

Abstract
Mucopolysaccharidosis type II (MPSII), or Hunter syndrome, arises from a deficiency in iduronate 2-sulfatase (IDS), and it is characterized by progressive somatic and neurological involvement. The MPSII mouse model reproduces the features of MPSII patients. Systemic administration of the AAV2/5CMV-hIDS vector in MPSII mouse pups results in the full correction of glycosaminoglycan (GAG) accumulation in visceral organs and in the rescue of the defects and GAG accumulation in the central nervous system (CNS). Remarkably, in treated MPSII animals, this CNS correction arises from the crossing of the blood-brain barrier by the IDS enzyme itself, not from the brain transduction. Thus, we show here that early treatment of MPSII mice with one systemic injection of AAV2/5CMV-hIDS results in prolonged and high levels of circulating IDS that can efficiently and simultaneously rescue both visceral and CNS defects for up to 18 months after therapy.
Main Text
Iduronate-2-sulfatase (IDS) is the enzyme that removes the sulfate group from dermatan and heparan sulfate, and its inactivity results in MPSII (MIM 309900), or Hunter syndrome, a rare X-linked inborn error of metabolism. The incidence of MPSII has been estimated at around 1 in 162,000 live births. The deficiency of IDS has been shown to be due to point mutations or deletions in the 24 kb IDS gene.1 As with all of the human sulfatases, IDS needs to be activated by sulfatase modifying factor 1 (SUMF1)2,3 (MIM 607939).
MPSII occurs in both severe and mild forms that cover a broad spectrum of symptoms with slow to rapid pathological progression. The severe form is characterized by progressive somatic and neurological involvement, with an onset between the second and fourth years of age and an early mortality. Facial features, hepatosplenomegaly, short stature, skeletal deformities, joint stiffness, severe retinal degeneration, and hearing impairment are coupled to an incremental deterioration of the neurological system.4
The MPSII (idsy/−) mouse model displays the features of Hunter syndrome.5 Affected mice develop skeleton deformities, particularly of the craniofacial bones, as well as alopecia, and they showed irregular gait, an abnormal walking pattern, and poor locomotor and exploratory abilities in the open-field test. IDS activity is not detectable in any of their tissues, and the affected animals show a progressive accumulation of glycosaminoglycans (GAGs) in many organs and in their urine. In addition, MPSII mice present neuropathological defects. The onset of the gross morphological phenotype is manifested at 3–4 months of age, it becomes progressively severe through their adult life, and MPSII mice usually die by about 60 weeks of age.6 We have previously used an AAV type 2/8 vector carrying human IDS cDNA under the control of the thyroxine-binding globulin liver-specific promoter (AAV2/8TBG-hIDS) to engineer liver secretion of IDS. This efficiently corrected the visceral organs through IDS uptake from the blood.6 It is now imperative to develop an efficient therapy to treat the CNS symptoms. Ideally, having circulating IDS that crosses the blood-brain barrier (BBB) would be highly preferred and would provide treatment of both visceral and CNS defects, simultaneously.
The BBB is formed by a layer of endothelial cells of the brain microvasculature that regulates the passage of molecules between the blood and the CNS. Tight junctions block the passage of most molecules, except some specific substances that cross the BBB via receptor-mediated transport.7
Enzyme-replacement therapy at high doses in mouse models of α-mannosidosis (MIM 609458), MPSVII (MIM 253220), and metachromatic leukodystrophy (MIM 250100) leads to enzyme delivery across the BBB, thereby partially reversing storage in brain tissues.8–10 Similarly, the intrahepatic administration of AAV2 vectors in MPSVII mice results in some correction of the CNS phenotype.11 We have previously shown that systemic administration of AAV2/8TBG-hIDS can lead to very high levels of circulating IDS, which partially cleared glycosaminoglycan (GAG) accumulation in the choroid plexus of idsy/− mice.6
To anticipate disease manifestation and to simultaneously treat the visceral and CNS defects of MPSII, we have again systemically administered the AAV type 2/5 vector carrying human IDS cDNA, but here under the control of the cytomegalovirus promoter (AAV2/5CMV-hIDS), in day 2 (p2) idsy/− mouse pups. We used this type of vector because it has been shown to provide efficient transduction of several tissues, such as skeletal muscle and lung.12,13 We observe full correction of the CNS symptoms and visceral defects. Furthermore, we provide evidence that correction of CNS storage and neuronal markers is due to the crossing of the BBB by IDS.
Groups of p2 idsy/− pups were injected in the temporal vein with 1 x 1011 AAV2/5CMV-hIDS viral particles, and the treated mice were sacrificed in two groups: after 1 month (T1; n = 8) or 18 months (T18; n = 4) of therapy. Control wild-type (WT) (n = 5) and MPSII (n = 5) mice were analyzed in parallel. The IDS activities were analyzed in the plasma of the mice at T1, T6, T12, and T18, and on average, they were up to 5-fold higher in the treated mice in comparison to the WT mice and up to 60-fold higher in the treated mice in comparison to the idsy/− control mice (Table 1). The untreated idsy/− mice died at 14 months. In contrast, the life span of the treated animals was prolonged up to and beyond 18 months. To monitor the systemic excretion of GAGs, we analyzed their accumulation in the urine of the mice at T1, T6, T12, and T18. GAGs were cleared in the urine of all of the treated mice for up to 18 months after the therapy, and their accumulation was almost fully normalized, as compared to WT levels (Table S1, available online). AAV vector preparation, plasma and urine collection, IDS activity assay, and quantitative analysis of GAG excretion were performed as previously described.6
Table 1.
IDS Activity in the Plasma
|
IDS Activity (nmol/4 h/mg) |
||||
|---|---|---|---|---|
| T1 | T6 | T12 | T18 | |
| WT | 320 ± 27 | 243 ± 32.1 | 220 ± 36.2 | 198 ± 18.9 |
| Idsy/− | 27 ± 1.5 | 24 ± 0.6 | 18 ± 1.1 | |
| Idsy/−+IDS | 2400 | |||
| Idsy/−+IDS | 2700 | |||
| Idsy/−+IDS | 1995 | |||
| Idsy/−+IDS | 2090 | |||
| Idsy/−+IDS | 2405 | |||
| Idsy/−+IDS | 2820 | |||
| Idsy/−+IDS | 1900 | |||
| Idsy/−+IDS | 2340 | |||
| Idsy/−+IDS | 2300 | 1410 | 909 | 820 |
| Idsy/−+IDS | 1900 | 950 | 659 | 420 |
| Idsy/−+IDS | 2435 | 1680 | 1409 | 1219 |
| Idsy/−+IDS | 2230 | 1735 | 1380 | 1022 |
IDS activity in the plasma measured at different times after therapy (T1, T6, T12, and T18: 1, 6, 12, and 18 months after the injection, respectively) of WT (n = 3), Idsy/− (n = 3), and AAV2/5CMV-hIDS-injected Idsy/− mice.
Next, we tested IDS activity in homogenized tissues from all of the control and treated mice. In the treated mice, there was a remarkable rescue of IDS activity that reached levels comparable to or higher than the IDS activities measured in the WT tissues; this was seen for all of the evaluated tissues from the treated mice that were sacrificed at T1 and T18 (Figure 1A). Surprisingly, a moderate rescue of IDS activity was evident in brain homogenates of T1 and T18 mice (Figure 1B). This led us to predict that we were measuring IDS activity of the enzyme that had crossed the BBB. Indeed, much higher IDS activity would be expected if there had been transduction of AAV2/5CMV-hIDS in the brain.
Figure 1.
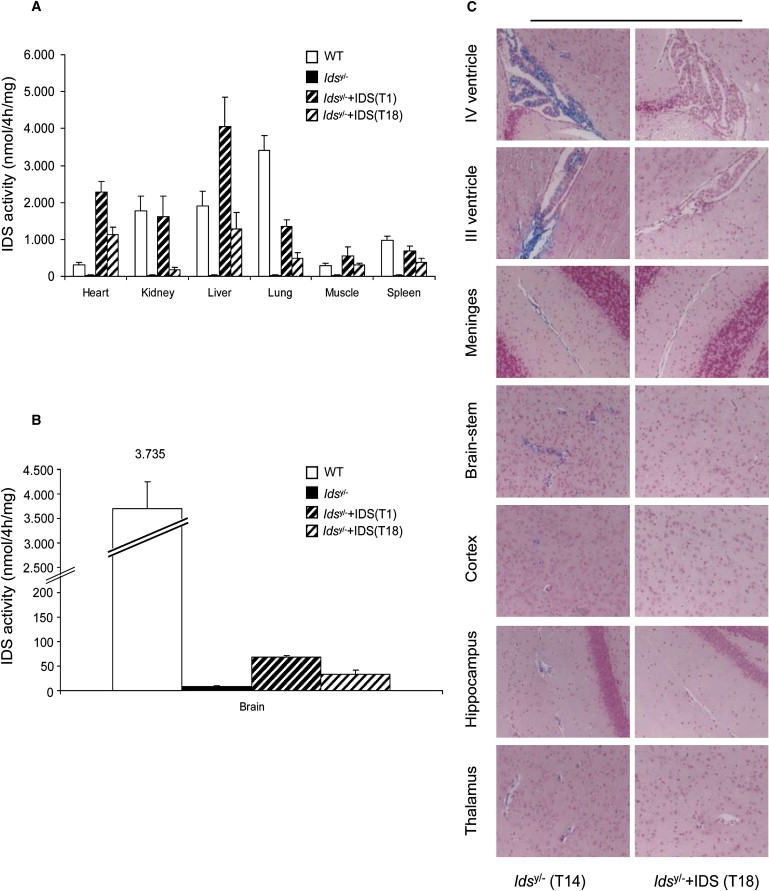
Increase in IDS Activity and Rescue of GAG Accumulation in Idsy/− Mice after Temporal Vein AAV2/5CMV-hIDS Injection
(A) P2 Idsy/− mice sacrificed 1 (T1) and 18 (T18) months after therapy. IDS activities in the treated mice were measured in protein extracts of: WT (n = 5), Idsy/− (n = 5), T1 Idsy/−+IDS (n = 8), and T18 Idsy/−+IDS (n = 4) mice.
(B) IDS measured in brain homogenates of the same groups of control and treated mice. Error bars indicate standard deviations. p < 0.05 (Student's t test).
(C) Alcian Blue-stained sections for GAG accumulation in different regions of brains of T14 Idsy/− control mice and T18 AAV2/5CMV-hIDS-injected Idsy/− mice. Magnification: 20×.
GAG accumulation was fully cleared in the visceral tissues of both of these treated groups (T1 and T18) (Figure S1) and, remarkably, also in all of the brain areas analyzed: the choroid plexus in the IV and III ventricles, as well as the meninges, brainstem, cortex, hippocampus, and thalamus (Figure 1C and Figure S2). This clearly showed that even if we succeeded in only a partial rescue of the IDS activity in the brain with this therapy, this constant low level of IDS that had crossed the BBB was sufficient to totally clear the GAG CNS storage. Alcian blue staining was performed as previously described.6
Then, to investigate this crossing of the BBB by IDS, we analyzed human IDS expression in cDNAs from T1 and T18 tissues. No positive expression signals were detected in the brain samples of the control (WT and idsy/−) or treated (T1 and T18) mice. In contrast, there was clear amplification of IDS expression in the liver and lungs of both treated groups. For controls, we also amplified mouse Ids transcripts (Figure 2A and Figure S3). For further confirmation of these results, p2 WT mice were injected with 1 x 1011 AAV2/5CMV-LacZ viral particles. LacZ expression and β-galactosidase activity were seen only in the liver and lungs, not in the brain (Figure 2B and data not shown). This indicates that both of the hIDS and LacZ transgenes from the AAV2/5 vectors were not expressed in the brains of the injected mice and that, thus, the measured IDS activity was due to IDS uptake from the bloodstream and its crossing of the BBB. Similarly, by using real-time PCR, we could not detect any AAV2/5 viral genomic DNA in the brains of the treated mice, only in their livers, further ruling out crossing of the BBB by AAV2/5 and transduction of the brains (Figure 2C). By contrast, efficient crossing of the BBB was achieved by intravenous injections of the AAV9 vector.14 Reverse transcriptase-polymerase chain reaction (RT-PCR) experiments were performed through amplifying the cDNAs prepared from the brains, livers, and lungs of WT mice, Idsy/− mice, and T1 and T18 AAV2/5CMV-hIDS-injected Idsy/− mice (Superscript III Reverse Transcriptase Kits; Invitrogen).
Figure 2.
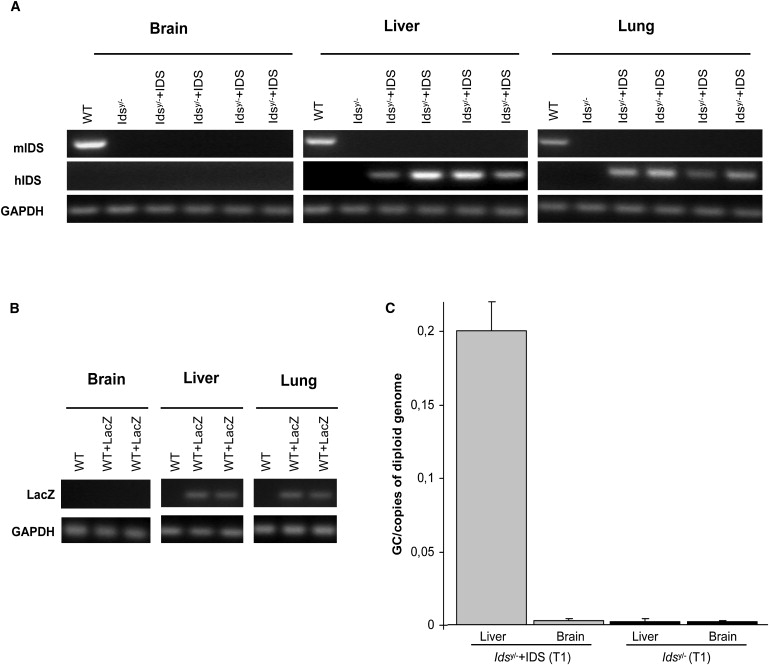
IDS Protein Crossing of the BBB in AAV2/5CMV-hIDS -Injected Idsy/− Mice
(A) RT-PCR of IDS from brains, livers, and lungs of T18 WT, T14 Idsy/−, and T18 Idsy/−+IDS mice, for amplification of human and mouse IDS mRNA.
(B) WT mice injected at p2 in the temporal vein with AAV2/5CMV-LacZ and sacrificed after 1 month. RT-PCR of LacZ mRNA from brains, livers, and lungs of T1 WT and T1 WT+LacZ mice.
(C) Real-time PCR for amplification of viral genomic DNA in brains and livers of T1 nontreated Idsy/− mice (n = 4) and T1 AAV2/5CMV-hIDS-injected Idsy/− mice (n = 4). Error bars indicate standard deviations. p < 0.05 (Student's t test). The primers used were: forward primer BGH: 5′-TCTAGTTGCCAGCCATCTGTTGT-3′, reverse primer BGH: 5′-TGGGAGTGGCACCTTCCA-3′, and the TaqMan probe 5′-6FAM-TCCCCCGTGCCTTCCTTGACC-TAMRA-3′ (LightCycler 480 Probes Master Kit, Roche). As standard, the diluted gene transfer vector was used (from 1 × 107 copies to 1 copy per reaction). To obtain the vector copy number for each cell, we divided the values by the number of cells corresponding to 100 ng of genomic mouse DNA.
The progressive accumulation of GAGs in lysosomes of neurons leads to severe vacuolization. This was cleared in neurons of the brainstem, cortex, hippocampus, and thalamus of treated mice (Figure S4). Interestingly, diffuse neurodegeneration in the thalamus, cerebral cortex, and brainstem of untreated animals was clearly seen, through observation of reduced neuronal density (decreased anti-NeuN signal) and increased ubiquitin-expressing neurons, as compared to the WT brain areas. This was also associated with the triggering of apoptosis, as shown by TUNEL-positive signals in neurons of the thalamus, cerebral cortex, and brainstem of MPSII mice (Figure 3A and Figures S5 and S6). In addition, in the white matter, there were α-synuclein aggregates (Figure 3B), which have previously been shown to accumulate in neurons during neurodegenerative disorders, such as Parkinson disease (MIM 168600) and Alzheimer disease (MIM 104300), and also during lysosomal storage disorders, such as Sandhoff disease (MIM 268800), Tay-Sachs disease (MIM 272800), metachromatic leukodystrophy, and beta-galactosialidosis15 (MIM 256540). Finally, in the treated mice, there were also increased numbers of activated microglia (GFAP immunohistochemistry) and massive infiltration of activated macrophages (CD68 positivity) (Figure 3A and Figures S5 and S6).
Figure 3.
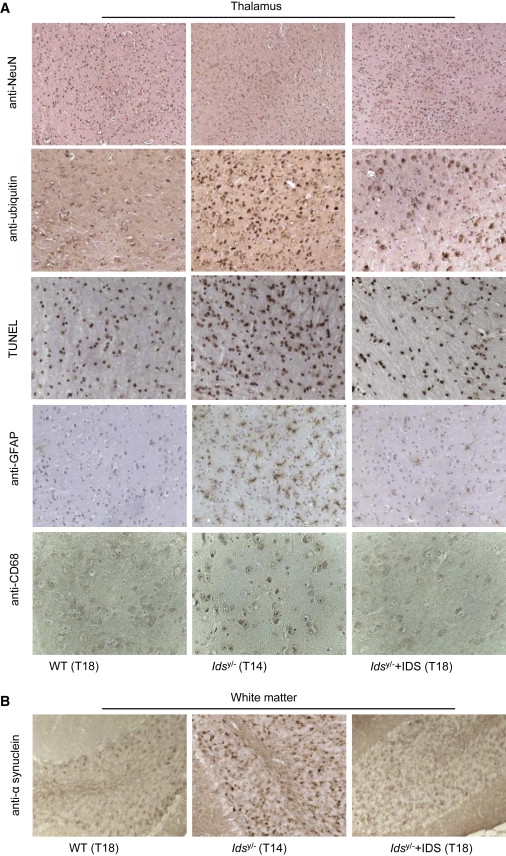
Rescue of Brain Defects in AAV2/5CMV-hIDS-Injected Idsy/− Mice
(A and B) Immunohistochemistry of different brain-specific markers (monoclonal anti-NeuN, diluted 1:100, Chemicon International; polyclonal anti-ubiquitin, diluted 1:50, Abcam; TUNEL, Chemicon International Staining Kit; monoclonal anti-GFAP, diluted 1:200, Sigma-Aldrich; and monoclonal anti-CD68, diluted 1:250, AbD Serotec) in the thalamus and white matter (monoclonal anti-α-synuclein, diluted 1:250, Abcam) of brain sections in T18 WT, T14 Idsy/−, and T18 AAV2/5CMV-hIDS-injected Idsy/− mice. Magnification: 10× (anti-NeuN sections); 40× (anti-CD68 sections); or 20× (all others).
Remarkably, in the T18 AAV2/5CMV-hIDS-injected Idsy/− mice, there was clear amelioration or complete correction of neurodegeneration (normalized staining of NeuN, ubiquitin, TUNEL, and α-synuclein), astrogliosis (normalized GFAP staining), and inflammation (normalized CD68 staining) (Figures 3A and 3B and Figures S5 and S6). Furthermore, in one-month-old MPSII mice, we observed increases in ubiquitin-expressing neurons and astroglysosis in the thalamus, including an increase in α-synuclein aggregates as well; these markers were normalized in the group of T1 AAV2/5CMV-hIDS-injected Idsy/− mice (Figures S7A and S7B). All of these data demonstrate that with this therapy, we can prevent the development of the CNS defects, along with the somatic manifestations. Indeed, normalized horizontal and vertical activities in the open-field test measured in T18 AAV2/5CMV-hIDS-injected Idsy/− mice confirmed their improved gross motor phenotype (Figures S8A and S8B). The open-field test was performed as previously described.6
All in all, this report clearly demonstrates that with only one intravenous systemic injection of AAV2/5CMV-hIDS vector particles administered to newborn p2 idsy/− mice, the mice do not develop the systemic and CNS disease phenotypes up to 18 months of age. We have clearly shown that the vector was not transduced in the brains of the treated animals and that the correction of the CNS symptoms was due to the crossing of the BBB by IDS. Although at present we do not know how IDS crosses the BBB, the long half-life of circulating IDS, as shown by its slow clearance from the blood (high plasma levels in treated animals), probably allows the enzyme to cross the BBB. Interestingly, chemically modified β-glucuronidase showed slower plasma clearance, and the modified enzyme efficiently crossed the BBB after its systemic infusion in MPSVII mice.16 Whether high circulating levels of IDS are required for its delivery through receptor-mediated uptake or via different routes, such as pinocytosis or transcytosis, remains an open question.
MPSII patients affected by the severe form of the disease ideally need to be treated with one administration of the vector that can rescue both visceral and CNS defects. In conclusion, with these findings, we believe to have demonstrated that high circulating levels of the IDS enzyme, coupled with very early treatment of the mice, provide an effective therapy for MPSII in all of the affected tissues. Potentially, modulation of the physical parameters of the enzymes, such as their stability, and early vector administration might be useful approaches for the treatment of other lysosomal-storage disorders.
Acknowledgments
The authors thank A. Auricchio, A. Ballabio, N. Brunetti, and M. E. Haskins for critical reading of the manuscript; S. Abbondante for technical suggestions; M. Doria for providing reagents; E. Nusco and R. Salvia for technical support; and AAV vector core for vector preparation. We are grateful for financial support from the Telethon Foundation and from the MPS Society. We thank SEMM (European School of Molecular Medicine) for support to V.A.P.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NCBI Protein Database, http://www.ncbi.nlm.nih.gov/protein/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
References
- 1.Hopwood J.J., Bunge S., Morris C.P., Wilson P.J., Steglich C., Beck M., Schwinger E., Gal A. Molecular basis of mucopolysaccharidosis type II: mutations in the iduronate-2-sulphatase gene. Hum. Mutat. 1993;2:435–442. doi: 10.1002/humu.1380020603. [DOI] [PubMed] [Google Scholar]
- 2.Cosma M.P., Pepe S., Annunziata I., Newbold R.F., Grompe M., Parenti G., Ballabio A. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113:445–456. doi: 10.1016/s0092-8674(03)00348-9. [DOI] [PubMed] [Google Scholar]
- 3.Dierks T., Schmidt B., Borissenko L.V., Peng J., Preusser A., Mariappan M., von Figura K. Multiple Sulfatase Deficiency Is Caused by Mutations in the Gene Encoding the Human C(alpha)-Formylglycine Generating Enzyme. Cell. 2003;113:435–444. doi: 10.1016/s0092-8674(03)00347-7. [DOI] [PubMed] [Google Scholar]
- 4.Neufeld E.F., Muenzer J. The mucopolysaccharidoses. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The metabolic and molecular basis of inherited disease. Mc Graw-Hill; New York: 2001. pp. 3421–3452. [Google Scholar]
- 5.Muenzer J., Lamsa J.C., Garcia A., Dacosta J., Garcia J., Treco D.A. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): a preliminary report. Acta Paediatr. Suppl. 2002;91:98–99. doi: 10.1111/j.1651-2227.2002.tb03115.x. [DOI] [PubMed] [Google Scholar]
- 6.Cardone M., Polito V.A., Pepe S., Mann L., D'Azzo A., Auricchio A., Ballabio A., Cosma M.P. Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum. Mol. Genet. 2006;15:1225–1236. doi: 10.1093/hmg/ddl038. [DOI] [PubMed] [Google Scholar]
- 7.Hawkins B.T., Davis T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 8.Roces D.P., Lullmann-Rauch R., Peng J., Balducci C., Andersson C., Tollersrud O., Fogh J., Orlacchio A., Beccari T., Saftig P., von Figura K. Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: a preclinical animal study. Hum. Mol. Genet. 2004;13:1979–1988. doi: 10.1093/hmg/ddh220. [DOI] [PubMed] [Google Scholar]
- 9.Vogler C., Levy B., Grubb J.H., Galvin N., Tan Y., Kakkis E., Pavloff N., Sly W.S. Overcoming the blood-brain barrier with high-dose enzyme replacement therapy in murine mucopolysaccharidosis VII. Proc. Natl. Acad. Sci. USA. 2005;102:14777–14782. doi: 10.1073/pnas.0506892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matzner U., Lullmann-Rauch R., Stroobants S., Andersson C., Weigelt C., Eistrup C., Fogh J., D'Hooge R., Gieselmann V. Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leukodystrophy. Mol. Ther. 2009;17:600–606. doi: 10.1038/mt.2008.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sferra T.J., Backstrom K., Wang C., Rennard R., Miller M., Hu Y. Widespread correction of lysosomal storage following intrahepatic injection of a recombinant adeno-associated virus in the adult MPS VII mouse. Mol. Ther. 2004;10:478–491. doi: 10.1016/j.ymthe.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 12.Hildinger M., Auricchio A., Gao G., Wang L., Chirmule N., Wilson J.M. Hybrid vectors based on adeno-associated virus serotypes 2 and 5 for muscle-directed gene transfer. J. Virol. 2001;75:6199–6203. doi: 10.1128/JVI.75.13.6199-6203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Auricchio A., O'Connor E., Weiner D., Gao G.P., Hildinger M., Wang L., Calcedo R., Wilson J.M. Noninvasive gene transfer to the lung for systemic delivery of therapeutic proteins. J. Clin. Invest. 2002;110:499–504. doi: 10.1172/JCI15780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki K., Iseki E., Togo T., Yamaguchi A., Katsuse O., Katsuyama K., Kanzaki S., Shiozaki K., Kawanishi C., Yamashita S. Neuronal and glial accumulation of alpha- and beta-synucleins in human lipidoses. Acta Neuropathol. (Berl.) 2007;114:481–489. doi: 10.1007/s00401-007-0264-z. [DOI] [PubMed] [Google Scholar]
- 16.Grubb J.H., Vogler C., Levy B., Galvin N., Tan Y., Sly W.S. Chemically modified beta-glucuronidase crosses blood-brain barrier and clears neuronal storage in murine mucopolysaccharidosis VII. Proc. Natl. Acad. Sci. USA. 2008;105:2616–2621. doi: 10.1073/pnas.0712147105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.