

Abstract
We report the case of a congenital myasthenic syndrome due to a mutation in AGRN, the gene encoding agrin, an extracellular matrix molecule released by the nerve and critical for formation of the neuromuscular junction. Gene analysis identified a homozygous missense mutation, c.5125G>C, leading to the p.Gly1709Arg variant. The muscle-biopsy specimen showed a major disorganization of the neuromuscular junction, including changes in the nerve-terminal cytoskeleton and fragmentation of the synaptic gutters. Experiments performed in nonmuscle cells or in cultured C2C12 myotubes and using recombinant mini-agrin for the mutated and the wild-type forms showed that the mutated form did not impair the activation of MuSK or change the total number of induced acetylcholine receptor aggregates. A solid-phase assay using the dystrophin glycoprotein complex showed that the mutation did not affect the binding of agrin to α-dystroglycan. Injection of wild-type or mutated agrin into rat soleus muscle induced the formation of nonsynaptic acetylcholine receptor clusters, but the mutant protein specifically destabilized the endogenous neuromuscular junctions. Importantly, the changes observed in rat muscle injected with mutant agrin recapitulated the pre- and post-synaptic modifications observed in the patient. These results indicate that the mutation does not interfere with the ability of agrin to induce postsynaptic structures but that it dramatically perturbs the maintenance of the neuromuscular junction.
Introduction
Congenital myasthenic syndromes (CMS [MIM 608931]) are a heterogeneous group of disorders caused by genetic defects affecting neuromuscular transmission and leading to muscle weakness accentuated by exertion. CMSs are classified on the basis of their pathophysiology into presynaptic compartment, synaptic basal lamina, and postsynaptic compartment CMSs. So far, 11 disease-causing genes coding for proteins that have a key role at the neuromuscular junction (NMJ) have been identified: CHAT (MIM 118490), CHRNA1 (MIM 100690), CHRNB1 (MIM 100710), CHRND (MIM 100720), CHRNE (MIM 100725), COLQ (MIM 603033), RAPSN (MIM 601592), SCN4A (MIM 603967), MUSK (MIM 601296), DOK7 (MIM 610285), and LAMB2 (MIM 150325).1–4 However, for half of CMS patients, the genes underlying the disease have not yet been identified. Until now, the identified genes encode for membrane receptors, ion channels, enzymes, or structural proteins, but none has been found to code for a secreted neural factor. Besides the mutations recently described in the laminin β2 chain,4 the latest mutations causing a CMS were identified in MUSK and DOK7, genes encoding, respectively, the postsynaptic tyrosine kinase receptor MuSK and Dok-7, a protein interacting with the juxtamembrane intracellular domain of MuSK.5,6
During NMJ development, MuSK plays a central role in the organization of the postsynaptic scaffold. MuSK activation is required to recruit downstream signaling components7,8, which triggers the local aggregation and synthesis of postsynaptic acetylcholine receptors (AChRs) and several other postsynaptic proteins, such as the cytoskeletal protein rapsyn.9,10 MuSK is activated by neural agrin, which is released from motor neurons, and binds and activates the LRP4-MuSK complex.7,8 Around embryonic day (E) 14 in mice (corresponding to the 7th week of gestation in humans), motor nerve terminals reach primary myotubes and secrete both acetylcholine and neural agrin, which participate in the stabilization of synaptic AChR clusters and the dispersal of the aneural AChR clusters, respectively.11,12 Agrin is critical for synaptic differentiation because agrin-deficient mice die at birth from respiratory failure due to aberrant innervation and lack of postsynaptic differentiation.13
Agrin is a heparan sulfate proteoglycan that has been shown to bind to laminins via its amino-terminal domain14 and to interact via its carboxy-terminal part with LRP4 and α-dystroglycan.7,8,15 Agrin is encoded by AGRN (MIM 103320), located on chromosome 1p36.33. The complete cDNA is 7319 bp long (NM_198576) and comprises 36 exons. The carboxy-terminal end of agrin contains three laminin G-like (LG) domains, two of which are required for binding to α-dystroglycan.16 Agrin mRNA undergoes cell-specific alternative splicing at several sites. Importantly, the two sites found in the carboxy-terminal part of the protein, called A and B in chickens17 or y and z in all mammals, including humans18, contain amino acid inserts when synthesized by motor neurons but lack these when synthesized in non-neural cells, including skeletal muscle fibers.19 An amino acid insert at the B/z site of agrin is required for MuSK activation and for NMJ formation.12,16 Consistent with the in vivo findings, the amino acid insert renders agrin approximately 1000 times more potent in inducing AChR clustering than the isoform that lacks amino acid inserts.20
In this article, we report the case of a CMS patient carrying a homozygous mutation in AGRN. Agrin recombinant proteins, either normal or reproducing the mutation, were produced and applied both in cell-culture systems and by injection in vivo in rat soleus muscle. This allowed us to evaluate the pathogenic consequences of this human mutation by demonstrating its deleterious effect on NMJ maintenance. These unexpected results imply that inducing the formation of postsynaptic structures via agrin's N-terminal laminin-binding domain and C-terminal A/z and B/z inserts is not the only function of agrin, which also appears to play a crucial role in the maintenance of the NMJs via other regions of the protein.
Material and Methods
Patient Mutation Analysis and Muscle Biopsy
Clinical evaluation, muscle biopsies, and blood samples were obtained after informed written consent was secured in accordance with the protocol approved by local ethics committees.
Mutation Analysis
Venous blood samples were drawn from the patients and their unaffected relatives after obtaining their signed informed consent. Genomic DNA was isolated using a blood DNA extraction kit according to the manufacturer's recommendations (Promega, Mannheim, Germany). The 36 exons of AGRN and their flanking intronic regions were amplified by PCR and sequenced (see Table S1 online for primer sequences). PCR-amplified fragments were purified, fluorescently labeled with dideoxy terminators (BigDye Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems), and run on an Applied Biosystems model 3730XL DNA Analyzer. The GenBank reference numbers used for comparison of the mRNA sequence of the AGRN exons is NM_198576.2. Any variant was confirmed as a disease-causing mutation by analysis of cosegregation in affected family members, conservation of the residue among species and isoforms, and absence in at least 200 control chromosomes of healthy adults. Extensive sequencing for AGRN revealed frequent variants that were thus considered to be polymorphisms (see Table S2 for missense polymorphisms).
Patient Muscle Biopsy
A muscle biopsy specimen was taken from deltoid muscles by open biopsy. The NMJ zone was determined by the small twitch provoked by the tip of the scalpel on the surface of the muscle fascicles. We confirmed the presence of NMJs on a longitudinal strip of the biopsy specimen by using the classic Koelle method21 to reveal cholinesterase activity. ATPase staining of muscle fibers was performed on cryostat sections according to the method of Brooke and Kaiser.22 Whole mounts of specimens fixed with 4% paraformaldehyde were stained for AChR with FITC- or TRITC-labeled α-bungarotoxin (Molecular Probes, Leiden, The Netherlands), and for neurofilaments with a 168 kDa neurofilament antibody (2H3, Hybridoma Bank, IA). The specimens were observed by confocal microscopy (Carl Zeiss LSM510, Oberkochen, Germany). Fragmentation of the NMJs was calculated as indicated below for injected rat muscle; see also Figure S1.
In addition, part of the biopsy specimen was processed for immunocytochemical analysis of cryostat sections with TRITC-labeled α-bungarotoxin and with polyclonal antibodies raised against agrin and MuSK provided by one of us (M.R.). The sections were observed by fluorescence microscopy (Zeiss Axiophot, Oberkochen, Germany). Electron microscopy of NMJs was performed by conventional methods.
Experimental Expression of Mutated Agrin
Production of Wild-Type and Mutated Agrin Recombinant Proteins
Because coding sequences for chicken and human agrin are highly homologous, we used a construct coding for a chicken mini-agrin. The chicken mini-agrin was cloned into the pCEP-Pu expression vector and codes for the amino-terminal NtA region followed by one follistatin-like repeat (25 kDa in size), which was then fused in-frame with the carboxy-terminal part (95 kDa size) of chicken agrin.23 The recombinant protein was targeted to the secretory pathway by the signal peptide of BM40, and it included a carboxy-terminal His6 tag. The carboxy-terminus included the 4- and 8-amino-acid inserts at the A/y and B/z sites and thus represents the neural isoform. Site-directed mutagenesis was performed by Genecust (Evry, France) and yielded a construct containing the same c.5127G>C mutation as that identified in the patient. Recombinant wild-type and mutated chicken mini-agrins were purified from conditioned media of 293 EBNA cells stably transfected with pCEP-Pu/wild-type or mutated mini-agrin plasmids. Proteins were purified from culture medium with a Ni-column (Ni-NTA agarose, QIAGEN).
Administration of Agrin Recombinant Proteins in Myogenic Cells in Culture
C2C12 cells seeded at a density of 5 × 104 cells per 35 mm dish were cultured for 24 hr at 37°C and 5% CO2 in a proliferation Dulbecco's modified Eagle's medium (DMEM) supplemented with antibiotics and 20% fetal bovine serum. They were switched to a differentiation DMEM containing 5% horse serum and antibiotics and cultured for 4 days at 37°C and 5% CO2. After 4 days, wild-type or mutant agrin was added at different concentrations (three dishes per concentration) for 18 hr at 37°C. Cells were washed twice with pre-warmed PBS, fixed for 10 min in PBS containing 4% paraformaldehyde (PFA), and then washed three times in PBS containing 0.1 M glycine. They were then incubated in PBS containing 3% BSA (PBS-BSA) for 2 hr followed by an incubation with TRITC-αBGT (1/500, Molecular Probes) in PBS-BSA for 1 hr. After several washes, plates were mounted in Vectashield mounting medium (Abcys, Paris, France) and observed with a 40× objective lens under an Olympus IX70 inverted microscope (Olympus Europa, Hamburg) linked to a CDD camera (Princeton Cool SNAP Fx, Trenton, NJ). Ten fields per dish were analyzed. AChR cluster number and myotube area were obtained with ImageJ software (NIH, Bethesda). The experiment was repeated twice.
For quantification of surface AChR number, C2C12 myotubes were fixed in PBS-4%-paraformaldehyde for 10 min, rinsed in PBS, incubated with 20 nM 3H-α-bungarotoxin in PBS-0.2%-BSA (TRK603-50UC1, GE Healthcare) for 45 min at room temperature, and washed four times in PBS-0.2%-BSA followed by PBS. Cells were solubilized in 200 μl PBS-1%-SDS, and 800 μl of scintillation liquid (Ultimate Gold, Packard) was added to the cell lysate. 3H radioactivity bound per culture dish was determined in a β counter (1900 TR, Packard). All toxin data were corrected for nonspecific binding, determined in the presence of “cold” 1 μM α-bungarotoxin (B-1601, Invitrogen). As an internal control, the total protein content per dish was measured. Protein concentration was determined with a bicinchoninic acid protein assay (Pierce, Rockford, IL, USA).
Administration of Agrin Recombinant Proteins in 293T Cells Coexpressing MuSK and LRP4
293T cells, transfected with LRP4 (kindly provided by Dr. Lin Mei) and HA-tagged MuSK expression vectors (2 μg each) via Lipofectamine 2000 (Invitrogen), were switched to DMEM without serum for 3 hr prior to stimulation with various concentrations of wild-type or mutated agrin (0.1 to 5 nM) for 1 hr. Cells were rinsed with ice-cold PBS and extracted in lysis buffer.24 Lysates were pre-cleared by centrifugation and incubated with anti-HA antibody overnight. Antibody was captured with protein G-Sepharose beads, which subsequently were washed four times in lysis buffer. Bound proteins were eluted from the beads with SDS sample buffer, resolved by SDS-PAGE, and transferred to Immobilon-P membranes (Millipore). Membranes were blocked in Tris-buffered saline (TBS) containing 5% skim milk and incubated with 4G10 antibody for anti-phosphotyrosine detection in TBS + 3% skim milk and with HA antibody for MuSK detection in TBS + 1% skim milk. Phosphorylation was estimated with ImageJ software. Three different experiments were done in triplicate.
Affinity of Agrins for the Dystrophin-Associated Glycoprotein Complex
A solid-phase assay was used. Ninety-six-well plates were coated overnight at 4°C and stirred with 0.5 μg of protein (dystrophin-associated glycoprotein complex [DGC] or BSA) in a pH 9.6, 50 mM NaHCO3 solution. The wells were then washed three times for 5 min with 200 μl of buffer A (PBS, 0.05% Tween, 1 mM CaCl2, and 1 mM MgCl2) and next incubated for 1 hr at room temperature with 200 μl of buffer B (PBS, 0.05% Tween, 1 mM CaCl2, 1 mM MgCl2, and 3% BSA). After three 5 min washes with 200 μl of buffer A, the wells were incubated with serial dilutions of each agrin in buffer B for 3 hr. After three 5 min washes in buffer A, the wells were incubated for 60 min with 100 μl of a 1:30,000 dilution of the agrin antibody in buffer B. The wells were then washed three times for 5 min with 200 μl of buffer A and next incubated for 45 min with 100 μl of HRP-conjugated anti-rabbit antibody in buffer B. After three 5 min washes with 200 μl of buffer A, 100 μl of ABTS detection solution (Roche, Mannheim, Germany) was added to the wells, and the optical density was read at 405 nm after 20 min. The background value, given by the binding of agrin to BSA, was subtracted from the results with DGC. DGC was extracted from chicken muscle as previously described for the initial studies of the interactions between agrin and dystroglycan.25
Injection of Agrin Recombinant Proteins in Rat Soleus Muscle
All experiments on rats were performed in accordance with European Community guidelines for laboratory animal handling. Four-week-old OFA female rats (Charles River Laboratories) were anesthetized with 80 mg/kg ketamine and 15 mg/kg xylazin. Five micrograms of either wild-type or mutated mini-agrin diluted in 80 μl of 0.9% NaCl were injected in the soleus muscle in the vicinity of the NMJ zone with a 1 ml syringe with a 27 gauge needle. Three rats per experimental condition (i.e., vehicle alone, wild-type agrin, and mutated agrin) were studied. Morphological analyses were performed with confocal microscopy as described above for human biopsy specimens. The area of synaptic gutters per NMJ and the mean α-bungarotoxin fluorescence intensity within that area were measured with ImageJ software. We studied fragmentation of the NMJs by counting the number of well-individualized synaptic gutter fragments composing one given NMJ and calculating the mean number of fragments for all NMJs studied, as shown in Figure S1.
Results
Study of the Patient
Clinical Data
The index patient is a 42-year-old woman who reports having been unable to run since early childhood and having had right-lid ptosis “since forever,” this condition was corrected surgically at the age of 22 years. Our examination showed she had mild bilateral lid ptosis but that ocular pursuit and pupillary reactivity were normal. There was a mild facial weakness but normal function of the axial muscles. The glutei and psoas muscles were slightly weak (MRC4+/5), and the patient had some difficulty walking on her heels. Atrophy, scoliosis, and contractures were not found, but the thorax and pelvis were thin. During the study of nerve conduction, single stimuli elicited normal compound muscle action potentials (CMAPs) and produced no “double” responses. Repetitive stimulation at 3 Hz resulted in a clear decrement of the CMAPs evoked from the trapezius (16%) and from the tibialis anterior (29%) muscles (normal < 10%), but no potentiation was seen after high-frequency repetitive nerve stimulation at 30 Hz with recording over tibialis anterior and first interosseus dorsalis muscles. By needle electromyography, no abnormal potentials were seen at rest in the deltoid or tibialis anterior muscles, but during 20 voluntary contractions the interference pattern analysis ratio was found to be abnormal (1.48, normal < 1), indicative of an early recruitment of polyphasic motor-unit potentials, a common result in myasthenic syndromes as well as in mild myopathies. Cholinesterase inhibitors and 3,4-diaminopyridine (3,4-DAP) were ineffective. The weakness fluctuated; it worsened during menstrual periods and during her pregnancy, but overall, the outcome showed no deterioration of muscle function. Diplopia, bulbar symptoms, and dyspnea were never reported.
The disease inheritance is autosomal recessive (Figure 1). The patient was born to asymptomatic parents who came from the same Swiss village. The patient's grandparents and great-grandparents were cousins. The index patient has five siblings, among whom a 36-year-old brother also presented CMS with similar manifestations: he has had difficulties in running since early childhood and had a fluctuating right ptosis (corrected at the age of 34 years), normal ocular pursuit, and intermittent mild masticatory difficulties with no dyspnea. The muscle examination was normal, but he had a thin thorax and flat feet. The course of the disease had been stable. The edrophonium test was negative, and pyridostigmine and ambenonium were ineffective. 3,4-DAP was mildly beneficial but was not tolerated. An abnormal (10% to 14%) decrement was found during repetitive stimulation at 3 Hz over orbicularis oculi, trapezius, and tibialis anterior muscles. The other family members were not examined.
Figure 1.

Identification of the Mutation c.5125G>C in AGRN exon 29 and Hereditary Transmission
(A) Sequence chromatograms from normal (wild-type) and affected (proband III-5) individuals are shown.
(B) Family pedigree. The proband is indicated by an arrow. The expected nucleotide change p.Gly1709Arg transmitted in this consanguineous family is indicated below the symbols. ND: not determined.
Ephedrine was recently given to the two patients. Improvement of the clinical symptoms after administration of ephedrine (50 mg/day for 3 days and then 2 mg/kg each morning) led to a remarkable and sustained increase in muscle performance and endurance and improved general well-being.
Mutation Analysis
All the known genes involved in CMS (CHAT, CHRNA1, CHRNB1, CHRND, CHRNE, COLQ, RAPSN, SCN4A, MUSK, and DOK7) with the exception of LAMB2 were initially sequenced, and no mutations were found. As part of a screen for candidate genes in CMS patients, a missense transversion, c.5125G>C, in exon 29 of AGRN was identified in the family, and this transversion led to the substitution of a conserved hydrophobic glycine for the basic amino acid arginine in the C-terminal, laminin G-like 2 domain of agrin (p.Gly1709Arg; RefSeq: NM_198576.2). The index patient III-5 was homozygous for the amino acid change (Figure 1). Molecular testing of the family members confirmed the recessive inheritance of the substitution. Because the father of the proband (II-3) was deceased, his brother (II-5) and sister (II-7) were tested: his sister is heterozygous for the mutant allele, indicating that one mutant allele came from the father. The other allele came from the mother because she carries a single mutant allele. The affected brother (III-4) of the index patient harbors the same homozygous mutation, and the unaffected siblings harbor one mutant allele (III-1, III-2) or none (III-3). The identification of this first pathogenic amino acid substitution in AGRN indicates that a founder effect in this village is probable and that the parents of the proband are also consanguineous. The p.Gly1709Arg substitution was not detected in 200 normal alleles.
Morphological Analysis of Patient Muscle-Biopsy Specimen
ATPase activity staining on cryostat sections showed type I fiber predominance and type II fiber atrophy in which a few fiber grouping islets were compatible with denervation-reinnervation processes (not shown). Whole-mount observations revealed that the global architecture of NMJs was perturbed in the patient. Whereas whole-mount preparations from normal individuals showed a typical fork-shaped nerve terminal innervating a well-defined synaptic structure, the patient muscle-biopsy specimen presented pre- and postsynaptic alterations. Among the 21 NMJs observed in the biopsy specimen from the patient, only 1 had a normal shape (normal), nine showed fragmented and dispersed synaptic gutters with evanescent borders and absence of nerve terminal profiles that are characteristic of denervation (denervated), seven NMJs were in a process of remodeling with either partially denervated or reinnervating profiles (remodeling), and four showed very thin axons contacting small newly formed synaptic cups (neoformed) (Figure 2A; for a representation in percentage, see Figure 2B). Of particular interest, within the group of NMJs undergoing remodeling, the morphology of the ramifications of the nerve terminals was abnormal: the neurofilaments in the axon terminal had a disheveled appearance (Figure 2A; see insert of the NMJ in remodeling), whereas at control NMJs the axon terminal was more compact and therefore thinner (Figure 2A, see insert of the normal NMJ). Another striking feature of the NMJs of the patient was the fragmentation of the synaptic gutters into several discontinued postsynaptic gutters as quantified in Figure 2C (see also the Material and Methods and Figure S1). The partial denervation and reinnervation patterns seen in the confocal microscope was also observed by electron microscopy (Figure 3). In the selected NMJs, the morphology of the postsynaptic apparatus showed only slight abnormalities and enlarged subneural folds. Conversely, the presynaptic compartment was perturbed and showed either a complete absence of axon profiles and the replacement by Schwann cell processes (denervated NMJ) or a partial reinnervation (in remodeling) characterized by small axon terminals that contained normal amounts of synaptic vesicles and some “dense” myelin figures but that occupied only part of the postsynaptic compartment. Terminal Schwann cells encased the axon terminals but never invaded the synaptic cleft, at least in the NMJs examined, contrary to what can be observed in other types of CMSs.4,26
Figure 2.

Abnormal NMJs Observed in Patient Muscle-Biopsy Specimen
(A) Whole-mount preparations stained for AChR with α-bungarotoxin in red and stained for axon terminals with neurofilament antibody in green. The different types of NMJs can be classified into four categories: normal, denervated, remodeling, and neoformed. The axonal branch classically ends as a fork and innervates a well-defined synaptic structure (normal). No axon profile faces the fragmented postsynaptic compartment (denervated). A thin axon terminal reinnervates some but not all of the fragmented gutters of the NMJ (remodeling). A thin axon innervating newly formed small and simplified NMJs can be observed (neoformed). In the bottom insert, note the disheveled appearance of the neurofilament staining observed in the NMJ in remodeling. The scale bar represents 10 μm for the four low-magnification prints and 4 μm for the two inserts.
(B) The graph represents the classification of the NMJs observed in the patient muscle-biopsy specimen into four categories (expressed as a percentage of the 21 NMJs examined).
(C) The fragmentation of NMJs was significantly increased in the patient in comparison with control muscle biopsies, according to the Mann-Whitney test (control: n = 28; patient: n = 21; U = 48,500, p < 0.001). Colored squares represent medians, and boxes represent 25th to 75th percentiles.
Figure 3.
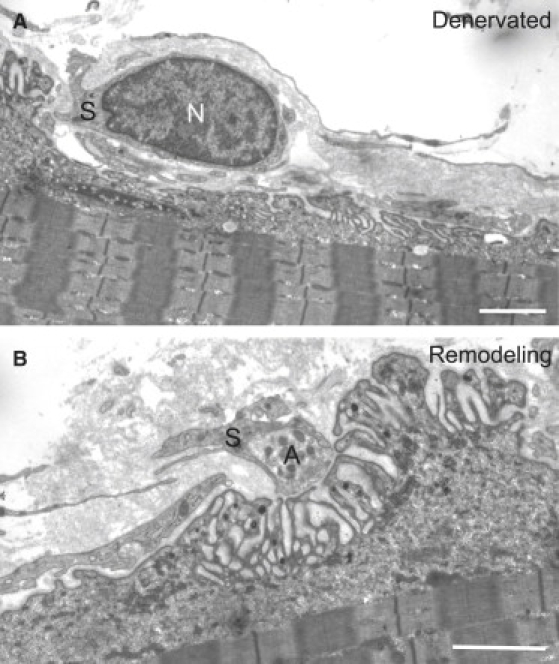
Electron Micrographs of Two NMJs from the Patient Biopsy Specimen
(A) An emptied NMJ is fully engulfed by a terminal Schwann cell (S) whose nucleus is clearly visible (N).
(B) A small reinnervating axon terminal (A) partially encased by Schwann cell processes (S) intrudes into the synaptic cleft, and there is pronounced reduction of the area of apposition between the axon terminal (A) and the postsynaptic membrane. The scale bar represents 2 μm in both prints.
To test whether the mutation in AGRN would affect expression of the protein, we next stained transverse cryostat sections with agrin and MuSK antibodies. As shown in Figure 4, both agrin and MuSK accumulated at the NMJs, and their expression levels were not different from those of controls. Altogether, these results indicate that agrin and its downstream targets are expressed and localized correctly in the patient's muscle but that perturbation of the overall NMJ organization has a marked effect on the presynaptic compartment.
Figure 4.
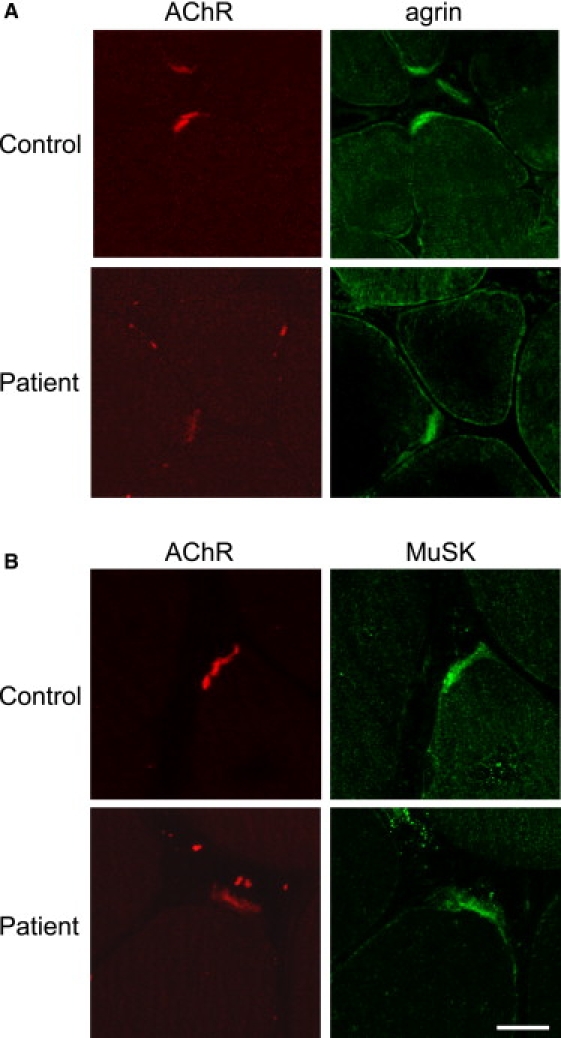
Cross-Sections of the Patient Biopsy Specimen Were Double Labeled for AChR in Red and for Agrin or MuSK in Green
Both agrin (A) and MuSK (B) colocalize with AChR at the NMJ, and their relative expression between the control and patient appears roughly similar. The scale bar represents 20 μm and applies to the eight prints.
Functional Evaluation of the Agrin Mutation
To investigate how the mutation would affect the NMJ as seen in the myasthenic syndrome observed in the patient, we produced mutated and wild-type recombinant agrin. To this end, we used a chick mini-agrin construct that encodes a fusion protein between a 25 kDa fragment of the amino-terminal end and a 95-kDa fragment of the carboxy-terminal end (Figure 5). The construct included the appropriate amino acid inserts at the A/y and B/z sites and thus encoded the neural form of agrin. Human and chick agrin are highly homologous and, most importantly, the Gly1709 residue of human agrin is conserved in chicks. Moreover, the chick mini-agrin allows the production of recombinant agrin23 and has been shown to be sufficient to substitute for agrin in other species and to rescue the phenotype in agrin knockout mice.12,27 We performed site-directed mutagenesis to reproduce the p.Gly1709Arg mutation of the patient. Wild-type and mutant recombinant agrin proteins were produced in 293-EBNA cells and purified as described in the Material and Methods section. For simplification, instead of “neural form of mini-agrin,” the word “agrin” will be used in the following sections.
Figure 5.
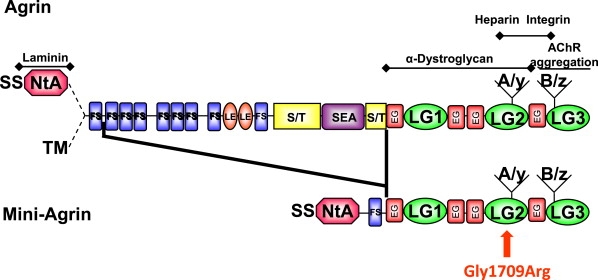
Protein Domains of the Recombinant Mini-Agrin and Localization of the p.Gly1709Arg Substitution
The neural mini-agrin is deleted from its central region. It contains the SS-NtA domain at the N-terminal region and the A/y and B/z inserts at the C-terminal region. The p.Gly1709Arg substitution is located in the laminin G-like 2 domain (LG2) in the region of agrin interaction with α-dystroglycan, heparin, and integrins.
The Mutation in Agrin Does Not Alter AChR Clustering in Cultured Myotubes
To test whether the mutation would affect the AChR-aggregating activity of agrin, we used C2C12 myotubes in culture. C2C12 myotubes express agrin but only the form that lacks inserts at the y and z sites. This form of agrin does not contribute to AChR clustering induced by the addition of agrin.28 C2C12 myotubes were cultured with wild-type or mutant neural agrin for 18 hr at different concentrations. AChR clusters were then visualized with fluorescent α-bungarotoxin, and the number of clusters of more than 5 μm in length were counted (Figure 6). No difference in clustering activity between wild-type and mutant agrin was noted with close dose-response curves and an EC50 (effective concentration needed to induce a half-maximal response) of about 0.05 nM. To check whether the mutation would affect the expression of AChR, we quantified surface and total AChRs by using 3H-α-bungarotoxin. The results indicated that, in our experimental conditions, wild-type and mutant agrin affected neither the total amount of AChR nor its recruitment to the membrane (data not shown). Altogether, these results suggest that the mutation does not significantly impair the AChR clustering function of agrin.
Figure 6.
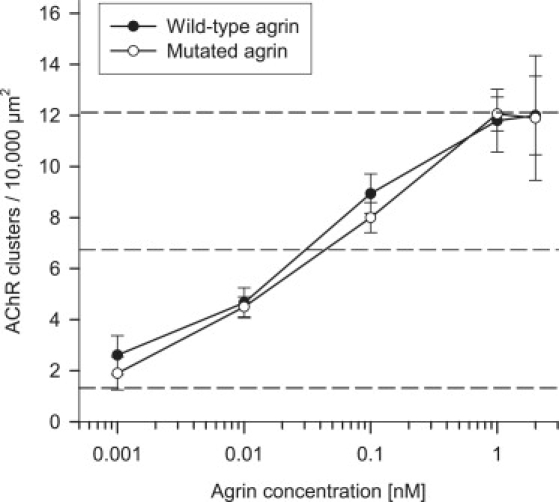
Effects of the Mutated Agrin on AChR Aggregation
Dose-response curves for AChR-aggregating activity of wild-type or mutant agrins. C2C12 myotubes were incubated for 18 hr with recombinant agrins at the concentrations indicated, and the number of AChR clusters per myotube was determined as described in the Material and Methods. Each data point represents results (mean ± SD) from triplicate myotube cultures; ten fields were studied per dish. Data shown represent the results of one representative experiment. The number of AChR clusters without agrin and the half-maximal and maximal number of clusters are indicated by dotted lines.
The Mutation in Agrin Does Not Alter MuSK Phosphorylation in Nonmuscle Cells Expressing Both LRP4 and MuSK
To determine whether the mutation affects the capacity of agrin to activate MuSK, 293T cells were cotransfected with both LRP4 and MuSK expression vectors. Because LRP4 is the agrin receptor, it confers to agrin the ability to phosphorylate MuSK in non-muscle cells.7 MuSK phosphorylation was evaluated in the presence of wild-type or mutated agrin at different doses ranging from 0.1 to 5 nM. As shown in Figure 7, the mutation does not inhibit the ability of agrin to activate MuSK.
Figure 7.
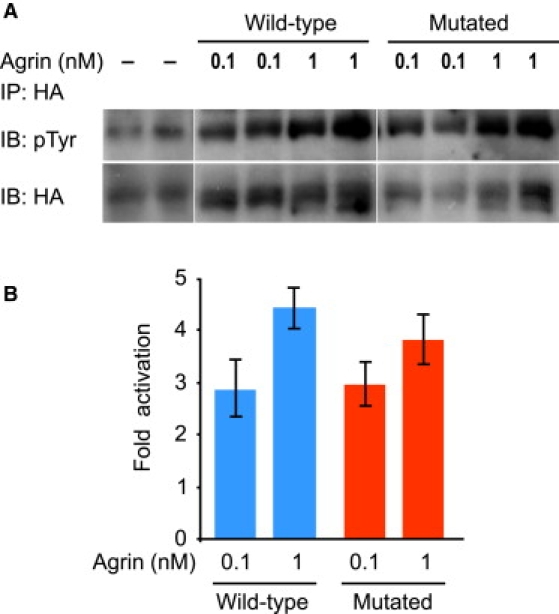
Effects of the Mutated Agrin on MuSK Phosphorylation
293T cells expressing both MuSK and LRP4 were incubated with wild-type and mutant agrin at different concentrations (0.1–5 nM). A representative experiment is shown with 0.1 and 1 nM agrin.
(A) HA-MuSK was immunoprecipitated (IP) with an anti-HA antibody (HA), and immunoblots (IB) for MuSK (HA) and phosphorylated MuSK (pTyr) were performed.
(B) Phosphorylation, estimated with ImageJ software, is expressed as n-fold activation. No significant differences between wild-type or mutant agrin were observed at any concentration studied.
The Mutation in Agrin Does Not Affect Its Binding to α-dystroglycan
Since the mutation is located in the region of agrin responsible for interactions with α-dystroglycan,16 the impact of the mutation on the affinity of agrin for purified DGC was evaluated in a solid phase assay (ELISA; Figure 8). The results showed that the mutation did not diminish the binding of agrin to DGC.
Figure 8.
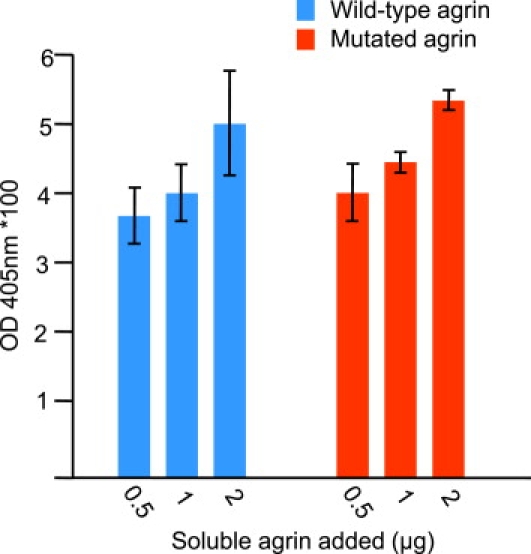
Evaluation of Agrin Binding to DGC in Solid-Phase Assay
Various amounts of wild-type and mutant agrin were added to 0.5 μg of DGC bound to 96-well plates. The amount of soluble agrin added is indicated in micrograms on the abscissa. The amount of bound agrin is expressed as absorbance ± SD.
Altogether, our results show that the mutation does not affect agrin's ability to activate MuSK and to cluster AChRs, nor does it affect the interaction with α-dystroglycan.
Effects of Mutated Agrin in Rat Soleus Muscle
To address in vivo the consequences of the mutation, we injected recombinant wild-type and mutant agrin proteins into rat soleus muscle to evaluate the formation of extrasynaptic AChR clusters and possible NMJ remodeling. Muscles were collected two weeks after injection, and whole muscles or cryostat sections were labeled for AChRs with α-bungarotoxin and labeled for agrin, MuSK, or neurofilament with the appropriate antibodies. As expected, agrin injection induced the formation of extrasynaptic AChR clusters that were never contacted by nerve endings, even when they were located in the vicinity of the synaptic zone (Figure 9A). When species-specific chick agrin antibodies were used for detection of injected chick agrin, we found that the extrasynaptic AChR clusters were associated with accumulation of chick agrin. Consistently, MuSK also accumulated at these sites (Figure S2). No obvious difference in the number, size, or shape of ectopic AChR clusters could be seen between muscles injected with mutant and wild-type agrin, indicating that AChR-inducing activity of agrin at these extrasynaptic sites was not significantly altered by the p.Gly1709Arg substitution. Thus, the in vivo paradigm of inducing extrasynaptic AChR aggregates was quite similar to what was observed for AChR clusters on cultured C2C12 myotubes.
Figure 9.
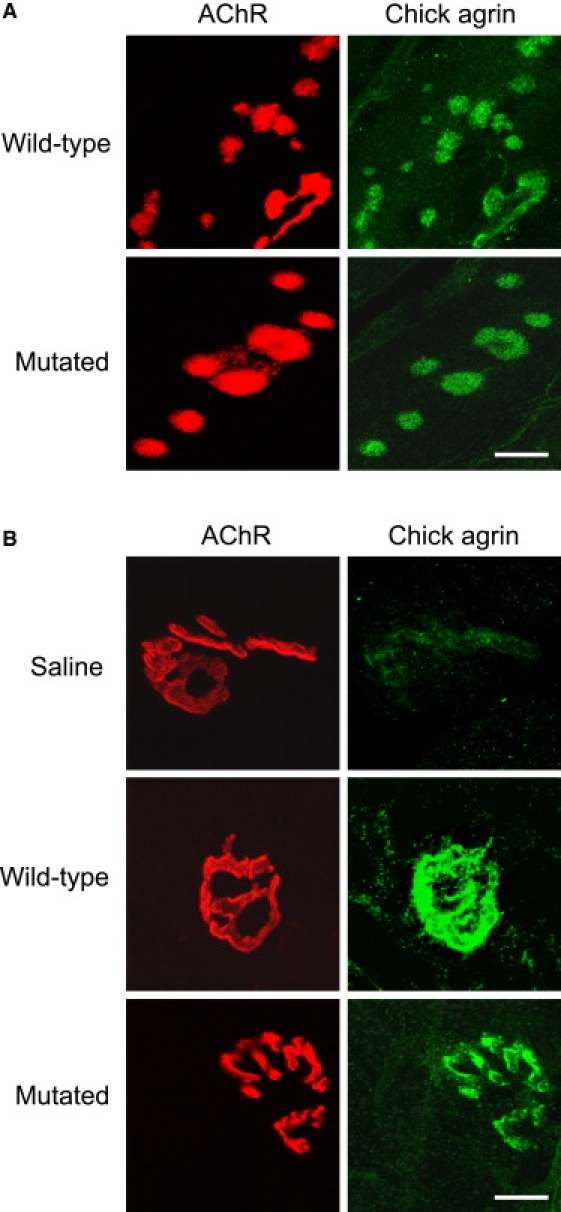
Chick Agrin Colocalizes with Extrasynaptic AChR Clusters and Endogenous NMJs in Both Wild-Type and Mutated Agrin-Injected Muscle
(A) Agrin-induced extrasynaptic clusters are double labeled for AChR in red and for chick agrin in green and observed by confocal microscopy. The calibration bar represents 4 μm.
(B) Native NMJs are double labeled for AChR in red and for chick agrin in green and observed by confocal microscopy. The calibration bar represents 10 μm. Note that chick agrin is present at NMJs in both wild-type and mutated-agrin-injected muscle, but not in saline-injected rat soleus muscle.
When we tested deposition of recombinant chick agrin at the endogenous NMJs, we observed that they were efficiently incorporated at the NMJ sites (Figure 9B). The staining was specific; it could not be detected upon saline injection. This accumulation of agrin is not directly correlated with the amount of protein injected, thus precluding the possibility of performing dose-response experiments. The amounts of agrin protein in the basement membrane of transgenic mice expressing small amounts of the transgene are still similar to those of mice that express much higher levels of the transgenic mRNA.29
The morphology of the pre- and postsynaptic compartments was evaluated in injected muscles by AChR and neurofilament staining (Figure 10A). First, the total area of the synaptic gutters of each NMJ was measured on “en face” views, and the mean fluorescent intensity of the α-bungarotoxin staining was measured. The presence of mutant agrin did not significantly alter the area of the primary gutters (mutant agrin = 95% ± 9%; wild-type agrin = 97% ± 8% of area in saline-injected control animals) or the mean intensity of AChR staining per NMJ (mutant agrin = 97% ± 13%; wild-type agrin = 101% ± 12% of mean intensity per NMJ in saline-injected control animals), consistent with the observation in cultured myotubes that the mutation does not affect AChR expression and recruitment to the membrane. NMJ morphologies were next classified according to the categorization used for human biopsies (see Figure 2A): (1) fork-shaped nerve terminal innervating a well-defined synaptic structure (normal); (2) absence of nerve terminal with fragmented AChR organization (denervated); (3) modified nerve terminals with fragmented AChR gutters corresponding to a remodeling process of either denervation or reinnervation (remodeling); and (4) newly formed NMJs (neoformed) with large-diameter axons associated with small AChR cups or gutters.
Figure 10.
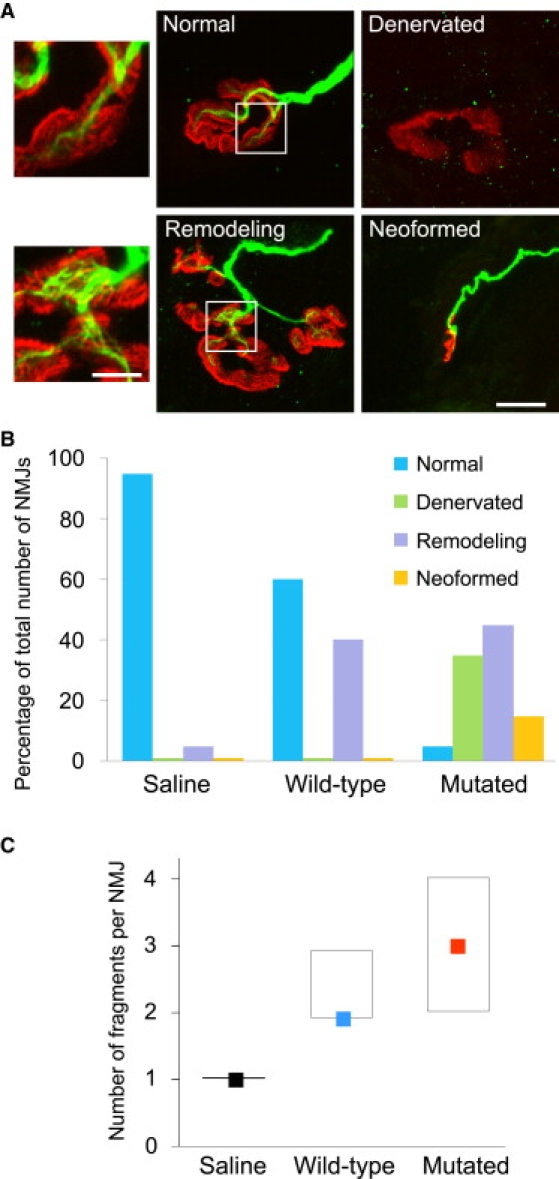
Mutated Agrin Induces Remodeling of Pre-existing NMJs and Their Fragmentation
(A) Rat soleus muscles were injected with saline, wild-type agrin, or mutated agrin. After 2 weeks, muscles were removed and stained for AChRs with α-bungarotoxin (red) and neurofilament antibodies (green). As for the patient biopsy specimen (see Figure 2), the different NMJs observed were divided into four categories: normal, denervated, remodeling, and neoformed. Note in the bottom insert the disheveled appearance of the stained neurofilaments observed in the NMJ in remodeling. This peculiar aspect of disassembly of terminal axons was specific of NMJs after injection of mutated agrin and was never observed after injection of wild-type agrin. The scale bar represents 10 μm for the four low-magnification prints and 4 μm for the two inserts.
(B) The graph represents the distribution of the NMJs in these four categories depending on the injection: saline, wild-type agrin, or mutated agrin. Note that the distribution for mutated agrin is similar to that observed in the patient muscle-biopsy specimen (Figure 2B).
(C) The fragmentation of NMJs was significantly increased in mutated agrin-treated muscles in comparison with the saline-treated and even the wild-type agrin-treated muscles according to Kruskal-Wallis test (saline: n = 42; wild-type agrin: n = 45; mutated agrin: n = 44; H = 52; p < 0.001). Colored squares represent medians, and boxes represent 25th to 75th percentiles.
Figure 10B shows the distribution of NMJs from the various injected muscles into these four categories. Saline-injected muscles mostly presented normal-shaped NMJs, with 5% remodeling and barely any denervated or neoformed NMJs. In muscles injected with wild-type agrin, 60% of the NMJs were normal, but 40% showed some remodeling, which was significantly higher than in controls. Denervated or neoformed NMJs were almost never seen. Conversely, in muscles injected with mutant agrin, only 4% of the NMJs were normal, 44% underwent remodeling, 34% were completely denervated, and 18% were classified as neoformed.
When the presynaptic compartment of the “remodeling” NMJs was analyzed more closely, major variations in the caliber and structure of the nerve terminals were observed. Peculiar aspects of the disassembly of terminal axons included a disheveled appearance of the stained neurofilaments (see lower insert in Figure 10A) and were observed in 70% of the remodeling NMJs after injection of mutated agrin, very similar to what was observed in the patient biopsy specimen, whereas such features were never observed in the remodeling NMJs after injection of wild-type agrin.
To further assess the changes observed on the postsynaptic side, we evaluated NMJ fragmentation by counting the number of individualized synaptic gutters (visualized with fluorescently labeled α-bungarotoxin) for each NMJ, except for neoformed NMJs (Figure S1), as was done for the patient muscle-biopsy specimen. Whereas normal NMJs are made of a single continuous gutter, denervated and remodeling NMJs comprise several of them. In comparison to injection with wild-type agrin or saline, the injection of mutant agrin induced a striking increase in the mean number of synaptic gutter fragments in individual NMJs (Figure 10C).
Altogether, these results demonstrate that the agrin p.Gly1709Arg substitution perturbs the organization of the NMJ, on both the pre- and postsynaptic sides, in a manner highly reminiscent of what was observed in the patient biopsies. Mutant agrin affects neither the size nor the total amount of AChRs at the NMJ, thus demonstrating that the perturbations induced by p.Gly1709Arg are not associated with a significant impairment of AChR expression at the postsynaptic membrane.
Discussion
We describe here a case of CMS associated with a homozygous missense mutation c.5125G>C in AGRN, which induces the expression of a functionally abnormal p.Gly1709Arg agrin protein. The CMS is of autosomal-recessive inheritance and occurred in the context of an inbred community. Both parents carry one mutated allele that has been transmitted to their two affected children. Both siblings present quite similar mild and stable symptoms that began in childhood with lid ptosis without diplopia, mild lower proximal limb weakness, and a thin thorax and pelvis. Fluctuations were reported, and abnormal decrement during repetitive nerve-stimulation studies was present. Both patients are unresponsive to cholinesterase inhibitors, and the index patient was unresponsive to 3,4-DAP, whereas her brother was slightly improved by this treatment but did not tolerate it. Both patients' conditions were remarkably improved after ephedrine administration. The three other children are asymptomatic. Among them, two carry one mutated allele but were not examined clinically or electrophysiologically.
NMJ alterations in the muscle-biopsy specimen of the index patient suggested that they could be related to the mutation. The injection of mutant agrin in rat soleus muscle clearly perturbs the organization of NMJ pre- and postsynaptic compartments. These perturbations are revealed by the fragmentation of the synaptic gutters and the disappearance and/or restructuring of the nerve terminals. The c.5125G>C mutation in AGRN is therefore sufficient to impair the NMJ, and the similarity of the features it induces to those observed in the muscle biopsies of the patient indicates that this mutation is indeed responsible for the CMS.
Interestingly, wild-type and mutant agrins have quite similar effects on AChR aggregation when applied to cultured muscle cells. In addition, in vivo, both mutated and wild-type chick agrins efficiently induce the formation of extrasynaptic AChR clusters in injected rat muscle, thus demonstrating that they can trigger the formation and the activity of the LRP4-MuSK complex.7,8 Likewise, wild-type and mutant agrins have the same ability to activate and phosphorylate MuSK. Finally, AChR levels are unaffected by the mutation, as evidenced by the absence of a decrease in synaptic AChR density or primary gutter area. Altogether, these observations indicate that the ability of neural agrin to induce AChR clustering, MuSK phosphorylation, and AChR expression is not affected.
The main effect of the injected mutant agrin is on the destabilization of pre-existing adult NMJs. The architecture of the NMJ is profoundly altered: synaptic gutters are highly fragmented, and axon terminal branches are greatly modified in comparison with muscles injected with wild-type agrin or saline. In addition, numerous NMJs are denervated, suggesting that their axon terminals have lost their capacity to regenerate, contrary to what occurs with the injection of wild-type agrin, when no denervated NMJs are observed. The possibility that these changes might be due to an immune response related to the expression of chick agrin in another species has already been dealt with by others and ruled out.30 Moreover, the protein-injection method used in this paper was also employed by others without triggering an immune response.31–33 The NMJ changes observed experimentally result from the injection of agrin doses above the physiological levels. However, our immunofluorescence images show that the injected NMJs probably incorporate normal amounts of agrin. This is consistent with previous work showing that the NMJ regulates the amount of agrin it incorporates, regardless of the expression level of the protein.12,29 Because injected agrin is efficiently incorporated into the NMJs, it can compete with the endogenous agrin, and given the high molecular excess of injected agrin, it might replace most of the endogenous molecules. Injected NMJs thus probably mainly contain injected agrin at physiological concentrations.
Whatever the underlying mechanism, one can observe a very similar partitioning of normal, denervated, remodeling and newly formed NMJs in the patient muscle-biopsy specimen and the rat muscle injected with mutant agrin. Overall, the number of NMJ fragments is increased 3-fold in mutated-agrin-treated muscle in comparison with muscles injected with wild-type agrin or saline. However, this fragmentation is accompanied by changes neither in mean synaptic area nor in mean AChR synaptic density. This implies that providing additional agrin, either mutated or wild-type, to NMJs already containing endogenous agrin could participate in the renewal of AChRs within the 15 day period of the experiment in the same manner as was shown with extrasynaptic clusters. Alternatively, agrin injected in the junctional area could generate AChR clusters near the NMJs, which could attract nerve terminals because of their proximity.
A possible mechanism for inducing NMJ fragmentation could involve the binding of agrin to molecules of the basal lamina. The p.Gly1709Arg substitution is located in the LG2 domain, which is involved in the binding of agrin to α-dystroglycan15,20, and the interaction between agrin and α-dystroglycan was shown to be involved in the structuring of the basal lamina through laminin interaction with the amino-terminal of agrin.34 However, α-dystroglycan was recently shown to be dispensable in restoration of synaptogenesis12 and this argues against a role of a modified interaction between α-dystroglycan and mutated agrin in the NMJ changes. Anyhow, the binding experiments showed that the mutation did not change the interaction between agrin and α-dystroglycan.
The fragmentation of synaptic gutters could also be the consequence of modifications of muscle cytoskeleton molecules, such as α-dystrobrevin35 or α-syntrophin.36 Mice deficient for these molecules have NMJs of frayed appearance, similar to what was noted in the patient biopsy specimen and the rat model.
Finally, for whatever reason, the axon terminals undergo major modifications (diameter decrease and neurofilament destructuring, if not complete disappearance) in the patient and the injected rats. These alterations are likely to induce neurotransmission dysfunction, which in turn would induce the destabilization of the synaptic gutters.37 Consistently, agrin was shown to bind the α3Na+/K+-ATPase and to regulate its Na+/K+-ATPase activity.38 α3Na+/K+-ATPase was shown by axotomy to be located in nerve terminals at the neuromuscular junction39 and to modulate Na+ pump physiology.40 Mutations in agrin could thereby interfere with neurotransmitter release and so destabilize NMJs.
In conclusion, the p.Gly1709Arg substitution we have identified does not seem to affect agrin's canonical function, which is to induce the formation of the postsynaptic compartment. Rather, it seems to perturb the maintenance of the NMJ.41
Our results thus indicate that agrin possesses functions in addition to those previously described to be mediated by its N-terminal laminin-binding domain and C-terminal A/z and B/z inserts. Further investigations will be needed to pin down the detailed molecular mechanisms involved in the destabilization of both the pre- and postsynaptic compartments of the NMJ by this mutation.
Supplemental Data
Supplemental Data include two figures and two tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Single Nucleotide Polymorphism, http://www.ncbi.nlm.nih.gov/SNP/
Acknowledgments
This work was supported by Assistance Publique-Hôpitaux de Paris (PHRC AOM 1036), Réseaux Inserm, ANR-Maladies Rares (#ANR-07-MRAR-001), Association Française contre les Myopathies, Comité Mixte Franco-Tunisien pour la Coopération Universitaire (CMCU Project #05G0809), and a Contrat d'Interface AP-HP Inserm (to D.H.).
References
- 1.Hantaï D., Richard P., Koenig J., Eymard B. Congenital myasthenic syndromes. Curr. Opin. Neurol. 2004;17:539–551. doi: 10.1097/00019052-200410000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Engel A.G., Sine S.M. Current understanding of congenital myasthenic syndromes. Curr. Opin. Pharmacol. 2005;5:308–321. doi: 10.1016/j.coph.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Müller J.S., Mihaylova V., Abicht A., Lochmüller H. Congenital myasthenic syndromes: Spotlight on genetic defects of neuromuscular transmission. Expert Rev. Mol. Med. 2007;9:1–20. doi: 10.1017/S1462399407000427. [DOI] [PubMed] [Google Scholar]
- 4.Maselli R.A., Ng J.J., Anderson J.A., Cagney O., Arredondo J., Williams C., Wessel H.B., Abdel-Hamid H., Wollmann R.L. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J. Med. Genet. 2009;46:203–208. doi: 10.1136/jmg.2008.063693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chevessier F., Faraut B., Ravel-Chapuis A., Richard P., Gaudon K., Bauché S., Prioleau C., Herbst R., Goillot E., Ioos C. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum. Mol. Genet. 2004;13:3229–3240. doi: 10.1093/hmg/ddh333. [DOI] [PubMed] [Google Scholar]
- 6.Beeson D., Higuchi O., Palace J., Cossins J., Spearman H., Maxwell S., Newsom-Davis J., Burke G., Fawcett P., Motomura M. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. doi: 10.1126/science.1130837. [DOI] [PubMed] [Google Scholar]
- 7.Kim N., Stiegler A.L., Cameron T.O., Hallock P.T., Gomez A.M., Huang J.H., Hubbard S.R., Dustin M.L., Burden S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang B., Luo S., Wang Q., Suzuki T., Xiong W.C., Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sobel A., Heidmann T., Hofler J., Changeux J.P. Distinct protein components from Torpedo marmorata membranes carry the acetylcholine receptor site and the binding site for local anesthetics and histrionicotoxin. Proc. Natl. Acad. Sci. USA. 1978;75:510–514. doi: 10.1073/pnas.75.1.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brockhausen J., Cole R.N., Gervásio O.L., Ngo S.T., Noakes P.G., Phillips W.D. Neural agrin increases postsynaptic ACh receptor packing by elevating rapsyn protein at the mouse neuromuscular synapse. Dev. Neurobiol. 2008;68:1153–1169. doi: 10.1002/dneu.20654. [DOI] [PubMed] [Google Scholar]
- 11.Misgeld T., Kummer T.T., Lichtman J.W., Sanes J.R. Agrin promotes synaptic differentiation by counteracting an inhibitory effect of neurotransmitter. Proc. Natl. Acad. Sci. USA. 2005;102:11088–11093. doi: 10.1073/pnas.0504806102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin S., Maj M., Bezakova G., Magyar J.P., Brenner H.R., Rüegg M.A. Muscle wide secretion of a miniaturized form of neural agrin rescues focal neuromuscular innervation in agrin mutant mice. Proc. Natl. Acad. Sci. USA. 2008;105:11406–11411. doi: 10.1073/pnas.0801683105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gautam M., Noakes P.G., Moscoso L., Rupp F., Scheller R.H., Merlie J.P., Sanes J.R. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- 14.Denzer A.J., Brandenberger R., Gesemann M., Chiquet M., Rüegg M.A. Agrin binds to the nerve-muscle basal lamina via laminin. J. Cell Biol. 1997;137:671–683. doi: 10.1083/jcb.137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gee S.H., Montanaro F., Lindenbaum M.H., Carbonetto S. Dystroglycan-alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 16.Scotton P., Bleckmann D., Stebler M., Sciandra F., Brancaccio A., Meier T., Stetefeld J., Rüegg M.A. Activation of muscle-specific receptor tyrosine kinase and binding to dystroglycan are regulated by alternative mRNA splicing of agrin. J. Biol. Chem. 2006;281:36835–36845. doi: 10.1074/jbc.M607887200. [DOI] [PubMed] [Google Scholar]
- 17.Tsim K.W., Rüegg M.A., Escher G., Kröger S., McMahan U.J. cDNA that encodes active agrin. Neuron. 1992;8:677–689. doi: 10.1016/0896-6273(92)90089-v. [DOI] [PubMed] [Google Scholar]
- 18.Rupp F., Ozçelik T., Linial M., Peterson K., Francke U., Scheller R. Structure and chromosomal localization of the mammalian agrin gene. J. Neurosci. 1992;12:3535–3544. doi: 10.1523/JNEUROSCI.12-09-03535.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bezakova G., Rüegg M.A. New insights into the roles of agrin. Nat. Rev. Mol. Cell Biol. 2003;4:295–308. doi: 10.1038/nrm1074. [DOI] [PubMed] [Google Scholar]
- 20.Gesemann M., Denzer A.J., Rüegg M.A. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J. Cell Biol. 1995;128:625–636. doi: 10.1083/jcb.128.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koelle G.B., Friedenwald J.A. A histochemical method for localizing cholinesterase activity. Proc. Soc. Exp. Biol. Med. 1949;70:617–622. doi: 10.3181/00379727-70-17013. [DOI] [PubMed] [Google Scholar]
- 22.Brooke M.H., Kaiser K.K. Some comments on the histochemical characterization of muscle adenosine-triphosphatase. J. Histochem. Cytochem. 1969;17:431–432. doi: 10.1177/17.6.431. [DOI] [PubMed] [Google Scholar]
- 23.Moll J., Barzaghi P., Lin S., Bezakova G., Lochmüller H., Engvall E., Müller U., Rüegg M.A. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 2001;413:302–307. doi: 10.1038/35095054. [DOI] [PubMed] [Google Scholar]
- 24.Fuhrer C., Sugiyama J.E., Taylor R.G., Hall Z.W. Association of muscle-specific kinase MuSK with the acetylcholine receptor in mammalian muscle. EMBO J. 1997;16:4951–4960. doi: 10.1093/emboj/16.16.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gesemann M., Cavalli V., Denzer A.J., Brancaccio A., Schumacher B., Rüegg M.A. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron. 1996;16:755–767. doi: 10.1016/s0896-6273(00)80096-3. [DOI] [PubMed] [Google Scholar]
- 26.Ishigaki K., Nicolle D., Krejci E., Leroy J.P., Koenig J., Fardeau M., Eymard B., Hantaï D. Two novel mutations in the COLQ gene cause endplate acetylcholinesterase deficiency. Neuromuscul. Disord. 2003;13:236–244. doi: 10.1016/s0960-8966(02)00243-2. [DOI] [PubMed] [Google Scholar]
- 27.Meier T., Marangi P.A., Moll J., Hauser D.M., Brenner H.R., Rüegg M.A. A minigene of neural agrin encoding the laminin-binding and acetylcholine receptor-aggregating domains is sufficient to induce postsynaptic differentiation in muscle fibres. Eur. J. Neurosci. 1998;10:3141–3152. doi: 10.1046/j.1460-9568.1998.00320.x. [DOI] [PubMed] [Google Scholar]
- 28.Pun S., Ng Y.P., Yang J.F., Ip N.Y., Tsim K.W. Agrin-deficient myotube retains its acetylcholine receptor aggregation ability when challenged with agrin. J. Neurochem. 1997;69:2555–2563. doi: 10.1046/j.1471-4159.1997.69062555.x. [DOI] [PubMed] [Google Scholar]
- 29.Meinen S., Barzaghi P., Lin S., Lochmüller H., Rüegg M.A. Linker molecules between laminins and dystroglycan ameliorate laminin-alpha2-deficient muscular dystrophy at all disease stages. J. Cell Biol. 2007;176:979–993. doi: 10.1083/jcb.200611152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hashemolhosseini S., Moore C., Landmann L., Sander A., Schwarz H., Witzemann V., Sakmann B., Brenner H.R. Electrical activity and postsynapse formation in adult muscle: gamma-AChRs are not required. Mol. Cell. Neurosci. 2000;16:697–707. doi: 10.1006/mcne.2000.0911. [DOI] [PubMed] [Google Scholar]
- 31.Bezakova G., Helm J.P., Francolini M., Lømo T. Effects of purified recombinant neural and muscle agrin on skeletal muscle fibers in vivo. J. Cell Biol. 2001;153:1441–1452. doi: 10.1083/jcb.153.7.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bezakova G., Lømo T. Muscle activity and muscle agrin regulate the organization of cytoskeletal proteins and attached acetylcholine receptor (AChR) aggregates in skeletal muscle fibers. J. Cell Biol. 2001;153:1453–1463. doi: 10.1083/jcb.153.7.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bezakova G., Rabben I., Sefland I., Fumagalli G., Lømo T. Neural agrin controls acetylcholine receptor stability in skeletal muscle fibers. Proc. Natl. Acad. Sci. USA. 2001;98:9924–9929. doi: 10.1073/pnas.171539698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishimune H., Valdez G., Jarad G., Moulson C.L., Müller U., Miner J.H., Sanes J.R. Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J. Cell Biol. 2008;182:1201–1215. doi: 10.1083/jcb.200805095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grady R.M., Zhou H., Cunningham J.M., Henry M.D., Campbell K.P., Sanes J.R. Maturation and maintenance of the neuromuscular synapse: Genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron. 2000;25:279–293. doi: 10.1016/s0896-6273(00)80894-6. [DOI] [PubMed] [Google Scholar]
- 36.Adams M.E., Kramarcy N., Krall S.P., Rossi S.G., Rotundo R.L., Sealock R., Froehner S.C. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol. 2000;150:1385–1398. doi: 10.1083/jcb.150.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cairns N.J., Lee V.M., Trojanowski J.Q. The cytoskeleton in neurodegenerative diseases. J. Pathol. 2004;204:438–449. doi: 10.1002/path.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hilgenberg L.G., Ho K.D., Lee D., O'Dowd D.K., Smith M.A. Alpha3Na+/K+-ATPase is a neuronal receptor for agrin. Cell. 2006;125:359–369. doi: 10.1016/j.cell.2006.01.052. [DOI] [PubMed] [Google Scholar]
- 39.Zahler R., Sun W., Ardito T., Zhang Z.T., Kocsis J.D., Kashgarian M. The alpha3 isoform protein of the Na+, K(+)-ATPase is associated with the sites of cardiac and neuromuscular impulse transmission. Circ. Res. 1996;78:870–879. doi: 10.1161/01.res.78.5.870. [DOI] [PubMed] [Google Scholar]
- 40.Blaustein M.P. Physiological effects of endogenous ouabain: Control of intracellular Ca2+ stores and cell responsiveness. Am. J. Physiol. 1993;264:C1367–C1387. doi: 10.1152/ajpcell.1993.264.6.C1367. [DOI] [PubMed] [Google Scholar]
- 41.Denzer A.J., Hauser D.M., Gesemann M., Rüegg M.A. Synaptic differentiation: The role of agrin in the formation and maintenance of the neuromuscular junction. Cell Tissue Res. 1997;290:357–365. doi: 10.1007/s004410050941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.