

Summary
Interleukin-10 (IL-10) has long been recognized to have potent and broad-spectrum anti-inflammatory activity, which has been unequivocally established in various models of infection, inflammation, and even in cancer. However, because of the marginal successes of the initial clinical trials using recombinant IL-10, some of the interest in this cytokine as an anti-inflammatory therapeutic has diminished. New work showing IL-10 production from regulatory T cells and even T-helper 1 T cells has reinvigorated the field and revealed the power of this cytokine to influence immune responses. Furthermore, new preclinical studies suggest that combination therapies, using antibodies to IL-10 along with chemotherapy, can be effective in treating bacterial, viral, or neoplastic diseases. Studies to understand IL-10 gene expression in the various cell types may lead to new therapeutics to enhance or inhibit IL-10 production. In this review, we summarize what is known about the regulation of IL-10 gene expression by various immune cells. We speculate on the promise that this cytokine holds to influence immune responses and mitigate immune pathologies.
Keywords: monocytes/macrophages, Th1/Th2/Th17 cells, cytokines, infectious disease, immunotherapies
Introduction
Interleukin-10 (IL-10) is a Type II cytokine and the ‘founding’ member of a family of cytokines that include IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29 (1). All of these cytokines have similar intron–exon genomic organization, bind to receptors with similar structures and in some cases shared components (2), and all activate Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathways. Despite these commonalities, the cytokines in this family have very different biological activities, which are largely determined by the cells producing the cytokine, the cells responding to them, and the immune environment in which they are released. A brief description of the IL-10-related cytokines is listed in Table 1. More detailed descriptions of each of these individual cytokines can be found elsewhere (3). In the present review, we focus on IL-10, the cytokine that has the most potent anti-immune and anti-inflammatory activity of all the family members.
Table 1.
The IL-10-related cytokines
| Member | Primary cell source | Receptor | Biological effect |
|---|---|---|---|
| IL-19 | Monocytes | IL-20R1/IL-20R2 | Immunoregulatory, skin development |
| IL-20 | Monocytes, keratinocytes | IL-20R1/IL-20R2, IL-22R1/IL-20R2 | Inflammation, hematopoiesis, skin development |
| IL-22 | T cells, NK cells, Th17 cells | IL-22R/IL-10R2 | Innate immunity, acute-phase responses |
| IL-24 | Monocytes, Th2 cells melanocytes | IL-20R1/IL-20R2, IL-22R1/IL-20R2 | Inflammation, proapoptotic, epidermal cell function |
| IL-26 | Monocytes, memory T cells | IL-20R1/IL-20R2 | Mucosal and cutaneous immunity |
| IL-28, IL-29 | DCs | IFNLR1/IL-10R2 | Antiviral immunity |
IL-10 was originally identified by Mosmann and colleagues (4, 5). Since this original description, the list of cells producing IL-10 has expanded rapidly, as have the number of cells that are capable of responding to this cytokine. The four major T-cell sources of IL-10 are T-helper type 2 (Th2) cells, subsets of regulatory T cells designated Tr1, Th1, and Th17 cells (6). CD8+ T cells also produce IL-10. Other important producers of IL-10 include monocytes and appropriately stimulated macrophages, as well as some subsets of dendritic cells (DCs). Human B cells are also a potentially important source of IL-10 (7), as are some granulocytes, including eosinophils and mast cells (8). Non-immune cell sources of IL-10 include keratinocytes, epithelial cells, and even tumor cells (9, 10).
It is difficult to determine which cells are the most important producers of IL-10. The recently developed IL-10/green fluorescence protein (GFP) reporter mice may provide us with this information (11), but at the present time, we must resort to conjecture regarding this important question. From our own perspective, it seems reasonable to assume that Th2 cells represent a very important source of antigen-specific IL-10. These cells exhibit epigenetic commitment to IL-10 production (described later in this article), which assures that clonal progeny cells will also retain high levels of IL-10 production over extended periods of time. Type 1 regulatory T cells have also emerged as important sources of IL-10 (12). There are now numerous examples in animal models where the presence of Tr1 cells can permit pathogen persistence, ameliorate or prevent autoimmunity, and even suppress graft or tumor rejection. Consequently, numerous studies to manipulate this subset of regulatory T cells are underway. There is no question that Th1 cells and more recently Th17 cells are capable of producing IL-10, and these cells represent an important source of IL-10 during infectious diseases (13, 14), but it seems more likely that this production may occur as a homeostatic mechanism to prevent uncontrolled T-cell activation (6). This IL-10 production in Th1 and Th17 cells may occur via a ‘strength of stimulus’ mechanism, which may also be true of B cells. In our hands, the production of IL-10 from myeloid DCs is fairly modest relative to macrophages, and plasmacytoid DCs do not seem to be important sources of IL-10 (15). The stimulus used to induce cytokine production from DCs may also dictate the amount of IL-10 produced by them. In general, Toll-like receptor 2 (TLR2) ligands seem to be better stimuli than TLR4 ligands for IL-10 production by DCs (16). It should be emphasized that even though some populations of DCs may not produce large amounts of IL-10, these cells are extremely sensitive to the inhibitory effects of IL-10, whether produced by the DCs themselves or by neighboring cells (see tumor-infiltrating DCs below). The production of IL-10 from macrophages may also depend on the stimulus used to induce it. For example, bacterial flagellin, which interacts with TLR5, seems to be a particularly poor inducer of IL-10 relative to TLR2 and TLR4 ligands. The hyper-induction of IL-10 that has been associated with regulatory macrophages may also occur by a ‘strength of signal’ mechanism that results in the activation of the mitogen-activated protein kinase (MAPK) extracellular signal-regulated kinase (ERK), resulting in high levels of IL-10 production. IL-10 production by mast cells in the skin may serve to limit local pathology in the skin during contact dermatitis or ultraviolet irradiation (17). Thus, there are several potentially important cellular sources of IL-10, and it is quite likely that the relative importance of each cell type may vary with the type, strength, and location of stimuli.
The human IL-10 gene spans about 4.7 kb on chromosome 1q21–32 and contains five exons that are separated by four introns. The murine IL-10 gene is organized in a similar fashion with 5.1 kb spanning chromosome 1E4. There are several genes adjacent to IL-10 gene within a segment of approximately 200 kb that forms the IL-10 gene cluster (Fig. 1). Within this cluster in both human and mouse, the genes encoding IL-20 and IL-19 are found approximately 90 kb upstream of IL-10, whereas genes encoding Mapkapk2 and Dyrk3 are localized downstream of IL-10. Among these five genes, the transcription of the IL-10 gene is initiated in one direction, whereas the others proceed in the opposite orientation.
Fig. 1. A schematic of mouse chromosome 1, showing the IL-10 gene and its neighbors.
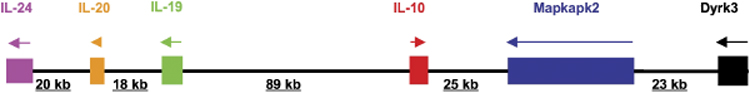
Arrows depict orientation of gene expression. The sizes of intervening sequences are designated below. Regulatory elements, such as insulators, enhancers, and silencers, are not shown.
The IL-10 receptor
IL-10 signals through a two-receptor complex consisting of two copies each of IL-10 receptor 1 (IL-10R1) and IL-10R2 (18). IL-10R1 binds IL-10 with a relatively high affinity (50–200 pM), and the recruitment of IL-10R2 to the receptor complex makes only a marginal contribution to ligand binding. However, the engagement of this second receptor to the complex enables signal transduction following ligand binding. Thus, the functional receptor consists of a dimer of heterodimers of IL-10R1 and IL-10R2 (Fig. 2). Most hematopoietic cells constitutively express low levels of IL-10R1, and receptor expression can often be dramatically upregulated by various stimuli. Non-hematopoietic cells, such as fibroblasts and epithelial cells, can also respond to stimuli by upregulating IL-10R1. The IL-10R2 is expressed on most cells, and therefore a large number of diverse cells have the ability to bind to and consume IL-10. This represents a problem for the therapeutic administration of recombinant IL-10 (rIL-10), because much of the cytokine can be diverted to non-immune cells thereby diminishing the effective dosage administered. This effect almost certainly contributed to the marginal success of some of the early clinical trials in which IL-10 was administered subcutaneously at sites distal to the sites of inflammation.
Fig. 2. The IL-10 receptor and a simplified version of signaling from this receptor.
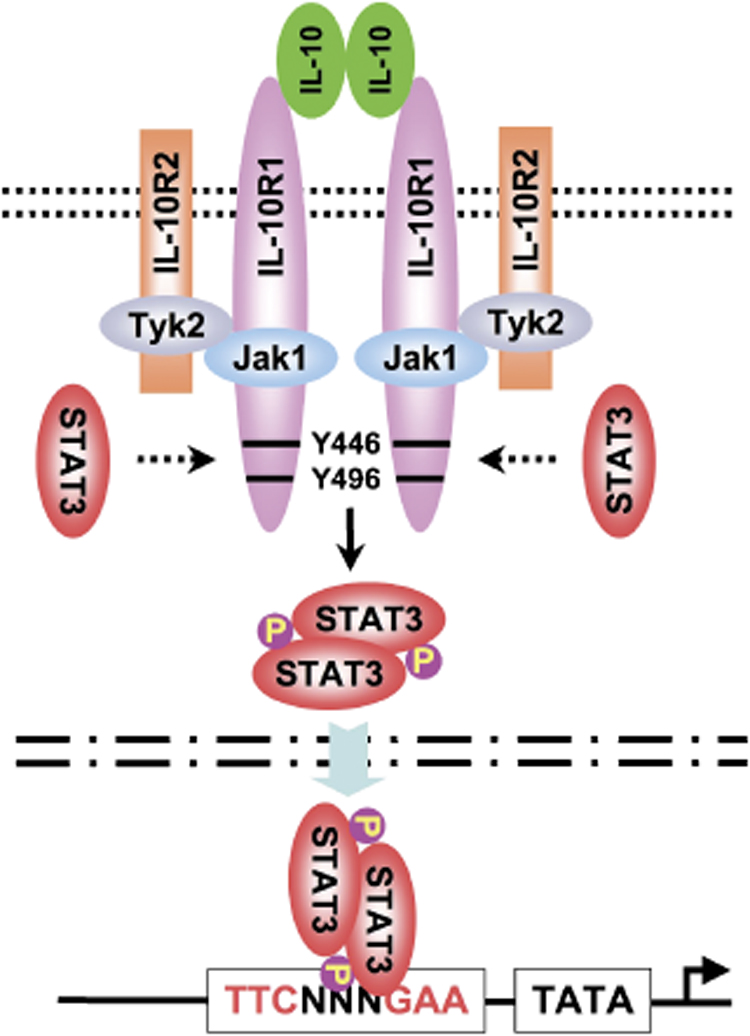
The functional receptor complex is composed of two subunits each of IL-10 R1 and IL-10R2. The Janus tyrosine kinases JAK1 and Tyk2 associate with the cytoplasmic tails of the receptor and phosphorylate tyrosine residues in IL-10R1, to which STAT3 is recruited. STAT3 homodimers translocate into the nucleus and bind to STAT elements in several immune response genes, including IL-10 itself and the SOCS genes.
The binding of IL-10 to the receptor complex activates the Janus tyrosine kinases, JAK1 and Tyk2, associated with IL-10R1 and IL-10R2, respectively, to phosphorylate the cytoplasmic tails of the receptors. This results in the recruitment of STAT3 to the IL-10R1 (2). The site of STAT3 recruitment is conserved between the human and murine receptor and is shared by other STAT3-recruiting receptors, including gp130, IL-20R1, and IL-22R1. The homodimerization of STAT3 results in its release from the receptor and translocation of the STAT homodimer into the nucleus, where it binds to STAT-binding elements in the promoters of various genes (18) (Fig. 2). One of these genes is IL-10 itself, which is positively regulated by STAT3 (19). STAT3 also activates the suppressor of cytokine signaling 3 (SOCS3), which controls the quality and quantity of STAT activation (20, 21). SOCS3 is induced by IL-10 and exerts negative regulatory effects on various cytokine genes. Importantly, the IL-10R does not have SOCS-binding sites and therefore is not subject to the regulation by SOCS3 that other cytokine receptors may be (20). SOCS proteins contain two major domains: a Src homology 2 (SH2) domain that binds to the substrate and a Socs box that complexes with elongins B and C, a cullin and Rbx2, to form a E3 ubiquitin ligase. It has been postulated that following SOCS binding to their substrates via the SH2 domain, the E3 activity directs the substrates for ubiquitination and the proteasome-mediated degradation (21). However, direct evidence in support of this degradation is lacking, and there is no evidence that SOCS3 can bind to IL-10 receptors and direct them for degradation. The phosphoinositide 3-kinase (PI3K) pathway is also activated by IL-10 (10). Although the roles of STAT3 and SOCS in IL-10 signal transduction have been well established (22), a complete understanding of the molecular mechanisms whereby IL-10 inhibits immune responses remains somewhat elusive. An engineered EpoR that activates STAT3 activates an anti-inflammatory response identical to IL-10, but other STAT3 activating receptors, such as the IL-6R, fail to exert anti-inflammatory responses (22). This finding suggests that STAT3 activation is required but not sufficient for the anti-inflammatory activity of IL-10. The differential regulation of signaling by SOCS proteins may also contribute to differences between IL-6 and IL-10R (22). Furthermore, the STAT3-dependent silencing of many cytokine genes in IL-10-treated macrophages is in stark contrast to the enhanced production of genes such as IL-1 receptor antagonist in these same cells. Several groups have demonstrated that IL-10 treatment results in diminished nuclear factor-κB (NF-κB) activation in response to a variety of different stimuli (10), although this effect may be cell-type specific. This may occur through the suppression of inhibitor of NF-κB (IκB) kinase (IKK) activity by IL-10, resulting in the retention of NF-κB subunits in the cytoplasm. IL-10 may also inhibit NF-κB translocation to the nucleus and DNA binding (23), and this inhibition may be specific to the p65 subunit, resulting in the preferential induction of p50 monomers and homodimers (24). The inhibition of NF-κB by IL-10 would explain the large number of immune response genes that are less responsive to stimuli following IL-10 treatment. Defective NF-κB activation would also prevent DC maturation and diminish antigen-presenting cell (APC) functionality, both of which have been attributed to IL-10. Furthermore, the inhibition of specific Rel family members, such as p65, may explain the ability of IL-10 to inhibit inflammatory genes but activate others. In human monocytes, IL-10 inhibition may not work at the level of NF-κB inhibition (25), and therefore other molecular mechanisms are likely to be involved. These possibilities may include an inhibition of MAPKs and/or an activation of an inhibitory PI3K/AKT pathway (10).
IL-10 bioactivity
The main biological function of IL-10 appears to be exerted on DCs and macrophages. IL-10 is a potent inhibitor of antigen presentation. It inhibits major histocompatibility complex class II expression as well as the upregulation of costimulatory molecules CD80 and CD86. IL-10 inhibits the differentiation of DCs from monocyte precursors, and it also inhibits DC maturation. Thus, many of the immuno-inhibitory characteristics of IL-10 can be traced to their effect on APCs to prevent the production of the Th1-associated cytokines IL-2 and interferon-γ (IFN-γ), and also the Th2-associated IL-4 and IL-5. Thus, the original description of IL-10 as a cross-regulator of Th1/Th2 immunity is probably no longer applicable. The other profound effect of IL-10 is to inhibit the production of proinflammatory cytokines and mediators from macrophages and DCs. The major inflammatory cytokines, IL-1, IL-6, IL-12, and tumor necrosis factor (TNF), are all dramatically repressed following exposure to IL-10. Inflammatory chemokines of both the CC and CXC type are also suppressed by IL-10, as is the production of macrophage matrix metalloproteases. IL-10 can further inhibit inflammation by increasing the release of IL-1 receptor antagonist by macrophages. IL-10 can also target naive CD4+ T cells, possibly via an inhibition of CD28 signaling pathway. IL-10 has little or no direct effects on activated or memory T cells due to the reduction of IL-10R upon T-cell activation (26).
It is important to also note that not all IL-10 bioactivity results in a suppression of immune responses. IL-10 can costimulate B-cell activation, prolong B-cell survival, and contribute to class switching in B cells. It can also costimulate natural killer (NK) cell proliferation and cytokine production (27). IL-10 can also act as a growth factor to stimulate the proliferation of certain subsets of CD8+ T cells (28–30). These stimulatory activities of IL-10 may be dose dependent, and this may explain the overproduction of inflammatory cytokines in humans treated with high doses of rIL-10 (31).
IL-10 transcriptional regulation
IL-10 is made by many different immune cells, and there are aspects of IL-10 gene regulation that are conserved among all these cells, while other mechanisms that appear to be cell specific. The promoters for IL-10 in all the cells producing IL-10 are essentially the same and therefore the transcription factors that initiate transcription are conserved. These transcription factors and their binding sites are reviewed individually below and depicted schematically in Fig. 3. In contrast to the conserved transcription factors, the signaling pathways that induce IL-10 are generally unique to each cell type. The chromatin encompassing the IL-10 locus may also be uniquely modified in each of the different cells, and the post-transcriptional modifications may also be cell specific.
Fig. 3. A diagram of regulatory elements in the murine and human IL-10 promoters.
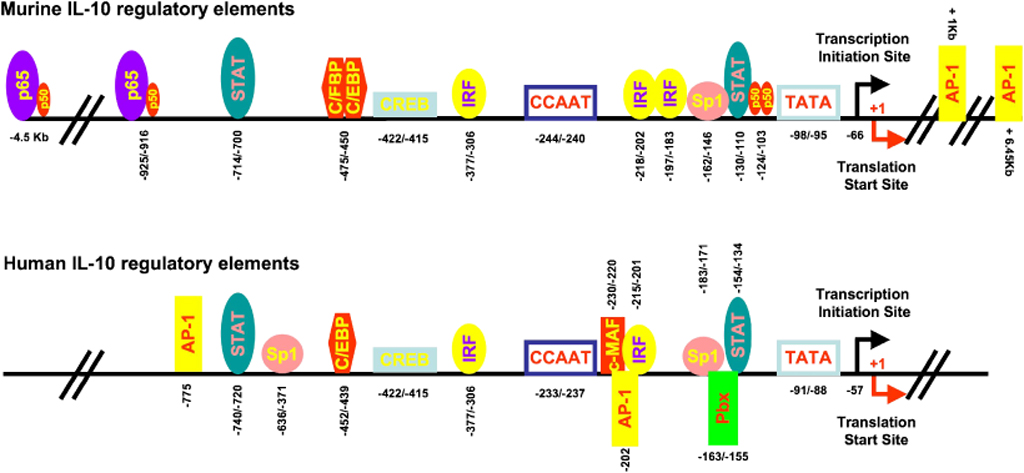
The binding sites for transcription factors (circles and boxes) in the murine (top) and human (bottom) IL-10 promoter with numbering relative to the translation start site. For details on each transcription factor, please consult text.
In the murine IL-10 gene, a TATA box is found between 98 and 95 bp upstream of the first methionine codon, while in the human IL-10 gene locus, its location is between 91 and 88 bp (Fig. 3). Note, for clarity and consistency we designate these locations relative to the translational start codon ATG. A CCAAT box is located between −233 and −237 bp in human IL-10 gene and between −244 and −240 bp (CCAGT) in the murine IL-10 gene. A predicated transcriptional initiation site for IL-10 gene – ACA is located 66 bp upstream of the first ATG for the murine gene and at 57 bp for the human gene.
The transcription of the IL-10 gene has been studied in detail in a variety of cells, including T cells, B cells, NK cells, monocytes, and macrophages. Transcription factors such as Sp1 and Sp3, STAT3, C/EBPβ and δ (CCATT/enhancer-binding protein β and δ), IRF-1 (IFN regulatory factor-1), c-Maf (cmusculoaponeurotic fibrosarcoma), AP-1 (activator protein-1), CREB (cAMP response element binding) protein, and NF-κB (family members, including p65-containing heterodimers as well as p50 homodimers, were all found to positively regulate IL-10 transcription. The binding of each of these transcription factors has been mapped to specific sites on the murine and human IL-10 promoters (Fig. 2). The role that each plays in IL-10 transcription is summarized briefly below.
Sp1
Sp1 is one of the first cellular transcription factors to be identified by virtue of its ability to bind to G-rich motifs in the SV-40 early promoter. The Sp1-binding site in the IL-10 promoter is located between −162 and −146 bp of the murine IL-10 gene (32–34). The element consists of six bases AGGAGG. The requirement for Sp1 in transcription of murine IL-10 gene was demonstrated convincingly in studies using Sp factor-deficient Drosophila SL2 cells. The addition of both Sp1 and Sp3 could restore transcription, and both factors bound to the same element. Importantly, while Sp1 uniformly drives transcription, in some settings Sp3 was shown to repress Sp1-mediated transcription. A similar AGGAGG-containing element is found in the human IL-10 promoter region, and this element is located within the region between −183 and −171 bp (35, 36). When this element was replaced with GAATTC, the IL-10 promoter activity was dramatically diminished. A second Sp1-binding site in human IL-10 promoter was identified between −636 and −631 bp (agacCCCGCCt gtc) (37). Human IL-10 promoter activity was abolished when a mutant Sp1 expression plasmid was introduced into cells. A single nucleotide polymorphism (SNP) of A–C at −627 bp renders the Sp1 elements fully repressive, possibly through interaction of Sp proteins with Ets family proteins that bind to a downstream Ets-like element (tgtAGGAAcca).
STATs
The STATs are cytoplasmic transcription factors that translocate to the nucleus to regulate gene expression in response to cytokines and growth factors. The canonical STAT-binding site contains the sequence TTCCNGGAA. In the human IL-10 gene, two putative STAT-binding sites have been reported (38, 39). The first one is located between −740 and −720 bp (CCAAG CACagTTGGG). A second one is found in the proximal promoter region between −154 and −134 bp (ATCCTGTgCCGGGAAACCTTG). In the murine IL-10 gene, the site corresponding to the first human IL-10 STAT site is found between −714 and −700 bp (TCATgCTGGGATCTG), whereas the second one is located at −130/−110 bp (ACCTTTgCCAGGAAGGCCCC). The proximal STAT-binding element has been shown to interact with STAT3 and mediate IFN-α-induced IL-10 production in a human B-cell line (40). Bream and colleagues (41) recently showed that STAT4 can induce IL-10 production in NK cells (42). STAT1 may play a negative regulatory role in IL-10 gene expression in monocytes (42). In T cells, however, both STAT1 and STAT3 are critical for IL-27-induced IL-10 gene expression, whereas STAT3 is essential for IL-6-induced IL-10 production (43).
C/EBP
The C/EBPs are a family of leucine zipper transcription factors that bind to DNA as homodimers or heterodimers. The C/EBP consensus sequence is ATTGCGCAAT. Two putative C/EBP sites were identified in the murine IL-10 promoter region at −475/ −450 bp relative to the translation start codon (TGGAGGAAACAATTATTTCTCAATCC). The downstream region of −463/ −450 bp (TTATTTCTCAATCC) appears to be a major C/EBP site on the promoter (44). In the human IL-10 promoter, the corresponding putative C/EBP site can be found in the region of −452/−439 (TTATTTCTCAATCC). It has been demonstrated that in the murine IL-10 gene both C/EBP-β and δ can interact with the downstream putative site to activate IL-10 gene transcription (45). In addition, C/EBP can synergize with Sp-1 to induce IL-10 production (46).
IRFs
The IRF family of transcription factors consist of nine members in both human and mice: IRF1, IRF2, IRF3, IRF4 (PIP, LSIRF, or ICSAT), IRF5, IRF6, IRF7, IRF8 (ICSBP), and IRF9 (ISGF3γ). The well-conserved N-terminal IRF DNA-binding domain (DBD) recognizes a DNA sequence corresponding to the IFN-stimulated response element (ISRE) (A/GNGAAANNGAAACT). Further analysis reveals that 5′-AANNGAAA-3′ is the consensus IRF recognition sequence. A putative IRF motif is found in the human IL-10 promoter region of −215/ −201 bp relative to the translation start codon: 5′-CAAAAATTGAAAACT-3′. In the murine IL-10 promoter region, several putative IRF motifs can also be spotted (47). There are 5′-GCTAAAAAGAAAAAA-3′ (−377/−306 bp), 5′-AAAAAAGGGAAAGGAAAAAA (−218/−202 bp with two overlapping motifs), and 5′-AAAAGAAAGAAATTA-3′ (−197/−183 bp). In a study using a human B-cell line, Ziegler-Heitbrock et al. (38) showed that IFN-α upregulated IL-10 production through an mechanism that was interdependent on the IRF and nearby STAT motifs, whereas lipopolysaccharide (LPS)-induced IL-10 transcription was independent of the IRF motif. Further analysis revealed that IRF-1 but not IRF-2 bound to the human IL-10 IRF motif. Recently, retinoic acid inducible gene-I (RIG-I)-mediated IRF-3 signaling has been attributed to Epstein–Barr virus-encoded small RNA-induced IL-10 production in promoting the growth of Burkitt’s lymphoma cells (48).
AP-1
AP-1 is a heterodimeric transcription factor composed of proteins belonging to the c-Fos, c-Jun, activating transcription factor (ATF), and Jun Dimerization Partner (JDP) family. AP-1 recognizes a DNA sequence defined as tissue plasminogen activator (TPA)-response element (TRE: 5′-TGAG/CTCA-3′) via a leucine zipper. Association of AP-1 activity with IL-10 production has been shown in T cells and monocyte/macrophages (49, 50). An AP-1-specific binding motif (5′-TGACTCA-3′) was identified in the non-coding region that is approximately 1 kb downstream of the IL-10 transcription initiation site (51). This element is the motif that binds to Jun/AP-1 protein and it is responsible for IL- 10 production in Th2 cells (52). An additional AP-1 site is located further downstream (approximately 6.45 kb) that binds AP-1 in the absence of nuclear factor of activated T cells (NFAT) and expresses intergenic RNA in a Th2-specific manner (53).
CREB
The CREB proteins bind to cAMP response elements that consist of 5′-TGAGC/CGTCA-3′. The human IL-10 promoter is responsive to cAMP stimulation in THP-1 cells and macrophages (44, 54, 55). Four putative CREB elements have been identified in the human IL-10 promoter region. They are CRE1 at −1272/ −1265 bp (5′-TGAtGTCA-3′), CRE2 at −1055/−1048 bp (5′-TGACtTCt-3′), CRE3 at −862/−855 bp (5′-TGAtGTaA-3′), and CRE4 at −411/−404 bp (50-ccACGTCA-3′). CRE1, CRE3, and CRE4, but not CRE2 are able to form DNA–protein complexes and slow migration in electrophoretic mobility shift assays. Antibody against CREB and ATF-1 can supershift and reduce DNA–protein complex formed by CRE1 and CRE4 but not CRE3. The complexes do not contain ATF-2, AP-1, or CREM-1. CRE1 and CRE4 are functional in reporter assays, affirming their importance in IL-10 regulation, and mutations in either CRE1 or CRE4 diminish cAMP-induced IL-10 promoter activity. An element that is identical to CRE4 can be found at −422/ −415 bp of the murine IL-10 promoter, and this element may be critical for adiponectin-induced IL-10 production. The IFN-α-mediated suppression of IL-10 may be due to CREB and AP-1 mediated mechanisms (50).
c-Maf
c-Maf is the cellular homolog of the avian viral oncogene v-mal, which belongs to the Maf family of basic region and leucine zipper transcription factors. The consensus Maf recognition sequence (MARE) has been defined as 5′-TGC(TGAG/CTCA) GCA-3′ or 5′-TGC(TGAGC/CGTCA)GCA-3′. This palindromic MARE sequence contains a TPA-responsive element [TGA(G/C) TCA] or a cAMP-responsive element [TGA(GC/CG)TCA]. The former is the binding sequence for AP-1 (Jun/Fos) family proteins, whereas the later is for the proteins of CREB/ATF family. The MARE core is flanked on both ends by extended sequence: 5′-TGC-MARE-GCA-3′, which is a unique signature of the binding site of Maf family proteins. A putative Maf-binding element was found in the human IL-10 promoter region at −233/−200 bp: 5′-TCATTTTTGC(TtACgaT)GCA AAAATTGAAAACTA-3′ (56). A second possible Maf binding sequence may be in the distal region of −491/−512 [5′-TGACTGC(ctAAGtTA)GCAAGGA-3′]. The proximal Maf site in humans contains an atypical TPA-response element, whereas the distal site contains an atypical cAMP-response element. The proximal putative site has been identified as a c-Maf-binding element for LPS-induced IL-10 production in macrophages. Whether the distal site is functional remains in question. A third possible candidate, which is conserved in the human and murine IL-10 genes, is in the region around the translation start site of+2/+14 bp: 5′-TGC(acAGCTCA)GCA-3′ in humans and 5′-TGC(ctgGCTCA)GCA-3′ in mouse. The sequence contains an atypical cAMP-response element. c-Maf was originally identified as Th2 transcription factor directing the expression of the IL-4 gene and thereby promoting Th2 differentiation (57, 58). IL-4 may enhance LPS-induced IL-10 production from macrophages via its activity on c-Maf (56).
NF-κB
Five DNA-binding members of NF-κB have been identified: p65 (RelA), c-Rel, RelB, p50, and p52. They all have a Rel homology domain (RHD) that imparts DNA binding. p105-derived p50 and p100-derived p52 lack trans-activation domains (59). The consensus sequence for NF-κB binding is . In an earlier study on human monocytederived macrophages, the overexpression of IκBα an inhibitor of p65 nuclear translocation which leads to a reduction in heterodimers formed between p65/other NF-κB proteins, had only minor effects on LPS-mediated IL-10 production, and no effect on IL-1-induced production (60). LPS-induced production of other inflammatory cytokines including IL-1β, IL-6, IL-8, and TNFα was significantly reduced by this, suggesting that NF-κB played little to no role in IL-10 biosynthesis. In LPS-treated macrophages derived from mouse bone marrow, a unique DNase I-sensitive site around −4.5 kb upstream of the IL-10 gene has been identified (61). Within this region, a putative NF-κB-binding site with p65 binding was revealed with a sequence of GGGGAATTCC. An identical sequence is conserved in a similar location of the human IL-10 gene, suggesting a role for this hypothetical region in IL-10 regulation. Another putative NF-κB-binding site in the murine IL-10 promoter region at −925/−916 bp has been recently identified: 5′-GaGAAGTCCC-3′. This element appears to be the regulatory motif responsible for PKR [double-stranded RNA (dsRNA)-dependent protein kinase]-mediated IL-10 induction in murine macrophages following stimulation with dsRNA (62). A putative NF-κB-binding site specific for p50–p50 homodimers has been identified in the proximal region of the promoter −124/−103 bp: 5′-GCCAGGAAgGCCCCACTGAGC-3′ (63). Preferential binding of p50 homodimers to this element specifically recruits p300, a coactivator with histone acetyltransferase activity, to initiate IL-10 gene transcription. The candidate for a putative p50 homodimer-binding site in the human IL-10 proximal promoter region can also be found in region of −110/−100: 5′-GaGGCcTCCC-3′. Given the controversial findings regarding a role for NF-κB in IL-10 gene expression (64), additional studies on individual Rel family members and IL-10 induction are clearly warranted.
Pbx1
Pre-B-cell leukemia transcription factor (Pbx) homeoproteins are cofactors for the Hox family of homeodomain-containing transcription factors that contribute to body pattern by promoting the expression of specific genetic programs. Pbx1 is required for hematopoiesis as well as for multiple developmental processes such as skeletal patterning and organogenesis (65). A binding site for Pbx1 in the human IL-10 promoter region has been located to the nucleotide of −163/−155 bp (−106/−98 bp relative to transcription initiation site): 5′-TTGATTGTGT-3′ (66). The consensus sequence for a Pbx-binding element is 5′-TGATTTAT-3′. Ma and colleagues (66) recently showed that Pbx1 together with another Hox cofactor, Pbx-regulating protein 1 (Prep-1), mediates the transcription of IL-10 genes in the macrophages following their interaction with apoptotic cells.
In summary, there are a variety of transcription factors that exhibit the potential to bind to the IL-10 promoter. This understanding explains, in part, the variety of different stimuli that can induce IL-10 production from immune cells. If we were asked to prioritize the importance of each transcription factor for IL-10 production, Sp1 and STAT3 would probably top the list. Some of the other transcription factors may be stimuli specific. For example, the stimulation of IL-10 production by cAMP would logically depend more on CREB. The contribution of the p65 subunit of NF-κB remains controversial and may be cell specific. Therefore, there certainly exist the potential to inhibit the production of inflammatory genes by targeting p65 without preventing IL-10 production in many cells.
Epigenetic modifications of IL-10 gene locus
The influence of epigenetics on IL-10 gene expression has been studied in a variety of different cells, and in all of these cells, epigenetic mechanisms appear to play significant roles in IL-10 gene regulation. However, in non-dividing cells, such as macrophages and DCs, these mechanisms confer transient regulation of gene expression, whereas in committed T cells, epigenetic mechanisms may stabilize gene expression and maintain it over several generations of daughter cells.
Nucleosome-poor regions are generally DNase I hypersensitive (HS) sites, which are often located at the regions close to protein-bound regulatory elements. These nucleosome-poor regions can be enhancers, locus control regions, matrix attachment regions, or insulator/boundary elements. Naive resting T cells exhibit essentially no HS sites in the neighborhood of the IL-10 gene (53). In Th1 and Th2 cells, a constitutive HS site (HS III) is found in a region immediately 3′ (downstream) of the last exon of the IL-10 gene. A HS site in the proximal region of IL-10 promoter is found only in Th2 cells (67). A phorbol myristate acetate (PMA)/ionomycin-inducible HS site common to both Th1 and Th2 cells has been located in a region further 3′ (downstream) of HS III. Two PMA/ionomycin-inducible HS sites specific to Th1 cells are located within intron 3 (HS I) and intron 4 (HS II). In IL-10-producing regulatory T cells, three HS sites have been identified upon activation. There are HSS−0.12 that is located at 0.12 kb of 5′-flanking region, HSS+1.65, and HSS+2.98, which are located respectively at 1 65 kb and 2.98 kb of 3′-downstream region of the IL-10 gene. In Th2 cells, there are HSS−0.12, HSS−2.0, and HSS+2.88. In macrophages, HSS+1.65, HSS−0.12, and HSS−2.0 as well as HSS−4.5 are found. HSS−4.5 is specifically present in LPS-stimulated macrophages and contains an NF-κB-binding motif (61). It appears that HSS−0.12 is present among all IL-10-producing cells examined. All of the HS sites have an open chromatin configuration, as characterized by constitutive hyperacetylation at histones H4 and H3.
In IL-10-producing T cells, GATA-3 expression is correlated with IL-10 production. GATA-3 appears to function as a promoter to enhance histone hyperacetylation around the HS sites, as suggesting by the observation that ectopic expression of GATA-3 does not increase IL-10 promoter activity in reporter assays. GATA-3 is specifically recruited to the sites of −86/−61 bp that are in the vicinity region of HSS−0.12. HSS+6.4 is another site specific for GATA-3 binding to remodel chromatin. Of note, HSS16.4 also contains AP-1 site for Jun protein (51). GATA-3 acts as a direct regulator to instruct chromatin remodeling at IL-10 gene locus, which is independent of IL-4.
In macrophages, histone phosphorylation rather than acetylation appears to be an important element in the control of IL-10 gene expression (34). Phosphorylation of serine 10 of histone H3 (H3S10) correlates temporally with the initiation of IL-10 gene transcription in macrophages. H3S10 phosphorylation is downstream of ERK activation, and multiple ERK activators can enhance macrophage IL-10 production (33). Macrophages stimulated with TLR ligands in the presence of FcγR cross-linking exhibit a rapid activation of ERK, which results in a dynamic enhancement of H3S10 phosphorylation at nucleosomes associated with the IL-10 promoter. H3S10 phosphorylation is transient and vanishes within 60min after stimulation. These changes in histone phosphorylation promote the rapid recruitment of Sp1 to the IL-10 promoter, which is followed by minor changes in histone H3 acetylation. It appears as though H3S10 phosphorylation in macrophages is sufficient for transcriptional activation of the IL-10 gene, because IL-10 mRNA levels are not dramatically affected by inhibitors of histone deacetylase. We have correlated histone phosphorylation with DNase sensitivity and showed that the IL-10 promoter becomes much more accessible following H3S10 phosphorylation.
Post-transcriptional regulation of IL-10
Similar to the majority of cytokine genes, IL-10 mRNA contains a long segment of 3′-untranslated region (UTR) (702 bp for the murine IL-10 and 1033 bp for the human one) that plays a role in mRNA instability (68). IL-10 mRNA becomes more stable after the 3′-UTR is deleted, extending the half-life from 1 h to >12 h. IL-10 mRNA contains class II AU-rich elements (AREs) in its 3′-UTR, which are classified based on the number and spacing of the canonical AUUUA pentamer. Class II AREs have a cluster of 4–7 overlapping pentamers within the U-rich context, typically an extended UUAUUUAUU nonamer. IL-10 3′-UTR contains six AUUUA pentamers, four of which are surrounded by U residues to form octameric motifs that are very similar to the nonamer. Tristetraprolin (TTP), an RNA-binding protein, is known to bind nonamers and trigger the rapid degradation of mRNA. IL-10 mRNA decay rate is reduced in the primary macrophages obtained from TTP knockout mice relative to control littermates (69). The half-life for IL-10 mRNA in these mice is increased from 1.5 to 3 h. Stability of mRNA is not the only mechanism for post-transcriptional regulation of IL-10. IL-10 production can also be regulated at the translational level. In LPS-stimulated macrophages, activation of adenosine receptor induced IL-10 production without affecting IL-10 gene transcription and mRNA stability. Instead, adenosine-mediated signals specifically relieve the translational repressive effects of the IL-10 mRNA 3′-UTR that contains the GUAUUUAUU nonamer (70). This post-transcriptional mechanism has been reported only in macrophages. Another IL-10-dependent mRNA destabilizing factor has also been identified in monocytes, and this may be a mechanism to rapidly terminate IL-10 production in these cells (71, 72).
Polymorphisms in the IL-10 promoter
There have been several studies showing strong correlations between disease prevalence and polymorphisms in the IL-10 gene. However, definitive ‘cause-and-effect’ studies to demonstrate how these polymorphisms contribute to pathology are less well developed. Polymorphisms in the IL-10 gene promoter have been associated with diseases, such as graft-versus-host disease (GVHD) and survival after hematopoietic cell transplantation (73). Five SNPs tagging the promoter haplotypes of the IL-10 gene have been widely studied. They are −3575, −2763, −1082, −819, and −592, which are defined relative to their positions in the IL-10 gene sequence upstream of the transcription initiation site. The lowest incidence of severe acute GVHD and remission is associated with homozygosity for the haplotype of T-C-A-T-A at (−3575)-( − 2763)-( − 1082)-( − 819)-( − 592), possibly due to the overproduction of IL-10, thereby inducing tolerance in donor T cells to foreign antigen in the recipients. Polymorphisms in the IL-10 promoter that have been linked to more severe forms of asthma have been associated with reduced levels of IL-10 production (74). The molecular effects of these SNPs in the IL-10 gene promoter region, however, are not precisely understood. The study of the SNP of A to C at −571 (a site with Sp1-binding motif) suggests that changes in IL-10 promoter activity may be one of the mechanisms underlying the alteration of IL-10 production (37). In Drosophila SL2 cells, which are devoid of Sp proteins, Sp1 functions as a positive regulator when a human IL-10 gene reporter construct with A allele is transfected, but Sp1 becomes a repressor when a C allele reporter construct is transfected. The A–C change converts the Sp1 element into a repressor, through the interaction of Sp proteins with Ets family proteins. Ets-1 has been shown to repress IL-10 gene expression in T cells (75, 76). The molecular mechanisms underlying this observation require further study.
IL-10 and the potential for therapeutics
Numerous studies using mice that are genetically deficient in IL-10 have illustrated the importance of this cytokine in limiting autoimmune pathologies. Mice lacking IL-10 or treated with blocking anti-receptor antibodies succumb to what would normally be sublethal doses of LPS (77). Furthermore, some bacterial and parasitic infections that are normally self-contained result in lethal autoimmune mortality in IL-10-deficient mice (78, 79). Virtually every murine model of autoimmunity, including experimental autoimmune encephalitis, rheumatoid arthritis, and inflammatory bowel disease, disease is dramatically exacerbated in mice lacking IL-10. These studies illustrate the power of IL-10 in limiting an over-exuberant immune response and preventing autoimmunity. Other mouse models in which IL-10 is overexpressed have illustrated the immunosuppressive power of this cytokine. Bacteria that are cleared in a normal host can cause lethal infections in mice in which IL-10 is genetically overexpressed. Viruses encode IL-10 homologs to suppress immunity and persist in an otherwise immunocompetent host, and there is now evidence that tumors and tumor-associated macrophages produce IL-10 to contribute to the immunosuppressive environment of the tumor. Thus, manipulating host IL-10 responses holds great promise, but this is a two-edged sword that is not without considerable risk. Blocking IL-10 introduces the risk of autoimmunity, whereas inducing IL-10 overexpression can lead to immunosuppression.
rIL-10 as a therapeutic
Human clinical trials on the use of rIL-10 to treat inflammatory diseases were initiated more than a decade ago. Some of the earliest trials were done in patients with psoriasis, and the initial studies performed on small numbers of patients were quite encouraging. Unlike many other recombinant cytokines, IL-10 was relatively well tolerated, and the subcutaneous injection of IL-10 beneath psoriatic lesions had significant clinical benefits. In one small study involving 10 patients, there were significant decreases in the size of psoriatic areas as well as the severity index following rIL-10 administration. In subsequent studies on larger numbers of patients with more severe forms of psoriasis, however, the administration of rIL-10 resulted in only temporary clinical improvement. A similar experience was observed in patients with Crohn’s disease (CD) who were treated with rIL-10. The Crohn’s Disease Cooperative Group concluded that the subcutaneous administration of rIL-10 (8 µg/kg) to adults with CD was well tolerated and resulted in ‘a tendency toward clinical improvement, but not remission’ (80). These trends toward modest clinical improvements were largely repeated in subsequent studies in both CD and rheumatoid arthritis. Furthermore, high doses of IL-10 were actually associated with enhanced inflammatory responses (31). There was some speculation that the subcutaneous administration of rIL-10 was at least partially responsible for the modest improvements observed in some of the trials, because IL-10 was not delivered locally to the area of inflammation. Although it should be emphasized that in the psoriasis studies described above, the rIL-10 was administered directly beneath the psoriatic lesions of the skin. This speculation was perhaps based on earlier studies in which the delivery of IL-10 to murine central nervous system by an adenovirus vector prevented experimental autoimmune encephalitis, whereas systemic administration was ineffective (81). The local delivery of IL-10 to sites of inflammation remains a major challenge, and exciting new experimental approaches are being undertaken in this area. Encouraging studies using Lactococcus lactis to deliver IL-10 to the gastrointestinal tract are beginning to show some promise. Early studies in mice resulted in a 50% decrease in experimental colitis (82), and in Phase I human clinical trials the genetically engineered bacteria were well tolerated in a small group of patients (83). Similar approaches with other bacterial delivery systems are being undertaken. Recently, a rIL-10 gene has been fused to the Type III secretion apparatus of Shigella, and the secreted cytokine appeared to inhibit bacterially induced inflammatory responses (84). Other potentially interesting new approaches have included the delivery of biologicals to specific areas of the gastrointestinal tract via the oral administration of gelatin nanoparticles. The IL-10 gene has been delivered to the gastrointestinal tract using this technique. These nanoparticles preferentially accumulate in the large intestine where the IL-10 gene expression could be detected by reverse transcriptase polymerase chain reaction. In a murine acute colitis model, this approach resulted in the restoration of colon length, suppression of inflammatory responses, and increased body weight (85).
Despite what must be considered modest clinical effects of rIL-10, there remain reasons to be optimistic about the prospects for IL-10 therapy. The local delivery of IL-10 is an area that continues to grow, and the use of rIL-10 in combination with other cytokines, growth factors, or therapeutics continues to hold potential. Finally, our understanding of IL-10 gene expression may allow us to manipulate specific immune cells to ‘program’ them to home to lesions and preferentially produce IL-10 rather than inflammatory cytokines.
Antibodies to IL-10 and its receptors
While rIL-10 holds some promise as an anti-inflammatory therapeutic, substantially more progress has been made in blocking the effects of IL-10 using antibodies to the cytokine or its receptor. These studies have been undertaken to enhance immune responses to infection and improve vaccines. Thus, the prevention of immune suppression or the reversal of immune biasing by transiently blocking IL-10 is an important emerging area of study.
One of the major areas of IL-10 intervention has been in the area of anti-viral responses and vaccinations against viral infections. One of the defining characteristics of persistent viral infections is the lack of functional anti-viral effector T cells. One way that this can occur is via the production of IL-10 to suppress cellular immune responses. It has long been appreciated that viruses can encode viral homologs of IL-10, which can bind to mammalian receptors for IL-10 and suppress immune responses to allow viral persistence. Other viruses appear to preferentially induce host IL-10 production to the same effect. In persistent lymphocytic choriomeningitis virus (LCMV) infection, for example, there is a pronounced increase in murine IL-10 levels during infection, and blockade of the IL-10 receptor resulted in a rapid resolution of infection (86). Blocking IL-10 activity may also improve anti-viral vaccine strategies. In recent studies using a DNA vaccination approach to LCMV, the blocking of IL-10 improved vaccine responses and provided protection against subsequent infection (87). New combination approaches to enhance immunity and enable therapeutic vaccination against intracellular pathogens may include the blockade of IL-10 and other immune-inhibitory molecules, such as programmed death-1 (PD-1) (87).
The therapeutic manipulation of IL-10 is not restricted to enhancing anti-viral responses. The addition of antibodies to the IL-10R resulted in enhanced murine immune responses to Mycobacterium avian (88) and importantly improved the effectiveness of chemotherapy when added late in a chronic infection (89). This protective effect may be due to the specific enhancement of Th1 responses following IL-10R blockade. Early studies demonstrated that immunization of mice with a nominal protein antigen, ovalbumin, in the presence of antibody to block the IL-10R gave rise to Th1 responses to that antigen (90). Conversely, we subsequently demonstrated that overexpressing IL-10 in APCs can preferentially induce Th2 responses (91).
IL-10 has been associated with tumor progression, and studies to block IL-10 in cancer continue to show great promise. In many different types of cancers, tumor IL-10 levels correlate with disease severity (92). Because IL-10 works primarily at the level of APCs, it is perhaps not surprising that tumor-associated DCs and macrophages exhibit profound phenotypic and functional alterations. Tumor-associated DCs are defective in antigen presentation and fail to activate cytotoxic T cells (93). Although there are undoubtedly many tumor-derived factors that contribute to DC dysfunctionality, the high levels of IL-10 derived from both the tumor and host cells is emerging as one of the most important immunosuppressive cytokines in cancer. Tumor-associated macrophages, for example, produce minimal amounts of the immunostimulatory cytokines IL-12 (92) and TNF (Fig. 4), but they constitutively produce relatively high levels of IL-10 (Fig. 4). Regulatory T cells producing IL-10 have also been isolated from tumors (94). IL-10 can prevent DC maturation, and it can inhibit DC differentiation from mononuclear cell precursors (95). Blocking the effects of IL-10 in many cases can reverse DC dysfunction and restore immune responsiveness. In vitro studies using small interfering RNA to silence IL-10 in human DCs have resulted in enhanced IL-12 production and improved cytotoxic T-cell responses to tumor epitopes (96).
Fig. 4. The production of cytokines from tumor-associated macrophages.
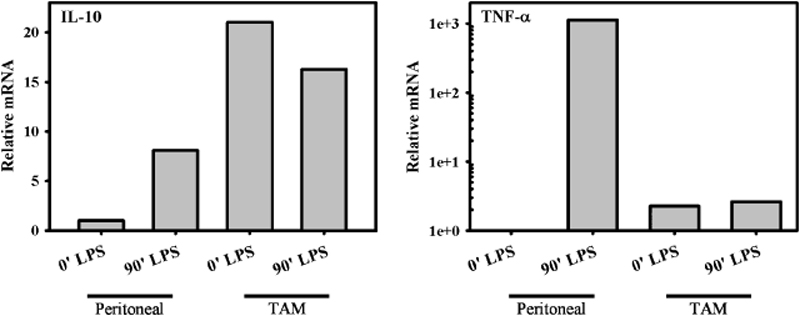
The relative levels of mRNA for IL-10 (left) and TNF (right) from tumor-associated macrophages were determined by reverse transcriptase polymerase chain reaction and were compared with peritoneal macrophages from normal non-tumor-bearing mice. mRNA levels were determined 2 h after in vitro stimulation with LPS and are compared with unstimulated cells (0′).
The basic studies on the role of IL-10 and immunosuppression described above have given rise to some exciting new combination therapies in which tumors are treated with cytotoxic therapeutics in combination with antibodies to block IL-10. The rationale behind these studies is that the antigens released from dying tumor cells can be processed and presented by DCs if their functionality is restored by blocking IL-10. In fact, several groups have suggested that the death of tumor cells may be precisely what induces host IL-10 production and therefore DC unresponsiveness (97). Other combination therapies include not only the addition of antibodies to block the immunosuppressive IL-10R but also immunostimulatory TLR ligands to activate DCs and promote DC maturation. In small animal models, these approaches are beginning to bear fruit (98). Continued studies in this area hold the potential to lead to the long-awaited dream of effective vaccinations against cancer.
Summary
There can be little doubt that IL-10 exerts a profound effect on immune responses. This conclusion is based on a vast number of studies using transgenic mice expressing IL-10 in various cellular compartments, on IL-10-deficient knockout mice, on studies utilizing monoclonal antibodies to IL-10 or its receptor, and on studies utilizing natural or genetically modified viruses that express IL-10-like molecules. These observations have given rise to the hope that the reliable manipulation of immune responses by controlling IL-10 levels may some day become a reality. There can be no doubt, however, that manipulating this cytokine carries a high degree of risk due to the fine balance between immunopathology and immuno-suppression. Further-more, the myriad activities of IL-10 that include both immunosuppressive as well as immunostimulatory activities suggest that optimal doses of IL-10 must be defined before we can achieve the desired effects. Our understanding of the regulation of IL-10 in various immune cells paves the way to augment or suppress IL-10 without affecting other immune activities. Studies on combination therapies to temporarily suppress IL-10 to augment immune responses hold particular promise. The hope is that one day we can augment immune responses or prevent immunopathology by making small alterations in the levels of IL-10 production or in the cellular location where it is produced.
References
- 1.Commins S, Steinke JW, Borish L. The extended IL-10 superfamily: IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29. J Allergy Clin Immunol. 2008;121:1108–1111. doi: 10.1016/j.jaci.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 2.Donnelly RP, Sheikh F, Kotenko SV, Dickensheets H. The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J Leukoc Biol. 2004;76:314–321. doi: 10.1189/jlb.0204117. [DOI] [PubMed] [Google Scholar]
- 3.Sabat R, Wallace E, Endesfelder S, Wolk K. IL-19 and IL-20: two novel cytokines with importance in inflammatory diseases. Expert Opin Ther Targets. 2007;11:601–612. doi: 10.1517/14728222.11.5.601. [DOI] [PubMed] [Google Scholar]
- 4.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–2095. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore KW, Vieira P, Fiorentino DF, Trounstine ML, Khan TA, Mosmann TR. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI. Science. 1990;248:1230–1234. doi: 10.1126/science.2161559. [DOI] [PubMed] [Google Scholar]
- 6.O’arra A, Vieira P. T(H)1 cells control themselves by producing interleukin-10. Nat Rev Immunol. 2007;7:425–428. doi: 10.1038/nri2097. [DOI] [PubMed] [Google Scholar]
- 7.Fillatreau S, Gray D, Anderton SM. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol. 2008;8:391–397. doi: 10.1038/nri2315. [DOI] [PubMed] [Google Scholar]
- 8.Ryan JJ, et al. Mast cell homeostasis: a fundamental aspect of allergic disease. Crit Rev Immunol. 2007;27:15–32. doi: 10.1615/critrevimmunol.v27.i1.20. [DOI] [PubMed] [Google Scholar]
- 9.Moore KW, de Waal MR, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 10.Williams LM, Ricchetti G, Sarma U, Smallie T, Foxwell BM. Interleukin-10 suppression of myeloid cell activation – a continuing puzzle. Immunology. 2004;113:281–292. doi: 10.1111/j.1365-2567.2004.01988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamanaka M, et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity. 2006;25:941–952. doi: 10.1016/j.immuni.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 12.Allan SE, et al. D41 T-regulatory cells: toward therapy for human diseases. Immunol Rev. 2008;223:391–421. doi: 10.1111/j.1600-065X.2008.00634.x. [DOI] [PubMed] [Google Scholar]
- 13.Jankovic D, et al. Conventional T-bet(+)Foxp3(−) Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204:273–283. doi: 10.1084/jem.20062175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4(+)CD25(−)Foxp3(−) Th1 cells are the source of IL-10-mediated immune suppression. J Exp Med. 2007;204:285–297. doi: 10.1084/jem.20061886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boonstra A, et al. Macrophages and myeloid dendritic cells, but not plasmacytoid dendritic cells, produce IL-10 in response to MyD88- and TRIF-dependent TLR signals, and TLR-independent signals. J Immunol. 2006;177:7551–7558. doi: 10.4049/jimmunol.177.11.7551. [DOI] [PubMed] [Google Scholar]
- 16.Dillon S, et al. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest. 2006;116:916–928. doi: 10.1172/JCI27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grimbaldeston MA, Nakae S, Kalesnikoff J, Tsai M, Galli SJ. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol. 2007;8:1095–1104. doi: 10.1038/ni1503. [DOI] [PubMed] [Google Scholar]
- 18.Donnelly RP, Dickensheets H, Finbloom DS. The interleukin-10 signal transduction pathway and regulation of gene expression in mononuclear phagocytes. J Interferon Cytokine Res. 1999;19:563–573. doi: 10.1089/107999099313695. [DOI] [PubMed] [Google Scholar]
- 19.Staples KJ, et al. IL-10 induces IL-10 in primary human monocyte-derived macrophages via the transcription factor Stat3. J Immunol. 2007;178:4779–4785. doi: 10.4049/jimmunol.178.8.4779. [DOI] [PubMed] [Google Scholar]
- 20.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 21.O’Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 23.Schottelius AJ, Mayo MW, Sartor RB, Baldwin AS., Jr Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J Biol Chem. 1999;274:31868–31874. doi: 10.1074/jbc.274.45.31868. [DOI] [PubMed] [Google Scholar]
- 24.Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: a role for p50. Clin Exp Immunol. 2004;135:64–73. doi: 10.1111/j.1365-2249.2004.02342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denys A, et al. Evidence for a dual mechanism for IL-10 suppression of TNF-alpha production that does not involve inhibition of p38 mitogen-activated protein kinase or NF-kappa B in primary human macrophages. J Immunol. 2002;168:4837–4845. doi: 10.4049/jimmunol.168.10.4837. [DOI] [PubMed] [Google Scholar]
- 26.Akdis CA, Blaser K. Mechanisms of interleukin- 10-mediated immune suppression. Immunology. 2001;103:131–136. doi: 10.1046/j.1365-2567.2001.01235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai G, Kastelein RA, Hunter CA. IL-10 enhances NK cell proliferation, cytotoxicity and production of IFN-gamma when combined with IL-18. Eur J Immunol. 1999;29:2658–2665. doi: 10.1002/(SICI)1521-4141(199909)29:09<2658::AID-IMMU2658>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 28.Santin AD, et al. Interleukin-10 increases Th1 cytokine production and cytotoxic potential in human papillomavirus-specific CD8(+) cytotoxic T lymphocytes. J Virol. 2000;74:4729–4737. doi: 10.1128/jvi.74.10.4729-4737.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rowbottom AW, Lepper MA, Garland RJ, Cox CV, Corley EG. Interleukin-10-induced CD8 cell proliferation. Immunology. 1999;98:80–89. doi: 10.1046/j.1365-2567.1999.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Groux H, Bigler M, de Vries JE, Roncarolo MG. Inhibitory and stimulatory effects of IL-10 on human CD8+ T cells. J Immunol. 1998;160:3188–3193. [PubMed] [Google Scholar]
- 31.Lauw FN, Pajkrt D, Hack CE, Kurimoto M, van Deventer SJ, van der PT. Proinflammatory effects of IL-10 during human endotoxemia. J Immunol. 2000;165:2783–2789. doi: 10.4049/jimmunol.165.5.2783. [DOI] [PubMed] [Google Scholar]
- 32.Brightbill HD, Plevy SE, Modlin RL, Smale ST. A prominent role for Sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J Immunol. 2000;164:1940–1951. doi: 10.4049/jimmunol.164.4.1940. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, Edwards JP, Mosser DM. Dynamic and transient remodeling of the macrophage IL-10 promoter during transcription. J Immunol. 2006;177:1282–1288. doi: 10.4049/jimmunol.177.2.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lucas M, Zhang X, Prasanna V, Mosser DM. ERK activation following macrophage FcgammaR ligation leads to chromatin modifications at the IL-10 locus. J Immunol. 2005;175:469–477. doi: 10.4049/jimmunol.175.1.469. [DOI] [PubMed] [Google Scholar]
- 35.Ma W, et al. The p38 mitogen-activated kinase pathway regulates the human interleukin- 10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J Biol Chem. 2001;276:13664–13674. doi: 10.1074/jbc.M011157200. [DOI] [PubMed] [Google Scholar]
- 36.Norkina O, et al. Acute alcohol intake induces SOCS1 and SOCS3 and inhibits cytokine-induced STAT1 and STAT3 signaling in human monocytes. Alcohol Clin Exp Res. 2008;32:1565–1573. doi: 10.1111/j.1530-0277.2008.00726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinke JW, Barekzi E, Hagman J, Borish L. Functional analysis of −571 IL-10 promoter polymorphism reveals a repressor element controlled by sp1. J Immunol. 2004;173:3215–3222. doi: 10.4049/jimmunol.173.5.3215. [DOI] [PubMed] [Google Scholar]
- 38.Ziegler-Heitbrock L, Lotzerich M, Schaefer A, Werner T, Frankenberger M, Benkhart E. IFN-alpha induces the human IL-10 gene by recruiting both IFN regulatory factor 1 and Stat3. J Immunol. 2003;171:285–290. doi: 10.4049/jimmunol.171.1.285. [DOI] [PubMed] [Google Scholar]
- 39.Unterberger C, et al. Role of STAT3 in gluco-corticoid-induced expression of the human IL-10 gene. Mol Immunol. 2008;45:3230–3237. doi: 10.1016/j.molimm.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 40.Benkhart EM, Siedlar M, Wedel A, Werner T, Ziegler-Heitbrock HW. Role of Stat3 in lipopolysaccharide-induced IL-10 gene expression. J Immunol. 2000;165:1612–1617. doi: 10.4049/jimmunol.165.3.1612. [DOI] [PubMed] [Google Scholar]
- 41.Grant LR, et al. Stat4-dependent, T-bet-independent regulation of IL-10 in NK cells. Genes Immun. 2008;9:316–327. doi: 10.1038/gene.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.VanDeusen JB, et al. STAT-1-mediated repression of monocyte interleukin-10 gene expression in vivo. Eur J Immunol. 2006;36:623–630. doi: 10.1002/eji.200535241. [DOI] [PubMed] [Google Scholar]
- 43.Stumhofer JS, et al. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol. 2007;8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- 44.Brenner S, Prosch S, Schenke-Layland K, Riese U, Gausmann U, Platzer C. cAMP-induced Interleukin-10 promoter activation depends on CCAAT/enhancer-binding protein expression and monocytic differentiation. J Biol Chem. 2003;278:5597–5604. doi: 10.1074/jbc.M207448200. [DOI] [PubMed] [Google Scholar]
- 45.Csoka B, et al. A2A adenosine receptors and C/EBPbeta are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood. 2007;110:2685–2695. doi: 10.1182/blood-2007-01-065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu YW, Tseng HP, Chen LC, Chen BK, Chang WC. Functional cooperation of simian virus 40 promoter factor 1 and CCAAT/ enhancer-binding protein beta and delta in lipopolysaccharide-induced gene activation of IL-10 in mouse macrophages. J Immunol. 2003;171:821–828. doi: 10.4049/jimmunol.171.2.821. [DOI] [PubMed] [Google Scholar]
- 47.Cuesta N, Salkowski CA, Thomas KE, Vogel SN. Regulation of lipopolysaccharide sensitivity by IFN regulatory factor-2. J Immunol. 2003;170:5739–5747. doi: 10.4049/jimmunol.170.11.5739. [DOI] [PubMed] [Google Scholar]
- 48.Samanta M, Iwakiri D, Takada K. Epstein–Barr virus-encoded small RNA induces IL-10 through RIG-I-mediated IRF-3 signaling. Oncogene. 2008;27:4150–4160. doi: 10.1038/onc.2008.75. [DOI] [PubMed] [Google Scholar]
- 49.Kremer KN, Kumar A, Hedin KE. Haplotype-independent costimulation of IL-10 secretion by SDF-1/CXCL12 proceeds via AP-1 binding to the human IL-10 promoter. J Immunol. 2007;178:1581–1588. doi: 10.4049/jimmunol.178.3.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu X, et al. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 51.Wang ZY, Sato H, Kusam S, Sehra S, Toney LM, Dent AL. Regulation of IL-10 gene expression in Th2 cells by Jun proteins. J Immunol. 2005;174:2098–2105. doi: 10.4049/jimmunol.174.4.2098. [DOI] [PubMed] [Google Scholar]
- 52.Rooney JW, Hodge MR, McCaffrey PG, Rao A, Glimcher LH. A common factor regulates both Th1- and Th2-specific cytokine gene expression. EMBO J. 1994;13:625–633. doi: 10.1002/j.1460-2075.1994.tb06300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones EA, Flavell RA. Distal enhancer elements transcribe intergenic RNA in the IL-10 family gene cluster. J Immunol. 2005;175:7437–7446. doi: 10.4049/jimmunol.175.11.7437. [DOI] [PubMed] [Google Scholar]
- 54.Platzer C, Fritsch E, Elsner T, Lehmann MH, Volk HD, Prosch S. Cyclic adenosine mono-phosphate-responsive elements are involved in the transcriptional activation of the human IL-10 gene in monocytic cells. Eur J Immunol. 1999;29:3098–3104. doi: 10.1002/(SICI)1521-4141(199910)29:10<3098::AID-IMMU3098>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 55.Park PH, Huang H, McMullen MR, Bryan K, Nagy LE. Activation of cyclic-AMP response element binding protein contributes to adiponectin-stimulated interleukin-10 expression in raw 264.7 macrophages. J Leukoc Biol. 2008;83:1258–1266. doi: 10.1189/jlb.0907631. [DOI] [PubMed] [Google Scholar]
- 56.Cao S, Liu J, Song L, Ma X. The protooncogene c-Maf is an essential transcription factor for IL-10 gene expression in macrophages. J Immunol. 2005;174:3484–3492. doi: 10.4049/jimmunol.174.6.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim JI, Ho IC, Grusby MJ, Glimcher LH. The transcription factor c-Maf controls the production of interleukin-4 but not other Th2 cytokines. Immunity. 1999;10:745–751. doi: 10.1016/s1074-7613(00)80073-4. [DOI] [PubMed] [Google Scholar]
- 58.Ho IC, Lo D, Glimcher LH. c-Maf promotes T helper cell type 2 (Th2) and attenuates Th1 differentiation by both interleukin 4-dependent and -independent mechanisms. J Exp Med. 1998;188:1859–1866. doi: 10.1084/jem.188.10.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 60.Bondeson J, Browne KA, Brennan FM, Fox-well BM, Feldmann M. Selective regulation of cytokine induction by adenoviral gene transfer of IkappaBalpha into human macrophages: lipopolysaccharide-induced, but not zymosan-induced, proinflammatory cytokines are inhibited, but IL-10 is nuclear factor-kappaB independent. J Immunol. 1999;162:2939–2945. [PubMed] [Google Scholar]
- 61.Saraiva M, et al. Identification of a macrophage-specific chromatin signature in the IL-10 locus. J Immunol. 2005;175:1041–1046. doi: 10.4049/jimmunol.175.2.1041. [DOI] [PubMed] [Google Scholar]
- 62.Chakrabarti A, Sadler AJ, Kar N, Young HA, Silverman RH, Williams BR. Protein kinase R-dependent regulation of interleukin-10 in response to double-stranded RNA. J Biol Chem. 2008;283:25132–25139. doi: 10.1074/jbc.M804770200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cao S, Zhang X, Edwards JP, Mosser DM. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem. 2006;281:26041–26050. doi: 10.1074/jbc.M602222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wessells J, et al. BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J Biol Chem. 2004;279:49995–50003. doi: 10.1074/jbc.M404246200. [DOI] [PubMed] [Google Scholar]
- 65.Neuteboom ST, Murre C. Pbx raises the DNA binding specificity but not the selectivity of antennapedia Hox proteins. Mol Cell Biol. 1997;17:4696–4706. doi: 10.1128/mcb.17.8.4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chung EY, et al. Interleukin-10 expression in macrophages during phagocytosis of apoptotic cells is mediated by homeodomain proteins Pbx1 and Prep-1. Immunity. 2007;27:952–964. doi: 10.1016/j.immuni.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Im SH, Hueber A, Monticelli S, Kang KH, Rao A. Chromatin-level regulation of the IL10 gene in T cells. J Biol Chem. 2004;279:46818–46825. doi: 10.1074/jbc.M401722200. [DOI] [PubMed] [Google Scholar]
- 68.Anderson P. Post-transcriptional control of cytokine production. Nat Immunol. 2008;9:353–359. doi: 10.1038/ni1584. [DOI] [PubMed] [Google Scholar]
- 69.Stoecklin G, et al. Genome-wide analysis identifies interleukin-10 mRNA as target of tristetraprolin. J Biol Chem. 2008;283:11689–11699. doi: 10.1074/jbc.M709657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nemeth ZH, et al. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J Immunol. 2005;175:8260–8270. doi: 10.4049/jimmunol.175.12.8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown CY, Lagnado CA, Vadas MA, Goodall GJ. Differential regulation of the stability of cytokine mRNAs in lipopolysaccharide-activated blood monocytes in response to interleukin-10. J Biol Chem. 1996;271:20108–20112. doi: 10.1074/jbc.271.33.20108. [DOI] [PubMed] [Google Scholar]
- 72.Kishore R, Tebo JM, Kolosov M, Hamilton TA. Cutting edge: clustered AU-rich elements are the target of IL-10-mediated mRNA destabilization in mouse macrophages. J Immunol. 1999;162:2457–2461. [PubMed] [Google Scholar]
- 73.Lin MT, et al. Relation of an interleukin-10 promoter polymorphism to graft-versus-host disease and survival after hematopoietic-cell transplantation. N Engl J Med. 2003;349:2201–2210. doi: 10.1056/NEJMoa022060. [DOI] [PubMed] [Google Scholar]
- 74.Lim S, Crawley E, Woo P, Barnes PJ. Haplotype associated with low interleukin-10 production in patients with severe asthma. Lancet. 1998;352:113. doi: 10.1016/S0140-6736(98)85018-6. [DOI] [PubMed] [Google Scholar]
- 75.Grenningloh R, Kang BY, Ho IC. Ets-1, a functional cofactor of T-bet, is essential for Th1 inflammatory responses. J Exp Med. 2005;201:615–626. doi: 10.1084/jem.20041330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li JC, Lau AS. A role for mitogen-activated protein kinase and Ets-1 in the induction of interleukin-10 transcription by human immunodeficiency virus-1 Tat. Immunology. 2007;121:337–348. doi: 10.1111/j.1365-2567.2007.02580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berg DJ, et al. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartz-man reaction but not endotoxin tolerance. J Clin Invest. 1995;96:2339–2347. doi: 10.1172/JCI118290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hunter CA, et al. IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi. J Immunol. 1997;158:3311–3316. [PubMed] [Google Scholar]
- 79.Gazzinelli RT, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 80.Schreiber S, et al. Crohn’s Disease IL-10 Cooperative Study Group. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Gastroenterology. 2000;119:1461–1472. doi: 10.1053/gast.2000.20196. [DOI] [PubMed] [Google Scholar]
- 81.Cua DJ, Hutchins B, LaFace DM, Stohlman SA, Coffman RL. Central nervous system expression of IL-10 inhibits autoimmune encephalomyelitis. J Immunol. 2001;166:602–608. doi: 10.4049/jimmunol.166.1.602. [DOI] [PubMed] [Google Scholar]
- 82.Steidler L, et al. Treatment of murine colitisby Lactococcus lactis secreting interleukin-10. Science. 2000;289:1352–1355. doi: 10.1126/science.289.5483.1352. [DOI] [PubMed] [Google Scholar]
- 83.Braat H, et al. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin Gastroenterol Hepatol. 2006;4:754–759. doi: 10.1016/j.cgh.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 84.Chamekh M, Phalipon A, Quertainmont R, Salmon I, Sansonetti P, Allaoui A. Delivery of biologically active anti-inflammatory cytokines IL-10 and IL-1ra in vivo by the Shigella type III secretion apparatus. J Immunol. 2008;180:4292–4298. doi: 10.4049/jimmunol.180.6.4292. [DOI] [PubMed] [Google Scholar]
- 85.Bhavsar MD, Amiji MM. Oral IL-10 gene delivery in a microsphere-based formulation for local transfection and therapeutic efficacy in inflammatory bowel disease. Gene Ther. 2008;15:1200–1209. doi: 10.1038/gt.2008.67. [DOI] [PubMed] [Google Scholar]
- 86.Ejrnaes M, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med. 2006;203:2461–2472. doi: 10.1084/jem.20061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brooks DG, Lee AM, Elsaesser H, McGavern DB, Oldstone MB. IL-10 blockade facilitates DNA vaccine-induced T cell responses and enhances clearance of persistent virus infection. J Exp Med. 2008;205:533–541. doi: 10.1084/jem.20071948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roque S, Nobrega C, Appelberg R, Correia-Neves M. IL-10 underlies distinct susceptibility of BALB/c and C57BL/6 mice to Mycobacterium avium infection and influences efficacy of antibiotic therapy. J Immunol. 2007;178:8028–8035. doi: 10.4049/jimmunol.178.12.8028. [DOI] [PubMed] [Google Scholar]
- 89.Silva RA, Pais TF, Appelberg R. Blocking the receptor for IL-10 improves antimycobacterial chemotherapy and vaccination. J Immunol. 2001;167:1535–1541. doi: 10.4049/jimmunol.167.3.1535. [DOI] [PubMed] [Google Scholar]
- 90.Castro AG, et al. Anti-interleukin 10 receptor monoclonal antibody is an adjuvant for T helper cell type 1 responses to soluble antigen only in the presence of lipopolysaccharide. J Exp Med. 2000;192:1529–1534. doi: 10.1084/jem.192.10.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anderson CF, Mosser DM. Cutting edge: biasing immune responses by directing antigen to macrophage Fc gamma receptors. J Immunol. 2002;168:3697–3701. doi: 10.4049/jimmunol.168.8.3697. [DOI] [PubMed] [Google Scholar]
- 92.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 93.Matsuda M, et al. Interleukin 10 pretreatment protects target cells from tumor- and allo-specific cytotoxic T cells and downregulates HLA class I expression. J Exp Med. 1994;180:2371–2376. doi: 10.1084/jem.180.6.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 95.Cirone M, et al. Suppression of dendritic cell differentiation through cytokines released by primary effusion lymphoma cells. Immunol Lett. 2008;120:37–41. doi: 10.1016/j.imlet.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 96.Chhabra A, Chakraborty NG, Mukherji B. Silencing of endogenous IL-10 in human dendritic cells leads to the generation of an improved CTL response against human melanoma associated antigenic epitope MART-1 27–35. Clin Immunol. 2008;126:251–259. doi: 10.1016/j.clim.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hatfield P, et al. Optimization of dendritic cell loading with tumor cell lysates for cancer immunotherapy. J Immunother. 2008;31:620–632. doi: 10.1097/CJI.0b013e31818213df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vicari AP, et al. Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J Exp Med. 2002;196:541–549. doi: 10.1084/jem.20020732. [DOI] [PMC free article] [PubMed] [Google Scholar]