

Abstract
Recent progress in cataloguing common genetic variation has made possible genome-wide studies that are beginning to elucidate the causes and consequences of our genetic differences. Approaches that provide a mechanistic understanding of how genetic variants function to alter disease susceptibility and why they were substrates of natural selection would complement other approaches to human-genome analysis. Here we use a novel cell-based screen of bacterial infection to identify human variation in Salmonella-induced cell death. A loss-of-function allele of CARD8, a reported inhibitor of the proinflammatory protease caspase-1, was associated with increased cell death in vitro (p = 0.013). The validity of this association was demonstrated through overexpression of alternative alleles and RNA interference in cells of varying genotype. Comparison of mammalian CARD8 orthologs and examination of variation among different human populations suggest that the increase in infectious-disease burden associated with larger animal groups (i.e., herds and colonies), and possibly human population expansion, may have naturally selected for loss of CARD8. We also find that the loss-of-function CARD8 allele shows a modest association with an increased risk of systemic inflammatory response syndrome in a small study (p = 0.05). Therefore, a by-product of the selected benefit of loss of CARD8 could be increased inflammatory diseases. These results demonstrate the utility of genome-wide cell-based association screens with microbes in the identification of naturally selected variants that can impact human health.
Introduction
Genome-wide association studies of clinical phenotypes have successfully identified common variants that correlate with an increased risk for the development of human diseases, including macular degeneration (MIM #610698),1–3 diabetes (MIM #125853),4,5 and inflammatory bowel disease (MIM #266600).6,7 Such studies challenge researchers to experimentally validate these associations and to elucidate the molecular and cellular mechanisms whereby the genetic variants alter disease susceptibility. Dense genotyping data have also made possible a second kind of genome-wide study, screens for signatures of natural selection.8,9 These studies use sequence analysis to highlight chromosomal regions that might have been targets of natural selection, but they provide no information about which traits have been under selection and how the identified regions have affected the fitness of humans now or in the past. Thus, although genome-wide association studies and scans for selection have identified loci important for disease and understanding our evolutionary past, there is a growing need for approaches that (1) provide mechanistic information for how variants impact disease pathogenesis and (2) identify genetic variation in traits subject to natural selection.
One set of traits that has certainly been shaped by natural selection is the host response to microorganisms.10,11 Pathogens that have caused significant epidemics and/or increased in frequency as human populations have increased in density are likely to have left lasting effects both on gene frequencies and human health. Although Salmonellae infection has been greatly reduced by modern waste disposal and water treatment, this Gram-negative enteric pathogen still causes major outbreaks, is currently the most common cause of bacterial gastroenteritis in the United States, and is estimated to cause 1.3 billion cases of disease annually worldwide.12,13 Salmonellae, particularly the human-specific species S. typhi, which causes more severe systemic illness, was probably a significant menace during times of increasing population density in particular; for example, one such time was the rise of agriculture (4000–10000 years ago14), and a more recent example is the Industrial Revolution. Polymorphisms that have increased in frequency as a result of past epidemic selection could still affect human health because genes involved in protection against pathogens are likely to be important to all inflammatory diseases, including autoimmunity.15 By examining variation in human cell-based measures of infectious-disease susceptibility and severity, we can begin to link variation affecting human disease and variation identified as being the subject of natural selection.
Here we present a screen for identifying genetic variation involved in cellular phenotypes of bacterial infection. In contrast to clinical association studies involving naturally infected human populations, this approach is not restricted to pathogens that currently infect large populations, and it reduces variation due to differences in pathogen exposure and availability of treatment. In this paper, we apply this approach to rapid cell death induced by Salmonella. In macrophages and dendritic cells, this has been shown to be a proinflammatory cell death dependent on caspase-1 (MIM ∗147678).16,17 Caspase-1 knockout mice are more susceptible to S. typhimurium18 and other bacterial infections,19,20 demonstrating the relevance of this intermediate phenotype to bacterial infection at the organismal level. Using our screening approach, we have identified a nonsynonymous CARD8 allele (MIM ∗609051) that increases susceptibility to Salmonella-induced cell death. In addition to demonstrating the validity of this association through overexpression and RNA interference, we provide evidence of past natural selection on this gene, suggesting a link between evolution and a trait still important for multiple aspects of human health.
Material and Methods
Cells
HapMap lymphoblastoid cells (LCLs) were provided by the Coriell Institute. A total of 173 LCLs from CEU subjects (Centre d'Etude du Polymorphisme Humain Utah residents with ancestry from northern and western Europe) and YRI subjects (from Yoruba in Ibadan, Nigeria) were assayed because some were not available from Coriell at the time of order. Cells were maintained in RPMI 1640 media supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 100 U/ml penicillin-G, and 100 μg/ml streptomycin.
Bacterial Strains
S. typhimurium 14028s was obtained from ATCC. prgH and ssaT deletions were constructed with lambda red21 and verified by PCR. The fliC/fljB mutant was previously described.22 For green fluorescent protein (GFP) tagging, strains were electroporated with pMMB67GFP, the kind gift of James Bliska.23 All GFP-tagged strains were grown in LB media with 100 μg/ml ampicillin.
Infection Assay
Cells were passaged at least twice after being received from Coriell. Three days prior to assay, cells were split to 150,000/ml. The day of assay, cells were washed with RPMI supplemented with 1% FBS and resuspended in RPMI 1640 (without phenol red) supplemented with 10% FBS. Overnight, bacterial cultures were subcultured 1:33 and grown for 2 hr 40 min at 37°C. Bacterial invasion was conducted for 1 hr at a multiplicity of infection (MOI) of 30. We chose the timing of infection and MOI to allow for a high-intermediate level of cell death and invasion with a normal distribution of variation. After infection, we added gentamicin for 1 hr to obtain a concentration of 50 μg/ml. We then split each infected culture into three subcultures to allow for measurement at 3.5, 24, and 48 hr (time from beginning of invasion). Seventy-five minutes prior to measurement, we added isopropyl β-D-1-thiogalactopyranoside (IPTG) to obtain a concentration of 1.5 mM. A portion of the supernatant was kept for cytokine measurement. Cells were stained with 7-AAD (eBioscience), and cell death and GFP-expressing bacteria were measured on a Guava Easycyte Plus flow cytometer. Each LCL was assayed on three sequential passages separated by 3 days. Percent Salmonella-induced cell death was background subtracted for uninfected cell death. Pilot measurements of LDH release gave results similar to those obtained for 7-AAD staining (not shown). As an additional measure of caspase-1 activation, IL1β was measured by ELISA (R&D Systems), but no increase over baseline was detected even with prestimulation with LPS. This is possibly due to the lack of expression of the appropriate TLRs for increasing pro-IL1β expression.
For inhibition with Ac-YVAD-FMK (Calbiochem), cells were incubated with the indicated concentration for 30 min prior to infection. For measurement of caspase-1 activity with FAM-YVAD-FLICA (Immunochemistry Technologies, LLC), cells were stained for 1 hr according to the manufacturer's instructions.
Overexpression and RNAi
For transient overexpression, 5 × 105 LCLs were transfected with the Amaxa Nucleofection Kit V according to the manufacturer's instructions. Cells were assayed 6–8 hr after transfection. Tests with GFP showed that more than 50% of viable, transfected cells showed expression. HA-CARD8 (A) and HA-CARD8 (T) expression vectors were the kind gift of Jose Fernandez-Luna.24 For detection of overexpressed CARD8, immunoblotting was performed with a rabbit CARD8 antibody used at a 1:1000 dilution (Abcam).
For stable knockdown, LCLs were transduced with pGIPz-based lentiviruses containing nonsilencing or CARD8 directed shRNA (CARD8-1:V2LHS_96279 and CARD8-2: V2LHS_96282; Open Biosystems). Lentiviruses were packaged with the Lenti-X system (Clontech). LCLs were selected for at least 2 weeks prior to assay with puromycin (0.1–0.5 μg/ml, depending on the sensitivity of the particular LCL). Knockdown was verified by two-step real-time RT-PCR with validated probes from Applied Biosystems (Hs00209095_m1) and normalized to an amplification of 18S rRNA.
Computational Analysis
Descriptive statistics, linear regression, and IC50 calculation were performed with GraphPad Prism 5. Linkage disequilibrium at the CARD8 locus was examined with HaploView.25 Genome-wide association analysis was conducted with PLINK, developed by Shaun Purcell.26 Analysis was carried out with QFAM-parents and QFAM-total with adaptive permutation under default settings. The QFAM procedures implemented in PLINK use linear regression to test for association while employing permutation of within- and between-family components separately to control for family structure. Although protection from stratification is not as complete as with a strictly within-family test (such as the TDT), consideration of parental phenotypes in QFAM results in a significant increase in power.27
After we pruned for frequencies and genotyping quality, 2,869,783 SNPs remained for association testing. Because of our small sample size, very low p values would only be obtained if the SNP had a very large genotypic effect. Because we did not know what effect size to expect and the low heritability of the trait suggested a large fraction of nongenetic variation for this phenotype, we used a relatively modest p value filter of 0.01 for our most powerful test, QFAM-parents on combined CEU-YRI. This filter alone gives 23,840 SNPs. The use of this less stringent filter is expected to result in greater sensitivity but would also result in a greater number of false positives. To reduce false positives, we therefore ran the QFAM-parents analysis on CEU and YRI populations separately; we reasoned that for SNPs present in both populations, we should see some evidence of association in both populations separately with the same direction of effect. Because these separate population analyses employ even fewer trios and consequently have less power, a less stringent p value filter of 0.1 was used. Finally, the QFAM-total filter (p < 0.05) allowed us to retain SNPs that showed an association even when we did not control for stratification.
Filtering of PLINK results with these p-value filters resulted in 2136 SNPs. SNPs that are truly modifying the cell-death phenotype will be correlated with the phenotype, as reflected in the PLINK p value, but they also will have a functional affect on the genes in which they are located or on genes that are nearby. Therefore, we further filtered our candidate SNP list to include only SNPs with a high probability of having a functional affect. In the complete Hapmap dataset, 34,895 SNPs were nonsynonymous (16,610), transcriptional regulatory (15,873), over splice sites (2787), or within microRNAs (34). Applying both p value and SNP-characteristic filters resulted in 20 SNPs for the cell-death phenotype. These filtering steps were carried out with the GWAS Analyzer database tool (our unpublished data). GWAS Analyzer serves as a database to store and integrate the HapMap genotypes and pedigrees, the phenotypic data generated by high-throughput human in vitro susceptibility testing (Hi-HOST, Figure 1A), and the p values generated by association analysis carried out with PLINK. In addition to allowing for easy retrieval of data queried by phenotype, SNP, or LCL ID, the web-based interface allows for filtering of PLINK results on the basis of p value and SNP characteristics.
Figure 1.
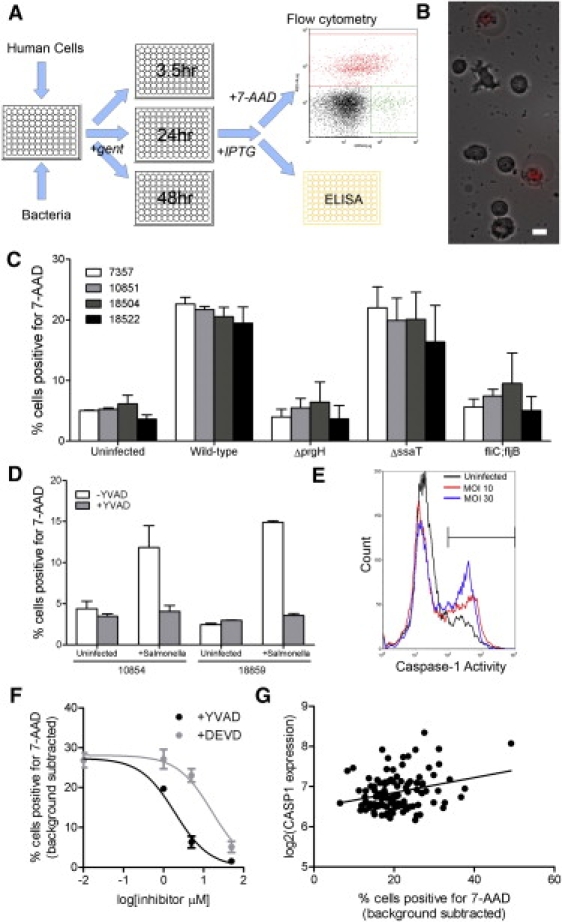
Hi-HOST Reveals that LCLs Undergo Salmonella-Induced Cell Death
(A) A schematic diagram showing the steps involved in carrying out the Hi-HOST assay. LCLs are infected in 96-well plates with bacteria tagged with an inducible GFP plasmid. After waiting an hour to allow for invasion, one adds gentamicin for an additional hour to kill extracellular bacteria and then splits LCLs into plates for each time point. Adding IPTG 75 min prior to each time point induces GFP expression. Supernatants are collected so that cytokine release can be measured, and cells are stained with 7-AAD and analyzed by flow-cytometry so that viability and intracellular bacteria can be measured.
(B) Phase image of LCLs overlaid with 7-AAD fluorescence (red) 2 hr after infection with S. typhimurium. The scale bar represents 10 μm.
(C) Effects of bacterial mutations on Salmonella-induced cell death. Four different LCLs were infected at an MOI of 30 with wild-type, ΔprgH, ΔssaT, or fliC;fljB S. typhimurium. Cell death is greatly reduced in all LCLs with the prgH and fliC;fljB mutants, and the observed variation among the four LCLs is similar to that in uninfected cells. Data shown are the means and standard deviation from two sets of experiments.
(D) Inhibition of cell death with the caspase-1 inhibitor, Ac-YVAD-cmk. Two LCLs were treated with 50 μM Ac-YVAD-cmk beginning 30 min prior to infection and assayed for cell death. Data shown are the means and standard deviation from two sets of experiments.
(E) Fluorescence-labeled inhibitor of caspase (FLICA) measurement of caspase-1 2 hr after S. typhimurium infection. The fraction of cells labeled by FAM-YVAD-fmk increases with Salmonella infection. Data shown are for LCL 7357 but are representative of several LCLs tested.
(F) Dose-response curves of inhibition of cell death in LCL 7357 via the caspase inhibitors Ac-YVAD-cmk and Ac-DEVD-cmk. Cells were treated with 0, 1, 5, or 50 μM of each inhibitor 30 min prior to infection and assayed for cell death. The percentage of cell death in uninfected cells was subtracted from the amount measured at 3 hr after the initial infection. The IC50 for YVAD is 1.9 μM, and that for DEVD is 16.1 μM. Similar values were obtained when an additional LCL (18853) was assayed (not shown). Data points are the means and standard error of the mean (SEM) for three separate experiments.
(G) Linear regression of Salmonella-induced cell death versus log2 (CASP1 expression) in combined CEU-YRI parents shows that higher levels of CASP1 are correlated with increased levels of cell death (Pearson r = 0.28; p = 0.002).
Expression levels in LCLs were obtained from the GENEVAR dataset.28 For CASP1, levels of probe GI-15431333-A were examined. For CARD8, levels of probe GI_7662403-S were examined.
We determined the presence or absence of CARD8 in mammalian genomes by using the UCSC Genome Browser29 and Ensembl release 49.30 Individual genomes were queried with “CARD8” in UCSC Genome Browser and Ensembl. If no hits were obtained, individual genomes were searched with the human 431 amino-acid CARD8 isoform via protein BLAST of peptides and ab initio peptides in Ensembl. The phylogeny used in the analysis was obtained from Prasad et al., 2008.31 Animal group size was determined from internet resources, including Wikipedia and the Animal Diversity Web. Assignments of group size have the caveat that current social structure might not reflect the structure during most of the organisms' evolutionary histories, and this might be especially true of domesticated and commensal organisms. Humans were classified as a “small group” because we have lived in small hunter-gatherer groups throughout most of our evolution. Correlation of animal group size and CARD8 was done via Pagel's method32 as implemented in Mesquite.33 Tests were conducted with the 15 species with higher confidence (>5× coverage to call absence; presence requiring at least half of protein sequence found) and also on all 24 mammalian species. A few species are intermediate, having groups larger than the family-sized packs of dogs (alpha pair and previous year's offspring) but less than the size of wild horse herds, for example. Specifically, the animals with intermediate-sized groups are (1) chimpanzees, which form troops of approximately 10 but then come together occasionally in dynamic “fusion-fission” groups, (2) European rabbits, which form small colonies that range in size from a single territorial breeding pair and offspring to larger groups of 6–10 adults, and (3) elephants, which form small herds of related females and offspring while males are primarily solitary after maturity. When we re-ran our analysis and switched the categorization of these three species with intermediate-sized groups to the “large group/herd” category, we obtained a p value of 0.04 for the 25-mammal tree. For the 15-mammal tree (which does not include rabbits because of their low genome coverage), we obtained a p value of 0.006.
The branch-site test of positive selection34 was carried out with PAML35 for the five complete primate CARD8 sequences aligned by MEGA4.36 Human Genome Diversity Project (HGDP) allele frequencies were obtained with SPSmart.37
Case-Control Studies for Sepsis and Systemic Inflammatory Response Syndrome
Healthy control subjects were recruited from the Seattle area and enrolled as described.38 The systemic inflammatory response sydrome (SIRS) cohort was enrolled from the intensive care units (ICUs) of Harborview Medical Center (Seattle, WA). Inclusion criteria included admission to an ICU for more than 24 hr and presence of three of four criteria for SIRS.39 Exclusions included admission for major trauma, chronic treatment with anti-inflammatory medications, history of cancer, massive transfusion, presence of an advanced directive against resuscitation, and an age of <18 or >90 years. The trauma cohort employed in the sepsis study has been previously described.40 In brief, patients admitted to the Harborview Medical Center ICU after sustaining major traumatic injury were enrolled, and DNA samples were collected. All subjects were followed for the presence of SIRS criteria and for the development of a microbiologically documented or clinically suspected infection. For both cohorts, we limited our analyses to subjects of European descent to minimize confounding due to population-specific allele frequencies. These studies were approved by the Division of Human Subjects Research, University of Washington (Seattle, WA). Genotyping for rs2043211 was conducted with a Taqman-based allelic discrimination assay (C_11708080_1) from Applied Biosystems. We achieved high-quality genotypic calls on >98% of the subjects tested, and genotype frequencies did not deviate from Hardy-Weinberg equilibrium.
Analysis was conducted with the Cochran-Armitage trend test41,42 as implemented in the COIN package for R43 and described in the MAXTest vignette (see Web Resources). On the basis of the genotypic medians for cell death in parental CEU and YRI LCLs, scores of 0, 0.75, and 1 were assigned for the AA, AT, and TT genotypes for an additive model with partial dominance of the T allele.
Results
Hi-HOST Reveals Variation in Cell Death after Salmonella Infection
In order to identify genetic variation that influences susceptibility to bacterial infection, we developed a high-throughput assay, Hi-HOST (Figure 1A), to measure multiple infection parameters. Standard cell-based infection assays typically involve bacterial enumeration through lysis of host cells and counting of bacterial colony-forming units. The method is not amenable to large-scale phenotyping and is not sensitive enough to detect small differences. Hi-HOST uses bacteria tagged with inducible GFP and flow cytometry to allow for rapid and accurate measurement of bacterial invasion, intracellular survival and replication, host cell death, and host cytokine production from a single set of infection assays. HapMap LCLs (Epstein-Barr virus (EBV)-transformed B cells) were used for phenotyping because they have been genotyped at more than 3 million SNPs.44 The utility of these cells for the examination of human variation in gene expression,28,45 chemotherapeutic sensitivity,46 and HIV invasion47 has been demonstrated. Importantly, Salmonella typhimurium infects B cells both in vitro and in vivo48, demonstrating the physiological relevance of this infection model.
One outcome of S. typhimurium infection measured in Hi-HOST is Salmonella-induced cell death. A caspase-1-dependent, proinflammatory form of cell death termed “pyroptosis” rapidly occurs in Salmonella-infected macrophages and dendritic cells49, and we found that a similar process occurs in LCLs. In LCLs, S. typhimurium triggered cell death within 2 hr of the start of infection, and the dying cells underwent a characteristic rounding and enlargement, consistent with the osmotic changes and proinflammatory release of cellular contents seen in pyroptosis (Figure 1B). Similar to what is reported for macrophages,16,22,50 Salmonella-induced cell death in LCLs was completely abolished when cells were infected with a SPI-1 type-III secretion mutant (ΔprgH) and partially abolished when infected with a flagellin mutant (fliC;fljB) (Figure 1C). Furthermore, pharmacological inhibition of caspase-1 by the peptide-based inhibitor Ac-YVAD-cmk also prevented cell death (Figure 1D), and a fluorescent derivative of this inhibitor labeled a greater fraction of cells after infection (Figure 1E). On the basis of published in vitro IC50 values, YVAD is highly specific to caspase-1. The IC50 of this drug measured with the canonical proapoptotic effector caspase-3 is at least 10,000× greater, and even the related inflammatory caspase-5 has an IC50 200× greater.51 To further examine the specificity of cell death in LCLs, we measured the effect of an additional caspase inhibitor, DEVD. This peptide strongly inhibits caspase-3, but it also inhibits caspase-1 with an IC50 only 20× greater than that of YVAD.51 Dose-response curves of YVAD and DEVD in inhibiting Salmonella-induced cell death show that the in vivo IC50 of DEVD is about 8× higher than that of YVAD (Figure 1F). This profile of being strongly inhibited by YVAD and moderately so by DEVD is unique to caspase-1.51
Finally, mining of publicly available genome-wide expression analysis of the HapMap LCLs uncovered a clear correlation between the level of CASP1 expression and the amount of LCL cell death (Figure 1G). Cells with more CASP1 mRNA had higher levels of Salmonella-induced cell death. This was true when LCLs were examined together (Pearson r = 0.28, p = 0.002) or as separate populations (for CEU, r = 0.31, p = 0.02; for YRI, r = 0.32, p = 0.01). We also attempted to measure the amount of activated caspase-1 by immunoblotting with human-specific p10 and p20 subunit antibodies. Although pro-CASP1 was readily detectable, we were unable to detect specific cleavage products generated upon S. typhimurium infection (not shown). The fraction of caspase-1 that is processed in LCLs could be a very small fraction of the total caspase-1, or the cleaved protein might undergo rapid turnover. Thus, evidence based on morphology, bacterial mutants, pharmacologic inhibition, and gene expression all indicates that Salmonella-induced cell death in LCLs is most likely dependent on the proinflammatory protease caspase-1.
The amount of Salmonella-induced cell death in 173 CEU and YRI LCLs demonstrated significant variation (range = 6.5% to 49%; Figure 2A and Table 1). Measurements were highly repeatable: analysis of variance (ANOVA) of three replicates performed on different passages for all LCLs demonstrated that 89% and 92% of the total variance was attributable to differences among individuals in CEU and YRI populations. The large interindividual variation could be due to genetic or environmental factors. The family structure of the CEU and YRI populations allows for estimation of narrow-sense (additive) heritability by parent-offspring regression.52 The heritability of Salmonella-induced cell death was 14.9% in the combined CEU-YRI dataset (Figure 2B). Heritability of traits can vary between populations, but similar values were obtained when the CEU and YRI data were plotted individually (20.8% and 12.8%; not shown). For comparison, the heritability of viable invasion of S. typhimurium as measured in Hi-HOST is 54% (Figure 2C).
Figure 2.
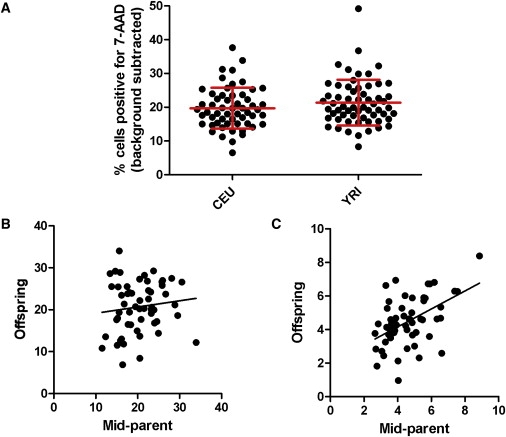
Variation in Salmonella-Induced Cell Death among CEU and YRI LCLs
(A) Scatter plots of cell death in CEU and YRI parental LCLs. Each dot represents the mean percentage of each LCL that stained positive for 7-AAD (uninfected cell death subtracted from background) assayed in three separate experiments. Mean and standard deviation of each population is displayed in red.
(B) Parent-offspring regression for cell death in combined CEU-YRI populations. Heritability is estimated at 14.9%.
(C) Parent-offspring regression for S. typhimurium viable invasion, measured as the percentage of LCLs containing GFP-Salmonella at 3.5 hr, in combined CEU-YRI populations. Heritability is estimated at 54%.
Table 1.
Population Statistics of Hapmap LCLs Assayed for Salmonella-Induced Cell Death
| CEU | YRI | Combined | p Valuea (CEU versus YRI) | |
|---|---|---|---|---|
| Mean | 19.70% | 21.40% | 20.56% | 0.17 |
| Standard Dev | 6.06% | 6.79% | 6.47% | 0.4 |
| p value normality testb | 0.07 | 0.25 | ||
| Repeatabilityc | 89% | 92% | ||
| Heritabilityd | 20.80% | 12.80% | 14.90% |
Mean, standard deviations, and normality test values are for parents only so that the independence of samples is maintained.
p value from unpaired two-tailed t test.
D'Agostino & Pearson normality test. A single outlier, 18523, was four standard deviations above the mean and was left out of this analysis.
Repeatability is defined as the between-individual component of variance calculated from ANOVA of three separate assays for each LCL.
Additive heritability estimated from the slope of parent-offspring linear regression of trios.
Family-Based Association Analysis Identifies SNPs that Correlate with Variation in Cell Death
Next, using family-based association analysis as implemented in PLINK26,27, we tested the hypothesis that common genetic variation contributed to heritability of Salmonella-induced cell death. The procedures employed use linear regression of phenotype on genotype and, subsequently, adaptive permutation to control for family structure. Our relatively modest set of 173 LCLs does not have sufficient power to identify true positives solely on statistical grounds. We therefore took a multistep approach, which we felt would maximize the probability of identifying true positives. Four sets of analyses were run within PLINK: (1) family-based association with parental phenotypes (QFAM-parents), which reduces false positives by controlling for between-family stratification, on the combined CEU-YRI data; (2) QFAM-parents on the CEU data alone; (3) QFAM-parents on YRI; and (4) total association taking into account family structure but not controlling for stratification (QFAM-total). QFAM-parents on CEU-YRI data has greater power than the same analysis on CEU and YRI data separately, but the individual population analysis allows for determining whether the same SNP correlates with cell death in two different populations. The QFAM-total test does not control for stratification, but the size of the genotypic effect is better reflected in p values obtained from this test than from the QFAM-parents test. By filtering on the basis of uncorrected p values from these tests, we obtained a set of 2136 SNPs that correlated with Salmonella cell death in both populations, controlled for between-family stratification, and resulted in a large effect on the phenotype. A second filtering step included only those SNPs that have a functional effect on the gene in which the SNP is located or on a nearby gene. This included SNPs that cause nonsynonymous changes, correlate with transcript levels, or overlap with splice sites or microRNAs. This functional-candidate-variant approach, similar to that used by Stranger et al., 200728, reduced our candidate list from 2136 to 20 SNPs (Table 2; complete results are available for further analysis and data mining with a newly developed web-based tool, the GWAS Analyzer). Because the statistical tests were chosen prior to analysis but p value filters were set post-hoc to give a manageable number of candidates, we then focused on performing additional experiments on SNPs from our candidate list to determine whether this approach was uncovering true positives.
Table 2.
SNPs Associated with Salmonella-Induced Cell Death
| Marker Name | QFAMpboth p Value | QFAMpCEU p Value | QFAMpYRI p Value | QFAMtboth p Value | SNP Type | Functional Effecta | Within Gene | Gene Expression Affectedb |
|---|---|---|---|---|---|---|---|---|
| rs12912 | 0.000002 | 0.01472 | 0.00003 | 0.002218 | genomic | Expression CEU, YRI, ASN | Hs. 397369c | |
| rs243327 | 0.000018 | 0.02666 | 0.0005328 | 0.01004 | intronic | Expression CEU, YRI, ASN | C16orf75 | C16orf75 |
| rs268671 | 0.00004508 | 0.0008454 | 0.02074 | 0.0457 | nonsynonymous | nonsynonymous | PRX | |
| rs243324 | 0.00009442 | 0.02393 | 0.00195 | 0.01683 | intronic | Expression CEU, YRI, ASN | C16orf75 | C16orf75 |
| rs2943512 | 0.000329 | 0.01142 | 0.01701 | 0.0004216 | nonsynonymous | nonsynonymous | MUC5B | |
| rs7099565 | 0.0007904 | 0.04287 | 0.01149 | 0.02011 | nonsynonymous | nonsynonymous | TRUB1 | |
| rs4802073 | 0.001498 | 0.02872 | 0.02316 | 0.03941 | intronic | Expression CEU | C19orf47 | C19orf47 |
| rs753852 | 0.001523 | 0.01223 | 0.04502 | 0.03563 | intronic | Expression CEU, YRI, ASN | ANKRD11 | SPG7 |
| rs683866 | 0.002736 | 0.03405 | 0.0455 | 0.01078 | nonsynonymous | nonsynonymous | KIAA0367 | |
| rs9630856 | 0.002853 | 0.07961 | 0.02528 | 0.005746 | intronic | Expression CEU | SERPINB2 | SERPINB10 |
| rs530978 | 0.003101 | 0.08761 | 0.0245 | 0.04131 | nonsynonymous | nonsynonymous | KIAA0367 | |
| rs4680 | 0.003585 | 0.06614 | 0.04163 | 0.01608 | nonsynonymous | nonsynonymous | COMT | |
| rs6509365∗ | 0.00421∗ | 0.083∗ | 0.02272∗ | 0.04947∗ | intronic∗ | Expression CEU, YRI, ASN∗ | CARD8∗ | CARD8∗ |
| rs188384 | 0.0052 | 0.05263 | 0.02943 | 0.01565 | intronic | Splice site SNP | CCDC50 | |
| rs3177676 | 0.007466 | 0.01323 | 0.0865 | 0.01018 | nonsynonymous | nonsynonymous | C1orf105 | |
| rs1014390 | 0.007859 | 0.0328 | 0.07482 | 0.03516 | intronic | Expression YRI, Expression CEU, YRI, ASN | JMJD6 | MXRA7 |
| rs6509366∗ | 0.008496∗ | 0.09732∗ | 0.04492∗ | 0.03857∗ | intronic∗ | Expression CEU, YRI, ASN∗ | CARD8∗ | CARD8∗ |
| rs1729659 | 0.009015 | 0.05243 | 0.09547 | 0.02354 | synonymous | Expression CEU, YRI, ASN | KIAA1841 | LOC339804 |
| rs557352 | 0.009863 | 0.09281 | 0.06316 | 0.04339 | nonsynonymous | nonsynonymous | KIAA0367 | |
| rs9807444 | 0.009885 | 0.07428 | 0.08632 | 0.01725 | genomic | Expression CEU,Y RI, ASN | TUBB6 | |
| rs2043211∗ | 0.01336∗ | 0.1283∗ | 0.0531∗ | 0.03723∗ | nonsynonymous∗ | Expression CEU, YRI, ASN∗ | CARD8∗ | CARD8∗ |
SNPs were filtered with PLINK results from QFAM-parents CEU+YRI < 0.01, QFAM-parents CEU < 0.1, QFAM-parents YRI < 0.1, and QFAM-total CEU+YRI < 0.05.
rs2043211 falls just outside the initial filtering criteria but is in LD with the other two asterisked CARD8 SNPs.
In addition to CARD8, three other genes on the list (SERPINB2, COMT, and KIAA0367) have been reported to be involved in caspase function or cell death.
Functional SNP filtering criteria that were fulfilled by the SNP.
Genes whose expression is correlated with the SNP on the basis of the GENEVAR data.22
Hs. 397369 is found on Illumina arrays but is no longer matched with a gene in NCBI (Build 36) or Ensembl (release 49).
A CARD8 Loss-of-Function Allele Is Unable to Inhibit Salmonella-Induced Cell Death
Two of the 20 candidate SNPs are within CARD8. Previous work has demonstrated that stable expression of CARD8 in THP-1 monocytes causes caspase-1 inhibition on the basis of decreased IL1β ELISA secretion and that this effect is probably through a direct physical interaction with caspase-1.53 Other reported functions of CARD8 include regulation of apoptosis54 and NF-KB-inhibitory properties.53,55 Because the two candidate SNPs are both within an intron, we analyzed the linkage disequilibrium (LD) of CARD8 to determine whether a more obvious causal variant was being inherited together with these SNPs. The two SNPs are in strong LD with each other and several other CARD8 SNPs (Figure 3). One of these SNPs, rs2043211, is predicted to replace an early residue with a stop codon (C10X) and cause a nonsense mutation in some CARD8 isoforms. Consistent with this, there is a strong correlation between the derived nonsense allele and reduced expression levels of CARD8 from the GENEVAR dataset28 (p = 3.72 × 10−5; Figure 4A), which we suspect is due to nonsense-mediated mRNA decay.56
Figure 3.
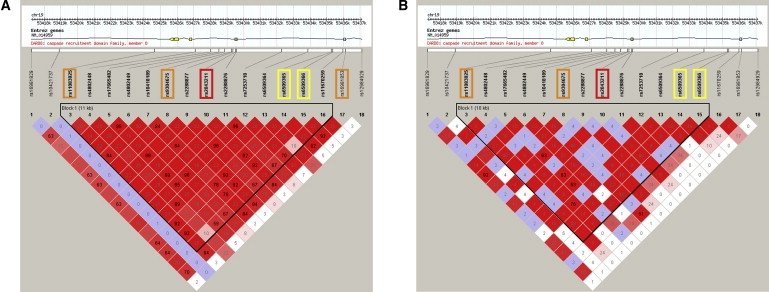
Linkage Disequilibrium around rs2043211 in CEU and YRI Populations
(A) CEU and (B) YRI populations. Boxes are colored on the basis of D′/LOD; red represents high LD. Numbers within boxes are r2 values. The two SNPs identified by the screen (rs6503965 and rs6503966) are highlighted in yellow, rs2043211 is highlighted in red, and other SNPs in strong LD with rs6503966 (r2 > 0.8) are highlighted in orange. Only 20 kb of the CARD8 gene (41.4 kb) is shown. Images were exported from HaploView.25
Figure 4.
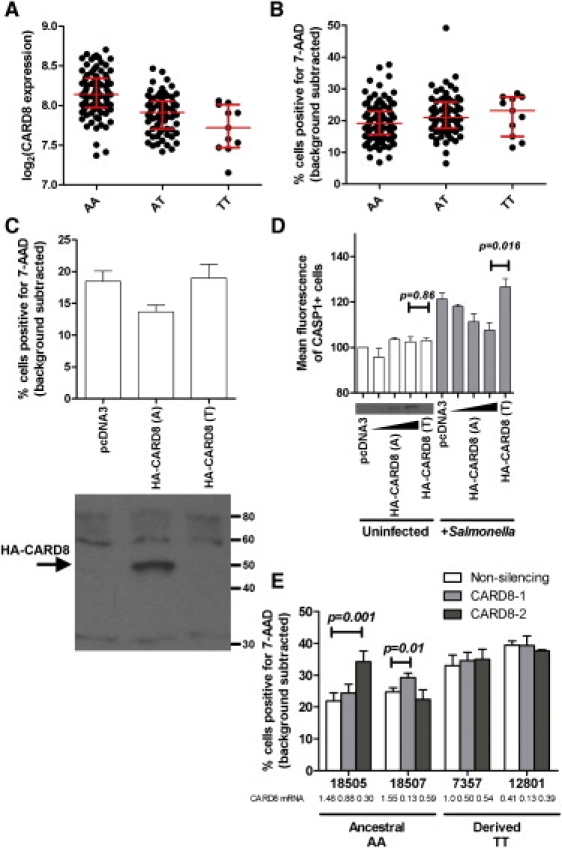
A Polymorphism in CARD8 Causes Increased Cell Death in Response to Salmonella
(A) The derived allele of rs2043211 correlates with decreased CARD8 expression levels. A is the ancestral allele, and T is the derived nonsense allele (designated based on the + strand of chromosome 19, as is the convention for the HapMap Project; CARD8 coding sequence is on the – strand). Expression levels for LCLs are from Stranger et al., 2007.28 All LCLs are displayed, and when family structure is controlled for in PLINK (QFAM-parents)26, a p value of 3.72 × 10−5 is obtained for the correlation between rs2043211 genotype and CARD8 expression. Median and interquartile ranges are displayed in red.
(B) The derived allele of rs2043211 correlates with increased cell death. All LCLs were assayed for S. typhimurium cell death in three separate experiments, and mean values for each are shown. Background-subtracted values reflect the subtraction of uninfected cell death. All LCLs are displayed, and when family structure is controlled for in PLINK (QFAM-parents), a p value of 0.013 is obtained for the correlation between rs2043211 genotype and Salmonella-induced cell death. Median and interquartile ranges are displayed in red.
(C) Overexpression of the ancestral CARD8 allele inhibits cell death. LCL 7357 was transfected with 2 μg empty vector or ancestral or derived alleles of CARD8. 6 hr after transfection, cells were infected with S. typhimurium at an MOI of 30, and cell death was measured by 7-AAD staining 2 hr after the initial infection. Only the ancestral allele inhibits the amount of detected cell death. The p value from repeated-measures ANOVA is 0.002. Similar results were obtained for LCL 18507 (not shown). Expression of HA-CARD8 was verified with a rabbit CARD8 antibody. Error bars show the SEM for three separate experiments.
(D) Overexpression of the ancestral CARD8 allele reduces caspase-1 activity in Salmonella-infected cells. LCL 7357 was transfected with 2 μg empty vector or ancestral or derived alleles of CARD8. For CARD8 ancestral dose-response measurements, 0.5, 1, and 2μg were used. Western blot inset demonstrates increasing HA-CARD8 protein detected with CARD8 antibody. 6 hr after transfection, cells were infected with S. typhimurium at an MOI of 30, and caspase-1 activation was measured by FLICA. No difference was noted in caspase-1 activation for uninfected cells, but infected cells show that the ancestral allele inhibits caspase-1 activation. The p values on the graph are from two-tailed t tests. Errors bars are SEM for assays conducted four separate times (except HA-CARD8 [A] 0.5 μg and 1 μg, which were assayed twice).
(E) RNAi against CARD8 causes increased cell death in LCLs with the ancestral allele. Each LCL was stably transduced with lentivirus expressing nonsilencing shRNA or two different CARD8 shRNAs. Knockdown was quantified in stably transduced lines via real-time RT-PCR, and expression levels normalized to LCL 7357 stably transduced with nonsilencing shRNA are shown. Each LCL was assayed in three separate experiments, and means with SEM are displayed. The p values on the graph are from paired two-tailed t tests. All other comparisons of nonsilencing versus CARD8 RNAi constructs give p > 0.05.
Cells with the derived rs2043211 allele exhibit modestly increased levels of Salmonella-induced cell death (p = 0.013; Figure 4B). This is consistent with the predicted loss-of-function nature of the polymorphism and the reported activity of CARD8 as an inhibitor of caspase-1: cells with the derived allele do not have as much functional CARD8, resulting in more active caspase-1 and more cell death. Although this is strong correlative evidence of the effect of rs2043211 on Salmonella-induced cell death, we performed experimental verification of the effect of the SNP on the phenotype to prove its functional significance.
To measure the effects of the alternative rs2043211 alleles, we carried out transient transfections with two different CARD8 alleles. Overexpression of ancestral CARD8, but not the derived nonsense allele, reduced the fraction of cells that underwent cell death (Figure 4C). As a second measure of caspase-1 activation in these cells, we used a fluorescently labeled inhibitor of caspase-1 (FLICA57). Infection with S. typhimurium caused a clear increase in the mean fluorescence of the population of cells labeled by the fluorescent caspase-1 inhibitor. Increased fluorescence is indicative of greater amounts of active caspase-1 within Salmonella-infected cells. This increase in mean fluorescence is blocked in a dose-dependent manner by expression of the ancestral rs2043211 allele (Figure 4D). In contrast, transfection of the derived allele, containing the stop codon, is unable to block the Salmonella-dependent increase in measured caspase-1 activity. Therefore, CARD8 appears to inhibit caspase-1 activation in LCLs, consistent with the reported inhibition of IL1β secretion from monocytes.53
To examine the role of CARD8 in a loss-of-function experiment, we stably transduced cells of varying rs2043211 genotypes with two different RNAi vectors directed against CARD8. If CARD8 is an inhibitor of Salmonella-induced cell death, as the overexpression experiments suggest, reducing expression of the gene should result in an increase in cell death. Furthermore, this effect should only be seen in cells with the ancestral allele, because those with the derived allele are predicted to already have impaired CARD8 function. The effectiveness of knockdown, as assessed by quantitative RT-PCR, was variable among the cell lines. However, increased cell death (compared to that of cells transduced with a nonsilencing shRNA vector) was detected only in cells that had the ancestral CARD8 genotype and displayed a large reduction of CARD8 transcripts (Figure 4E). No effect on cell death was detected in cells with the derived, nonsense genotype, although we cannot rule out the possibility that greater levels of knockdown would result in an effect. Due to alternative splicing, the effect of rs2043211 may not be a complete protein null58, but our attempts to detect endogenous protein from LCLs with multiple CARD8 antibodies were not successful. However, the data strongly suggest that inhibiting production of CARD8 transcripts results in higher levels of Salmonella-induced cell death and that cells with the derived allele are resistant to the effects of CARD8 knockdown. The data are most consistent with a scenario in which CARD8 acts as an inhibitor of cell death and the derived allele results in reduced CARD8 function.
Evidence for Adaptive Evolution of CARD8
The role of CARD8 in the host response to infection prompted us to test the hypothesis that CARD8 has been subjected to adaptive evolution. Others have noted that CARD8 is absent in several mammalian species, including mice and cows.58 Because group size and infectious-disease burden have been correlated in mammals that live in larger groups59, we hypothesized that loss of CARD8 could have been selected for in these animals. Animals that live in herds or large colonies demonstrate a significant correlation with absence and presumed evolutionary loss of CARD8 (Figure 5A; p value = 0.002 for Pagel's correlation method32,33). Notably, this correlation is robust despite some ambiguity in the categorization of animal group size and the determination of CARD8 presence or absence as a result of low coverage of some genomes (Figure S1 in the Supplemental Data; see also Material and Methods). The correlation suggests that loss of CARD8 might have been selected for; a stronger caspase-1 inflammatory response would be more effective in combating a greater infectious disease burden.
Figure 5.
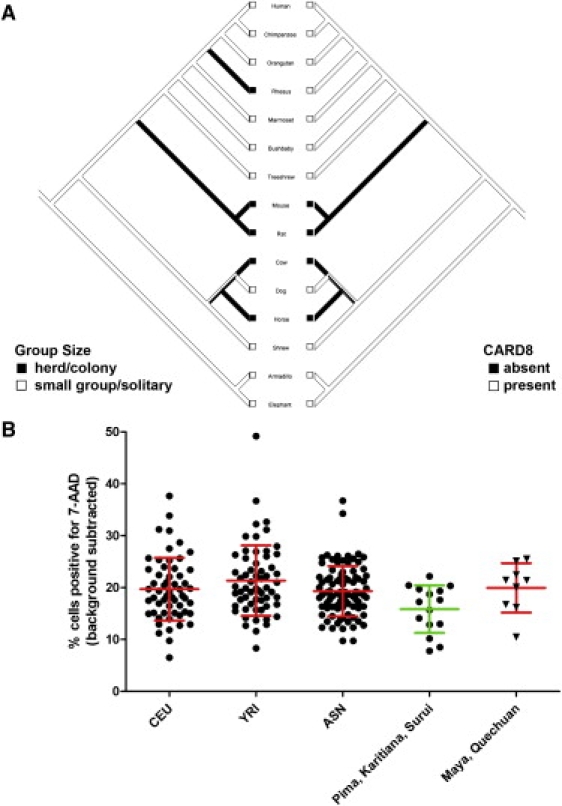
Evidence of Natural Selection Involving CARD8 and Cell Death in Mammals and Humans
(A) Absence of CARD8 in a 15-mammal phylogenetic tree correlates with large group size. A mirror tree of 15 mammalian species displays color-coded group size on the left and the presence of CARD8 on the right. Details regarding putative orthologs can be found in Table S1. From Pagel's test of independent evolution32, a p value of 0.002 is obtained from 1000 simulations. This tree contains only species where the presence or absence of CARD8 could be determined with high confidence (>5× coverage to call absence; presence requiring at least half of protein sequence found). An additional mirror tree of 24 mammalian species is presented as Figure S1. Image was exported from Mesquite.33
(B) Reduced Salmonella-induced cell death in humans from hunter-gatherer versus agricultural populations. Cells from Native American individuals were grouped into populations that had traditionally lived as hunter-gatherers (Pima, Karitiana, and Surui) and those that had lived in denser agricultural communities (Maya and Quechuan) at the time of contact with Europeans. The aggregated hunter-gatherer populations exhibit on average 4.2% less cell death (a 21% reduction) than the CEU, YRI, ASN, and agricultural Native American groups. Error bars show means and standard deviations for each population. One-way ANOVA gives a p value of 0.017 for the means' being different among the populations. The data suggest adaptation of inflammatory caspase activation in response to changes in human societies.
In sequenced primates, CARD8 is uniformly present, consistent with a small-group lifestyle. The two exceptions to this social structure in our analysis are rhesus monkeys, which live in large overlapping troops that can number in the hundreds, and humans, who underwent a relatively recent transition from small groups of hunter-gatherers to large, agriculture-based societies. Comparison of dN/dS ratios in the five primates with complete CARD8 sequences demonstrates evidence of positive selection only in the rhesus monkey lineage (p value = 3.8 × 10−3 according to the branch-site test of positive selection34). CARD8 in rhesus monkeys might be undergoing positive selection for loss of function, although proof of this would require functional assays of the rhesus monkey CARD8 gene.
This signature of selection is not present in human CARD8, but the introduction and increase in frequency of rs2043211 could be a step toward loss of the gene. Gene frequencies for the strongly linked SNP rs6509366 (see Figure 3) from the HGDP-CEPH Diversity Panel60 demonstrate that the two geographic regions that acquired agriculture most recently (∼4000 years ago14) have the lowest frequency: 0.18 in America and 0.211 in Africa versus 0.347 for the complete set. Furthermore, the two lowest population frequencies in this dataset are found in a hunter-gatherer population in America (Pima, 0.04) and Africa (San, 0.1). Do these population differences in CARD8 allele frequency indicate that phenotypic variation in caspase-1 activation is subject to natural selection in humans? If so, LCLs from hunter-gatherer populations should display a reduced level of caspase-1 activation. We therefore assayed publicly available Native American LCLs for cell death in response to Salmonella. Although each population has undergone its own unique demographic history, we grouped the 24 LCLs as primarily hunter-gatherer (Pima, Karitiana, and Surui) or agricultural (Maya and Quechuan) for our analysis. The Maya of Mexico and Quechuan-speaking people of the Andes (including the Inca) practiced high-density terraced farming methods for many crops, including corn, cotton, and potatoes. Additionally, the Inca domesticated several animal species, including llamas and guinea pigs. The mean cell death for the LCLs derived from primarily agricultural Native Americans was very similar to the mean cell death for CEU, YRI, and ASN LCLs. In contrast, the mean cell death for the hunter-gatherer LCLs is approximately 4% less than that for CEU, YRI, ASN, or agricultural Native American populations (a relative 21% decrease; p value = 0.017 for one-way ANOVA; Figure 5B). Although we assayed nearly all the publicly available LCLs derived from Native American populations, the total number is still small, and confirming the significance of these results will require replication with additional cells or populations. Still, the result suggests population differentiation toward a more optimal level of caspase-1 activation depending on the subsistence method of the population.
Variation in CARD8 Is Associated with Acute Inflammatory Disease
Although a more robust inflammatory response is probably adaptive to survival under conditions with a higher infectious-disease burden, an enhanced ability to fight off bacterial infection may not come without a price. Despite this advantage, the CARD8 nonsense allele has not reached fixation. We hypothesized that losing CARD8 inhibition of caspase-1 could be detrimental if it led to an inappropriately robust immune response. Acutely, this could manifest as a higher incidence or severity of inflammation in the setting of a severe infection. To test this, we genotyped individuals who were admitted to an ICU and met criteria for SIRS39 and compared their CARD8 genotype frequencies with those of healthy controls (Table 3). Patients with SIRS have physiological changes in response to an inflammatory stimulus that may be infectious (for example, pneumonia) or noninfectious (for example, pancreatitis). We tested for an association between CARD8 genotype and SIRS by using a genotypic model dictated by the effect of the CARD8 allele on cell death in LCLs. Because the median genotypic values for AA, AT, and TT in the parental CEU and YRI populations were 19.2, 20.4, and 20.8, we used an additive model with partial dominance of the T allele (genotypic scores of 0, 0.75, and 1) in the Cochran-Armitage trend test. We found that genotypes with the T allele were associated with the presence of SIRS (p value = 0.050). This value was just slightly lower than the values obtained for simple additive (scores of 0, 0.5, 1) and T-dominant (scores of 0, 1, 1) models, which gave p values of 0.058 and 0.055, respectively. The overrepresentation of the ancestral CARD8 allele in the healthy controls suggests that a functional CARD8 gene might be protective against SIRS.
Table 3.
Association between CARD8 rs2043211 and Risk of SIRS
| SIRS Cases versus Healthy Controls | AA | AT | TT | p Valuea |
|---|---|---|---|---|
| SIRS (n = 319) | 135 (42.3%) | 149 (46.7%) | 35 (11.0%) | 0.050 |
| Healthy Controls (n = 567) | 278 (49.0%) | 238 (42.0%) | 51 (9.0%) | |
| HapMap CEU parents (n = 60) | 29 (48.3%) | 26 (43.3%) | 5 (8.3%) |
Genotypes are relative to the + strand of chromosome 19. CARD8 coding sequence is on the − strand.
For comparison, the genotypes of the 60 HapMap CEU parents is shown but was not used in the case-control study.
p value is for the Cochran-Armitage trend test using a genotypic model with scores of 0, 0.75, and 1 for the three genotypes.
Discussion
The experimental validation and evolutionary analysis of the CARD8 SNP demonstrate the utility of the Hi-HOST approach in identifying functional genetic variation in cellular phenotypes that have probably been the target of natural selection. Natural selection modifies traits at the level of organisms within populations. However, an organism's quantitative traits are the aggregate of many intermediate phenotypes at the organ, tissue, cellular, and molecular levels. Thus, adaptation of populations might be manifest in the variation of cellular phenotypes, such as cell death in response to pathogens, that play a role in the overall survival of individuals. Hi-HOST is a useful approach in understanding the variation of important cellular phenotypes, and the relevance of variants identified in this way can be put into the broader context of human health through clinical association studies.
Our data support the idea that populations acquire a level of caspase-1 response adapted to their environment and that one way this is achieved is through the presence or absence of functional CARD8. It is likely that each species has undergone fine-tuning through natural selection to strike the optimal balance between the risks of too much or too little caspase-1 inflammatory response. Too little response could be ineffective at warding off bacterial infections, whereas too much response could predispose a species to SIRS or chronic autoimmune disease. For many human populations, this balance probably underwent a shift as the risk of death from highly transmissible human “crowd diseases” (such as S. typhi) has grown to outweigh the risk of overreacting to bacterial strains incidental to trauma.
Animal models provide further evidence for the consequences of too little or too much caspase-1 response. CASP1 knockout mice have increased susceptibility to bacterial infection.18 Conversely, these mice are resistant to endotoxic shock from LPS challenge61, and the caspase-1 inhibitor YVAD has been shown to decrease inflammation and mortality in rat models of endotoxic shock.62,63 Surprisingly, the mechanism by which caspase-1 inhibition protects against sepsis is apparently independent of its pro-inflammatory cleavage of IL-1 β and IL-18. Unlike CASP1−/− mice, IL-1β and IL-1β /IL-18 double-knockout mice are not protected from death due to E. coli sepsis.64 A key difference noted among the mice in these studies was the presence of extensive B lymphocyte apoptosis in wild-type and IL-1β knockout mice but not in CASP1−/− mice. The authors suggest that prevention of lymphocyte apoptosis might be protective during sepsis. This echoes the ideas of Hotchkiss et al.65, who have documented apoptosis of B cells during sepsis in humans66, and the finding that prevention of lymphocyte cell death in a mouse sepsis model improves survival.67 We suggest that the higher incidence of SIRS in individuals with the derived, nonsense CARD8 allele might be mediated by increased caspase-1 activity, which could result in more B lymphocyte cell death. Therefore, the cell-death read-out in the LCLs we used could have further relevance as an in vitro model of lymphocyte cell death during systemic infection.
Our data suggest that individuals carrying the CARD8 nonsense allele are at higher risk of developing SIRS, possibly as an acute consequence of an excessive caspase-1 response. The association between CARD8 and risk of SIRS is weak, but a more informative evaluation could perhaps be made if we examined subcategories of SIRS patients. One obvious distinction is between sepsis (SIRS with a verified or strongly suspected source of infection) versus SIRS without infection. In a cohort of patients admitted to a level-one trauma center after sustaining major traumatic injury, we compared CARD8 genotype frequencies in patients meeting criteria for SIRS with or without clinically suspected or microbiologically confirmed infection (i.e, a comparison of sepsis versus SIRS only). We found no significant association between CARD8 genotypes and the development of sepsis in these trauma patients (p = 0.53; Table S2). A possibly more useful criterion in defining subgroups of SIRS that may show a stronger association with CARD8 genotype could be made if we had a clearer understanding of which SIRS etiologies lead to caspase-1 activation. Sepsis, for example, is probably highly variable in caspase-1 activation depending on the causative bacteria. It will be interesting to determine in future, larger studies whether a more significant association and larger effect can be detected if cases in the study are restricted to those with measurable caspase-1 activation (perhaps on the basis of IL-1β levels) or to etiologies known to result in caspase-1 activation.
Other published evidence suggests that CARD8 genotype could also have chronic consequences on human health. An inappropriately strong caspase-1 response could result in autoimmune disease. Consistent with this, anakinra, a drug that blocks IL-1 (which is cleaved into its active form by caspase-1) is used to treat rheumatoid arthritis.68 rs2043211 does not show an association with the incidence of rheumatoid arthritis or seven other common diseases (including Crohn's disease) on the basis of mining of the Wellcome Case-Control Consortium data.69 However, two recent studies show that the CARD8 nonsense allele is associated with increased severity of rheumatoid arthritis (MIM #180300).24,70 Decreased inhibition of NF-κB has been proposed as the mechanism of action for this association24, but our results show that loss of caspase-1 inhibition may be an additional mechanism. Interestingly, CARD8 is not the only potential inhibitor of caspase-1 to have undergone a loss-of-function mutation during human evolution. CASP12 (MIM ∗608633) has also acquired a nonsense mutation, and in this case, the allele has increased to fixation in European and Asian populations.71 Therefore, the loss of regulators of inflammatory responses may be a common evolutionary mechanism for protecting against negative infectious disease outcomes.
The CARD8 validation experiments provide evidence that our p value and functional SNP filters are useful in prioritizing polymorphisms identified by association studies and has prompted us to pursue validation of the other SNPs associated with the cell-death phenotype. One of the 20 SNPs (rs9630856) lies in SERPINB2 (MIM ∗173390) and correlates with the expression of the adjacent SERPINB10 (MIM ∗602058) gene (GENEVAR dataset28). SERPINB2 is a serine-protease inhibitor that has been implicated in cell death in response to bacteria72–74, and one member of the SERPINB family, CrmA, has even been demonstrated to directly inhibit caspase-1.74,75 Another associated SNP is the V158T polymorphism in COMT (catechol-O-methyl-transferase [MIM +116790]; rs4680), an enzyme that inactivates catecholamines and has been linked to cognitive function and schizophrenia.75 Not only does this SNP correlate with cell death (QFAM-parents of CEU and YRI p value = 0.0036), but the more active valine variant correlates with lower caspase-1 expression (QFAM-parents of CEU and YRI p value = 0.016). Others have shown in lymphocytes that catecholamines can function in an autocrine fashion to increase cell death, partially through transcriptional regulation of caspases.76 We hypothesize that the greater COMT activity demonstrated by the valine variant might decrease catecholamine levels and thus result in less CASP1 transcription and less cell death upon Salmonella infection. Thus, not only can the Hi-HOST approach identify SNPs important in human phenotypic variation, but it might also lead to the discovery of new branches intersecting the pathways being screened. Although not all of the 18 additional candidate SNPs are likely to be validated, it is plausible that a significant fraction regulate Salmonella-induced cell death. Our ongoing studies are aimed at discovering additional SNPs that truly regulate this process and the numerous other Hi-HOST phenotypes.
Therefore, this work provides proof-of-principle that in vitro human phenotyping with microbes could develop as functional tests of susceptibility and outcomes for acute and chronic inflammatory disease. The approach is adaptable for any invasive pathogen, and we have applied it to other Gram-negative bacteria, including Yersinia spp. (not shown). Such tests may be practically important in the future in combination with individual genotyping, particularly for diseases attributable to multiple complex genotypes.
Supplemental Data
Supplemental Data include one figure and two tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Animal Diversity Web, http://animaldiversity.ummz.umich.edu/
Data and the GWAS Analyzer software, http://nwrce.org/gwas-analyzer
GENEVAR dataset, http://www.sanger.ac.uk/humgen/genevar/
MAXTest vignette, http://cran.r-project.org/web/packages/coin/vignettes/MAXtest.pdf
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
SPSmart, http://spsmart.cesga.es/
Wikipedia, http://www.wikipedia.org/
Acknowledgments
We thank James Bliska, Jose Fernandez-Luna, and Seamus Martin for kindly providing reagents. This project was funded by an award from the National Institute of Allergy and Infectious Diseases to the Northwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (U54 AI057141). D.C.K was funded by the UW Bacterial Pathogenesis Training Grant and is a Wyeth Fellow of the Life Sciences Research Foundation.
References
- 1.Edwards A.O., Ritter R., Abel K.J., Manning A., Panhuysen C., Farrer L.A. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 2.Haines J.L., Hauser M.A., Schmidt S., Scott W.K., Olson L.M., Gallins P., Spencer K.L., Kwan S.Y., Noureddine M., Gilbert J.R. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 3.Klein R.J., Zeiss C., Chew E.Y., Tsai J.Y., Sackler R.S., Haynes C., Henning A.K., SanGiovanni J.P., Mane S.M., Mayne S.T. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saxena R., Voight B.F., Lyssenko V., Burtt N.P., de Bakker P.I., Chen H., Roix J.J., Kathiresan S., Hirschhorn J.N., Daly M.J. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 5.Sladek R., Rocheleau G., Rung J., Dina C., Shen L., Serre D., Boutin P., Vincent D., Belisle A., Hadjadj S. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 6.Hampe J., Franke A., Rosenstiel P., Till A., Teuber M., Huse K., Albrecht M., Mayr G., De La Vega F.M., Briggs J. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 7.Rioux J.D., Xavier R.J., Taylor K.D., Silverberg M.S., Goyette P., Huett A., Green T., Kuballa P., Barmada M.M., Datta L.W. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabeti P.C., Varilly P., Fry B., Lohmueller J., Hostetter E., Cotsapas C., Xie X., Byrne E.H., McCarroll S.A., Gaudet R. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voight B.F., Kudaravalli S., Wen X., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooke G.S., Hill A.V. Genetics of susceptibility to human infectious disease. Nat. Rev. Genet. 2001;2:967–977. doi: 10.1038/35103577. [DOI] [PubMed] [Google Scholar]
- 11.Quintana-Murci L., Alcais A., Abel L., Casanova J.L. Immunology in natura: clinical, epidemiological and evolutionary genetics of infectious diseases. Nat. Immunol. 2007;8:1165–1171. doi: 10.1038/ni1535. [DOI] [PubMed] [Google Scholar]
- 12.Coburn B., Grassl G.A., Finlay B.B. Salmonella, the host and disease: A brief review. Immunol. Cell Biol. 2007;85:112–118. doi: 10.1038/sj.icb.7100007. [DOI] [PubMed] [Google Scholar]
- 13.Maki D.G. Coming to grips with foodborne infection—Peanut butter, peppers, and nationwide salmonella outbreaks. N. Engl. J. Med. 2009;360:949–953. doi: 10.1056/NEJMp0806575. [DOI] [PubMed] [Google Scholar]
- 14.Jobling M.A., Hurles M.E., Tyler-Smith C. Garland Science; New York: 2004. Human Evolutionary Genetics: Origins, Peoples, and Disease. [Google Scholar]
- 15.Hill N.J., King C., Flodstrom-Tullberg M. Recent acquisitions on the genetic basis of autoimmune disease. Front. Biosci. 2008;13:4838–4851. doi: 10.2741/3043. [DOI] [PubMed] [Google Scholar]
- 16.Hersh D., Monack D.M., Smith M.R., Ghori N., Falkow S., Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. USA. 1999;96:2396–2401. doi: 10.1073/pnas.96.5.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Velden A.W., Velasquez M., Starnbach M.N. Salmonella rapidly kill dendritic cells via a caspase-1-dependent mechanism. J. Immunol. 2003;171:6742–6749. doi: 10.4049/jimmunol.171.12.6742. [DOI] [PubMed] [Google Scholar]
- 18.Lara-Tejero M., Sutterwala F.S., Ogura Y., Grant E.P., Bertin J., Coyle A.J., Flavell R.A., Galan J.E. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J. Exp. Med. 2006;203:1407–1412. doi: 10.1084/jem.20060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsuji N.M., Tsutsui H., Seki E., Kuida K., Okamura H., Nakanishi K., Flavell R.A. Roles of caspase-1 in Listeria infection in mice. Int. Immunol. 2004;16:335–343. doi: 10.1093/intimm/dxh041. [DOI] [PubMed] [Google Scholar]
- 20.Sansonetti P.J., Phalipon A., Arondel J., Thirumalai K., Banerjee S., Akira S., Takeda K., Zychlinsky A. Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity. 2000;12:581–590. doi: 10.1016/s1074-7613(00)80209-5. [DOI] [PubMed] [Google Scholar]
- 21.Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miao E.A., Alpuche-Aranda C.M., Dors M., Clark A.E., Bader M.W., Miller S.I., Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 23.Pujol C., Bliska J.B. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect. Immun. 2003;71:5892–5899. doi: 10.1128/IAI.71.10.5892-5899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fontalba A., Martinez-Taboada V., Gutierrez O., Pipaon C., Benito N., Balsa A., Blanco R., Fernandez-Luna J.L. Deficiency of the NF-kappaB inhibitor caspase activating and recruitment domain 8 in patients with rheumatoid arthritis is associated with disease severity. J. Immunol. 2007;179:4867–4873. doi: 10.4049/jimmunol.179.7.4867. [DOI] [PubMed] [Google Scholar]
- 25.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 26.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purcell S., Sham P., Daly M.J. Parental phenotypes in family-based association analysis. Am. J. Hum. Genet. 2005;76:249–259. doi: 10.1086/427886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stranger B.E., Nica A.C., Forrest M.S., Dimas A., Bird C.P., Beazley C., Ingle C.E., Dunning M., Flicek P., Koller D. Population genomics of human gene expression. Nat. Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hubbard T.J., Aken B.L., Beal K., Ballester B., Caccamo M., Chen Y., Clarke L., Coates G., Cunningham F., Cutts T. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prasad A.B., Allard M.W., Green E.D. Confirming the phylogeny of mammals by use of large comparative sequence datasets. Mol. Biol. Evol. 2008;25:1795–1808. doi: 10.1093/molbev/msn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pagel M. Detecting correlated evolution on phylogenies: A general method for the comparative analysis of discrete characters. Proc. R. Soc. Lond. B. 1994;255:37–45. [Google Scholar]
- 33.Maddison, W.P., and Maddison, D.R. (2008) Mesquite: A modular system for evolutionary analysis. Version 2.5. http://mesquiteproject.org.
- 34.Zhang J., Nielsen R., Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 35.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 36.Tamura K., Dudley J., Nei M., Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 37.Amigo J., Salas A., Phillips C., Carracedo A. SPSmart: Adapting population based SNP genotype databases for fast and comprehensive web access. BMC Bioinformatics. 2008;9:428. doi: 10.1186/1471-2105-9-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wurfel M.M., Gordon A.C., Holden T.D., Radella F., Strout J., Kajikawa O., Ruzinski J.T., Rona G., Black R.A., Stratton S. Toll-like receptor 1 polymorphisms affect innate immune responses and outcomes in sepsis. Am. J. Respir. Crit. Care Med. 2008;178:710–720. doi: 10.1164/rccm.200803-462OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levy M.M., Fink M.P., Marshall J.C., Abraham E., Angus D., Cook D., Cohen J., Opal S.M., Vincent J.L., Ramsay G. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med. 2003;29:530–538. doi: 10.1007/s00134-003-1662-x. [DOI] [PubMed] [Google Scholar]
- 40.Shalhub S., Junker C.E., Imahara S.D., Mindrinos M.N., Dissanaike S., O'Keefe G.E. Variation in the TLR4 gene influences the risk of organ failure and shock posttrauma: a cohort study. J. Trauma. 2009;66:115–122. doi: 10.1097/TA.0b013e3181938d50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Armitage P. Tests for linear trends in proportions and frequencies. Biometrics. 1955;11:375–386. [Google Scholar]
- 42.Cochran W.G. Some methods for strengthening the common chi-squared tests. Biometrics. 1954;10:417–451. [Google Scholar]
- 43.Hothorn T., Hornik K., van de Wiel M.A., Zeileis A. Implementing a class of permutation tests: The coin package. J. Stat. Software. 2008;28:1–23. [Google Scholar]
- 44.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spielman R.S., Bastone L.A., Burdick J.T., Morley M., Ewens W.J., Cheung V.G. Common genetic variants account for differences in gene expression among ethnic groups. Nat. Genet. 2007;39:226–231. doi: 10.1038/ng1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang R.S., Duan S., Bleibel W.K., Kistner E.O., Zhang W., Clark T.A., Chen T.X., Schweitzer A.C., Blume J.E., Cox N.J. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc. Natl. Acad. Sci. USA. 2007;104:9758–9763. doi: 10.1073/pnas.0703736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loeuillet C., Deutsch S., Ciuffi A., Robyr D., Taffe P., Munoz M., Beckmann J.S., Antonarakis S.E., Telenti A. In vitro whole-genome analysis identifies a susceptibility locus for HIV-1. PLoS Biol. 2008;6:e32. doi: 10.1371/journal.pbio.0060032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosales-Reyes R., Alpuche-Aranda C., Ramirez-Aguilar Mde L., Castro-Eguiluz A.D., Ortiz-Navarrete V. Survival of Salmonella enterica serovar Typhimurium within late endosomal-lysosomal compartments of B lymphocytes is associated with the inability to use the vacuolar alternative major histocompatibility complex class I antigen-processing pathway. Infect. Immun. 2005;73:3937–3944. doi: 10.1128/IAI.73.7.3937-3944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fink S.L., Cookson B.T. Pyroptosis and host cell death responses during Salmonella infection. Cell. Microbiol. 2007;9:2562–2570. doi: 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- 50.Franchi L., Am A., Body-Malapel M., Kanneganti T.D., Ozoren N., Jagirdar R., Inohara N., Vandenabeele P., Bertin J., Coyle A. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 51.Garcia-Calvo M., Peterson E.P., Leiting B., Ruel R., Nicholson D.W., Thornberry N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- 52.Falconer D.S., Mackay T.F.C. Longmans Green; Harlow, Essex, U.K.: 1996. Introduction to Quantitative Genetics. [Google Scholar]
- 53.Razmara M., Srinivasula S.M., Wang L., Poyet J.L., Geddes B.J., DiStefano P.S., Bertin J., Alnemri E.S. CARD-8 protein, a new CARD family member that regulates caspase-1 activation and apoptosis. J. Biol. Chem. 2002;277:13952–13958. doi: 10.1074/jbc.M107811200. [DOI] [PubMed] [Google Scholar]
- 54.Pathan N., Marusawa H., Krajewska M., Matsuzawa S., Kim H., Okada K., Torii S., Kitada S., Krajewski S., Welsh K. TUCAN, an antiapoptotic caspase-associated recruitment domain family protein overexpressed in cancer. J. Biol. Chem. 2001;276:32220–32229. doi: 10.1074/jbc.M100433200. [DOI] [PubMed] [Google Scholar]
- 55.Bouchier-Hayes L., Conroy H., Egan H., Adrain C., Creagh E.M., MacFarlane M., Martin S.J. CARDINAL, a novel caspase recruitment domain protein, is an inhibitor of multiple NF-kappa B activation pathways. J. Biol. Chem. 2001;276:44069–44077. doi: 10.1074/jbc.M107373200. [DOI] [PubMed] [Google Scholar]
- 56.Amrani N., Sachs M.S., Jacobson A. Early nonsense: mRNA decay solves a translational problem. Nat. Rev. Mol. Cell Biol. 2006;7:415–425. doi: 10.1038/nrm1942. [DOI] [PubMed] [Google Scholar]
- 57.Grabarek J., Amstad P., Darzynkiewicz Z. Use of fluorescently labeled caspase inhibitors as affinity labels to detect activated caspases. Hum. Cell. 2002;15:1–12. doi: 10.1111/j.1749-0774.2002.tb00094.x. [DOI] [PubMed] [Google Scholar]
- 58.Bagnall R.D., Roberts R.G., Mirza M.M., Torigoe T., Prescott N.J., Mathew C.G. Novel isoforms of the CARD8 (TUCAN) gene evade a nonsense mutation. Eur. J. Hum. Genet. 2008;16:619–625. doi: 10.1038/sj.ejhg.5201996. [DOI] [PubMed] [Google Scholar]
- 59.Altizer S., Nunn C.L., Thrall P.H., Gittleman J.L., Antonovics J., Cunningham A.A., Dobson A.P., Ezenwa V., Jones K.E., Pedersen A.B. Social organization and parasite risk in mammals: Integrating theory and empirical studies. Annu. Rev. Ecol. Syst. 2003;34:517–547. [Google Scholar]
- 60.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 61.Li P., Allen H., Banerjee S., Franklin S., Herzog L., Johnston C., McDowell J., Paskind M., Rodman L., Salfeld J. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 62.Boost K.A., Hoegl S., Hofstetter C., Flondor M., Stegewerth K., Platacis I., Pfeilschifter J., Muhl H., Zwissler B. Targeting caspase-1 by inhalation-therapy: Effects of Ac-YVAD-CHO on IL-1 beta, IL-18 and downstream proinflammatory parameters as detected in rat endotoxaemia. Intensive Care Med. 2007;33:863–871. doi: 10.1007/s00134-007-0588-0. [DOI] [PubMed] [Google Scholar]
- 63.Mathiak G., Grass G., Herzmann T., Luebke T., Zetina C.C., Boehm S.A., Bohlen H., Neville L.F., Hoelscher A.H. Caspase-1-inhibitor ac-YVAD-cmk reduces LPS-lethality in rats without affecting haematology or cytokine responses. Br. J. Pharmacol. 2000;131:383–386. doi: 10.1038/sj.bjp.0703629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sarkar A., Hall M.W., Exline M., Hart J., Knatz N., Gatson N.T., Wewers M.D. Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin-1beta and interleukin-18. Am. J. Respir. Crit. Care Med. 2006;174:1003–1010. doi: 10.1164/rccm.200604-546OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hotchkiss R.S., Coopersmith C.M., Karl I.E. Prevention of lymphocyte apoptosis–a potential treatment of sepsis? Clin. Infect. Dis. 2005;41(Suppl 7):S465–S469. doi: 10.1086/431998. [DOI] [PubMed] [Google Scholar]
- 66.Hotchkiss R.S., Swanson P.E., Freeman B.D., Tinsley K.W., Cobb J.P., Matuschak G.M., Buchman T.G., Karl I.E. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit. Care Med. 1999;27:1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 67.Hotchkiss R.S., Tinsley K.W., Swanson P.E., Chang K.C., Cobb J.P., Buchman T.G., Korsmeyer S.J., Karl I.E. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc. Natl. Acad. Sci. USA. 1999;96:14541–14546. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bresnihan B., Alvaro-Gracia J.M., Cobby M., Doherty M., Domljan Z., Emery P., Nuki G., Pavelka K., Rau R., Rozman B. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41:2196–2204. doi: 10.1002/1529-0131(199812)41:12<2196::AID-ART15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 69.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kastbom A., Verma D., Eriksson P., Skogh T., Wingren G., Soderkvist P. Genetic variation in proteins of the cryopyrin inflammasome influences susceptibility and severity of rheumatoid arthritis (the Swedish TIRA project) Rheumatology (Oxford) 2008;47:415–417. doi: 10.1093/rheumatology/kem372. [DOI] [PubMed] [Google Scholar]
- 71.Saleh M., Vaillancourt J.P., Graham R.K., Huyck M., Srinivasula S.M., Alnemri E.S., Steinberg M.H., Nolan V., Baldwin C.T., Hotchkiss R.S. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004;429:75–79. doi: 10.1038/nature02451. [DOI] [PubMed] [Google Scholar]
- 72.Losick V.P., Isberg R.R. NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 2006;203:2177–2189. doi: 10.1084/jem.20060766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moffitt K.L., Martin S.L., Walker B. The emerging role of serine proteases in apoptosis. Biochem. Soc. Trans. 2007;35:559–560. doi: 10.1042/BST0350559. [DOI] [PubMed] [Google Scholar]
- 74.Park J.M., Greten F.R., Wong A., Westrick R.J., Arthur J.S., Otsu K., Hoffmann A., Montminy M., Karin M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis–CREB and NF-kappaB as key regulators. Immunity. 2005;23:319–329. doi: 10.1016/j.immuni.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 75.Craddock N., Owen M.J., O'Donovan M.C. The catechol-O-methyl transferase (COMT) gene as a candidate for psychiatric phenotypes: Evidence and lessons. Mol. Psychiatry. 2006;11:446–458. doi: 10.1038/sj.mp.4001808. [DOI] [PubMed] [Google Scholar]
- 76.Jiang J.L., Peng Y.P., Qiu Y.H., Wang J.J. Effect of endogenous catecholamines on apoptosis of Con A-activated lymphocytes of rats. J. Neuroimmunol. 2007;192:79–88. doi: 10.1016/j.jneuroim.2007.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.