

Abstract
The matrix metalloproteinases MMP9 and MMP13 catalyze the degradation of extracellular matrix (ECM) components in the growth plate and at the same time cleave and release biologically active molecules stored in the ECM, such as VEGFA. In mice, ablation of Mmp9, Mmp13, or both Mmp9 and Mmp13 causes severe distortion of the metaphyseal growth plate. We report that mutations in either MMP9 or MMP13 are responsible for the human disease metaphyseal anadysplasia (MAD), a heterogeneous group of disorders for which a milder recessive variant and a more severe dominant variant are known. We found that recessive MAD is caused by homozygous loss of function of either MMP9 or MMP13, whereas dominant MAD is associated with missense mutations in the prodomain of MMP13 that determine autoactivation of MMP13 and intracellular degradation of both MMP13 and MMP9, resulting in a double enzymatic deficiency.
Introduction
Different families of proteases mediate ECM turnover and cartilage remodelling in the growth plate, in particular ADAM-TSs, cathepsins, and matrix metalloproteinases (MMPs).1,2 These enzyme families are known to differ in expression pattern and substrate specificity, but their function in the growth plate remains poorly defined. Classified by homologous functional domains and common activation mechanisms, the MMP family currently comprises 23 ECM-degrading endopeptidases in the human.3 Many MMPs are able to cleave the most abundant proteoglycans and collagens of skeletal tissues in vitro; therefore, they are presumed to play important roles in bone formation and growth. However, mice with targeted ablation of Mmp genes are, with few exceptions, viable and show only a mild or no skeletal phenotype.2 The expression patterns of MMP9 (MIM ∗120361) and MMP13 (MIM ∗600108) at the chondro-osseous junction within the human growth plate show only partial overlap.4 MMP9 is predominantly secreted by chondroclasts, osteoclasts, and endothelial cells, whereas MMP13 is produced by hypertrophic chondrocytes and osteoblasts. Nevertheless, loss of either Mmp9 or Mmp13 causes similar alterations of growth plate architecture in mice. The hypertrophic zone of these animals is markedly expanded as a result of reduced ECM remodelling, prolonged chondrocyte survival, and delayed vascular invasion.5–7 Mmp9 synergizes with Mmp13 in the coordinated decomposition of cartilage matrix, as indicated by a more severe phenotype in double-null mice.6 On the basis of these observations, ECM remodelling by the two enzymes is proposed to be a rate-limiting process in normal skeletal morphogenesis. Remarkably however, the skeletal changes in the knockout mice are only apparent during the growth phase, as even the more pronounced phenotype of a combined inactivation of Mmp9 and Mmp13 resolves spontaneously when longitudinal bone growth ceases.7
Most of the phenotypic features described in Mmp9 and Mmp13 knockout mice mimic the clinical picture of metaphyseal anadysplasia (MAD [MIM 309645]), a human skeletal dysplasia characterized by severe skeletal changes that, in contrast with the progressive course of most other skeletal dysplasias, resolve spontaneously with age (Figures 1A–1D). Patients attain a normal stature in adolescence and show improvement or complete resolution of varus deformity of the legs and rhizomelic micromelia.8 Inheritance of MAD is controversial in the sense that a single but different mode of transmission was claimed to be applicable; in particular, X-linked recessive, autosomal-recessive, and autosomal-dominant transmission have all been proposed.8–12 The presented data resolve this controversy, adding MAD to the list of disorders with more than one pattern of inheritance and a variety of disease mechanisms.
Figure 1.
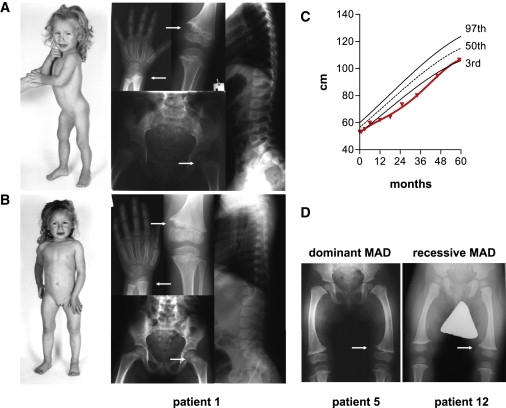
Clinical and Radiographic Presentation of MAD
(A and B) Patient 1 (index patient IV.2 of family A) at the age of (A) 2 yrs, 3 mo and (B) 4 yrs, 3 mo. Radiographs demonstrate spontaneously regressive metaphyseal irregularities (arrows) of radius, ulna, tibia, and femur; the spine is normal.
(C) The growth curve of patient 1 (red line) shows a transient growth deceleration maximal in the second year of life.
(D) Genua vara and metaphyseal fraying in dominant (patient 5, index patient IV.2 of family B at the age of 1 yr, 8 mo) and recessive (patient 12, index patient II.2 of family E in Figure 2B at the age of 1 yr, 1 mo) MAD are indistinguishable. Patient numbers refer to Table 1 and Figure 2.
We identified mutations in MMP9 and MMP13 as the molecular basis of MAD. Loss of function of either MMP9 or MMP13 is associated with recessive disease, whereas dominant missense mutations in the prodomain of MMP13 cause intracellular autocatalysis, similar to the effect of a single amino acid substitution described in spondyloepimetaphyseal dysplasia, Missouri type (SEMDMO [MIM 602111]), another skeletal dysplasia with dominant inheritance.13,14 Key clinical and radiographic features of SEMDMO and MAD are highly similar;13 accordingly, activating MMP13 prodomain mutations found in both disorders have the same consequences at the molecular level.14 The dominant effect of autoactivated proMMP13 was resolved by the observation that both mutant and wild-type proteins are degraded in the endoplasmatic reticulum.14 Our results confirm these findings and provide evidence that the transcatalytic mechanism also targets proMMP9, explaining the more severe phenotype of dominant disease by double enzymatic deficiency. Furthermore, our data indicate a functional link between MMP13 and MMP9 in endochondral ossification, as impaired MMP9 protein function, caused by direct inactivation (in recessive disease due to MMP9 loss of function), impaired activation (in recessive disease due to MMP13 loss of function), or transcatalytic degradation (in dominant disease caused by MMP13 gain of function) appears to be a common downstream step in the pathogenesis of MAD.
Subjects and Methods
Subjects
All subjects were ascertained by physician-initiated referral. We collected blood and isolated DNA from cases diagnosed at the Children's Hospital of Mainz and Freiburg University under a protocol approved by the ethics review board Rheinland-Pfalz, or from cases referred through the ESDN (European Skeletal Dysplasia Network) and SKELNET (German Skeletal Dysplasia Network). Informed consent was obtained for all subjects. Each family member was assessed by clinical and radiographic criteria. Control samples and primary cells were obtained from ancestry-, sex-, and age-matched healthy individuals.
Genomic Analysis and Mutation Detection
The exons including intron-exon boundaries of MMP9 and MMP13 were amplified by polymerase chain reaction (PCR) via standard protocols with primers listed in Table S1 or ones that had been published previously.14 Sequences of amplified PCR products were determined on an ABI3100 capillary sequencer (Applied Biosystems). Sequence data were processed with ABI software and analyzed with Sequencher (Genecodes). We compared amplicon sequences against the cDNA reference sequence for MMP9 and MMP13, with nucleotide numbering starting from the first transcribed nucleotide of the reference sequence. Mutations and polymorphisms were confirmed in two independently amplified PCR products via bidirectional sequencing.
Allele-Frequency Analysis
In 114 normal controls of European descent, exon 2 of MMP13 was amplified and sequenced unidirectionally to screen for dominant-MAD-associated mutations. For MMP13 c.300T>C, detected in families B and C, the sequence change falls within a BspHI restriction endonuclease cleavage site, ablating the recognition sequence in the mutant allele. PCR products of exon 2, derived from 114 normal controls of Japanese and Korean descent, were cleaved with BspHI (New England Biolabs). Only cleavage products of 222 bp and 207 bp, expected for wild-type MMP13, were seen in controls; affected individuals showed one uncleaved PCR product of 429 bp in addition. Restriction analysis for the recessive-MAD-associated sequence change in MMP13 was done by digesting the PCR product of exon 5 with BslI (MBI Fermentas). Controls showed only uncleaved wild-type alleles at 379 bp; patients showed two bands at 162 bp and 217 bp, resulting from a new recognition sequence introduced by the homozygous c.722C>A mutation in family D. Exon 1 of MMP9 was analyzed by restriction with NlaIII (New England Biolabs). PCR products from individuals affected by the homozygous c.21T>A mutation lack the recognition site and show a single band at 293 bp, whereas only the cleavage products at 102 bp and 191 bp were detected in unaffected controls.
RNA Extraction and Quantitative Real-Time PCR
Total RNA from fibroblasts and cell lines was extracted with Trizol (Invitrogen), followed by DNaseI treatment (Roche) and column purification (QIAGEN). cDNA was synthesized from 1 μg total RNA with Superscript III reverse transcriptase (Invitrogen) and oligo dT16 primers. Quantitative PCR was performed on an ABI 7300 Real-Time PCR System (Applied Biosystems). Delta Ct relative quantification, PCR efficiency correction, and multiple reference gene normalization (UBC, HMBS, PDHA) were calculated with qBase.15
Cellular Transformation and Expression of MMP9 and MMP13 cDNAs
HEK293T human embryonic kidney cells, NIH 3T3, and primary fibroblasts derived from skin biopsies (after the second passage) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 2 mM L-glutamine, 2 U/ml penicillin, and 2 mg/ml streptomycin in a humidity-controlled incubator at 37°C with 5% CO2. Full-length MMP9 and MMP13 cDNAs were generated by PCR with the use of Pfu proofreading polymerase (Stratagene) with cDNA from normal human fibroblasts as template and were inserted into pCRII-TOPO (Invitrogen). Sequence-verified inserts were subcloned into pcDNA3.1 and into pcDNA3.1V5-HIS-TOPO (Invitrogen). We introduced mutations in MMP13 by PCR-mediated site-directed mutagenesis as described previously.16 The nucleotide sequences of the mutants were verified by sequence analysis of the complete corresponding cDNAs. Minigene constructs were generated by subcloning PCR products of the MMP9 promoter, including exon 1, into pUC19, with the SapI site in exon 1 used to generate a complete MMP9 open reading frame fused to the endogenous 5′-regulatory region. The MMP9 c.21T>A mutation was introduced by using DNA of patient 12 as template in the PCR, and wild-type constructs were generated with DNA from unaffected controls. Blunt-ended fragments of Minigene_1 (including MMP9 cDNA and 1623 bp upstream of the transcriptional start) and Minigene_2 (including MMP9 cDNA and 741 bp upstream of the transcriptional start) replaced the CAG promoter in the HincII-EcoRV-digested pCAGIG17 vector, which allows detection of transfected cells on the basis of their green fluorescence when illuminated at 478 nm. Expression constructs were transiently transfected with the use of FuGENE6 (Roche) and a standard calcium phosphate protocol.18 Cells and supernatants were harvested 48 hr after transfection; prior to RNA and protein extraction, cells were washed with PBS. Lysates were prepared by incubation in 1 ml lysis buffer (20 mM Tris-HCl, pH 8, 137 mM NaCl, 10% glycerol, 1 mM NaF, 1 mM Na3VO4, 0.1% NP-40) plus complete protease inhibitor (Roche) for 20 min on ice, followed by centrifugation at 14,000 × g and at 4°C for 10 min. For the analysis of secreted MMP proteins, the medium was changed to serum-free Optimem I (Invitrogen) 24 hr prior to collection, conditioned supernatants concentrated by centricon Plus-20 columns (Millipore). Total protein concentration in lysates and supernatants was determined by the Bradford microassay procedure (Bio-Rad), equal protein amounts (50–100 μg) precipitated by acetone, fractionated by SDS-PAGE, and transferred to nitrocellulose membranes (GE Healthcare). Immunoblots were probed with antibodies against human MMP13 (MAB13424 and AB8114, Millipore), human MMP9 (MAB3309, Millipore), the V5 (V8012, Sigma) and HIS (11922416001, Roche) epitope tags, and β-actin (AV40173, Sigma). Appropriate secondary antibodies (Dako) were used for visualization with enhanced chemiluminescence (GE Healthcare).
Concentration and Activity of MMP9 and MMP13
Concentration of MMP9 and MMP13 was measured in plasma (NH4-heparinate) collected under standardized conditions, serum, and conditioned supernatants via a flurorogenic substrate activity assay (Fluorokine E, R&D). MMP proteins were specifically captured in an antibody-coated microtiter plate from 100-fold diluted samples. Wells were washed four times, then activated with 1 mM of p-aminophenylmercuric acetate (APMA) and incubated for 20 hr in the dark at 37°C. MMP actitviy was detected by exciting the cleaved flurorogenic peptide substrate ES001 at 320 nm and reading the emission at 405 nm, endpoint mode (Mithras Multimode reader, Berthold). The concentrations of MMPs were extrapolated from standard curves generated with recombinant human MMP9 and MMP13 (R&D). Collagenase activity in conditioned supernatants was quantified with fluorescently labeled native type II collagen (Millipore) and gelatin (Molecular Probes). For fibrillar type II collagen, diluted samples were activated with 1 mM APMA for 1 hr, then incubated with the substrate for 2 hr at 35°C in the dark, before the fluorescence extracted from degradation products was excited at 490 nm and measured at 520 nm. Degradation of gelatin was measured after activation of diluted samples for 16 hr with 1 mM APMA and incubation for 2 hr at ambient temperature in the dark. Kinetic determination of fluorescence intensity was used in calculating specific activity, given that collagenase activity is directly proportional to the intensity of fluorescence generated by the cleavage of substrates. Units were defined as cleaved μg of collagen per minute. Total protein concentration of whole-cell lysates and conditioned supernatants was measured by the Bradford microassay procedure (Bio-Rad). Specific collagenolytic activity (U per mg protein) of the samples was determined by the ratio of total collagenase units and total protein content.
Protein Structure Modeling
The atomic coordinates of the X-ray crystal structure and NMR structure of MMP13 domains19 and a model of proMMP13 were obtained from the Protein Data Bank (PDB 1UC1, 1FM1, 1XUC); a prodomain model was based on the structures of proMMP1 (PDB 1SU1), proMMP2 (PDB 1CK7), and proMMP3 (PDB 1SLM) with the use of the semi-automated modeling server SWISS-MODEL20 as described previously.14 Figures were created with MBT21 and the molecular graphics program PyMOL.
Statistics
We tested significance between individual groups by using the two-tailed unpaired Student's t test (p < 0.05). Data are expressed as the mean ± SDM.
Results
We investigated the molecular basis of MAD in five families (Table 1). All patients fulfilled the minimal clinical diagnostic criteria of MAD, namely normal length at birth and transitory bowing of the legs. In dominant disease, bowing and rhizomelic shortening was coincident with growth deceleration in the second year of life. Height dropped to a range of −1 SD to −4 SD; significant catch-up growth and spontaneous regression of bow legs in late childhood was noted (Figures 1A–1C). Radiographic examination demonstrated extensive metaphyseal widening and fraying in all cases (Figures 1A, 1B, and 1D). Follow-up examination documented spontaneous regression and disappearance of the metaphyseal alterations over several years. Pedigrees of unrelated families of diverse ethnic origin suggested two genetically distinct forms of MAD: the clinically more severe type with reduced stature (MAD type 1; Spranger type) shows autosomal-dominant inheritance over several generations in three kindreds (Figure 2A), whereas the milder type with normal stature (MAD type 2; Maroteaux type), observed in consanguineous families, is inherited as an autosomal-recessive trait (Figure 2B). The radiographic presentation of both types was indistinguishable; no discriminating feature could be defined on the basis of our collective observations and previous reports8–12 (Figure 1D).
Table 1.
Summary of Clinical and Molecular Findings in MAD Patients
| Patient | Age at Ascertain-ment | Sex | Family, Individual | Inheritance | Mutation | Origin | Maximum Growth Deceleration in the 2nd Yr of Life | Stature at Specified Age | Bowed Legs in Infancy | Micro-melia | Laboratory Parameters |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2y6m | F | A, IV.2 | dominant | MMP13 p.F55S | Germany | −3.89 SD | 6y, −2.35 SD | severe | yes | normal |
| 2 | 39y | F | A, III.2 | dominant | MMP13 p.F55S | Germany | −3.17 SD | 39y, −1.72 SD | severe | yes | normal |
| 3 | 65y | F | A, II.4 | dominant | MMP13 p.F55S | Germany | n. d. | 65y, −2.58 SD | severe,persistent | yes | normal |
| 4 | 15y | M | B, IV.2 | dominant | MMP13 p.M72T | Germany | −2.29 SD | 18y, +1.60 SD | yes | yes | normal |
| 5 | 12y | F | B, IV.3 | dominant | MMP13 p.M72T | Germany | −2.51 SD | 15y, −0.89 SD | yes | yes | normal |
| 6 | 42y | M | B, III.3 | dominant | MMP13 p.M72T | Germany | n. d. | 42y, +1.2 SD | yes | yes | normal |
| 7a | 62y | M | B, II.1 | dominant | MMP13 p.M72T | Germany | n. d. | 62y, +0.72 SD | yes | yes | n. d. |
| 8 | 3y | F | C, II.1 | dominant | MMP13 p.M72T | Japan | −2.9 SD | 6y, −2.89 | yes | yes | normal |
| 9 | 4m | F | C, II.2 | dominant | MMP13 p.M72T | Japan | −2.78 SD | 3y1m, −1.54 SD | yes | yes | normal |
| 10 | 28y | F | C, I.2 | dominant | MMP13 p.M72T | Japan | n. d. | 32y, −1.9 SD | yes | yes | normal |
| 11 | 4m | M | D, II.1 | recessive | MMP13 p.H213N | Morocco | −1.35 SD | 3y2m, −1.0 SD | yes | mild | normal |
| 12 | 14m | F | E, II.1 | recessive | MMP9 p.M1K | Pakistan | +0.23 SD | 7y, −0.23 SD | yes | no | normal |
| 13 | 2y | M | E, II.2 | recessive | MMP9 p.M1K | Pakistan | −0.15 SD | 5y, −0.77 SD | yes | no | normal |
Abbreviations are as follows: F, female; M, male; n.d., not determined; y, year; m, month; SD, standard deviation.
Patient 7 was deceased at the time of ascertainment; mutation analysis was performed with DNA extracted from archival pathologic specimen. Laboratory parameters included calcium, phosphate, alkaline phosphatase, vitamin D, and intact parathyroid hormone.
Figure 2.
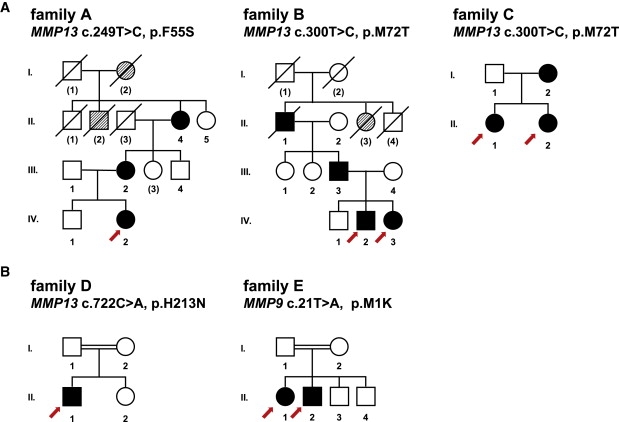
Pedigrees of MAD Families
Abridged pedigrees of familes with (A) dominant and (B) recessive MAD are shown. Numbers of individuals correspond to Table 1; numbers in parentheses indicate individuals from whom no DNA was available. Symbol notations are as follows: squares, males; circles, females; open symbols, unaffected individuals; filled symbols, affected mutation carrier of the nucleotide and predicted amino acid change indicated on top; hatched symbols, likely affected individuals. Symbols with a slash through them indicate deceased individuals. Index patients are marked with a red arrow.
The similarity between mouse MMP knockouts and human MAD prompted us to consider MMP9 and MMP13 as possible candidates. Analysis of both genes by direct sequencing of genomic DNA demonstrated heterozygous single-nucleotide exchanges within exon 2 of MMP13 in all patients affected by dominant MAD, whereas MMP9 sequence was normal in coding and regulatory regions. The changes in MMP13 segregated with the disease phenotype in familial cases and were not present among 228 alleles of ancestry-matched unaffected individuals. In family A, a c.249T>C transition predicted a single amino acid substitution, p.F55S, in the prodomain of MMP13 (Figure S1). In proximity to the first sequence change, a c.T300>C transition results in the amino acid substitution p.M72T that is also located in the prodomain of MMP13 (Figure S1). This most prevalent mutation was detected in all affected individuals of families B and C. Disease-associated sequence changes in dominant MAD are in the same highly conserved domain as p.F56S (Figure S2), a missense mutation in proMMP13 reported as the cause of SEMDMO in a single large kindred.13,14 Thus, both diseases are allelic and may represent a clinical spectrum.
Recessive MAD proved to be genetically heterogeneous, as we identified homozygous sequence changes in either in MMP13 or MMP9. In family D, a c.722C>A transition in exon 5 of MMP13 changes the highly conserved histidine 213 of the catalytic domain to asparagine in the predicted open reading frame of proMMP13 (Figure S3). In the second recessive kindred, family E, MMP13 was normal, but a c.21T>A transversion in exon 1 altered the start codon of proMMP9, substituting methionine with lysine (Figure S4). As the next AUG providing a putative aberrant initiation site in the mRNA sequence is located 177 nucleotides downstream, the mutation is likely to ablate translation of a functional proMMP9 protein. In both families, homozygous mutations segregated with the MAD phenotype, while the parents were heterozygous and the unaffected sibs were heterozygous or homozygous wild-type; the mutations were not seen in 228 alleles of unaffected controls.
To elucidate the effect of MAD-associated mutations on MMP protein level and function, we quantified both proteins in patients' plasma and cells by immunoblot and collagenolytic activity. Circulating MMP13 activity in patients with dominant MAD was barely detectable (Figure 3A). Immunoblot analysis of extracts from cultured normal fibroblasts revealed a band at the size expected for proMMP13, but this band was absent in the patient's cell samples (Figure 3C). We then transfected wild-type and mutagenized expression constructs of human MMP13 cDNA in HEK cells and compared secreted MMPs in conditioned supernatants. The catalytic domain of MMPs contains within the sequence HExxHxxGxxH three conserved histidines that ligate the active site Zn2+ (Figure 4A and Figure S3). The glutamate residue of the catalytic motif activates a zinc-bound H2O molecule, providing the nucleophile that cleaves peptide bonds.3 The recessive-MAD-associated MMP13 mutation, p.H213N, substitutes the third histidine of the consensus sequence with asparagine, thus preventing zinc ion ligation and correct conformation of the catalytic core. Accordingly, secretion and protein stability were not affected, but the secreted protein had no detectable collagenolytic activity (Figures 4B and 4C).
Figure 3.
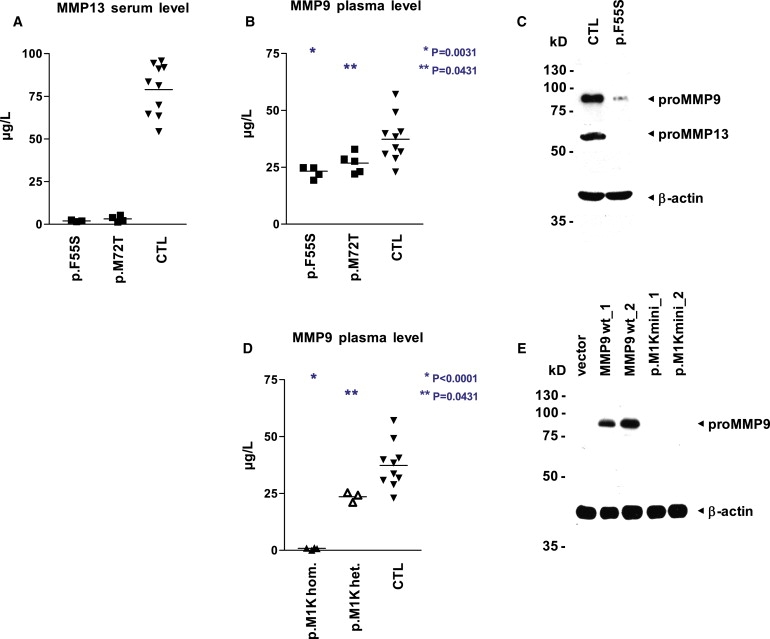
Concentration and Activity Levels of MMP9 and MMP13 in MAD Patients
(A and B) MMP13 activity is not detectable in the serum (A), and MMP9 activity is reduced in the plasma (B), of patients with dominant MAD (MMP13 p.F55S, n = 3; MMP13 p.M72T, n = 3) in comparison to unaffected controls (n = 10). Specific MMP activity was quantified via a fluorogenic peptide substrate cleavage assay. Samples were measured in quadruplicates, and p values were obtained with a two-tailed Student's t test for the comparison of means (horizontal line).
(C) Both proMMP13 and proMMP9 are strongly reduced in immunoblot analysis of whole-cell lysates from early-passage fibroblasts of patient 1 with dominant MAD (MMP13 p.F55S) as compared with an age-matched unaffected control; equal loading is demonstrated by reprobing with an antibody directed against β-actin.
(D) The plasma of patients with recessive MAD lacks MMP9 activity (MMP9 c.21T>A, p.M1K, n = 2), and heterozygous carriers (n = 2) show a significant reduction in comparison to unaffected controls (n = 10). MMP9 activity was quantified via a fluorogenic peptide substrate cleavage assay. Samples were measured in triplicates and p values were obtained with a two-tailed Student's t test for the comparison of means (horizontal line).
(E) No lower-sized bands indicative of initiation at downstream start codons are visible in immunoblot analysis of whole-cell lysates from fibroblasts transfected with minigene constructs bearing the recessive-MAD-associated mutation MMP9 c.21T>A, p.M1K. In wild-type constructs, proMMP9 is readily detectable with an antibody directed against the carboxy terminus. Transfection efficiency was similar, as monitored by fluorescence microscopy of EGFP expressed from an internal ribosome entry site downstream of MMP9; the promoter region includes 1644 bp (construct_1) and 762 bp (construct_2) upstream of the MMP9 start codon.
Figure 4.
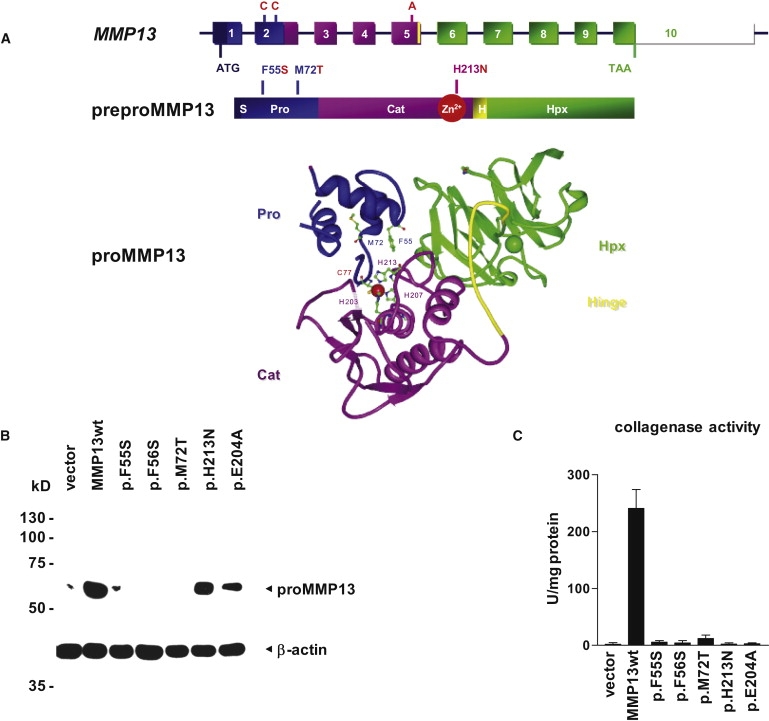
Functional Analysis of Disease-Associated MMP13 Mutations in HEK Cells
(A) Schematic diagram of the genomic structure of the MMP13 gene, illustrating the position of MAD-associated mutations and protein domains. MMP13 encodes a 471 amino acid (aa) preproprotein including a 19 aa signal sequence (S) removed upon secretion, a 84 aa prodomain (Pro) maintaining latency of the proenzyme, and a 166 aa catalytic domain (Cat) connected by an 11 aa hinge peptide (H) to the 191 aa hemopexin-like domain (Hpx). The contribution of MMP13 exons to each protein domain is indicated by color. Within the catalytic domain, a conserved motif contains three histidine residues that ligate the active site Zn2+. The prodomain regulates enzymatic activity through the molecular interaction between the thiol group of cysteine 77 with the zinc ion in the catalytic center, modeled in the ribbon diagram of proMMP13 structure. In addition to the zinc-binding amino acids C77 (the “switching” cysteine), H203, H207, and H213, the atomic structure and relative positions of disease-associated F55 and M72 are shown. For simplicity, only the Zn2+ of the catalytic core and the central Ca2+ of the hemopexin-like domain are represented, as red and green spheres, respectively.
(B) All dominant-MAD-associated mutations cause a strong reduction of the proMMP13-specific signal in immunoblot analysis of whole-cell lysates from human embryonic kidney cells transfected with expression constructs bearing disease-associated MMP13 mutations; there is no apparent difference between the individual dominant-MAD- and SEMDMO-associated mutations. Protein stability is normal for the mutation p.H213N associated with recessive MAD and the missense mutation p.E204A ablating enzymatic activity. Transfection efficiency and mRNA stability was similar for all constructs, as analyzed in parallel by quantitative reverse transcriptase-PCR (not shown); equal protein loading is documented by reprobing the blot with an antibody directed against β-actin.
(C) Collagenase activity for native collagen type II is reduced to background levels by all disease-associated MMP13 missense mutations, similar to the catalytically inactive mutant p.E204A. Conditioned supernatants of transfected cells were activated with 1 mM APMA for 1 hr. No collagenolysis was detected without activation. Relative collagenase activity was normalized to recombinant MMP13 and total protein content. Bars represent the mean of triplicates, and error bars indicate the SDM.
In family E bearing the homozygous MMP9 mutation, MMP13 levels in plasma were normal, but MMP9 reactivity was absent as anticipated (Figure 3D). Given that heterozygous carriers also showed a reduced concentration (Figure 3D), we concluded that the p.M1K start codon mutation abolished translation and that this effect was not compensated for by aberrant initiation at a non-AUG or AUG encoded in the mRNA. The analysis of minigene constructs comparing the expression levels of wild-type and mutant human MMP9 under the control of two endogenous promoter fragments fully supported these conclusions (Figure 3E).
Although the molecular pathology of recessive MAD disease could thus be attributed to impaired synthesis of MMP9 protein or synthesis of catalytically inactive MMP13, an explanation for the drastic decrease of MMP13 levels observed in dominant MAD was less evident. Surprisingly, not only MMP13, but also MMP9, was significantly reduced in the plasma of these patients in spite of normal genomic MMP9 sequences (Figure 3B). For the MMP13 p.F55S mutation, MMP9 reduction was more pronounced in immunoblot analysis of skin fibroblasts (Figure 3C). The prodomains of diverse proteases serve as autoinhibitory intramolecular chaperones to prevent activation during synthesis and protein folding;22–24 therefore, prodomain mutations may cause autoactivation.14,25,26 To test for possible autocatalytic activation of proMMP13 in MAD, we compared protein stability and enzymatic activity of all pathogenic missense proMMP13 mutants. A strong reduction of intact proMMP13 was evident in immunoblot analysis of conditioned medium or cell lysates from HEK cells transfected with constructs bearing disease-associated mutations (Figures 4A–4C). The degree of destabilization conferred by the different prodomain mutations (p.F55S, p.F56S, p.M72T) appeared to be similar, judged by the effect on the level and enzymatic activity of secreted MMP13 protein (Figure 4C), arguing against a genotype-phenotype correlation based on the extent of autoactivation.
Notably, we observed that the dominant-negative effect of autoactivated MMP13 extended to MMP9 as substrate, given that no MMP9 was produced by cells cotransfected with mutant MMP13 and wild-type MMP9 expression constructs (Figure 5A). Reduced protein levels of mutant MMPs were reflected by a loss of proteolytic activity for native collagen and gelatin in conditioned supernatants of cell lines and fibroblasts (Figure 4C and Figure 5A). In transfected cells, stability of all disease-associated proMMP13 mutants was restored by additional inactivation of the catalytic center by site-directed mutagenesis. These compound transfectants had normal amount and activity of secreted MMP9, proving that in dominant MAD, loss of both MMP13 and MMP9 protein stability and function was caused solely by aberrant intracellular activation of the catalytic domain of MMP13 (Figures 5B and 5D). Secretion of proMMP9 also increased when the mutant MMP13 transcript was specifically suppressed by siRNA in fibroblasts, whereas the level of proMMP9 in control cells remained unchanged (data not shown). Altogether, our experiments indicate that heterozygosity for critical MMP13 mutations is enough to induce the premature activation of proMMP13 and that prematurely activated MMP13 in the endoplasmic reticulum degrades both itself (autocatalytic degradation) and the structurally normal MMP9 (transcatalytic degradation).
Figure 5.
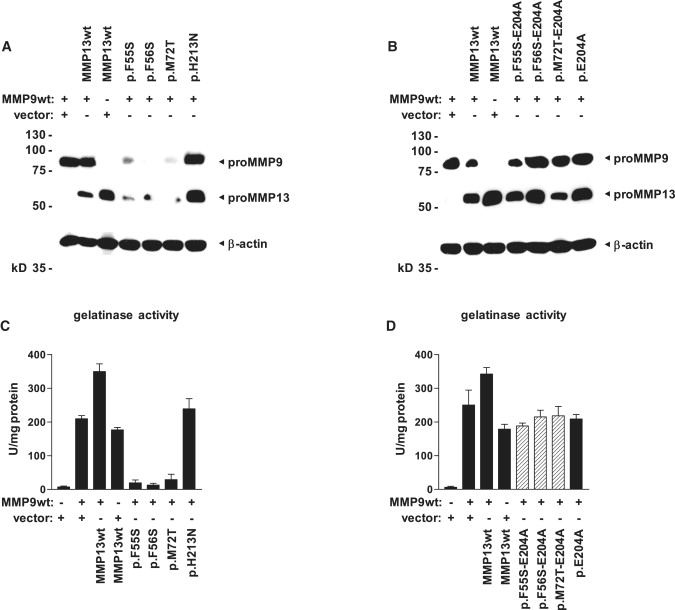
Transcatalytic Intracellular Degradation of MMP9 by Autoactivated MMP13
(A) Dominant-MAD-associated MMP13 mutations cause a strong reduction of both MMP13- and MMP9-specific signals in immunoblot analysis of whole-cell lysates from human embryonic kidney cells cotransfected with expression constructs bearing disease-associated MMP13 mutations and wild-type MMP9; loss-of-function mutations do not affect the stability of either protein.
(B) Dominant MMP13 mutations lead to autoactivation and intracellular degradation of both MMP13 and MMP9; MMP stability is restored by additional inactivation of the catalytic center through the missense mutation p.E204A. Constructs bearing compound mutations were analyzed by immunoblotting.
(C) Transcatalysis reduces enzymatic activity of secreted MMPs. Dominant MMP13 mutations cause a strong decrease of gelatinase activity in conditioned supernatants of cotransfected cells, whereas MMP9 activity is not altered by the recessive-MAD-associated mutant p.H213N. Bars represent the mean of triplicates, and error bars indicate the SDM.
(D) Additional ablation of enzymatic activity in dominant MMP13 mutants by site-directed mutagenesis brings secreted gelatinase activity back to levels measured in MMP9 wild-type transfectants. Conditioned supernatants were activated with 1 mM APMA for 16 hr. Bars represent the mean of triplicates, and error bars indicate the SDM.
Discussion
MMPs are translated in a prepro form that contains a signal peptide targeting to the secretory pathway and a prodomain maintaining an inactive enzymatic state of proMMPs within the cell (Figure 4A). The prodomain regulates enzymatic activity through a mechanism called “cysteine switch,” describing the molecular interaction between the thiol group of a specific cysteine in the prodomain with the zinc ion in the catalytic center27 (Figure 4A and Figure S1). Enzymatic activity is unleashed only when this interaction is disrupted, either by modification of the free thiol residue or by cleavage of the prodomain by another proteinase. Importantly, conformational changes of the proMMP affecting the thiol-Zn2+ interaction may cause intramolecular autolytic cleavage of the prodomain, thereby irreversibly activating the catalytic domain.28,29 Amino acid substitutions in proximity of the highly conserved consensus prodomain sequence PRCxxPD can release the cysteine switch, resulting in premature intracellular autoactivation and autodegradation of the enzyme.14,25,28,29 Our observations in patients and in patient-derived primary cells confirm the pathogenic relevance of the MMP cysteine-switch mechanism that had so far been studied only in heterologous expression systems.14,25 We speculated that different degrees of MMP13 autoactivation might explain the apparent phenotypic differences between the phenotype of MAD and that of SEMDMO, but this was not supported by our in vitro data. The metaphyseal features of both conditions are identical, including the spontaneous resolution of symptoms, whereas the supposedly discriminating radiographic changes in the epiphyses and the spine are minor; in fact, the clinical and radiographic delineation of SEMDMO was based on a single family, and experts disagree about the degree of vertebral involvement (J.S., S.U., A.S.-F., unpublished data).30 For the time being, we would tentatively propose that SEMDMO may be a distinct disorder but could form part of the spectrum of dominant MAD.
When the data presented here are combined with the data from mouse experiments, the following pattern emerges: in spite of partially different expression patterns, inactivation of either MMP9 or MMP13 (by gene ablation in mice or by mutations in human recessive MAD) leads to an identical phenotype of disorganization of the growth plate, seen as metaphyseal dysplasia on radiographs. The double knockout (in mice) and the combined deficiency (in human dominant MAD) are only slightly more severe than the individual enzyme deficiencies, suggesting that these enzymes may act in the same pathway or cascade. Is a common pathogenetic mechanism responsible for the similar phenotype? A number of MMPs can cleave the prodomain of other MMP zymogens in vitro, and a general role as activating proprotein convertases was proposed for the MMPs.28 Notably, active MMP13 effectively converts proMMP9 in vitro,31 and activation of proMMP9 during myofibroblast differentiation requires the presence of active MMP13.32 In cocultured articular chondrocytes, MMP13 was identified as the major upstream proteinase controlling the conversion and activity of proMMP9 secreted by macrophages, supporting a general role of an MMP-dependent activation cascade in the pathogenesis of arthritis.33 Given that our data suggest that MMP13 can target MMP9 intracellularly, it seems likely that MMP9 is a physiological substrate of MMP13 also in the extracellular matrix of the growth plate. A sequential, rather than parallel or divergent, action of MMP13 and MMP9 might explain the scarcely additive effect of the combined deficiency.
What could be the downstream target(s) of MMP9? In the growth plate, increased degradation of collagens and proteoglycans coincides with hypertrophic chondrocyte differentiation and immediately precedes vascular invasion. A large body of evidence supports that angiogenesis is the crucial step in bone formation.5,34–37 Whereas ECM remodelling is essential for vascular invasion to occur, terminal chondrocyte differentiation, apoptosis, and matrix remodelling depend on angiogenic factors, such as VEGFA. MMP9 has been shown to regulate the bioavailability of VEGFA in the hypertrophic zone of the growth plate, and blocking Vegfa activity mimics the Mmp9 knockout phenotype.34,36 Thus, we suggest that there exists an activation cascade from MMP13 over MMP9 to VEGFA and that this cascade is disturbed by isolated loss of either MMP13 or MMP9 function or by autocatalytic and transcatalytic inactivation of MMP13 and MMP9, leading to reduced release of VEGFA and to impaired angiogenesis in endochondral ossification (Figure S5).
Supplemental Data
Supplemental Data include five figures and one table and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensembl.org
ESDN, http://www.esdn.org
ExPASy Proteomics, http://www.expasy.org
MBT, http://mbt.sdsc.edu
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
Protein Data Bank, http://www.rcsb.org
PyMOL Molecular Graphics, http://www.pymol.org
SKELNET, http://www.skelnet.de
Acknowledgments
The authors are grateful to the affected individuals and their families for participation in the study. This work was underwritten in part by grants from the Bundesministerium für Bildung und Forschung (SKELNET) and the European Union (FP6, EuroGrow). E.L. and B.Z. are supported by individual grants from Deutsche Forschungs-gemeinschaft (La 1381/1-3).
References
- 1.Cawston T.E., Wilson A.J. Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract. Res. Clin. Rheumatol. 2006;20:983–1002. doi: 10.1016/j.berh.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Page-McCaw A., Ewald A.J., Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brinckerhoff C.E., Matrisian L.M. Matrix metalloproteinases: A tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3:207–214. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- 4.Haeusler G., Walter I., Helmreich M., Egerbacher M. Localization of matrix metalloproteinases, (MMPs) their tissue inhibitors, and vascular endothelial growth factor (VEGF) in growth plates of children and adolescents indicates a role for MMPs in human postnatal growth and skeletal maturation. Calcif. Tissue Int. 2005;76:326–335. doi: 10.1007/s00223-004-0161-6. [DOI] [PubMed] [Google Scholar]
- 5.Vu T.H., Shipley J.M., Bergers G., Berger J.E., Helms J.A., Hanahan D., Shapiro S.D., Senior R.M., Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inada M., Wang Y., Byrne M.H., Rahman M.U., Miyaura C., López-Otín C., Krane S.M. Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc. Natl. Acad. Sci. USA. 2004;101:17192–17197. doi: 10.1073/pnas.0407788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stickens D., Behonick D.J., Ortega N., Heyer B., Hartenstein B., Yu Y., Fosang A.J., Schorpp-Kistner M., Angel P., Werb Z. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiedemann H.-R., Spranger J. Chondrodysplasia metaphysaria (Dysostosis metaphysaria) - ein neuer Typ? Z. Kinderheilk. 1970;108:171–186. [PubMed] [Google Scholar]
- 9.Maroteaux P., Verloes A., Stanescu V., Stanescu R. Metaphyseal anadysplasia: a metaphyseal dysplasia of early onset with radiological regression and benign course. Am J Med Genet. 1991;1:4–10. doi: 10.1002/ajmg.1320390103. [DOI] [PubMed] [Google Scholar]
- 10.Le Merrer M., Maroteaux P. Metaphyseal anadysplasia type II: a new regressive metaphyseal dysplasia. Pediatr. Radiol. 1998;28:771–775. doi: 10.1007/s002470050463. [DOI] [PubMed] [Google Scholar]
- 11.Nishimura G., Ikegawa S., Saga T., Nagai T., Aya M., Kawano T. Metaphyseal anadysplasia: evidence of genetic heterogeneity. Am J Med Genet. 1999;82:43–48. doi: 10.1002/(sici)1096-8628(19990101)82:1<43::aid-ajmg9>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 12.Slama M., Mathieu M., Dehouck I., al Hosri J., Vanthournout I., Baratte B., Grumbach Y. Metaphyseal anadysplasia in two sisters. Pediatr. Radiol. 1999;29:372–375. doi: 10.1007/s002470050610. [DOI] [PubMed] [Google Scholar]
- 13.Patel A.C., McAlister W.H., Whyte M.P. Spondyloepimetaphyseal dysplasia: clinical and radiologic investigation of a large kindred manifesting autosomal dominant inheritance, and a review of the literature. Medicine. 1993;72:326–342. [PubMed] [Google Scholar]
- 14.Kennedy A.M., Inada M., Krane S.M., Christie P.T., Harding B., López-Otín C., Sánchez L.M., Pannett A.A., Dearlove A., Hartley C. MMP13 mutation causes spondyloepimetaphyseal dysplasia, Missouri type (SEMDMO) J Clin Invest. 2005;115:2832–2842. doi: 10.1172/JCI22900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hellemans J., Mortier G., De Paepe A., Speleman F., Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lausch E., Hermanns P., Farin H.F., Alanay Y., Unger S., Nikkel S., Steinwender C., Scherer G., Spranger J., Zabel B. TBX15 mutations cause craniofacial dysmorphism, hypoplasia of scapula and pelvis, and short stature in Cousin syndrome. Am J Hum Genet. 2008;83:649–655. doi: 10.1016/j.ajhg.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuda T., Cepko C.L. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A. 2004;101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spangenberg C., Lausch E.U., Trost T.M., Prawitt D., May A., Keppler R., Fees S.A., Reutzel D., Bell C., Schmitt S. ERBB2-mediated transcriptional up-regulation of the alpha5beta1 integrin fibronectin receptor promotes tumor cell survival under adverse conditions. Cancer Res. 2006;66:3715–3725. doi: 10.1158/0008-5472.CAN-05-2823. [DOI] [PubMed] [Google Scholar]
- 19.Gomis-Rueth F.X., Gohlke U., Betz M., Knäuper V., Murphy G., López-Otín C., Bode W. The helping hand of collagenase-3 (MMP-13): 2.7 A crystal structure of its C-terminal haemopexin-like domain. J. Mol. Biol. 1996;264:556–566. doi: 10.1006/jmbi.1996.0661. [DOI] [PubMed] [Google Scholar]
- 20.Schwede T., Kopp J., Guex N., Peitsch M.C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moreland J.L., Gramada A., Buzko O.V., Zhang Q., Bourne P.E. The Molecular Biology Toolkit (MBT): a modular platform for developing molecular visualization applications. BMC Bioinformatics. 2005;6:21. doi: 10.1186/1471-2105-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moss M.L., Bomar M., Liu Q., Sage H., Dempsey P., Lenhart P.M., Gillispie P.A., Stoeck A., Wildeboer D., Bartsch J.W. The ADAM10 prodomain is a specific inhibitor of ADAM10 proteolytic activity and inhibits cellular shedding events. J. Biol. Chem. 2007;282:35712–35721. doi: 10.1074/jbc.M703231200. [DOI] [PubMed] [Google Scholar]
- 23.Golubkov V.S., Chekanov A.V., Shiryaev S.A., Aleshin A.E., Ratnikov B.I., Gawlik K., Radichev I., Motamedchaboki K., Smith J.W., Strongin A.Y. Proteolysis of the membrane type-1 matrix metalloproteinase prodomain: implications for a two-step proteolytic processing and activation. J. Biol. Chem. 2007;282:36283–36291. doi: 10.1074/jbc.M706290200. [DOI] [PubMed] [Google Scholar]
- 24.Bitar A.P., Cao M., Marquis H. The metalloprotease of Listeria monocytogenes is activated by intramolecular autocatalysis. J. Bacteriol. 2008;190:107–111. doi: 10.1128/JB.00852-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freimark B.D., Feeser W.S., Rosenfeld S.A. Multiple sites of the propeptide region of human stromelysin-1 are required for maintaining a latent form of the enzyme. J. Biol. Chem. 1994;269:26982–26987. [PubMed] [Google Scholar]
- 26.O'Neil H.S., Forster B.M., Roberts K.L., Chambers A.J., Bitar A.P., Marquis H. The propeptide of the metalloprotease of Listeria monocytogenes controls compartmentalization of the zymogen during intracellular infection. J. Bacteriol. 2009;191:3594–3603. doi: 10.1128/JB.01168-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Wart H.E., Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA. 1990;87:5578–5582. doi: 10.1073/pnas.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagase H. Activation mechanisms of matrix metalloproteinases. Biol. Chem. 1997;378:151–160. [PubMed] [Google Scholar]
- 29.Ra H.J., Parks W.C. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007;26:587–596. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Superti-Furga A., Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. A. 2007;143:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 31.Knäuper V., Smith B., López-Otin C., Murphy G. Activation of progelatinase B (proMMP-9) by active collagenase-3 (MMP-13) Eur. J. Biochem. 1997;248:369–373. doi: 10.1111/j.1432-1033.1997.00369.x. [DOI] [PubMed] [Google Scholar]
- 32.Han Y.P., Yan C., Zhou L., Qin L., Tsukamoto H. A matrix metalloproteinase-9 activation cascade by hepatic stellate cells in trans-differentiation in the three-dimensional extracellular matrix. J. Biol. Chem. 2007;282:12928–12939. doi: 10.1074/jbc.M700554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dreier R., Grässel S., Fuchs S., Schaumburger J., Bruckner P. Pro-MMP-9 is a specific macrophage product and is activated by osteoarthritic chondrocytes via MMP-3 or a MT1-MMP/MMP-13 cascade. Exp Cell Res. 2004;297:303–312. doi: 10.1016/j.yexcr.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 34.Gerber H.P., Vu T.H., Ryan A.M., Kowalski J., Werb Z., Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 35.Engsig M.T., Chen Q.J., Vu T.H., Pedersen A.C., Therkidsen B., Lund L.R., Henriksen K., Lenhard T., Foged N.T., Werb Z. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J. Cell Biol. 2000;151:879–889. doi: 10.1083/jcb.151.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zelzer E., McLean W., Ng Y.S., Fukai N., Reginato A.M., Lovejoy S., D'Amore P.A., Olsen B.R. Skeletal defects in VEGF(120/120) mice reveal multiple roles for VEGF in skeletogenesis. Development. 2002;129:1893–1904. doi: 10.1242/dev.129.8.1893. [DOI] [PubMed] [Google Scholar]
- 37.Zelzer E., Mamluk R., Ferrara N., Johnson R.S., Schipani E., Olsen B.R. VEGFA is necessary for chondrocyte survival during bone development. Development. 2004;131:2161–2171. doi: 10.1242/dev.01053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.