

Abstract
BSND encodes barttin, an accessory subunit of renal and inner ear chloride channels. To date, all mutations of BSND have been shown to cause Bartter syndrome type IV, characterized by significant renal abnormalities and deafness. We identified a BSND mutation (p.I12T) in four kindreds segregating nonsyndromic deafness linked to a 4.04-cM interval on chromosome 1p32.3. The functional consequences of p.I12T differ from BSND mutations that cause renal failure and deafness in Bartter syndrome type IV. p.I12T leaves chloride channel function unaffected and only interferes with chaperone function of barttin in intracellular trafficking. This study provides functional data implicating a hypomorphic allele of BSND as a cause of apparent nonsyndromic deafness. We demonstrate that BSND mutations with different functional consequences are the basis for either syndromic or nonsyndromic deafness.
Main Text
Antenatal Bartter syndrome comprises a genetically and phenotypically heterogeneous group of salt-losing nephropathies.1,2 Affected individuals with Bartter syndrome type IV (MIM 602522) suffer from increased urinary chloride excretion, elevated plasma renin activity, hyperaldosteronism, and hyperprostaglandinuria as well as sensorineural deafness. Typically, this disorder manifests in the second gestational trimester with the development of prenatal renal salt wasting, polyhydramnios, premature delivery, nephrocalcinosis, and life-threatening volume depletion in the neonate.3–5
We ascertained Pakistan families segregating nonsyndromic deafness with IRB approval at the National Institutes of Health and at the University of the Punjab. We mapped DFNB73 to chromosome 1p32 in a genome-wide linkage analysis of families PKDF393 and PKDF606 by using 388 fluorescently labeled microsatellite markers (STRs) spaced an average of 10 cM apart (ABI Prism Linkage Mapping Set, v2.5 Applied Biosystems). Subsequently, we screened ∼700 Pakistani families by using STRs in the DFNB73 linkage interval and found two additional families segregating deafness (PKDF067 and PKDF815) linked to DFNB73. The four DFNB73-linked families were further genotyped with STR markers in the 1p32 region (Figures 1 and 2). Meiotic breakpoint information defined an interval of 4.04 cM containing 26 predicted and annotated genes, and among them was BSND (MIM #606412; NM_057176.2) encoding barttin.6 We PCR-amplified and sequenced the four exons of BSND from affected members of the four DFNB73 families that were analyzed in this study. Three of these families, PKDF393, PKDF606, and PKDF067, were found to be segregating the c.35T>C allele of BSND resulting in a substitution of threonine for a highly conserved isoleucine (p.I12T) (Figure 2B). In a fourth family, PKDF815, 25 affected members enrolled in this study. Twenty-two affected individuals are homozygous for the c.35T>C (p.I12T) mutation, and three (VI:15, VI:16, and VI:17) are compound heterozygotes [c.35T>C (p.I12T)/c.10G>T (p.E4X)] (Figure 1B). We did not detect carriers of either mutation in 384 chromosomes from Pakistani hearing control individuals.
Figure 1.
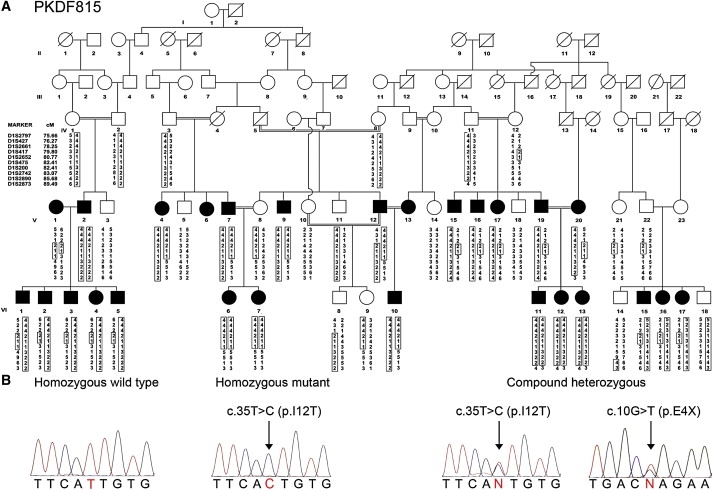
Mutations of BSND Result in Nonsyndromic and Syndromic Hearing Loss in Family PKDF815
(A) Affected individuals V:15-17, V:19, and VI:2-5 have distal and proximal breakpoints at D1S2661 and D1S475, respectively, defining the smallest linkage interval to ∼4 cM. Individuals VI:15-17 are segregating prelingual profound deafness along with elevated renin and nephrocalcinosis (Table 1).
(B) Electropherograms of amplimers from genomic DNA templates illustrate homozygosity for the c.35T>C mutant allele among affected family members segregating isolated hearing loss. Affected family members segregating deafness along with nephrocalcinosis and elevated renin are compound heterozygous for c.35T>C and c.10G>T.
Figure 2.
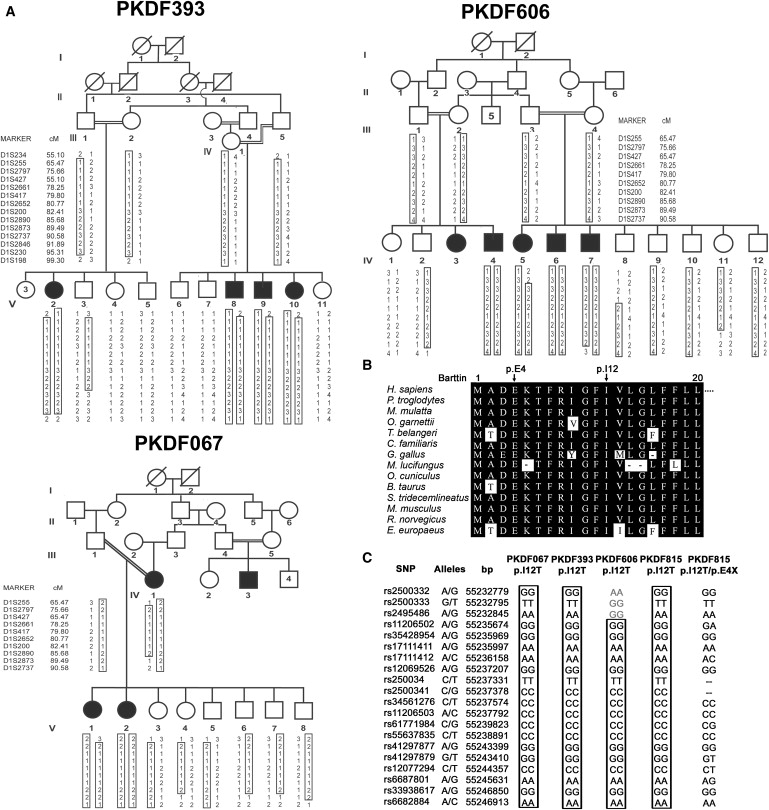
A Highly Conserved Isoleucine Residue of Barttin when Mutated Causes Nonsyndromic Hearing Loss, DFNB73
(A) Pedigrees of three Pakistani families cosegregating nonsyndromic hearing loss with genetic markers linked to DFNB73. Affected members of all three pedigrees are homozygous for the p.I12T mutant allele of BSND. The unaffected members are homozygous or heterozygous for the wild-type allele. All obligate carriers are heterozygous.
(B) Amino acid sequence alignment of barttin orthologs for the amino terminus. Identical residues are colored in black. The isoleucine residue at the 12th position is conserved among a variety of species.
(C) Haplotype analysis on chromosome 1p32.3 in affected members of the four families (PKDF067, PKDF393, PKDF606, and PKDF815). For each SNP, the distance on the physical map is provided (UCSC genome browser, March 2006 assembly). The p.I12 allele resides between SNPs rs2500341 and rs34561276. Homozygotes for p.I12T share a common haplotype. Further SNP genotyping of the affected members of the four families that were homozygous for p.I12T confirmed that these families carry a common ancestral haplotype in the region surrounding the mutation, whereas the p.E4X mutation arose on a different haplotype.
We obtained a detailed clinical history for each affected individual to determine the neonatal course, any developmental delays, signs of polyuria, polydipsia, and salt craving. For all affected individuals, we performed a physical examination to evaluate motor skills, body weight, stature, and morphology and determined serum and urinary chemistry values to define aspects of metabolic and renal function. We performed renal sonography to determine the presence or absence of nephrocalcinosis. On a selected number of affected individuals of the DFNB73 families, we conducted an audiometric evaluation by measuring the threshold of hearing at 250–8000 Hz for pure-tone air conduction and bone conduction.
All affected members of families PKDF393, PKDF606, and PKDF067 are deaf, but did not show other signs or symptoms of Bartter syndrome type IV. They were born from full-term pregnancies with reportedly normal birth weight and length. Polyhydramnios was not identified during gestation. Nor have the affected individuals displayed problems with polydipsia, polyuria, nocturnal enuresis, muscle weakness and fatigue, growth or motor retardation, or signs of developmental delay. The compound heterozygous (p.I12T/p.E4X) members of family PKDF815 manifest nephrocalcinosis, elevated renin levels, and electrolyte levels, suggesting borderline, not clinically significant metabolic alkalosis and hypokalemia (Table 1, and Table S1 available online). Homozygous p.I12T individuals display elevated renin levels and hypocalciuria, but neither metabolic alkalosis nor nephrocalcinosis.
Table 1.
Clinical and Biochemical Features of Wild-Type, Carriers, and Compound Heterozygotes of Family PKDF815
| Features | Reference Ranges | Family Members |
||||||
|---|---|---|---|---|---|---|---|---|
| V:22 | V:23 | VI:14 | VI:15 | VI:16 | VI:17 | VI:18 | ||
| Genotypes | Ta/+ | Xb/+ | +/+ | X/T | X/T | X/T | X/+ | |
| Present age (y) | 53 | 40 | 17 | 20 | 16 | 19 | 24 | |
| Hearing loss | hearing | hearing | hearing | deaf | deaf | deaf | hearing | |
| Other symptoms of BSIVc | no | no | no | no | no | no | no | |
| Sd Na (mmol/L) | 136–148 | 138 | 143 | 143 | 139 | 141 | 141 | 137 |
| S K (mmol/L) | 3.6–5.0 | 4.6 | 4.0 | 4.8 | 3.5 | 3.6 | 3.5 | 4.2 |
| S Cl (mmol/L) | 104–114 | 105 | 107 | 106 | 98 | 102 | 103 | 102 |
| S HCO3 (mmol/ L) | 17.5–27.5 | 27.1 | 31.0 | 27.4 | 29.5 | 29.5 | 31.2 | 26.8 |
| S Mg (mg/dl) | 1.9–2.5 | 2.1 | 2.4 | 2.3 | 2.2 | 2.0 | 2.1 | 2.2 |
| S Ca (mg/dl) | 8.6–10.5 | 8.7 | 8.9 | 9.7 | 9.4 | 10.1 | 9.8 | 9.2 |
| S Creatinine (mg/dl) | 0.85–1.35 | 0.9 | 0.6 | 0.9 | 0.7 | 0.5 | 0.6 | 0.9 |
| Pe Renin (ng/ml/hr) | 0.15–2.33 | 0.199 | 0.29 | 5.22 | 11.64 | 23.42 | 12.74 | 0.83 |
| S Aldosterone (ng/dl) | 1–16;f 4–31 | 3.2 | >1.1f | 12.0 | 11.5 | 12.0 | 4.2 | 5.8 |
| S Osmolality (mosm/Kg) | 275–295 | 289 | 294 | 287 | 283 | 285 | 284 | 284 |
| Ug Na (mmol/L) | 30–150 | 32.9 | 15.5 | 152.2 | 37.5 | 17.3 | 12.5 | 65.3 |
| U K (mmol/L) | 20–67 | 9.44 | 2.5 | 94.66 | 11.95 | 6.33 | 3.22 | 28.41 |
| U Cl (mmol/L) | 46–168 | 37.0 | 14.2 | 176.1 | 49 | 18.2 | 10.3 | 76.8 |
| U Mg (mg/dl) | 2.1 | 0.8 | 22.4 | 3.8 | 2.8 | 1.1 | 6.5 | |
| U Ca (mg/dl) | 2.0 | <2.0 | 13.0 | 6.5 | 1.5 | <2.0 | 7.4 | |
| U osmolality (mosm/Kg) | 50–1400 | 186 | 70 | 891 | 187 | 103 | 53 | 308 |
| Nephrocalcinosis | absent | absent | absent | present | present | present | absent | |
p.I12T.
p.E4X.
Other symptoms of BSIV are polyhydramnion, premature birth, low birth weight, failure to thrive, polyuria, polydipsia, and nocturnal enuresis.
“S” denotes serum.
“P” denotes plasma.
There are two normative values for serum aldosterone: 1–16 ng/dl for blood drawn while the patient is supine and 4–31 ng/dl for blood drawn while the patient is standing.
Urinary (U) values were determined from spot samples.
Audiometric evaluation showed severe hearing loss across all frequencies, without differences among homozygotes for p.I12T and compound heterozygotes (p.I12T/p.E4X) (Figure S1). These data indicate that homozygosity for p.I12T results in hearing loss. However, p.I12T in combination with a likely loss-of-function nonsense mutation, p.E4X, additionally leads to mild, asymptomatic metabolic abnormalities, in agreement with a partial loss of function of p.I12T barttin. For mutations of BSND, our data in combination with published reports indicate that there is a wide phenotypic continuum from nonsyndromic deafness with elevated renin of uncertain clinical significance to salt-losing nephropathies.3–6
Barttin is an accessory subunit of two human ClC-K channels, ClC-Ka (MIM #602024) and ClC-Kb (MIM #602023), that are essential for chloride reabsorption along the distal nephron and for endolymph formation in the inner ear.7–13 Barttin promotes exit of ClC-K channels from the endoplasmic reticulum,7,8,10 increases protein stability,9 and converts human ClC-K channels from a nonconducting into an active state.10 We used heterologous expression in mammalian cells to test for impairments of these functions by p.I12T.
We coexpressed wild-type (WT) or mutant p.I12T barttin with human ClC-Ka or ClC-Kb in HEK293T cells and measured anion currents through patch-clamp recordings10,12 (Figure 3). In cells expressing ClC-Ka or ClC-Kb together with barttin, anion currents can be observed over the whole voltage range without time-dependent current relaxations (Figures 3A and 3C). Untransfected HEK293T cells or cells that express ClC-Ka, ClC-Kb, or barttin alone exhibit negligible currents under the same ionic conditons.10,12 Current amplitudes in cells expressing ClC-Ka/ClC-Kb channels together with p.I12T barttin are smaller than corresponding values for WT barttin, for standard internal and external [Cl−] (Figures 3B and 3D) as well as for reduced intra- and extracellular anion concentrations (Figure S2). The effect of p.I12T on ClC-Ka/barttin currents (ratio of mean current amplitudes at −100 mV = 24%) is more pronounced than for ClC-Kb/barttin (ratio of mean current amplitudes at −100 mV = 44%).
Figure 3.
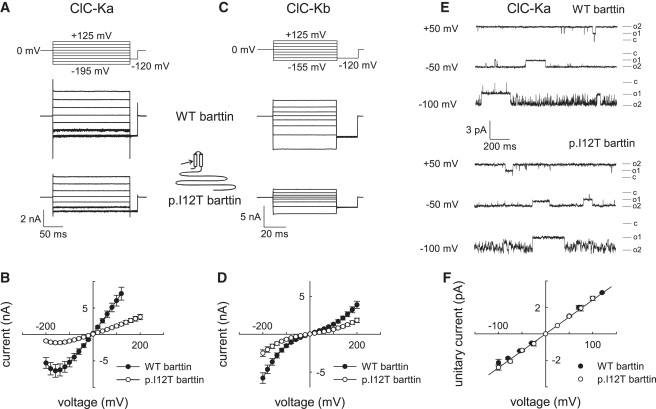
Functional Consequences of p.I12T on ClC-K/Barttin Channel Activity
(A–D) Representative recordings and current-voltage relationships of macroscopic currents from HEK293T cells coexpressing ClC-Ka (A and B) or ClC-Kb (C and D) and wild-type (WT) or p.I12T barttin with standard internal and external solutions. Whole-cell and single-channel patch-clamp recordings were performed with an Axopatch 200B amplifier (Molecular Devices).10,17 The extracellular solution contained 140 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES (pH 7.4), whereas the intracellular solution contained 120 mM NaCl, 2 mM MgCl2, 5 mM EGTA, and 10 mM HEPES (pH 7.4). The voltage dependence of instantaneous current amplitudes was determined with voltage steps (8–10 ms) from a holding potential of 0 mV so that potentials between −200 to +200 mV could be tested. Data points represent means ± SEM from between 16 and 26 cells.
(E and F) Representative single-channel recordings (E) and unitary current-voltage relationships (F) of ClC-Ka/barttin channels for WT or p.I12T barttin. Single-channel recordings were either performed in the inside-out configuration, with pipettes containing the standard extracellular solution and cells bathed in the standard intracellular solution, or in the cell-attached configuration with cells bathed in the standard extracellular solution and the pipettes containing a modified bath solution with 140 mM KCl, 4 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES (pH 7.4). Currents were sampled at 50 kHz and filtered at 500–1000 Hz. Because of the high open probability of ClC-Ka/barttin channels, unitary current amplitudes were manually determined from channel closures that exceeded 5 ms. Data points represent means ± SEM from three to ten measurements.
To distinguish whether p.I12T affects the function or the intracellular trafficking of ClC-K/barttin channels, we performed single-channel recordings and noise analysis. In agreement with the double-barreled structure of ClC channels,14 we observed two equally spaced subconductance states with a unitary conductance of 26 pS in patches expressing ClC-Ka/barttin (Figures 3E and 3F). Channels were open most of the time at positive potentials, whereas at negative potentials short open and short closed times resulted in a fast flickering behavior (Figure 3E). Ten out of twenty-one patches from cells expressing ClC-Ka/WT barttin contained channels with unitary conductances and gating as shown in Figure 3E. Such channels were neither observed in 12 patches from untransfected cells nor in 37 patches from cells expressing V166E rClC-K1.10 In cells expressing ClC-Ka and p.I12T barttin, we observed channels with properties identical to those of ClC-Ka/WT barttin channels in 11 out of 85 patches. Remaining patches either lacked single-channel events or contained endogenous channels. Hence, unitary currents of ClC-Ka/p.I12T barttin were indistinguishable from ClC-Ka/WT barttin (Figures 3E and 3F).
To confidently assign unitary channels observed in patch-clamp experiments to macroscopic ClC-Ka/barttin currents, we additionally employed a variation of stationary noise analysis (Figure S3).15–18 Steady-state variances were determined at voltages between −85 mV and −205 mV, divided by the product of the mean current amplitudes and the electrical driving force (V − Vr), and plotted against the macroscopic conductance (Figures S3B–S3D). The slope (−) of the fitted straight gives the number of channels N, and the y axis intercept provides the unitary conductance. Experiments were performed for two different anion compositions, and for both ionic conditions the unitary conductances of ClC-Ka/barttin channels were not different for WT and p.I12T barttin (Figure S3E). The number of channels was 8800 ± 2190 (n = 8) in cells transfected with WT barttin, whereas cells expressing p.I12T barttin contained only 3232 ± 1048 channels (n = 8). We next determined absolute open probabilities from macroscopic current amplitudes (I), unitary current amplitudes (i), and the number of channels (N) as p = I/Ni. Absolute open probabilities change with anion concentrations but are not different for ClC-Ka/WT barttin and ClC-Ka/p.I12T barttin (Figure S3F). In single-channel recordings as well as in noise analysis, ClC-Ka/p.I12T barttin channels displayed unaltered properties as compared to ClC-Ka/WT barttin channels (Figure 3 and Figure S3). Thus, the p.I12T mutation has no effect on permeation or gating of ClC-Ka/barttin channel but only reduces the number of channels in the surface membrane.
We were not able to determine unitary properties of ClC-Kb/barttin channels. These channels appear to be voltage independent, precluding noise analysis for determining unitary current amplitudes and absolute open probabilities. Moreover, we could not reliably identify unitary events in single-channel recordings. We therefore determined ratios of the current variance (σ2) and the macroscopic current amplitude (I). This value is identical to the product of the unitary current amplitude and the absolute probability of the channel to be closed (σ2/I = i(1 − p)).17 For all tested voltages, values are similar for ClC-Kb/WT barttin (σ2/I = −0.03 ± 0.007 pA; n = 9, at −105 mV) and ClC-Kb/p.I12T barttin (σ2/I = −0.027 ± 0.006 pA; n = 7, at −105 mV), in agreement with the notion that p.I12T also leaves ClC-Kb/barttin channel function unaffected.
To test whether these reduced current levels are due to reduced protein expression levels, we resolved YFP- and CFP-fusion proteins from cells expressing YFP-ClC-Ka or YFP-ClC-Kb together with CFP-labeled WT or mutant barttin, on reducing SDS-PAGE.12 We electrophoresed cleared lysates from transfected HEK293T cells on 6%–15% SDS polyacrylamide gradient gels and quantified CFP- or YFP-tagged proteins by scanning wet PAGE gels with a fluorescence scanner (Typhoon, GE Healthcare). We observed comparable expression levels for CFP-WT barttin and CFP-p.I12T barttin as well as for YFP-ClC-Ka and YFP-ClC-Kb coexpressed with WT or mutant barttin (Figure 4A). We conclude that p.I12T leaves protein amounts of pore-forming and accessory subunits of ClC-K/barttin channels unaffected.
Figure 4.
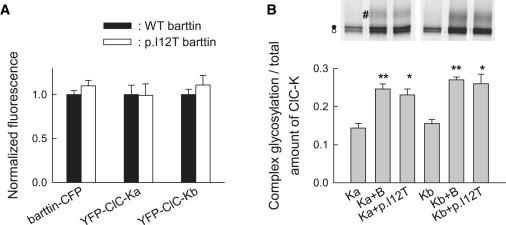
p.I12T Neither Affects the Number of Expressed ClC-K Channels nor the Glycosylation Status
(A) Fluorescent protein expression levels from HEK293T cells coexpressing WT or p.I12T barttin-CFP with YFP-ClC-Ka or YFP-ClC-Kb. For each experiment, SDS-PAGE gels (6%–15%) were prepared from the lysates of whole dishes of comparable cell number and transfection efficiency. Fluorescence was quantified with the ImageQuant software and normalized to corresponding fluorescence levels from cells coexpressing WT barttin. Data represent means ± SEM from eight or nine measurements.
(B) SDS-PAGE gel (6%–15%) of YFP-ClC-Ka or YFP-ClC-Kb either transfected alone or cotransfected with WT or mutant barttin in MDCKII cells shows complex-glycosylated (#) as well as core-glycosylated (●) or not glycosylated (◯) protein. The lower panel shows ratio of complex-glycosylated to total YFP-ClC-Ka and YFP-ClC-Kb fluorescence from cells coexpressing WT or mutant barttin. Mean ± SEM is from three or more experiments. Asterisks indicate levels of significance (∗∗p < 0.01 or ∗p < 0.05) for comparison with cells without barttin. Glycosylation levels were not different for cells expressing WT or p.I12T barttin.
To study channel trafficking, we used MDCKII cells, an established model for intracellular protein trafficking in epithelial cells, instead of HEK293T cells.9,10,12 WT barttin is necessary for export of ClC-K/barttin out of the endoplasmic reticulum.10,12 ClC-K channels are N-glycosylated in eukaryotic cells, and the presence of complex oligosaccharides can be used for monitoring the efficiency of the exit of the protein from the ER.12 p.I12T does not impair complex glycosylation of ClC-Ka or ClC-Kb, indicating normal ER exit (Figure 4B). We next studied the subcellular distribution of WT and p.I12T barttin and ClC-K by using confocal imaging of cells expressing fluorescent fusion proteins (Figure 5). MDCKII cells transfected with expression plasmids encoding barttin-CFP and YFP-ClC-Ka or YFP-ClC-Kb were scanned as living cells 1–3 days after transfection with a FluoView 1000 (Olympus) equipped with an argon-ion laser and a 60× objective with a NA of 1.35. CFP and YFP were excited with the 458 and 514 nm argon lines and photometrically detected with Olympus filters BA465-495 and BA535-565,19 respectively. When expressed alone, WT as well as p.I12T barttin insert themselves into the plasma membrane (Figure 5A). For WT and mutant barttin, colocalization of YFP and CFP fluorescence confirmed surface localization and effective binding of ClC-Ka and ClC-Kb with barttin (Figures 5B and 5C). Cells expressing p.I12T exhibit increased fluorescent staining of intracellular compartments, in agreement with impaired surface insertion of p.I12T barttin and ClC-K/p.I12T barttin. We conclude that p.I12T impairs late steps in trafficking of barttin and ClC-K/barttin.
Figure 5.
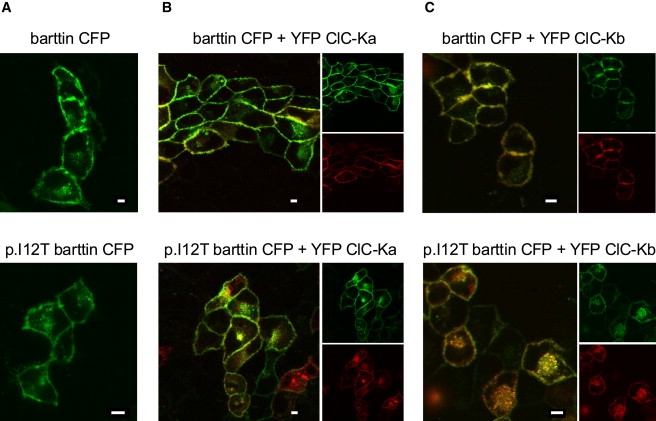
Subcellular Distribution of WT and Mutant Barttin
Fluorescent confocal images of living MDCK cells expressing WT or p.I12T mutant barttin-CFP fusion proteins either alone (A) or together with YFP ClC-Ka (B) or YFP ClC-Kb (C). The scale bar represents 5 μm. CFP is shown in green, and YFP is shown in red. There is an orange color where both proteins overlap.
Lack of either one of the two ClC-K isoforms impairs urinary concentration, but not hearing.11,20 These results indicate that in the inner ear, only one isoform is required and suggest that one ClC-K isoform can compensate for a genetic defect of the other one. In contrast, moderate impairment of the chaperone function of barttin by p.I12T causes nonsyndromic deafness. These findings demonstrate that barttin performs a rate-limiting step in ClC-K channel maturation. In the presence of dysfunctional barttin, the inner ear is more sensitive to a reduction in chloride conductances than the kidney. Moreover, p.I12T appear to have less pronounced effects on macroscopic current amplitudes of ClC-Kb/barttin than of ClC-Ka/barttin (Figure 3), and the residual ClC-Kb activity in kidney might contribute to the preserved renal function.
Several disease-causing BSND mutations have been identified and functionally analyzed. In all cases, there is a genotype-phenotype relationship in that the level of function of mutant barttin predicts the renal phenotype.12 Whereas nonsense mutations result in a pronounced renal phenotype,6 mutations that were identified in patients with late-onset or mild deterioration of the renal system5,21 have milder effects on barttin function than mutations causing early-onset renal symptoms.12 A small but significant reduction in ClC-K/barttin currents by p.I12T barttin selectively causes a hearing impairment, whereas there is a borderline renal salt-reabsorption deficit in compound heterozygotes p.12T/E4X. Our findings demonstrate that a pathogenic mutation of BSND can cause deafness with subclinical renal metabolic changes and thus defines BSND as a causal gene in nonsyndromic deafness DFNB73.
Acknowledgments
We thank the families for their participation and J. Ehrich, D. Ewers, E. Miranda, D. Nothmann, and D. Wojchiechowski, for helpful discussions. This study was supported by the Higher Education Commission and the Ministry of Science and Technology in Pakistan to S.R., the Intramural Program of the NICD/National Institutes of Health 1 ZO1 DC000039-12 to T.B.F. and the DFG (FA 301/8 and FA 301/10) to C.F.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3, http://frodo.wi.mit.edu/
UCSC, http://genome.ucsc.edu/
Accession Numbers
The GenBank accession number reported for Homo sapiens Bartter syndrome, infantile, with sensorineural deafness (Barttin) (BSND) mRNA is NM_057176.2.
References
- 1.Bartter F.C., Pronove P., Gill J.R., Jr., MacCardle R.C. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am. J. Med. 1962;33:811–828. doi: 10.1016/0002-9343(62)90214-0. [DOI] [PubMed] [Google Scholar]
- 2.Hebert S.C. Bartter syndrome. Curr. Opin. Nephrol. Hypertens. 2003;12:527–532. doi: 10.1097/00041552-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Jeck N., Reinalter S.C., Henne T., Marg W., Mallmann R., Pasel K., Vollmer M., Klaus G., Leonhardt A., Seyberth H.W., Konrad M. Hypokalemic salt-losing tubulopathy with chronic renal failure and sensorineural deafness. Pediatrics. 2001;108:E5. doi: 10.1542/peds.108.1.e5. [DOI] [PubMed] [Google Scholar]
- 4.Landau D., Shalev H., Ohaly M., Carmi R. Infantile variant of Bartter syndrome and sensorineural deafness: A new autosomal recessive disorder. Am. J. Med. Genet. 1995;59:454–459. doi: 10.1002/ajmg.1320590411. [DOI] [PubMed] [Google Scholar]
- 5.Shalev H., Ohali M., Kachko L., Landau D. The neonatal variant of Bartter syndrome and deafness: Preservation of renal function. Pediatrics. 2003;112:628–633. doi: 10.1542/peds.112.3.628. [DOI] [PubMed] [Google Scholar]
- 6.Birkenhäger R., Otto E., Schürmann M.J., Vollmer M., Ruf E.M., Maier-Lutz I., Beekmann F., Fekete A., Omran H., Feldmann D. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 2001;29:310–314. doi: 10.1038/ng752. [DOI] [PubMed] [Google Scholar]
- 7.Estevez R., Boettger T., Stein V., Birkenhager R., Otto E., Hildebrandt F., Jentsch T.J. Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature. 2001;414:558–561. doi: 10.1038/35107099. [DOI] [PubMed] [Google Scholar]
- 8.Waldegger S., Jeck N., Barth P., Peters M., Vitzthum H., Wolf K., Kurtz A., Konrad M., Seyberth H.W. Barttin increases surface expression and changes current properties of ClC-K channels. Pflugers Arch. 2002;444:411–418. doi: 10.1007/s00424-002-0819-8. [DOI] [PubMed] [Google Scholar]
- 9.Hayama A., Rai T., Sasaki S., Uchida S. Molecular mechanisms of Bartter syndrome caused by mutations in the BSND gene. Histochem. Cell Biol. 2003;119:485–493. doi: 10.1007/s00418-003-0535-2. [DOI] [PubMed] [Google Scholar]
- 10.Scholl U., Hebeisen S., Janssen A.G., Muller-Newen G., Alekov A., Fahlke Ch. Barttin modulates trafficking and function of ClC-K channels. Proc. Natl. Acad. Sci. USA. 2006;103:11411–11416. doi: 10.1073/pnas.0601631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kramer B.K., Bergler T., Stoelcker B., Waldegger S. Mechanisms of Disease: The kidney-specific chloride channels ClCKA and ClCKB, the Barttin subunit, and their clinical relevance. Nat. Clin. Pract. Nephrol. 2008;4:38–46. doi: 10.1038/ncpneph0689. [DOI] [PubMed] [Google Scholar]
- 12.Janssen A.G., Scholl U., Domeyer C., Nothmann D., Leinenweber A., Fahlke Ch. Disease-causing dysfunctions of barttin in Bartter syndrome type IV. J. Am. Soc. Nephrol. 2009;20:145–153. doi: 10.1681/ASN.2008010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rickheit G., Maier H., Strenzke N., Andreescu C.E., De Zeeuw C.I., Muenscher A., Zdebik A.A., Jentsch T.J. Endocochlear potential depends on Cl- channels: Mechanism underlying deafness in Bartter syndrome IV. EMBO J. 2008;27:2907–2917. doi: 10.1038/emboj.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dutzler R., Campbell E.D., Cadene M., Chait M.B., MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 15.Sigworth F.J., Zhou J. Ion channels. Analysis of nonstationary single-channel currents. Methods Enzymol. 1992;207:746–762. doi: 10.1016/0076-6879(92)07054-r. [DOI] [PubMed] [Google Scholar]
- 16.Sesti F., Goldstein S.A.N. Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J. Gen. Physiol. 1998;112:651–663. doi: 10.1085/jgp.112.6.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torres-Salazar D., Fahlke Ch. Neuronal glutamate transporters vary in substrate transport rate but not in unitary anion channel conductance. J. Biol. Chem. 2007;282:34719–34726. doi: 10.1074/jbc.M704118200. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Olivares J., Alekov A., Boroumand M.R., Begemann B., Hidalgo P., Fahlke Ch. Gating of human ClC-2 chloride channels and regulation by carboxy-terminal domains. J. Physiol. 2008;586:5325–5336. doi: 10.1113/jphysiol.2008.158097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papadopoulos S., Leuranguer V., Bannister R.A., Beam K.G. Mapping sites of potential proximity between the dihydropyridine receptor and RyR1 in muscle using a cyan fluorescent protein-yellow fluorescent protein tandem as a fluorescence resonance energy transfer probe. J. Biol. Chem. 2004;279:44046–44056. doi: 10.1074/jbc.M405317200. [DOI] [PubMed] [Google Scholar]
- 20.Schlingmann K.P., Konrad M., Jeck N., Waldegger P., Reinalter S.C., Holder M., Seyberth H.W., Waldegger S. Salt wasting and deafness resulting from mutations in two chloride channels. N. Engl. J. Med. 2004;350:1314–1319. doi: 10.1056/NEJMoa032843. [DOI] [PubMed] [Google Scholar]
- 21.Miyamura N., Matsumoto K., Taguchi T., Tokunaga H., Nishikawa T., Nishida K., Toyonaga T., Sakakida M., Araki E. Atypical Bartter syndrome with sensorineural deafness with G47R mutation of the beta-subunit for ClC-Ka and ClC-Kb chloride channels, barttin. J. Clin. Endocrinol. Metab. 2003;88:781–786. doi: 10.1210/jc.2002-021398. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.