identified all relevant genes based on their kME to the light yellow module. We included every gene that had a kME>0.63 (P<0.001, FDR correction), creating an expanded light yellow (#3) module with 826 genes. Hierarchical clustering of this larger meta-module based on TO showed submodules representing subtle variants of the original expression pattern, and we examined these submodules for relationships to interneuron development and specification.
The first submodule had an expression pattern that was very similar to that of the original light yellow module. The two most highly connected genes were transcription factors Dlx1 and Arx (Figure 3A) that are critical for the specification of interneuron subtypes and migration from the medial ganglionic eminence (Anderson et al, 1997a; Kitamura et al, 2002). The ME and the known roles of these hubs in development show that this module corresponds to the transcriptional program present in subpallially derived interneurons. The second submodule corresponded to cholecystokinin-positive interneurons (Figure 3B). These interneurons are derived from the caudal ganglionic eminence, and they tend to be born later than their counterparts from the medial ganglionic eminence (López-Bendito et al, 2004; Wonders and Anderson, 2006). In this submodule, Npas1 was highly connected (kME=0.91), which is consistent with recent data showing that Npas1 is expressed in a related subset of interneurons that express calretinin (Erbel-Sieler et al, 2004; Cobos et al, 2006). However, its functional role in this specific subset of interneurons has not been shown earlier. These data predict that Npas1 is a gene central to their development and mature function.
Figure 3.

Submodules of the light yellow module define distinct interneuron classes with common developmental origins. Expression within each of these submodules is shown in the heatmap and summarized with the module eigengene (described above). The visualization of these modules was performed using VisANT to plot the 250 strongest connections within each module. Genes that are positively correlated are connected by blue lines, whereas genes that are inversely correlated are connected by red lines. (A) The first submodule contains genes co-regulated in all telencephalic interneurons in this analysis, and Dlx1 and Arx are central genes, which is consistent with their known roles as important interneuron specifiers. (B) The second submodule contains genes regulated in cholecystokinin-positive interneurons, which are derived from the caudal ganglionic eminence. (C) The third submodule contains genes regulated in somatostatin- and parvalbumin-positive interneurons, which are both derived from the medial ganglionic eminence. Lhx6 is highly connected in this module, which is consistent with its role in the development of interneurons from the medial ganglionic eminence.
The third submodule had genes specifically regulated in both somatostatin- and parvalbumin-positive interneurons (Figure 3C). Interestingly, these interneuron populations are derived from the medial ganglionic eminence within the subpallium (Wonders and Anderson, 2006). Furthermore, Lhx6 was highly connected within this module (kME=0.96), and it has been shown to play an important role in migration of this class of interneurons from the subpallium to the neocortex (Alifragis et al, 2004; Liodis et al, 2007). In addition to observing genes known to be important for interneuron development, we identified another of the most highly connected genes, galectin-1 (Lgals1) (kME=0.95) within this module, which had no known role in these cell populations. Visualization of this submodule illustrates that galectin-1 and somatostatin are closely related (Figure 3C); because of its hub status, we hypothesized that Lgals1 would be a marker of somatostatin-positive interneurons. We tested this by immunostaining for both galectin-1 and somatostatin in wild-type adult mouse brain and found that galectin-1 was preferentially expressed in the somatostatin-positive interneurons of the cortex. We observed that nearly 80% of the galectin-1-positive cells were also somatostatin positive and a similar proportion of the somatostatin-positive cells were also galectin-1 positive (Figure 4A). As a control, galectin-1 was occasionally co-expressed with another marker of a different class of interneurons, parvalbumin, with only 3.2% of galectin-1-positive cells being parvalbumin positive and 8.5% of parvalbumin-positive cells being galectin-1 positive (Figure 4B). These data show that galectin-1 is highly enriched in the somatostatin-positive interneurons that are derived from the medial ganglionic eminence and may serve as a useful marker for this class of cells.
Figure 4.

Galectin-1 is preferentially expressed in somatostatin-positive interneurons. Photomicrographs of galectin-1 (Lgals1) expression in normal adult mouse cortex and counterstaining with interneuron markers. (A) Immunostaining of galectin-1 (red) and somatostatin (green) shows frequent colocalization. (B) Immunostaining of galectin-1 (red) and parvalbumin (green) shows rare colocalization. Quantification of colocalization is shown in the barplot to the right of each figure (±s.e.m.). Scale bar: 20 μm.
Physiological heterogeneity
Neurons differ greatly in their characteristic firing activity, and we hypothesized that some modules would be related to this fundamental neuronal phenotype. We tested this by comparing physiological parameters to the gene expression patterns found within the modules. We derived a mean firing rate for each neuronal population (from Sugino et al, 2006; Figure 1) and compared these values to each ME (Supplementary Table 8). After correction for multiple comparisons four MEs had significant correlations, including the blue (#2), yellow (#13), black (#1), and pink (#9) (Supplementary Figure 2). The blue (#2) module had the highest correlation with mean firing rate (r=0.61 P=6.2e−5), and we observed earlier an enrichment in proteins localized to mitochondria (P=1.9e−6) and involved in carboxylic acid metabolism (P=1.2e−4). This suggests that the coupling between neuronal activity and oxidative energy production (Kasischke et al, 2004) extends to the transcriptional level and identifies a key set of transcripts involved in this process.
Subcellular diversity and correspondence with the proteome
Neuronal morphology and metabolism are aspects of neuronal phenotypic diversity that we theorized might be reflected at the transcriptional level through variation in organellar composition. We tested this hypothesis by comparing modular organization to proteomic data from a large-scale analysis of subcellular organelles (Foster et al, 2006), and other studies that focused on specific neuronal features, such as the synaptosome (Schrimpf et al, 2005), the postsynaptic density (Collins et al, 2006), a presynaptic fraction (Phillips et al, 2005), and synaptic vesicles (Morciano et al, 2005). We observed significant overrepresentation of many subcellular components within a specific subset of modules (Figure 5A; Supplementary Tables 9 and 10), validating the predictions of the transcriptional network at the level of the proteome. For example, organelles involved in trafficking were overrepresented within the brown (#3) module, including the early endosomes, the ER, the golgi apparatus, the recycling endosomes, and the ER/golgi vesicles. These data support our earlier hypothesis that the brown (#3) module was associated with trafficking within the cell.
Figure 5.

Modules correspond to various aspects of the proteome. Comparison of module membership to the organelle proteome and synaptic proteome. (A) Table depicting modules with nominally significant (P<0.05) enrichment in specific components of either the organelle or synaptic proteome. Three modules have significant overlap with the organelle proteome after strict Bonferroni correction (P<0.004), including the brown (#3), blue (#2), and turquoise (#12) modules. The brown (#3) module overlaps significantly with multiple organelles that are involved in protein trafficking, such as the golgi (P=7.7e−4), recycling endosomes (P=7.4e−11), and the plasma membrane (P=3.0e−5). (B) Barplot showing the comparisons between module membership and the mitochondrial or synaptic proteomes. The y-axis represents the observed to expected ratio of enrichment of either mitochondrial or synaptic proteins (see key) within each module. The asterisk denotes significant enrichment within the module after Bonferroni correction (P<0.004). Mitochondrial proteins are 3.4 (P=6.4e−9) and 2.4 fold (P=5.6e−5) enriched in the blue (#2) and turquoise (#12) modules, respectively. However, the synaptic proteins are only significantly enriched over expected values in the turquoise module (P=2.4e−7).
We also observed that two modules, the blue (#2) (P=6.4e−9) and turquoise (#12) modules (P=5.6e−5) showed significant overrepresentation of mitochondrial proteins (Figure 5B). Although these two modules shared this similarity, they also had key differences. The turquoise (#12) module had a highly significant overlap with synaptic proteins in general (P=2.4e−7), but the blue (#2) module was not enriched in synaptic proteins (Figure 5B). Furthermore, the blue (#2) ME was significantly correlated with neuronal firing rate across different classes, unlike the turquoise (#12) ME (Supplementary Figure 2b). Therefore, these modules both correspond to the mitochondria, but they had distinct gene expression profiles and were related to different aspects of neuronal physiology, which led to the hypothesis that they represented two different mitochondrial populations.
Characterization of two classes of neuronal mitochondria
The existence of mitochondrial heterogeneity within neurons has been suggested earlier, with one population localized to the cell body and the other localized to synapses (Lai et al, 1977). However, no clear functional or transcriptional difference between these mitochondria has been shown. Therefore, we hypothesized that genes within these two modules reflect different mitochondrial populations and tested this hypothesis by determining whether hub genes within the modules could be used to differentiate between mitochondrial populations. Using similar methods to those described earlier in the interneuron module, we created submodules within the blue (#2) and turquoise (#12) modules to identify the most specific mitochondrial components. We assessed each submodule for enrichment in genes localized to the mitochondria using GO analysis and identified submodules within the blue (#2) (P=3.5e−26) and turquoise (#12) module (P=4.8e−27) that were enriched with mitochondrial genes. Given the correlations of the parent modules, we hypothesized that the turquoise submodule corresponded to synaptic mitochondria and that the blue submodule corresponded to non-synaptic mitochondria.
To validate this finding, we chose to examine a well-characterized subset of representative hub genes that were known to localize within mitochondria (Figure 6A). These genes included Phb and Fis1/Ttc11 in the non-synaptic mitochondrial module and Vdac2 and Uqcrfs1 in the synaptic mitochondrial module. We used cellular fractionation to obtain the free mitochondrial fraction and synaptosomal fraction from adult mouse brain (Dodd et al, 1981). The identity of these fractions was confirmed by showing enrichment of synaptophysin in the synaptosomal fraction and the presence of cytochrome (c) in all preparations (Figure 6B). By comparing the expression between these fractions, we observed that modular membership correctly predicted enrichment of mitochondrial proteins within each fraction. The expression of hub genes in the non-synaptic mitochondrial module (Phb and Fis1/Ttc11) was enriched in the mitochondrial fraction, whereas the genes in the synaptic mitochondrial module (Vdac2 and Uqcrfs1) were enriched in the synaptosomal fraction (Figure 6B). We further examined the localization of these proteins in primary culture using confocal microscopy. Phb, a gene in the non-synaptic mitochondrial module, was mainly colocalized with mitochondria in the cell body (Figure 6C), whereas Uqcrfs1, a gene in the synaptic mitochondrial module, was colocalized with mitochondria in the processes, as well as the cell body (Figure 6D). These data indicate for the first time that mitochondrial heterogeneity in neurons is reflected at the transcriptional level and provides a set of new markers to enable the study of mitochondrial populations and their physiological function within neurons.
Figure 6.

Submodules within the blue and turquoise modules represent different mitochondrial populations. We examined the expression of genes known to be localized to the mitochondria in submodules of both the blue and turquoise modules subcellular fractionation. (A) Table showing the most highly connected genes in the synaptic and non-synaptic mitochondrial submodules, sorted by intramodular connectivity. The genes that are in bold (Vdac2, Uqcrfs1, Phb, Fis1/Ttc11) denote those genes that we chose to experimentally validate. Although Phb and Fis1/Ttc11 are not within the most highly connected genes, they have a kME>0.75. (B) Representative western blots of the synaptosomal and mitochondrial fractions that show the relative enrichment of specific genes within one fraction that was predicted by the network. Control blots of synaptophysin (34 kDa) and cytochrome (c) (14 kDa) show appropriate enrichment in synaptic and mitochondrial fractions, respectively. (C) Three replicate western blots showing the ratios of synaptosomal or mitochondrial enrichment of each of the proteins (±s.e.m.). The genes in the non-synaptic mitochondrial module, Fis1/Ttc11 (17 kDa) and Phb (30 kDa), were enriched 47- and 6-fold in the free mitochondrial fraction versus the synaptosomal fraction, respectively. The genes in the synaptic mitochondrial module, Uqcrfs1 (25 kDa) and Vdac2 (38 kDa), were enriched 8- and 7-fold in the synaptosomal fraction versus the free mitochondrial fraction, respectively. (D) Primary hippocampal neurons after 3 weeks in vitro. MitoTracker (red) was used to label the mitochondria within a neuron, whereas Map2 (blue) was used to label the neuronal processes. Phb (green) is a hub in the non-synaptic mitochondrial module, and it co-localizes with mitochondria mainly within the cell body (arrows). (E) Uqcrfs1 (Green) is a hub in the synaptic mitochondrial module, and it co-localizes primarily with mitochondria in the neuronal processes (arrows), as well as those in the cell body. These data indicate that genes in the synaptic mitochondrial module are enriched in mitochondria that are localized to neuronal processes. Scale Bar: 20 μm.
Regulation of modular organization in vivo
We have shown that modules represent functionally related gene groupings that are related to important aspects of neuronal function. To provide a further experimental validation, we tested basic network predictions in two strains of knockout mice. Network theory predicts that the disruption of a highly connected gene, or hub, will preferentially affect genes within the same module because of their high degree of co-regulation (Albert et al, 2000; Jeong et al, 2001). Such a relationship has been observed in unicellular organisms, such as yeast (Carlson et al, 2006), but has not been shown earlier in multicellular tissues such as the brain.
To test the hypothesis that connectivity predicts co-regulation in vivo, we performed microarray expression analysis on the cortex of mutant mice that had a single large deletion of Dlx1 and Dlx2 (Anderson et al, 1997b) because Dlx1 was a highly connected gene in our analysis. Owing to the perinatal lethality of the mutation, we were only able to obtain gene expression data from mice killed at embryonic day 15.5. Genes were filtered based on reliable presence calls and assessed for differential expression using a Bayesian ANOVA (Baldi and Long, 2001). We hypothesized that that the light yellow (#6) module would be enriched for differentially expressed genes because Dlx1 is a hub within that module. We observed that the light yellow (#6) module had a significant overrepresentation of differentially expressed genes (P=5.3e−5) (Figure 7A), which included two known targets of Dlx1, Arx, and Dlx5 (Zerucha et al, 2000; Zhou et al, 2004; Cobos et al, 2005a). We then directly examined the relationship between connectivity and gene expression by comparing the TO that a gene shares with the deleted gene and its chance of being differentially expressed. We observed a significant relationship between connectivity and differential expression in knockout animals because genes in the top quartile of connectivity with Dlx1 were more likely to be differentially expressed than genes in lower quartiles (Figure 7C). These data show that Dlx1 and Dlx2 have functional roles in regulating the expression of genes within the light yellow module, which was predicted by their network centrality. This analysis also provides a set of genes for future exploration of interneuron development and physiology, many of which were not known to have a function in interneuron function.
Figure 7.

In vivo validation of network model. Validation of the network model using gene expression data from two separate knockout mice. (A) Barplot representing the observed to expected ratio of differentially expressed genes (P<0.01) by module in the Dlx1/2 knockout mice and (B) Rgs4 knockout mice. In both the cases, only the modules containing the deleted gene were significantly enriched in differentially expressed genes (P<0.05). (C) Relationship between a gene's topological overlap or connectedness with a ‘hub' gene (i.e. Dlx1/Dlx2 or Rgs4) and differential expression. Genes were ranked by connectivity within the module and the number that was differentially expressed within each quartile was counted and expressed as a percentage of total differentially expressed genes within the module. Error bars show the margin of error for the percentages. In both modules, there is a clear relationship in which genes that are highly connected to the deleted gene are more likely to be differentially expressed than other genes that are not as well connected. Nearly 60% of the genes that were differentially expressed in the enriched modules of either knockout strains were in the top quartile of connectivity with the deleted gene, which is significantly greater than other quartiles (P<0.005).
We tested the robustness of this relationship between connectivity and expression by examining the effects of a deletion of Rgs4, which is a regulator of G-protein signaling (Hepler et al, 1997; Ladds et al, 2007) that has been linked to schizophrenia (Mirnics et al, 2001; Chowdari et al, 2002). Although Rgs4 was not the most central gene, it was well connected in the green (#4) module (kME=0.80), providing a stringent validation of network predictions. We generated transgenic mice with a targeted deletion of Rgs4 (Supplementary Figure 3), and despite having no Rgs4 protein expression in the frontal cortex (Supplementary Figure 4), these mice grow to reproductive age, are fertile, and exhibited no gross abnormalities. In addition, there was no deviation from expected Mendelian frequencies when heterozygotes were interbred and no apparent difference between embryronic, neonatal, or adult lethality between knockout and wild-type strains (data not shown). We collected RNA from the frontal cortex of male adult knockout animals and performed microarray analysis to examine the effects of the Rgs4 deletion on gene expression. We observed that Rgs4 was called absent in all samples from knockout mice and present in all samples from wild-type mice, further verifying the deletion of Rgs4 (Supplementary Figure 4b). We then filtered genes for reliable presence calls and assessed for differential expression using a Bayesian ANOVA (Baldi and Long, 2001), as described for the Dlx1/Dlx2 mutant above. We then assessed whether genes differentially expressed between wild type and knockout mice were enriched in a specific module(s) or randomly distributed throughout the network. There was significant enrichment of differentially expressed genes in the green (#4) module, containing Rgs4 (P=0.004) (Figure 7B), but no enrichment in any other module. In addition, we examined the relationship between intramodular connectivity and gene expression, by comparing the extent of TO with the deleted hub gene and differential expression. We observed a clear relationship between the intramodular connectivity of a gene with Rgs4, in terms of TO, and its chance of being differentially expressed (Figure 7C), which was strikingly similar to what was observed in the Dlx1/Dlx2 mutant. These data provide further evidence that relationships detected by the network analysis reflect real relationships between genes and their co-regulation that are present in vivo, providing an additional level of experimental validation of network organization and predictions.
Discussion
The molecular basis of neuronal diversity can be examined from multiple perspectives. Here, we present an unbiased view of transcriptome organization in various neuronal subtypes and show that such data can be used for exploration of neuronal function and diversity. We provide systematic validation of the co-expression relationships observed in these data by comparing with other large, independent transcriptional and proteomic data sets. Furthermore, by elucidating the modular organization of gene co-expression, we show relationships between the transcriptome and several of the core elements that distinguish different neuronal populations, including basic cell types, developmental origins, physiological properties, and metabolic features of the cell. We also show that co-expression on the transcriptional level correlates with functional relationships on the protein level through the correspondence between modules and proteomic data. Finally, we extend these analyses by performing confirmatory, proof-of-principle experiments in vitro and in vivo. For example, we provide evidence for the existence of two transcriptionally distinct mitochondria classes in neurons for the first time, one that is synaptic and clustered in processes, and the other mainly in the cell body.
These data, though providing many specific biological insights, also allow us to contrast WGCNA with more standard analyses of differential gene expression. These approaches are clearly complementary and emphasize different aspects of neuronal cell biology. For example, genes that are differentially expressed between different cell types are likely to be marker genes of specific populations, and therefore, cell-type-specific gene expression is emphasized by analysis of differential expression. Hence, although many hubs within the interneuron submodules were known to be important in interneuron development, such as members of the Distalless family, Arx and Lhx6 (Kitamura et al, 2002; Panganiban and Rubenstein, 2002; Liodis et al, 2007), these results could have been expected knowing the lineage and phenotype similarities between these neurons. For this reason, some of these cell-type-specific expression patterns (Dlx1, Arx) were identified by Sugino et al (2006). However, in contrast to WGCNA, analysis of differential expression does little to organize the relationships between such markers and other genes that are differentially expressed between cell types. For example, there were hubs within the interneuron modules that were not known to be involved in interneuron function, including Lhx6 and galectin-1, a lectin involved in cell adhesion, which we show by immunohistochemistry to be highly enriched in somatostatin-positive interneurons.
The complementary nature of analysis of differential expression and WGCNA is further emphasized by our observation of only one cell-type-specific module using WGCNA. This is because WGCNA is emphasizing the major sources of variance in gene expression patterns that are shared between more than one cell types. WGCNA suggests that neuronal diversity is created by several quantitative, continuous characteristics and the intersection of these gene co-expression relationships correspond to the major phenotypic differences that are observed. Examination and elucidation of these shared gene expression patterns showed several other biological insights. For example, multiple modules correspond to basic neuronal characteristics, such as energy production, transport, mitochondria, and synaptic structure, showing that these basic aspects of neuronal cell biology are important sources of neuronal diversity, along with widely recognized lineage and developmental differences. We further show how one can use the modular organization of transcriptional network for functional discovery, identifying two mitochondrial modules that correspond to differential localization of mitochondria classes within neurons. Within modules, the extent of connectivity or correlation with the ME can be used as a measure of network centrality (Dong and Horvath, 2007), which allows for the identification of hub genes that are critical to modular function. We used this measure of connectivity to select targets for experimental manipulation that provide evidence showing that network position predicted by WGCNA is reflected in vivo by changes in gene expression. These findings provide further evidence that a systems perspective of the neuronal transcriptome highlights functional co-expression relationships that are involved in fundamental neuronal processes.
We focused on the interneuron module for extended examination of how neuronal diversity created by differentiation was reflected at the transcriptional level in the adult brain. We observed expression patterns that corresponded to known interneuron developmental programs, and although many of fluroescently labeled populations are known to be heterogeneous (Oliva et al, 2000; López-Bendito et al, 2004), these patterns were consistent between samples, suggesting that there is a predominant cell type within these populations. In addition, we observed that many of the genes central to these modules were known to be crucial for interneuron development. For example, Dlx1 and Arx were hubs of a submodule corresponding to telencephalic interneurons. Dlx1 and Dlx2 are functionally redundant and deleting both leads to failed interneuron development (Anderson et al, 1997a, 1997b; Qiu et al, 1997). Arx is necessary for normal interneuron development and function, and mutations in Arx cause defective interneuron migration, resulting in an epilepsy syndrome in humans and mice (Kitamura et al, 2002). Another example of a gene observed to be central in this analysis and interneuron function was Lhx6 (Alifragis et al, 2004; Liodis et al, 2007). Lhx6 was a hub within a module corresponding to parvalbumin- and somatostatin-positive interneurons, which are derived from the medial ganglionic eminence. In the course of this analysis, Liodis et al showed that mice lacking Lhx6 showed impaired migration of interneurons from the medial ganglionic eminence and decreased numbers of both parvalbumin- and somatostatin-positive interneurons (Liodis et al, 2007). However, not all of the hubs that we observed were known regulators of interneuron development. For example, Lgals1 was found to be a hub in the same module as Lhx6, but it had not been studied earlier in either interneuron subtypes. We found that it was a reliable marker for a somatostatin-positive interneuron population. This analysis predicts that hub genes identified in this analysis are involved in maintenance of differentiated populations of interneurons, despite the fact that some have clear roles in development. This is consistent with data showing that the expression of many developmental patterning genes are maintained into adulthood (Zapala et al, 2005), as well as examples of genes that have known roles in development and maintenance of neurons, such as Mef2 and NeuroD (Chae et al, 2004; Heidenreich and Linseman, 2004). Dlx1 is similar because interneurons in Dlx1 knockout mice undergo apoptosis during postnatal development (Cobos et al, 2005b). Therefore, it will be interesting to pursue the functions of the hub genes within these modules in the maintenance of interneuron function and phenotype in the adult animal.
Another aspect of neuronal heterogeneity highlighted by this analysis is the difference in basic organelle composition among neurons. We observed that the blue (#2) module was enriched in mitochondrial proteins by both GO analysis and comparison with proteomic data, and expression in this module correlates significantly with the firing rates of the neuronal populations. Although the expression of specific ion channels can dramatically impact neuronal activity (Toledo-Rodriguez et al, 2004), our data indicate that mitochondrial gene expression is associated with activity in a more global manner, which is consistent with the known connection between neuronal firing rate and cellular oxidative energy production (Kasischke et al, 2004). The turquoise (#12) module was also enriched for mitochondrial and synaptic proteins, but it was unrelated to firing rate. We confirmed the network-derived hypothesis that the turquoise (#12) and blue (#2) modules might represent synaptic and non-synaptic mitochondria by comparing protein expression in the synaptosomal versus free mitochondrial fractions and immunohistochemistry. These data provide a set of genes with which to mark these different mitochondrial classes and probe their function. Recently, it has been shown that synaptic mitochondria are more susceptible to calcium overload and mitochondrial permeability transition (Brown et al, 2006) because of increased expression of Cyclophilin D (Ppif) in mitochondria purified from synaptosomes (Naga et al, 2007). Ppif is present within the synaptic mitochondria submodule (kME=0.77), providing a systems context in which to interpret these physiological data. In addition, our analysis shows that Vdac2 (kME=0.94) and Slc25a5 (Ant2) (kME=0.90) are hubs within the synaptic mitochondria submodule and they are both thought to be involved in mitochondrial permeability transition (Crompton et al, 1998; Kokoszka et al, 2004; Baines et al, 2007). These two mitochondrial submodules that we have identified and studied contain a molecular blueprint for these different mitochondrial populations, which provides a new basis for understanding their roles in neuronal physiology. That these genes, Vdac2, Slc25a5, and Ppif, are hubs, reinforces the idea that susceptibility to mitochondrial permeability transition provides one key functional distinction between these two classes of mitochondria.
We have shown that modules correspond to several phenotypic aspects of neuronal diversity, but within these modules we are also able to identify central genes involved in regulating gene expression within the module. For example, Dlx1 is a central gene within the light yellow (#6) module, and we observed a highly significant enrichment of differentially expressed genes between Dlx1/Dlx2 knockout and control within the light yellow (#6) module (P=5.3e−5). In this case, differential expression is probably caused by the absence of interneurons in the cortex because of disrupted interneuron migration (Anderson et al, 1997a). However, gene expression in knockout animals was assessed at embryonic day 15.5, whereas the data used to construct the network were obtained in adult mice. Thus, this module seems to be stable throughout development, which has interesting implications for the relationship between developmental programs and adult neuronal function and is consistent with other data showing that developmental patterns of gene expression are relevant to adult neuronal function (Zapala et al, 2005).
Despite the robust relationship between connectivity and expression in the Dlx1/Dlx2 knockout mice, this relationship might be expected because these genes are critical transcription factors known to be involved in the differentiation of interneurons (Anderson et al, 1997a). Therefore, we tested this relationship in Rgs4 knockout mice because Rgs4 was a hub, but not as well connected as Dlx1/Dlx2. As was observed in the Dlx1/Dlx2 knockout, genes in the same module as Rgs4 were preferentially perturbed in the knockout mice, and there was a clear relationship between magnitude of TO with Rgs4 and the likelihood of being differentially expressed. These data illustrate that connectivity and module membership are powerful predictors of interaction in vivo, and the deletion of a gene can have specific effects on transcription even if the gene itself does not directly affect transcription, suggesting tight transcriptional regulation using complex feedback systems. Rgs4 is implicated in schizophrenia based on several lines of evidence (Mirnics et al, 2001; Chowdari et al, 2002), and this analysis provides candidate genes that may functionally interact with Rgs4, which could be important in delineating the mechanisms through which this gene functions in health and disease. These data support our hypothesis that central genes are important for modular organization, and therefore are likely to be important drivers of neuronal diversity, whether they are responsible for differentiation or more basic functional aspects of the cell. In addition, poorly characterized genes that are highly connected to hub genes with known function are likely to participate in similar biological processes, providing a means to annotate gene function.
In summary, we have used WGCNA to elucidate the organization of gene expression within neurons, which is organized into a hierarchical, scale-free network, containing modules of highly co-expressed genes. These modules correspond to multiple aspects of neuronal function and illustrate the relationships between gene expression and the basic variable features of neurons that give rise to anatomical and physiological diversity. We have systematically validated the identified co-expression relationships using published large-scale proteomic and genomic data sets, as well as by specific in vitro and in vivo experiments. These experiments provide examples of functional discovery by using the network to generate hypotheses that are then tested. Overall, these analyses provide a systems-level framework for understanding the molecular basis of neuronal diversity, which can be used to gain insight into the underlying mechanisms of important biological processes in health and disease.
Materials and methods
Microarray data and network construction
Raw data from all microarrays from Sugino et al (2006) (GSE2882: GSM63015–GSM63050) were imported into R (http://www.r-project.org/), scaled to the same average intensity, and normalized using the quantile normalization method from Bioconductor (http://www.bioconductor.org/) (Choe et al, 2005; Oldham et al, 2006, 2008). Genes with consistent presence in at least one cell type, high coefficient of variation (>0.21), and high connectivity (k>0.11) were selected for network construction and resulted in a network size of 4097 genes. The TO between these genes was calculated based on Zhang and Horvath (2005), where the TO between genes i and j is calculated from the adjacency matrix (a):
where we assume that the diagonal elements of a are equal to zero. These genes were clustered on based on their TO, and modules were identified using an automatic module detection algorithm (with 50 genes being the minimum module size) and assigned both a color and number (Langfelder et al, 2008). Similar modules were identified by calculating the Pearson correlation between MEs, and modules were combined according to Oldham et al (2008). Although the network initially included these genes, the network analysis was subsequently extended to all genes on the array based on their correlation with the module eigengene (see Supplementary information for detailed methods).
Module visualization
To visualize the pairwise relationships between genes, we used the software VisANT (http://visant.bu.edu/). Approximately 300 pairs of genes with the highest intramodular TO were depicted (Oldham et al, 2006). Each link corresponds to a TO value between the connected nodes. The ‘relaxing' algorithm was used to visualize the network structure.
Functional annotation
GO analysis was performed using the DAVID functional annotation tool (http://david.abcc.ncifcrf.gov/) (Dennis et al, 2003; Hosack et al, 2003). Each module was compared against the Mus musculus background for enrichment within the GO categories biological process, cellular compartment, and molecular function. Only level five GO categories with more than two genes were selected for inclusion in this work, and each term is associated with its EASE score in the text.
To determine whether any modules were related to firing rate, we calculated a mean firing rate from the current clamp plots in Sugino et al (2006) Figure 1. This was done by counting the number of action potentials in each plot and dividing by the total time that was shown in the plot, which resulted in a mean firing rate for each population in the analysis.
To determine whether modules corresponded to particular subcellular components, we accessed the Organelle Map Database (http://proteome.biochem.mpg.de/ormd/), which organizes 1405 proteins into 10 specific organelles based on correlation profiling (Foster et al, 2006). For each organelle, all protein identifiers were obtained and converted to Affymetrix identifiers. Within each module, the number of genes that corresponded to each specific organelle was counted, and the χ2-test was used to determine whether enrichment for a specific organelle was significant.
A similar analysis was performed on the synaptic compartment, which was not included in the Organelle Map Database. Synaptic proteins from four proteomic studies of different synaptic fractions were analyzed, including the synaptosome (Schrimpf et al, 2005), the postsynaptic density (Collins et al, 2006), the presynaptic fraction (Phillips et al, 2005), and the synaptic vesicles (Morciano et al, 2005). Enrichment analysis was performed as described above.
Immunohistochemistry
Adult C57/Bl6 mice were perfused, and their brains were fixed with 4% PFA and sectioned at 30 μm. The anti-galectin-1 antibody was a generous gift from Dr Linda Baum. The anti-somatostatin antibody (CURE.S6) was obtained from Animal Core of CURE, Digestive Diseases Division, UCLA. Anti-galectin-1 was used 1:250 and anti-somatostatin was used 1:50 overnight at 4°C. Donkey secondary antibodies conjugated to Alexa-488 and Alexa-594 were incubated for 1 h at room temperature.
Cellular fractionation
Brains from adult C57/Bl6 mice were homogenized using a glass dounce homogenizer in mitochondrial buffer (250 mM D-mannitol, 70 mM sucrose, 10 mM HEPES, pH 7.4), and the mitochondrial and synaptosomal fractions were collected according to Dodd et al (1981). Briefly, nuclei and debris were removed by centrifugation at 1000 g, and the supernatant was collected. This material was pelleted by centrifuging at 10 000 g. The pellet was then resuspended in 8 ml of mitochondrial buffer and layered over a 1.2 M sucrose gradient, which was subsequently spun at 39 000 r.p.m. (TH-641 Sorvall rotor) for 30 min. The interphase was collected and re-diluted to 8 ml, and the mitochondrial pellet was collected in a separate tube and re-suspended in mitochondrial buffer. The dilute interphase was then layered over a 0.8 M sucrose gradient, which was spun at 39 000 r.p.m. for 30 min. The synaptosomal pellet was then re-suspended in mitochondrial buffer. The synaptosomes were then lysed in 6 mM Tris–HCl pH 8.1. The mitochondrial and synaptosomal fractions were denatured in SDS loading buffer with DTT, and the samples were run on a 12% acrylamide gel, which was transferred to nitrocellulose membranes. The following antibodies were used to probe the membranes, anti-synaptohysin (Abcam), anti-cytochrome c (Cell Signaling), anti-Vdac2 (Santa Cruz), anti-Uqcrfs1 (Abcam), anti-Fis1/Ttc11 (Abcam), and anti-Phb (Abcam).
Immunocytochemistry
Primary hippocampal neuronal cultures were established according to established methods (Jahr and Stevens, 1987). Briefly, the hippocampus was dissected from P3 wild-type mice, digested in Papain (10 units/ml) (Worthington) for 35 min, and dissociated by pipetting through fire-polished Pasteur pipets. They were plated onto coverslips coated with poly-ornithine and laminin, and they were grown in neurobasal supplemented with B27. After 3 weeks in vitro, coverslips were incubated with 50 mM MitoTracker for 15 min, and then they were immediately fixed in 4% paraformaldehyde for 10 min. Antigen retrieval was performed by incubating coverslips in 0.1 M Tris–HCl and 5% urea pH 8.0 at 85°C for 20 min. Anti-Phb and anti-Uqcrfs1 were used at a dilution of 1:200 overnight at 4°C, and donkey secondary antibodies were used at 1:1000 for 1 h at room temperature.
In vivo validation of network predictions
Dlx1/Dlx2 knockout animals were obtained and RNA was obtained using Trizol (GibcoBRL) from wild type and mutant cortex at embryonic day 15.5 in duplicate. The obtained RNA was amplified and labeled using the GeneChip IVT Labeling kit (Affymetrix), and run on Affymetrix MOE430 2.0 microarrays. Data were normalized as described above and filtered for genes present in both the replicates of either mutant or wild-type samples. Differential expression was assessed using a Bayesian ANOVA (Baldi and Long, 2001) with a threshold of P<0.01.
Rgs4 knockout animals were generated using standard methods of embryonic stem cell electroporation, selection, and screenings (see Supplementary information for details). Briefly, a targeting vector containing homologous regions to the Rgs4 locus and LoxP sites flanking a neomycin resistance cassette and all coding regions except for exon 1 was linearized and electroporated into TL1 (129SvEvTac) cells (Supplementary Figure 3). Resistant colonies were selected using PCR and confirmed using a Southern blot (Supplementary Figure 4a), and these cells were injected into blastocysts. Congenic mice with floxed Rgs4 were bred with a CRE deletor line that led to the inheritance of the Rgs4 deletion. Frontal cortex was obtained from animals that were homozygous for the Rgs4 deletion and loss of Rgs4 was confirmed at both the mRNA (Supplementary Figure 4b) and protein level (Supplementary Figure 4c). Microarray analysis was performed as described above.
Data exploration resources
We have only been able to study a few functional insights from this co-expression network. Beyond the analyses presented in this manuscript, we have also included two resources to allow investigators to query the position of specific genes of interest and generate new hypotheses. The kME table contains the predicted module membership for all genes on the array, allowing direct annotation of specific genes based on the modular functions described here. The second is a network neighborhood tool that allows one to identify the genes that are co-regulated with a gene(s) of interest based on their TO. MultiTOM identifies the nearest neighbors of any expressed gene (Li and Horvath, 2007). We have included step-by-step instructions in the Supplementary information and the tool can be downloaded from http://www.genetics.ucla.edu/labs/horvath/MTOM/.
Steps for performing WGCNA.
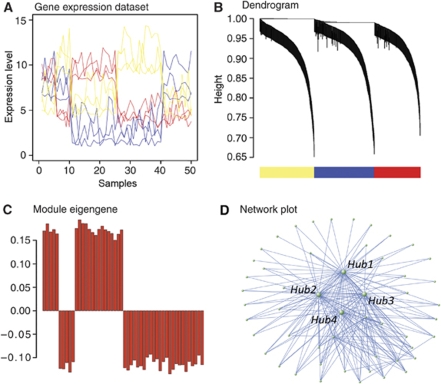
(A) Network construction–expression levels are shown for a subset of genes representing a hypothetical data set in which the genes measured have three distinct expression patterns (red, blue, and yellow), in which the y-axis is expression level and each point on the x-axis is an individual sample. The Pearson correlations between all genes within the data set are calculated and converted into a connection strength by scaling the correlation values to a power, which is empirically determined to best approximate scale-free topology (Zhang and Horvath, 2005) within the network. These connection strength values are then used to calculate the topological overlap (TO) between all genes, which is the degree of all shared connections by two genes (see Materials and methods). (B) Module definition—genes are clustered based on TO, and the result of this clustering is depicted in the dendrogram. Separate branches of the dendrogram comprise co-expressed genes with high TO (modules), and these modules are assigned different colors to emphasize the modular framework of the network. (C) The module eigengene—the first principal component or ME is calculated, based on singular value decomposition, to summarize the major vector of gene expression within each module. We account for the observation that genes may have significant co-expression relationships with more than one module by using the term kME, which is the Pearson correlation between the ME and each gene's expression level. The kME of a gene summarizes its relationship to each module, in which a higher value corresponds to more central position. (D) Network module visualization—we typically plot the 300 strongest connections (based on TO) within a module for illustrative purposes. This highlights the central most connected genes, or hubs, as well as their major relationships within the network. For more methodological details see the Supplementary information for methods, as well as Oldham et al (2006), Oldham et al (2008), Zhang and Horvath (2005), and http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork/.
Supplementary Material
This file contains supplementary information, instructions for use of MultiTOM, and supplementary figures and tables.
This file contains all of the genes and their calculated kME for all of the modules.
This file contains descriptions for all of the module eigengenes identified in this manuscript.
This file contains the genes and their connectivity within the three interneuron submodules shown in Figure 3.
This file contains the genes and their connectivity within the two mitochondrial submodules shown in Figure 6.
This file contains the topological matrix between all genes included in the original network.
This file contains the normalized gene expression data used in this study.
Supplementary figure 1b
Supplementary figure 1c
Supplementary figure 1d
Supplementary figure 1e
Supplementary figure 1f
Supplementary figure 1g
Supplementary figure 1h
Supplementary figure 1i
Supplementary figure 1j
Supplementary figure 1k
Supplementary figure 1l
Supplementary figure 1m
Supplementary figure 1n
Supplementary figure 1
Supplementary figure 2a
Supplementary figure 2b
Supplementary figure 2c
Supplementary figure 2d
Supplementary figure 2e
Supplementary figure 2f
Supplementary figure 3a
Supplementary figure 3b
Supplementary figure 3c
Acknowledgments
We thank Sugino et al for their generous sharing of the data and Sacha Nelson for his valuable comments on an earlier draft of this manuscript. We thank members of the Geschwind lab for their helpful input and Ezra Rosen specifically for testing the network explorer on this data set. We thank Dr Kelsey Martin and members of the Martin lab, Dr Carrie Heusner, and Rachel Jefferey, for valuable discussions and technical expertise. We thank Dr Linda Baum for generous gift of the galectin-1 antibody and the Animal Core of CURE, Digestive Diseases Division, UCLA for the generous gift of the somatostatin antibody (CURE.S6). For her excellent work on generating the microarray data for the Rubenstein lab, we thank Winnie Liang at the NIH Neuroscience Microarray, Translational Genomics Research Institute, Neurogenomics Division. We acknowledge support from NIH grants NIMH #R37 MH60233-06A1 (DHG, KW, MO), NINDS #U24 NS52108 (DHG, SH), NIGMS GM08042 (KW), Neurobehavioral Genetics Training Grant T32MH073526-01A1 (KW). Medical Scientist Training Program UCLA (KW), Aesculapians Fund of the UCLA School of Medicine (KW), the Miriam and Sheldon Adelson Program in Neural Repair and Rehabilitation (DHG, SH), and the JLRR from Nina Ireland, NIMH RO1 MH49428-01, RO1, and K05 MH065670 (JR, CS).
Footnotes
The authors declare that they have no conflict of interest.
References
- Albert R, Jeong H, Barabasi A (2000) Error and attack tolerance of complex networks. Nature 406: 378–382 [DOI] [PubMed] [Google Scholar]
- Alifragis P, Liapi A, Parnavelas JG (2004) Lhx6 regulates the migration of cortical interneurons from the ventral telencephalon but does not specify their gaba phenotype. J Neurosci 24: 5643–5648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL (1997a) Interneuron migration from basal forebrain to neocortex: dependence on dlx genes. Science 278: 474–476 [DOI] [PubMed] [Google Scholar]
- Anderson SA, Qiu M, Bulfone A, Eisenstat DD, Meneses J, Pedersen R, Rubenstein JL (1997b) Mutations of the homeobox genes dlx-1 and dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron 19: 27–37 [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD (2005) Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 45: 207–221 [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD (2007) Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9: 550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi A, Albert R (1999) Emergence of scaling in random networks. Science 286: 509–512 [DOI] [PubMed] [Google Scholar]
- Baldi P, Long AD (2001) A bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics 17: 509–519 [DOI] [PubMed] [Google Scholar]
- Bhardwaj N, Lu H (2005) Correlation between gene expression profiles and protein-protein interactions within and across genomes. Bioinformatics 21: 2730–2738 [DOI] [PubMed] [Google Scholar]
- Bonaventure P, Guo H, Tian B, Liu X, Bittner A, Roland B, Salunga R, Ma XJ, Kamme F, Meurers B, Bakker M, Jurzak M, Leysen JE, Erlander MG (2002) Nuclei and subnuclei gene expression profiling in mammalian brain. Brain Res 943: 38–47 [DOI] [PubMed] [Google Scholar]
- Brown MR, Sullivan PG, Geddes JW (2006) Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J Biol Chem 281: 11658–11668 [DOI] [PubMed] [Google Scholar]
- Carlson MR, Zhang B, Fang Z, Mischel PS, Horvath S, Nelson SF (2006) Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genomics 7: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae JH, Stein GH, Lee JE (2004) Neurod: the predicted and the surprising. Mol Cells 18: 271–288 [PubMed] [Google Scholar]
- Chen JG, Rasin MR, Kwan KY, Sestan N (2005) Zfp312 is required for subcortical axonal projections and dendritic morphology of deep-layer pyramidal neurons of the cerebral cortex. Proc Natl Acad Sci USA 102: 17792–17797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe SE, Boutros M, Michelson AM, Church GM, Halfon MS (2005) Preferred analysis methods for affymetrix genechips revealed by a wholly defined control dataset. Genome Biol 6: R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T, Deshpande SN, Thelma BK, Ferrell RE, Middleton FA, Devlin B, Levitt P, Lewis DA, Nimgaonkar VL (2002) Association and linkage analyses of rgs4 polymorphisms in schizophrenia. Hum Mol Genet 11: 1373–1380 [DOI] [PubMed] [Google Scholar]
- Cobos I, Broccoli V, Rubenstein JLR (2005a) The vertebrate ortholog of aristaless is regulated by dlx genes in the developing forebrain. J Comp Neurol 483: 292–303 [DOI] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JL (2005b) Mice lacking dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci 8: 1059–1068 [DOI] [PubMed] [Google Scholar]
- Cobos I, Long JE, Thwin MT, Rubenstein JL (2006) Cellular patterns of transcription factor expression in developing cortical interneurons. Cereb Cortex 16(Suppl 1): i82–i88 [DOI] [PubMed] [Google Scholar]
- Collins MO, Husi H, Yu L, Brandon JM, Anderson CN, Blackstock WP, Choudhary JS, Grant SG (2006) Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J Neurochem 97(Suppl 1): 16–23 [DOI] [PubMed] [Google Scholar]
- Crompton M, Virji S, Ward JM (1998) Cyclophilin-d binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem 258: 729–735 [DOI] [PubMed] [Google Scholar]
- Day A, Carlson MR, Dong J, O'Connor BD, Nelson SF (2007) Celsius: a community resource for affymetrix microarray data. Genome Biol 8: R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA (2003) David: database for annotation, visualization, and integrated discovery. Genome Biol 4: P3. [PubMed] [Google Scholar]
- Dodd PR, Hardy JA, Oakley AE, Edwardson JA, Perry EK, Delaunoy JP (1981) A rapid method for preparing synaptosomes: comparison, with alternative procedures. Brain Res 226: 107–118 [DOI] [PubMed] [Google Scholar]
- Dong J, Horvath S (2007) Understanding network concepts in modules. BMC Syst Biol 1: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake TA, Schadt EE, Lusis AJ (2006) Integrating genetic and gene expression data: application to cardiovascular and metabolic traits in mice. Mamm Genome 17: 466–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbel-Sieler C, Dudley C, Zhou Y, Wu X, Estill SJ, Han T, Diaz-Arrastia R, Brunskill EW, Potter SS, McKnight SL (2004) Behavioral and regulatory abnormalities in mice deficient in the npas1 and npas3 transcription factors. Proc Natl Acad Sci USA 101: 13648–13653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri AL, Lin W, Mavromatakis YE, Wang JC, Sasaki H, Whitsett JA, Ang SL (2007) Foxa1 and foxa2 regulate multiple phases of midbrain dopaminergic neuron development in a dosage-dependent manner. Development 134: 2761–2769 [DOI] [PubMed] [Google Scholar]
- Foster LJ, de Hoog CL, Zhang Y, Zhang Y, Xie X, Mootha VK, Mann M (2006) A mammalian organelle map by protein correlation profiling. Cell 125: 187–199 [DOI] [PubMed] [Google Scholar]
- Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, Horvath S (2007) Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm Genome 18: 463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidenreich KA, Linseman DA (2004) Myocyte enhancer factor-2 transcription factors in neuronal differentiation and survival. Mol Neurobiol 29: 155–166 [DOI] [PubMed] [Google Scholar]
- Hepler JR, Berman DM, Gilman AG, Kozasa T (1997) Rgs4 and gaip are gtpase-activating proteins for gq alpha and block activation of phospholipase c beta by gamma-thio-gtp-gq alpha. Proc Natl Acad Sci USA 94: 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood L, Heath JR, Phelps ME, Lin B (2004) Systems biology and new technologies enable predictive and preventative medicine. Science 306: 640–643 [DOI] [PubMed] [Google Scholar]
- Horvath S, Dong J (2008) Geometric interpretation of gene co-expression network analysis. PloS Computational Biology 4: e1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, Lee Y, Scheck AC, Liau LM, Wu H, Geschwind DH, Febbo PG, Kornblum HI, Cloughesy TF, Nelson SF, Mischel PS (2006) Analysis of oncogenic signaling networks in glioblastoma identifies aspm as a molecular target. Proc Natl Acad Sci USA 103: 17402–17407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosack DA, Dennis G Jr, Sherman BT, Lane HC, Lempicki RA (2003) Identifying biological themes within lists of genes with ease. Genome Biol 4: R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard A, Tamas G, Soltesz I (2005) Lighting the chandelier: new vistas for axo-axonic cells. Trends Neurosci 28: 310– 316 [DOI] [PubMed] [Google Scholar]
- Jahr CE, Stevens CF (1987) Glutamate activates multiple single channel conductances in hippocampal neurons. Nature 325: 522–525 [DOI] [PubMed] [Google Scholar]
- Jansen R, Greenbaum D, Gerstein M (2002) Relating whole-genome expression data with protein-protein interactions. Genome Res 12: 37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Mason SP, Barabási AL, Oltvai ZN (2001) Lethality and centrality in protein networks. Nature 411: 41–42 [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW (2004) Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305: 99–103 [DOI] [PubMed] [Google Scholar]
- Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, Matsuo M, Kamijo S, Kasahara M, Yoshioka H, Ogata T, Fukuda T, Kondo I, Kato M, Dobyns WB, Yokoyama M et al. (2002) Mutation of arx causes abnormal development of forebrain and testes in mice and x-linked lissencephaly with abnormal genitalia in humans. Nat Genet 32: 359–369 [DOI] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC (2004) The adp/atp translocator is not essential for the mitochondrial permeability transition pore. Nature 427: 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladds G, Goddard A, Hill C, Thornton S, Davey J (2007) Differential effects of rgs proteins on g alpha(q) and g alpha(11) activity. Cell Signal 19: 103–113 [DOI] [PubMed] [Google Scholar]
- Lai JC, Walsh JM, Dennis SC, Clark JB (1977) Synaptic and non-synaptic mitochondria from rat brain: isolation and characterization. J Neurochem 28: 625–631 [DOI] [PubMed] [Google Scholar]
- Langfelder P, Zhang B, Horvath S (2008) Defining clusters from a hierarchical cluster tree: the dynamic tree cut package for r. Bioinformatics 24: 719–720 [DOI] [PubMed] [Google Scholar]
- Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P (2004) Coexpression analysis of human genes across many microarray data sets. Genome Res 14: 1085–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445: 168–176 [DOI] [PubMed] [Google Scholar]
- Li A, Horvath S (2007) Network neighborhood analysis with the multi-node topological overlap measure. Bioinformatics 23: 222–231 [DOI] [PubMed] [Google Scholar]
- Liodis P, Denaxa M, Grigoriou M, Akufo-Addo C, Yanagawa Y, Pachnis V (2007) Lhx6 activity is required for the normal migration and specification of cortical interneuron subtypes. J Neurosci 27: 3078–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW (2006) Facs-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci 9: 443–452 [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Sturgess K, Erdélyi F, Szabó G, Molnár Z, Paulsen O (2004) Preferential origin and layer destination of gad65-gfp cortical interneurons. Cereb Cortex 14: 1122–1133 [DOI] [PubMed] [Google Scholar]
- Malecz N, McCabe PC, Spaargaren C, Qiu R, Chuang Y, Symons M (2000) Synaptojanin 2, a novel rac1 effector that regulates clathrin-mediated endocytosis. Curr Biol 10: 1383–1386 [DOI] [PubMed] [Google Scholar]
- McConnell SK (1991) The generation of neuronal diversity in the central nervous system. Annu Rev Neurosci 14: 269–300 [DOI] [PubMed] [Google Scholar]
- McConnell SK (1995) Strategies for the generation of neuronal diversity in the developing central nervous system. J Neurosci 15: 6987–6998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM (1997) Identification and characterization of the vesicular gaba transporter. Nature 389: 870–876 [DOI] [PubMed] [Google Scholar]
- McKay RD, Hockfield SJ (1982) Monoclonal antibodies distinguish antigenically discrete neuronal types in the vertebrate central nervous system. Proc Natl Acad Sci USA 79: 6747–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, Geschwind DH (2008) A systems level analysis of transcriptional changes in alzheimer's disease and normal aging. J Neurosci 28: 1410–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P (2001) Disease-specific changes in regulator of g-protein signaling 4 (rgs4) expression in schizophrenia. Mol Psychiatry 6: 293–301 [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD (2005) Fezl is required for the birth and specification of corticospinal motor neurons. Neuron 47: 817–831 [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD (2007) Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci 8: 427–437 [DOI] [PubMed] [Google Scholar]
- Morciano M, Burré J, Corvey C, Karas M, Zimmermann H, Volknandt W (2005) Immunoisolation of two synaptic vesicle pools from synaptosomes: a proteomics analysis. J Neurochem 95: 1732–1745 [DOI] [PubMed] [Google Scholar]
- Naga KK, Sullivan PG, Geddes JW (2007) High cyclophilin d content of synaptic mitochondria results in increased vulnerability to permeability transition. J Neurosci 27: 7469–7475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Horvath S, Geschwind DH (2006) Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci USA 103: 17973–17978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH (2008) Functional organization of the transcriptome in human brain. Nat Neurosci 11: 1271–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva AA Jr, Jiang M, Lam T, Smith KL, Swann JW (2000) Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in gabaergic interneurons. J Neurosci 20: 3354–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panganiban G, Rubenstein JLR (2002) Developmental functions of the distal-less/dlx homeobox genes. Development 129: 4371–4386 [DOI] [PubMed] [Google Scholar]
- Peri S, Navarro JD, Amanchy R, Kristiansen TZ, Jonnalagadda CK, Surendranath V, Niranjan V, Muthusamy B, Gandhi TK, Gronborg M, Ibarrola N, Deshpande N, Shanker K, Shivashankar HN, Rashmi BP, Ramya MA, Zhao Z, Chandrika KN, Padma N, Harsha HC et al. (2003) Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res 13: 2363–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips GR, Florens L, Tanaka H, Khaing ZZ, Fidler L, Yates JR III, Colman DR (2005) Proteomic comparison of two fractions derived from the transsynaptic scaffold. J Neurosci Res 81: 762–775 [DOI] [PubMed] [Google Scholar]
- Polleux F (2005) Genetic mechanisms specifying cortical connectivity: let's make some projections together. Neuron 46: 395–400 [DOI] [PubMed] [Google Scholar]
- Qiu M, Bulfone A, Ghattas I, Meneses JJ, Christensen L, Sharpe PT, Presley R, Pedersen RA, Rubenstein JL (1997) Role of the dlx homeobox genes in proximodistal patterning of the branchial arches: mutations of dlx-1, dlx-2, and dlx-1 and -2 alter morphogenesis of proximal skeletal and soft tissue structures derived from the first and second arches. Dev Biol 185: 165–184 [DOI] [PubMed] [Google Scholar]
- Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabási AL (2002) Hierarchical organization of modularity in metabolic networks. Science 297: 1551–1555 [DOI] [PubMed] [Google Scholar]
- Schrimpf SP, Meskenaite V, Brunner E, Rutishauser D, Walther P, Eng J, Aebersold R, Sonderegger P (2005) Proteomic analysis of synaptosomes using isotope-coded affinity tags and mass spectrometry. Proteomics 5: 2531–2541 [DOI] [PubMed] [Google Scholar]
- Shawlot W, Behringer RR (1995) Requirement for lim1 in head-organizer function. Nature 374: 425–430 [DOI] [PubMed] [Google Scholar]
- Stuart JM, Segal E, Koller D, Kim SK (2003) A gene-coexpression network for global discovery of conserved genetic modules. Science 302: 249–255 [DOI] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB (2006) Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat Neurosci 9: 99–107 [DOI] [PubMed] [Google Scholar]
- Toledo-Rodriguez M, Blumenfeld B, Wu C, Luo J, Attali B, Goodman P, Markram H (2004) Correlation maps allow neuronal electrical properties to be predicted from single-cell gene expression profiles in rat neocortex. Cereb Cortex 14: 1310–1327 [DOI] [PubMed] [Google Scholar]
- Wonders CP, Anderson SA (2006) The origin and specification of cortical interneurons. Nat Rev Neurosci 7: 687–696 [DOI] [PubMed] [Google Scholar]
- Yip AM, Horvath S (2007) Gene network interconnectedness and the generalized topological overlap measure. BMC Bioinformatics 8: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura Y, Callaway EM (2005) Fine-scale specificity of cortical networks depends on inhibitory cell type and connectivity. Nat Neurosci 8: 1552–1559 [DOI] [PubMed] [Google Scholar]
- Zapala MA, Hovatta I, Ellison JA, Wodicka L, Del Rio JA, Tennant R, Tynan W, Broide RS, Helton R, Stoveken BS, Winrow C, Lockhart DJ, Reilly JF, Young WG, Bloom FE, Lockhart DJ, Barlow C (2005) Adult mouse brain gene expression patterns bear an embryologic imprint. Proc Natl Acad Sci USA 102: 10357–10362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerucha T, Stühmer T, Hatch G, Park BK, Long Q, Yu G, Gambarotta A, Schultz JR, Rubenstein JL, Ekker M (2000) A highly conserved enhancer in the dlx5/dlx6 intergenic region is the site of cross-regulatory interactions between dlx genes in the embryonic forebrain. J Neurosci 20: 709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S (2005) A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4: Article17. [DOI] [PubMed] [Google Scholar]
- Zhou QP, Le TN, Qiu X, Spencer V, de Melo J, Du G, Plews M, Fonseca M, Sun JM, Davie JR, Eisenstat DD (2004) Identification of a direct dlx homeodomain target in the developing mouse forebrain and retina by optimization of chromatin immunoprecipitation. Nucleic Acids Res 32: 884–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains supplementary information, instructions for use of MultiTOM, and supplementary figures and tables.
This file contains all of the genes and their calculated kME for all of the modules.
This file contains descriptions for all of the module eigengenes identified in this manuscript.
This file contains the genes and their connectivity within the three interneuron submodules shown in Figure 3.
This file contains the genes and their connectivity within the two mitochondrial submodules shown in Figure 6.
This file contains the topological matrix between all genes included in the original network.
This file contains the normalized gene expression data used in this study.
Supplementary figure 1b
Supplementary figure 1c
Supplementary figure 1d
Supplementary figure 1e
Supplementary figure 1f
Supplementary figure 1g
Supplementary figure 1h
Supplementary figure 1i
Supplementary figure 1j
Supplementary figure 1k
Supplementary figure 1l
Supplementary figure 1m
Supplementary figure 1n
Supplementary figure 1
Supplementary figure 2a
Supplementary figure 2b
Supplementary figure 2c
Supplementary figure 2d
Supplementary figure 2e
Supplementary figure 2f
Supplementary figure 3a
Supplementary figure 3b
Supplementary figure 3c