

Abstract
Ichthyosis prematurity syndrome (IPS) is an autosomal-recessive disorder characterized by premature birth and neonatal asphyxia, followed by a lifelong nonscaly ichthyosis with atopic manifestations. Here we show that the gene encoding the fatty acid transport protein 4 (FATP4) is mutated in individuals with IPS. Fibroblasts derived from a patient with IPS show reduced activity of very long-chain fatty acids (VLCFA)-CoA synthetase and a specific reduction in the incorporation of VLCFA into cellular lipids. The human phenotype is consistent with Fatp4 deficiency in mice that is characterized by a severe skin phenotype, a defective permeability barrier function, and perturbed VLCFA metabolism. Our results further emphasize the importance of fatty acid metabolism for normal epidermal barrier function illustrated by deficiency of a member in the FATP family of proteins.
Main Text
Autosomal-recessive congenital ichthyosis (ARCI) is a heterogeneous group of keratinization disorders characterized by a defective barrier function.1,2 ARCI is associated with mutations in genes of importance for the intercellular lipid layer or cornified cell envelope.3 To date, seven genes are known to cause ARCI in humans: TGM1 (MIM ∗190195),4,5 ABCA12 (MIM ∗607800),6,7 the two lipoxygenase genes ALOXE3 (MIM ∗607206) and ALOX12B (MIM ∗603741),8 CGI-58 (ABHD5 [MIM ∗604780]),9,10 Ichthyin (NIPAL4 [MIM ∗609383]),11,12 and CYP4F22 (MIM ∗611495).13 Furthermore, linkage analysis has established at least three additional ARCI loci on chromosomes 9q33.3-q34.13,14,15 12p11.2-q13,16 and 19p13.1-p13.2 (MIM %604781).17 The 9q33.3-q34.13 loci is associated with the ichthyosis prematurity syndrome (IPS [MIM %608649]) or ichthyosis congenita type IV.18 Key features in IPS are complications in the second trimester of pregnancy resulting from polyhydramnion, with premature birth of a child with thick caseous desquamating epidermis, respiratory complications, and transient eosinophilia. After recovery during the first months of life, the symptoms are relatively benign and the patients suffer from a lifelong nonscaly ichthyosis with atopic manifestations (Figures 1A–1C). Ultrastructural analyses reveal membrane aggregations in the upper epidermal layers, and histological analysis of the skin shows a thickening of the epidermis (Figures 1D–1F). The prevalence of IPS is relatively high in a region across mid-Scandinavia, suggesting a founder effect in this population with one predominant ancestral mutation.14 We performed a homozygosity scan on a consanguineous family of North African origin comprising several individuals with IPS. Affected family members were found homozygous for a 76 kb genomic region on chromosome 9p that coincides with the IPS locus in the Scandinavian families. The region spans four genes, including the FATP4 (SLC27A4 [MIM ∗604194]) gene encoding a member of the fatty acid transport protein (FATPs or solute carrier family 27, SLC27) family. The FATP4 gene consists of 13 exons encoding a peptide of 643 residues with a predicted size of 72 kDa. FATP4 is highly conserved among widely divergent species and contains an N-terminal transmembrane (TM) region, an ER localization signal (ERx), and an AMP-binding domain.19 The FATP family comprises transmembrane proteins of importance for the uptake of exogenous fatty acids into cells.20 Members of this family also function as acyl-CoA synthetases with specificity for very long-chain fatty acids (VLCFA) or bile acids.21 Several FATPs, including FATP4, are present in the epidermis, and animal studies suggest an important role of these transporters for skin barrier function.22 Fatp4−/− mice die either in utero23 or in the neonatal period,24,25 showing a thickened, malformed skin with ineffective barrier properties and severe breathing difficulties secondary to the tight skin. The critical role of FATP4 is demonstrated by rescuing the lethal phenotype in Fatp4−/− mice by keratinocyte-specific transgenic expression of FATP4, resulting in viable mice with only a mild skin phenotype.26
Figure 1.
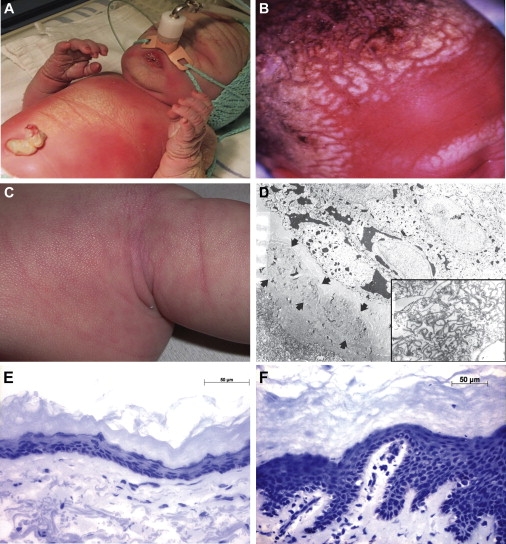
Key Features of IPS
(A and B) Premature birth of a child with IPS showing thick, caseous, desquamating skin, particularly of the head, and respiratory complications.
(C) Later, the phenotype becomes mild with a cobblestone-like surface. The pictures are published with permission from the parents.
(D) Electron microscopic analysis of superficial epidermis from an IPS patient showing accumulation of curved multilamellar membranes in stratum granulosum (×7,500); the inset shows the region at a higher magnification (×30,000).
(E and F) Histological analysis by hematoxylin and eosin (H&E) staining of skin from an IPS patient (E) showing thickening of the epidermis when compared to normal control (F).
Sequence analysis of FATP4 in affected members from the North African family, 1 Middle Eastern family, and 18 IPS families of Scandinavian origin revealed 7 different mutations (Figure 2A). The study complies with the declaration of Helsinki and is performed according to a protocol approved by the regional ethical committee at CEA and at the Universities of Uppsala and Bab El Oued. Sequencing of 120 healthy control individuals failed to reveal any of the described mutations (N = 240; >80% power with a polymorphism frequency of 0.01).27 All Scandinavian patients were found homozygous or compound heterozygous for a nonsense mutation (c.504c>a [p.C168X]) in exon 3, indicating a common ancestor for this mutation. A founder effect was confirmed by the presence of a conserved 786 kb region spanning the mutation (Figure S1 available online). The North African patient, daughter to consanguineous parents, was homozygous for an exon 5 acceptor splice site mutation (c.716-1g>a) predicting a truncated protein (Table S1). The mutations segregated with the disease in each family, with parents being heterozygous carriers for the respective mutations based on haplotype analysis. FATPs are required for cellular fatty acid uptake and for the activation of fatty acids. The AMP-binding domain (aa 103–536) of FATP4 comprise two distinct motifs: the ATP/AMP motif, involved in ATP binding and adenylate formation common to all adenylate-forming enzymes (aa 243–345), as well as the FATP/VLACS motif of the FATP and very long-chain acyl-CoA synthetase (VLACS) protein families (aa 500–551), involved in fatty acid binding. We identified two missense mutations (p.S247P and p.Q300R) located in the highly conserved AMP binding domain and one missense mutation situated within the C-terminal domain (p.R583H) (Figure 2B). The functional importance of the AMP-binding domain, as well as the C-terminal domain, has previously been demonstrated by base pair substitution of the FATP1 yeast ortholog, FAT1, and by using protein domain chimeras of FATP1, FATP4, and FATP6.28,29 These studies show that the ATP/AMP and FATP/VLACS motifs are important for transport and activation of fatty acids. Mutations in the ATP/AMP motif repress both fatty acid import and fatty acid activation, supporting the notion that the binding of ATP is required for these functions. Mutations in the FATP/VLACS motif results in an almost complete elimination of both transport and activation, indicating that this region is crucial for FATP function.
Figure 2.
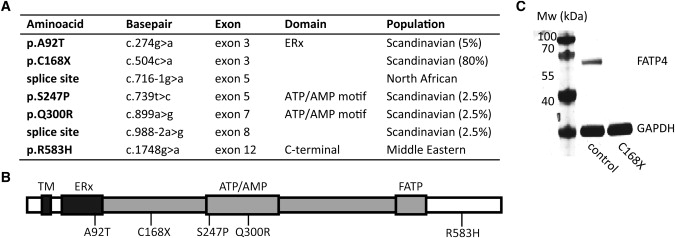
Overview of FATP4 Mutations and Western Analysis
(A) Positions of FATP4 mutations in IPS patients with frequencies in the Scandinavian patients.
(B) Schematic overview of FATP4 functional domains; the N-terminal transmembrane region (TM), the ER localization signal (ERx; aa 47–102), and the AMP binding domain (gray; aa 103–536) containing the ATP/AMP motif involved in ATP binding and adenylate formation (ATP/AMP; aa 243–345) and the conserved FATP motif of importance for fatty acid binding (FATP; aa 500–551). The numbers below the bar correspond to residues mutated in IPS.
(C) Protein blot showing FATP4 protein expression in a control individual and absent expression in a patient homozygous for p.C168X (MW: FATP4 72 kDa, GAPDH 37 kDa) with a monoclonal FATP4 antibody (Abnova, 1F4-1B10).
Western blot analysis of keratinocyte and fibroblast cell lines derived from an adult IPS patient and a control individual was performed with an antibody detecting the C-terminal part of FATP4. Cells from the patient homozygous for p.C168X did not show any detectable band whereas cells from the normal donor revealed a single band corresponding to the size of FATP4 (Figure 2C). Immunohistochemistry with the same antibody on normal skin showed a strong expression of FATP4 (Figure 3A), whereas no staining was detected in a skin biopsy from the IPS patient (Figure 3B), demonstrating absence of full-length FATP4.
Figure 3.
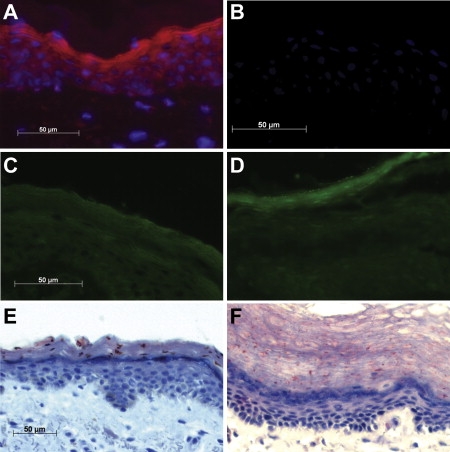
Analysis of Skin Biopsies
Analysis of skin biopsies from a control (A, C, E) and an IPS patient homozygous for p.C168X (B, D, F).
(A and B) Immunostaining with a FATP4 antibody (Abnova) shows strong expression in the normal epidermis but no expression in the patient specimen.
(C and D) Nile red staining shows an even distribution of lipids in the control biopsy whereas IPS is associated with an accumulation of lipids in the stratum granulosum that coincides with the normal expression pattern of FATP4 in (A).
(E and F) Staining of neutral lipids with Oil Red O shows few lipid droplets in normal skin but numerous droplets in the epidermis of the patient.
The fatty acid transport properties as well as the enzymatic activity of FATP4 suggest an important role of the protein for the formation of the skin barrier. To investigate for presence of skin lipids in epidermis, we stained neutral lipids with Nile red. We observed an equal staining of the epidermal layers in normal skin (Figure 3C) in contrast to an intense lipid staining restricted to stratum corneum (SC) in skin from an adult IPS patient (Figure 3D). The abnormal distribution of lipids between epidermal layers in IPS is consistent with the expression pattern of FATP4 in normal epidermis, which seems strongest in the outer layers (Figure 3A). Epidermal lipid distribution was also investigated with Oil Red O. Normal epidermis show few lipid droplets (Figure 3E) when compared to epidermis of an IPS patient exhibiting numerous small droplets (Figure 3F). These observations suggest a defect in lipid homeostasis and skin barrier formation in IPS, which is similar to that found in the Slc27a4wrfr mice.25
Murine fibroblasts with FATP4 deficiency exhibit a substantial reduction in VLCFA activation, suggesting that this enzyme is a major VLCFA-CoA synthetase.30 To investigate the biochemical effects of FATP4 mutations, we determined LCFA-CoA and VLCFA-CoA synthetase activity in lysates obtained from fibroblasts homozygous for the p.C168X mutation. With palmitic acid (C16:0) as substrate, acyl-CoA formation was identical in wild-type and mutant cells. In the presence of erucic acid (C22:1), however, acyl-CoA formation was reduced by 55% in mutated cells when compared to control cells (wild-type), suggesting a defect in VLCFA activation (Figure 4A). We next determined the incorporation of radiolabeled palmitic acid and erucic acid into neutral and polar lipids of cultured fibroblasts. As shown in Figures 4A–4D, mutated fibroblasts show a decreased incorporation of erucic acid into cholesterol esters (CE), triglycerides (TG), and phospholipids (PL) by 69%, 60%, and 37%, respectively. The incorporation of palmitic acid was unchanged in the CE (Figure 4B) and TG (Figure 4C) fractions and slightly increased (29%) in the PL fraction (Figure 4D). In contrast to observations in murine fibroblasts,30 p.C168X fibroblasts did not show a reduced incorporation of fatty acids into the phosphatitylethanolamine (PE) and phosphatidylcholin (PC) fractions. However, in accordance with this study, we observed reduced accumulation of radioactivity in the lysophophatitylcholin (LPC) fraction (Figure S2). Cellular neutral lipid accumulation was also investigated with BODIPY 493/503 and FA uptake by C1-BODIPY-C12, a fatty acid analog that mimics LCFA, respectively. Patient-derived and control keratinocytes show a similar lipid content and uptake of BODIPY (Figure S3). Taken together, our results demonstrate that FATP4 deficiency in human fibroblasts is associated with reduced VLCFA-CoA synthetase activity and a reduced incorporation of VLCFA into neutral and polar lipids.
Figure 4.
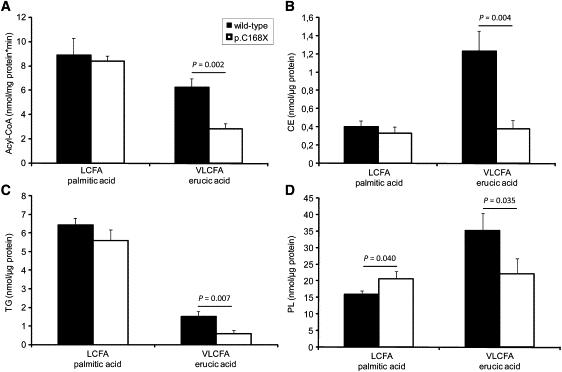
Fatty Acid Activation in Fibroblasts from a Control Individual and an IPS Patient Homozygous for p.C168X
(A) Acyl-CoA synthetase activities in lysates of wild-type (filled bars) and p.C168X (open bars) fibroblasts. Activation of VLCFA and LCFA were determined with erucic acid (22:1) and palmitic acid (16:0) as substrate, respectively.
(B–D) Incorporation of 3H-labeled erucic acid and palmitic acid (LCFA) into (B) cholesterol esters (CE), (C) triglycerides (TG), and (D) phospholipids (PL) of wild-type and p.C168X fibroblast. Cells were cultured in the presences of 50 μM radiolabeled fatty acids for 6 hr. Subsequently, lipid fractions were separated by TLC and the comigrating radioactivity was determined by liquid scintillation counting. Data show the mean ± standard deviation (SD) and are representative for three independent experiments done in triplicate. p values are generated with two-tailed Student's t test assuming equal variance.
Mice with a disruption of Fatp4 present with a defective skin barrier function resulting in a severe skin phenotype, which has been described as restrictive.24 Interestingly, adult mice can partially compensate for Fatp4 deficiency by other Fatps, and surviving Fatp4-deficient mice present with a milder phenotype from the age of 10 weeks.31 This suggests that Fatp4 is critical for the generation of the epidermal barrier during the embryogenic and neonatal period but of less importance for its maintenance where other FATPs may compensate. Similarly, the human IPS phenotype is severe in the neonatal period but symptoms become milder with age and are often benign in adults. Lipid transport and/or processing are pivotal for the skin barrier function. This is supported by mutations identified in different genes encoding proteins of lipid metabolism downstream of the FATPs in disorders of keratinization.1 Our findings provide, to our knowledge, the first example of a human inherited disease caused by deficiency of a member in the FATP family. These observations emphasize the critical role of fatty acid metabolism for normal epidermal barrier function. Exactly how the barrier defect in IPS might explain massive scaling in utero and prematurity with asphyxia at birth remains to be studied. This also applies to the intriguing connection between IPS and transient eosinophilia with atopic manifestations later in life.
Acknowledgments
The authors thank the individuals with IPS and their families for participating in this research. We would like to thank Florence Jobard for assistance in sequencing. The Norwegian patients were diagnosed and sampled in collaboration with Tobias Gedde-Dahl, who passed away in 2006. This work has been financially supported by the Swedish Research Council, The Swedish Society for Medical Research, The Torsten and Ragnar Söderbergs Fund, Uppsala University, and University Hospital. The work has also been financially supported by CEA Institut de Génomique. The authors declare no conflict of interest.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Akiyama M., Shimizu H. An update on molecular aspects of the non-syndromic ichthyoses. Exp. Dermatol. 2008;17:373–382. doi: 10.1111/j.1600-0625.2007.00691.x. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama M. Harlequin ichthyosis and other autosomal recessive congenital ichthyoses: The underlying genetic defects and pathomechanisms. J. Dermatol. Sci. 2006;42:83–89. doi: 10.1016/j.jdermsci.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Elias P.M., Williams M.L., Holleran W.M., Jiang Y.J., Schmuth M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: Inherited disorders of lipid metabolism. J. Lipid Res. 2008;49:697–714. doi: 10.1194/jlr.R800002-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huber M., Rettler I., Bernasconi K., Frenk E., Lavrijsen S.P., Ponec M., Bon A., Lautenschlager S., Schorderet D.F., Hohl D. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science. 1995;267:525–528. doi: 10.1126/science.7824952. [DOI] [PubMed] [Google Scholar]
- 5.Russell L.J., DiGiovanna J.J., Rogers G.R., Steinert P.M., Hashem N., Compton J.G., Bale S.J. Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat. Genet. 1995;9:279–283. doi: 10.1038/ng0395-279. [DOI] [PubMed] [Google Scholar]
- 6.Kelsell D.P., Norgett E.E., Unsworth H., Teh M.T., Cullup T., Mein C.A., Dopping-Hepenstal P.J., Dale B.A., Tadini G., Fleckman P. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am. J. Hum. Genet. 2005;76:794–803. doi: 10.1086/429844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lefevre C., Audebert S., Jobard F., Bouadjar B., Lakhdar H., Boughdene-Stambouli O., Blanchet-Bardon C., Heilig R., Foglio M., Weissenbach J. Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Hum. Mol. Genet. 2003;12:2369–2378. doi: 10.1093/hmg/ddg235. [DOI] [PubMed] [Google Scholar]
- 8.Jobard F., Lefevre C., Karaduman A., Blanchet-Bardon C., Emre S., Weissenbach J., Ozguc M., Lathrop M., Prud'homme J.F., Fischer J. Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum. Mol. Genet. 2002;11:107–113. doi: 10.1093/hmg/11.1.107. [DOI] [PubMed] [Google Scholar]
- 9.Fischer J., Faure A., Bouadjar B., Blanchet-Bardon C., Karaduman A., Thomas I., Emre S., Cure S., Ozguc M., Weissenbach J. Two new loci for autosomal recessive ichthyosis on chromosomes 3p21 and 19p12-q12 and evidence for further genetic heterogeneity. Am. J. Hum. Genet. 2000;66:904–913. doi: 10.1086/302814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lefevre C., Jobard F., Caux F., Bouadjar B., Karaduman A., Heilig R., Lakhdar H., Wollenberg A., Verret J.L., Weissenbach J. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am. J. Hum. Genet. 2001;69:1002–1012. doi: 10.1086/324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dahlqvist J., Klar J., Hausser I., Anton-Lamprecht I., Pigg M.H., Gedde-Dahl T., Jr., Ganemo A., Vahlquist A., Dahl N. Congenital ichthyosis: Mutations in ichthyin are associated with specific structural abnormalities in the granular layer of epidermis. J. Med. Genet. 2007;44:615–620. doi: 10.1136/jmg.2007.050542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lefevre C., Bouadjar B., Karaduman A., Jobard F., Saker S., Ozguc M., Lathrop M., Prud'homme J.F., Fischer J. Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum. Mol. Genet. 2004;13:2473–2482. doi: 10.1093/hmg/ddh263. [DOI] [PubMed] [Google Scholar]
- 13.Lefevre C., Bouadjar B., Ferrand V., Tadini G., Megarbane A., Lathrop M., Prud'homme J.F., Fischer J. Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Hum. Mol. Genet. 2006;15:767–776. doi: 10.1093/hmg/ddi491. [DOI] [PubMed] [Google Scholar]
- 14.Klar J., Gedde-Dahl T., Jr., Larsson M., Pigg M., Carlsson B., Tentler D., Vahlquist A., Dahl N. Assignment of the locus for ichthyosis prematurity syndrome to chromosome 9q33.3–34.13. J. Med. Genet. 2004;41:208–212. doi: 10.1136/jmg.2003.012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melin M., Klar J., Gedde-Dahl T., Jr., Fredriksson R., Hausser I., Brandrup F., Bygum A., Vahlquist A., Hellstrom Pigg M., Dahl N. A founder mutation for ichthyosis prematurity syndrome restricted to 76 kb by haplotype association. J. Hum. Genet. 2006;51:864–871. doi: 10.1007/s10038-006-0035-z. [DOI] [PubMed] [Google Scholar]
- 16.Mizrachi-Koren M., Geiger D., Indelman M., Bitterman-Deutsch O., Bergman R., Sprecher E. Identification of a novel locus associated with congenital recessive ichthyosis on 12p11.2-q13. J. Invest. Dermatol. 2005;125:456–462. doi: 10.1111/j.0022-202X.2005.23777.x. [DOI] [PubMed] [Google Scholar]
- 17.Virolainen E., Wessman M., Hovatta I., Niemi K.M., Ignatius J., Kere J., Peltonen L., Palotie A. Assignment of a novel locus for autosomal recessive congenital ichthyosis to chromosome 19p13.1-p13.2. Am. J. Hum. Genet. 2000;66:1132–1137. doi: 10.1086/302813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anton-Lamprecht I. Diagnostic ultrastructure of non-neoplastic diseases. In: Papadimitriou J.M., Spagnolo D.V., editors. The Skin, H.D. Churchill Livingstone; Edinburgh: 1992. pp. 459–550. [Google Scholar]
- 19.Milger K., Herrmann T., Becker C., Gotthardt D., Zickwolf J., Ehehalt R., Watkins P.A., Stremmel W., Fullekrug J. Cellular uptake of fatty acids driven by the ER-localized acyl-CoA synthetase FATP4. J. Cell Sci. 2006;119:4678–4688. doi: 10.1242/jcs.03280. [DOI] [PubMed] [Google Scholar]
- 20.Stahl A. A current review of fatty acid transport proteins (SLC27) Pflugers Arch. 2004;447:722–727. doi: 10.1007/s00424-003-1106-z. [DOI] [PubMed] [Google Scholar]
- 21.Gimeno R.E. Fatty acid transport proteins. Curr. Opin. Lipidol. 2007;18:271–276. doi: 10.1097/MOL.0b013e3281338558. [DOI] [PubMed] [Google Scholar]
- 22.Schmuth M., Ortegon A.M., Mao-Qiang M., Elias P.M., Feingold K.R., Stahl A. Differential expression of fatty acid transport proteins in epidermis and skin appendages. J. Invest. Dermatol. 2005;125:1174–1181. doi: 10.1111/j.0022-202X.2005.23934.x. [DOI] [PubMed] [Google Scholar]
- 23.Gimeno R.E., Hirsch D.J., Punreddy S., Sun Y., Ortegon A.M., Wu H., Daniels T., Stricker-Krongrad A., Lodish H.F., Stahl A. Targeted deletion of fatty acid transport protein-4 results in early embryonic lethality. J. Biol. Chem. 2003;278:49512–49516. doi: 10.1074/jbc.M309759200. [DOI] [PubMed] [Google Scholar]
- 24.Herrmann T., van der Hoeven F., Grone H.J., Stewart A.F., Langbein L., Kaiser I., Liebisch G., Gosch I., Buchkremer F., Drobnik W. Mice with targeted disruption of the fatty acid transport protein 4 (Fatp 4, Slc27a4) gene show features of lethal restrictive dermopathy. J. Cell Biol. 2003;161:1105–1115. doi: 10.1083/jcb.200207080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moulson C.L., Martin D.R., Lugus J.J., Schaffer J.E., Lind A.C., Miner J.H. Cloning of wrinkle-free, a previously uncharacterized mouse mutation, reveals crucial roles for fatty acid transport protein 4 in skin and hair development. Proc. Natl. Acad. Sci. USA. 2003;100:5274–5279. doi: 10.1073/pnas.0431186100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moulson C.L., Lin M.H., White J.M., Newberry E.P., Davidson N.O., Miner J.H. Keratinocyte-specific expression of fatty acid transport protein 4 rescues the wrinkle-free phenotype in Slc27a4/Fatp4 mutant mice. J. Biol. Chem. 2007;282:15912–15920. doi: 10.1074/jbc.M701779200. [DOI] [PubMed] [Google Scholar]
- 27.Collins J.S., Schwartz C.E. Detecting polymorphisms and mutations in candidate genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DiRusso C.C., Darwis D., Obermeyer T., Black P.N. Functional domains of the fatty acid transport proteins: Sstudies using protein chimeras. Biochim. Biophys. Acta. 2008;1781:135–143. doi: 10.1016/j.bbalip.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zou Z., DiRusso C.C., Ctrnacta V., Black P.N. Fatty acid transport in Saccharomyces cerevisiae. Directed mutagenesis of FAT1 distinguishes the biochemical activities associated with Fat1p. J. Biol.0020Chem. 2002;277:31062–31071. doi: 10.1074/jbc.M205034200. [DOI] [PubMed] [Google Scholar]
- 30.Jia Z., Moulson C.L., Pei Z., Miner J.H., Watkins P.A. Fatty acid transport protein 4 is the principal very long chain fatty acyl-CoA synthetase in skin fibroblasts. J. Biol. Chem. 2007;282:20573–20583. doi: 10.1074/jbc.M700568200. [DOI] [PubMed] [Google Scholar]
- 31.Herrmann T., Grone H.J., Langbein L., Kaiser I., Gosch I., Bennemann U., Metzger D., Chambon P., Stewart A.F., Stremmel W. Disturbed epidermal structure in mice with temporally controlled fatp4 deficiency. J. Invest. Dermatol. 2005;125:1228–1235. doi: 10.1111/j.0022-202X.2005.23972.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.