

Abstract
Autosomal-dominant progressive external ophthalmoplegia (adPEO) is a mitochondrial disorder that is characterized by accumulation of multiple mitochondrial DNA (mtDNA) deletions in postmitotic tissues. The disorder is heterogeneous, with five known nuclear disease genes that encode the proteins ANT1, Twinkle, POLG, POLG2, and OPA1. Defects in these proteins affect mtDNA maintenance, probably leading to stalled replication forks, consequent mtDNA deletion formation, and progressive respiratory chain deficiency. Here we present a large adPEO family with multiple mtDNA deletions, whose disease was not explained by mutations in any of the known adPEO loci. We mapped the disease locus in this family to chromosome 8q22.1-q23.3. The critical linkage region contained the RRM2B gene, which encodes the small subunit of the ribonucleotide reductase p53R2, which has previously been shown to be essential for the maintenance of mtDNA copy number. Mutation screening of RRM2B revealed a heterozygous nonsense mutation in exon 9 (c.979C→T [p.R327X]) in all affected individuals that was absent in 380 control chromosomes. The same mutation was found to segregate in another adPEO family. The mutant mRNA escaped nonsense-mediated decay and resulted in a protein with truncation of 25 highly conserved C-terminal amino acids essential for the interaction with the ribonucleotide reductase subunit R1. We conclude that dominant-negative or gain-of-function mutations in RRM2B are a cause of multiple mtDNA deletions and adPEO.
Main Text
Autosomal-dominant progressive external ophthalmoplegia (adPEO) (MIM 157640; 609283; 609286; 610131) is characterized by multiple mitochondrial DNA deletions in postmitotic tissues and respiratory-chain-deficient ragged-red fibers with abnormal mitochondria in the muscle biopsy.1 Its most common clinical features include adult-onset weakness of the external eye muscles and exercise intolerance. Additional symptoms are variable and may include cataracts, hearing loss, sensory axonal neuropathy, optic atrophy, ataxia, depression, hypogonadism, and Parkinsonism.2–6
adPEO is a genetically heterogeneous disorder. We and others have previously reported underlying mutations in five genes encoding both subunits of DNA polymerase gamma (POLG [MIM 174763] and POLG2 [MIM 604983]), the DNA helicase Twinkle (C10ORF2 [MIM 606075]), the adenine nucleotide translocator ANT1 (SLC25A4 [MIM 103220]), and optic atrophy 1 (OPA1 [MIM 605290]).7–11 POLG and Twinkle are essential parts of the mtDNA replisome12 in the mitochondrial nucleoid, whereas ANT1 may affect mtDNA maintenance by regulating the intramitochondrial adenine pools via its function as the ADP/ATP translocator in the inner mitochondrial membrane. Defects in these proteins are believed to disturb mtDNA replication, thus leading to stalled replication forks and consequently mtDNA deletion formation.13 In addition to the dominant trait, recessively inherited conditions caused by mutations in the same genes have also been described, often resulting in mtDNA copy-number depletion and/or in a more severe disease with early onset.14–17 OPA1 participates in mitochondrial inner-membrane fusion, and the mechanism by which its defects cause mtDNA deletions could involve nucleoid segregation or mtDNA turnover.
Several adPEO families have been identified in which mutations in SLC25A4, POLG, POLG2, C10ORF2, or OPA1 are not responsible for the disease phenotype. To identify the underlying gene in one such family, we recruited members of a large four-generation North American family of European origin that has 13 affected individuals and an autosomal-dominant inheritance pattern (family 1, Figure 1). The diagnosis was based on the finding of ophthalmoparesis and ptosis with the onset of symptoms or signs late in the second decade or in adult life. Some affected family members had restricted eye movements without ptosis. Commonly, patients were unaware of limitations in eye movements until they were discovered in routine medical examinations. In addition, some patients experienced easy muscle fatigue with exercise. The patients who underwent muscle biopsy exhibited cytochrome c oxidase-deficient skeletal muscle fibers (Figure 2A) and had multiple mtDNA deletions in muscle (Figure 2B), detected by long-range polymerase chain reaction (PCR) as described previously.18 mtDNA deletions were not present in patients' lymphocytes. Informed consent was obtained from all participating patients and unaffected family members as approved by the Institutional Review Board of the University of Texas Southwestern Medical Center. We also obtained 190 unrelated anonymous DNA samples of European origin, which were used as controls.
Figure 1.
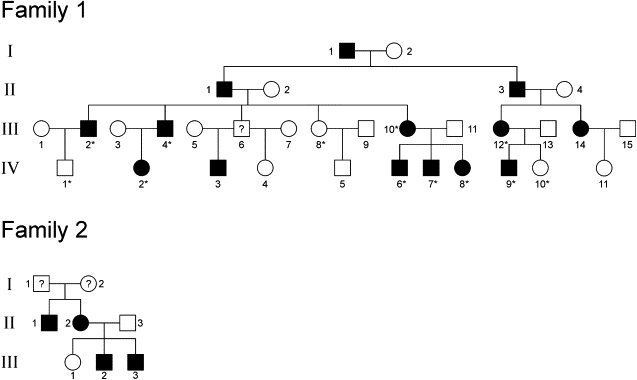
The adPEO Pedigrees
The reduced pedigrees of the families show the participating members and their immediate relatives. The members of family 1 whose genotype data were used in the linkage analyses are marked with an asterisk (∗). The individuals marked with a question mark (?) were not clinically or genetically examined.
Figure 2.
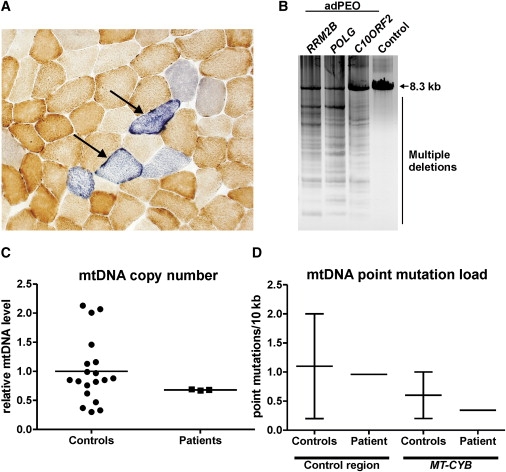
Analyses of Patient Muscle Biopsy for Mitochondrial Respiratory Chain Activity and mtDNA Quality and Quantity
(A) Histochemical staining of frozen muscle section of a patient (family 1, IV:6) for cytochrome c oxidase (brown) and succinate dehydrogenase (blue) reveals abnormal mitochondrial respiratory chain activities, as indicated by arrows. Approximately 10% of the muscle fibers are affected.
(B) Long-PCR analysis shows multiple mtDNA deletions in a muscle sample from an adPEO patient (family 1, IV:6). An 8.3 kb fragment of mtDNA was amplified. Similar multiple deletions are present in the muscle of adPEO patients that have a mutation in RRM2B, POLG, or C10ORF2, but not in a control sample.
(C) mtDNA copy number is normal in muscle samples of adPEO patients (family 1, IV:6, IV:7, IV:8) as determined by quantitative real-time PCR as the ratio of the mitochondrial MT-CYB gene to the nuclear APP gene. Values are shown relative to the average of all control samples. The results were similar when the gene encoding mitochondrial 12S RNA was analyzed instead of MT-CYB.
(D) mtDNA point mutation load is normal in a muscle sample of a patient (family 1, IV:6) as compared to the control range38 in both the control region of mtDNA and the MT-CYB gene.
DNA was extracted from whole blood or muscle biopsy via standard procedures. A genome-wide scan for linkage was performed with an Illumina Infinium HumanLinkage-12 panel containing 6090 SNPs. The genotype data of nine patients and three unaffected family members of family 1 (Figure 1) were included in the multipoint logarithm of the odds (LOD) score analysis, which was performed with the MERLIN program. The analysis was conducted assuming an autosomal-dominant mode of inheritance with full penetrance and no phenocopies. A disease allele frequency of 0.001 was used in the analysis. The allele frequency for all markers was assumed to be equal. The 16 Mb region between SNPs RS874643 and RS1019603 on chromosome 8 (8q22.1-q23.3) was identified as the region of significant linkage (see Figure S1 available online). A maximum multipoint LOD score of 3.6 occurred at 17 SNP markers between RS2022922 and RS3110040. No linkage was found in chromosomes 10q24, 4q35, 15q25, 17q23, or 3q28-q29 where the previously identified adPEO disease genes reside (Figure S1).
The positive linkage region comprises 59 known and predicted genes. One of the genes, RRM2B, has previously been shown to have a role in the maintenance of mtDNA copy number19 and was therefore subjected to direct sequencing as a first priority. The genomic structure of RRM2B consists of nine exons spanning ∼35 kb on chromosome 8. The full-length mRNA is 4955 bp and encodes a 351 aa protein, p53R2. Primers were designed to amplify all RRM2B exons, splice sites, and the flanking sequences (Table S1). Sequencing of the amplified fragments was initially performed for one affected individual (family 1, III:1). Sequence analysis revealed a nonsense mutation in the last exon of RRM2B, exon 9: c.979C→T (p.R327X), which changes an arginine residue to a premature termination codon, predicting a protein with truncation of the last 25 amino acids (Figure 3B). This mutation segregated with the disease phenotype in the examined family members and was not found in 380 control chromosomes of European origin.
Figure 3.
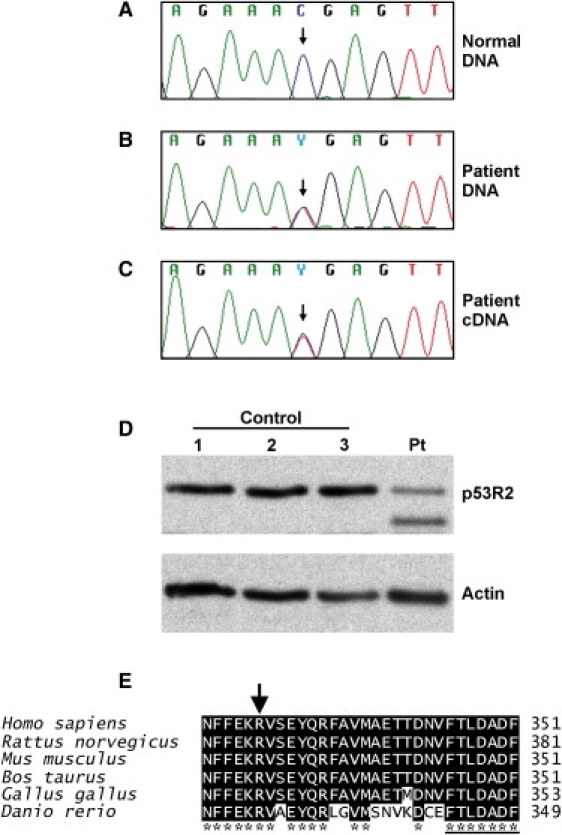
Gene Sequence Analysis of RRM2B and Protein Analysis of p53R2
(A) Partial DNA sequence of normal RRM2B gene.
(B) Partial DNA sequence of patient's RRM2B gene showing the heterozygous c.979C→T mutation.
(C) RRM2B cDNA of patient (family 1, IV:7) myoblasts also contains the heterozygous c.979C→T mutation. We extracted total RNA from the cells with an RNeasy kit (QIAGEN). The RNA was used to generate cDNA with random primers and M-MLV Reverse Transcriptase (Promega) followed by amplification of part of the RRM2B cDNA with primers listed in Table S1 and Phusion polymerase (Finnzymes).
(D) Western analysis of the p53R2 protein in myoblasts shows a truncated p53R2 protein present only in the patient sample (Pt) and not in the control samples (1–3). Ten micrograms of total cellular protein lysate was analyzed by western blot with p53R2 antibody (ab8105 rabbit polyclonal, Abcam). Actin was used as a loading control (sc-1616 goat polyclonal, Santa Cruz Biotechnology).
(E) The C-terminal amino acids of the p53R2 protein are highly conserved. The R327 residue is indicated by an arrow, and the heptapeptide is underlined.
We also collected DNA samples of patients from six PEO families with an unidentified gene defect and screened those for mutations in RRM2B by direct sequencing. We identified the same c.979C→T mutation in patients of an adPEO family of Hungarian origin (family 2, Figure 1). Patient III:2 in this family was a 40-year-old male who had suffered from exercise intolerance and bilateral ptosis since his twenties. Neurological investigation revealed external ophthalmoplegia, hypoacusis, decreased reflexes, and mild gait ataxia. This patient had depressive mood, anxiety, moderate cognitive dysfunction, and alcohol abuse. His brother, mother, and maternal uncle who also suffered from external ophthalmoplegia had the same mutation. Genotyping of additional intragenic SNPs revealed that families 1 and 2 do not share a common disease haplotype and that the mutations therefore arose from two separate events in the two families. No RRM2B mutations were identified in five other unrelated adPEO patients that we tested.
DNA replication and repair require a balanced supply of deoxynucleoside triphosphates (dNTPs). In all cells, the major supply of dNTPs comes from the de novo reduction of ribonucleoside diphosphates to deoxyribonucleoside diphosphates by the cytoplasmic enzyme ribonucleotide reductase (RNR).20 In cycling cells, RNR is a heterodimeric tetramer that consists of R1 and R2 subunits. In postmitotic cells, mitochondria are partially supplied for dNTPs by their own deoxyribonucleotide salvage pathway, but nuclear DNA repair and mtDNA replication also require the nucleosides produced by the RNR tetramer, which is composed of R1 and p53R2 subunits.21–24 RRM2B encodes p53R2. The p53-inducible subunit p53R2 was initially reported to contribute to nuclear DNA damage response,25,26 and its candidacy for tumor suppression has been evaluated in several mutational analyses of different cancer types.27–29 However, the contribution of p53R2 to the DNA damage response has been questioned because its transcriptional induction upon DNA damage is not rapid enough for prompt DNA repair.20,26 Instead, ATM-mediated phosphorylation has been suggested to regulate the DNA repair activity of p53R2 posttranslationally.30 Despite the unclear role of p53R2 in nuclear DNA repair, it is certainly essential for the maintenance of mtDNA copy number in differentiated cells by reducing ribonucleotides in the cytoplasm:24 homozygous loss-of-function mutations in RRM2B are a cause of severe mtDNA depletion syndrome (MDS) in infants,19 and Rrm2b knockout mice die after weaning and present with reduced mtDNA copy number.19,31 We studied whether the mtDNA copy number was affected in the skeletal muscle of adPEO patients of family 1 (IV:6, IV:7, and IV:8) by using real-time qPCR with TaqMan probes for a portion of the mitochondrial MT-CYB gene and the nuclear APP gene as described14 and found that the mtDNA level in the skeletal muscle of three patients of family 1 was comparable to the postmortem samples of control individuals of 19–65 years of age (Figure 2C).
Some of the RRM2B mutations underlying MDS lead to early stop codons,19,32 suggesting that the heterozygous mutation carriers could have both wild-type and truncated p53R2 protein. However, previously reported western analysis of tissues from an MDS patient homozygous for a Q284X mutation showed no protein, suggesting that the mutated mRNA was subject to nonsense-mediated decay.19 Therefore, haploinsufficiency of RRM2B is most likely not pathological because the heterozygous mutation carriers, i.e., the parents of the MDS patients, have been reported to be healthy.19 In our patients, the R327X nonsense mutation predicted a truncated protein of 326 amino acids lacking the 25 most C-terminal, highly conserved amino acids. We cultured myoblasts of one of the patients in family 1 (IV:7) and studied the presence of the mutation at the RNA level (Figure 3C). The sequencing of the PCR product revealed that the c.979C→T mutation was present in the patient's RRM2B cDNA and that the mutant transcript thus escaped nonsense-mediated decay. Nonsense mutations in the last exon of a gene can escape nonsense-mediated decay because this process usually requires the presence of an exon-intron junction downstream of the mutation. We then studied whether the mutant protein was present in the patient cells by western blot and p53R2 antibody (Figure 3D). We identified a truncated variant of p53R2 in the patient sample in addition to the full-length p53R2 protein, showing that the mutant protein was stable. This suggested that the mutant protein can compete with the wild-type in binding to the heterodimeric RNR.
The C terminus of the small subunit of RNR is believed to be required for the interaction between the homodimers of R1 and the small subunit (R2 or p53R2) in the heterotetramer that represents the active form of the enzyme. However, the precise manner of interaction is not known because the C terminus of the small subunit is not visible in the crystal structures of RNR. The truncated p53R2 protein lacking the C-terminal 25 amino acids is thus predicted to affect the interaction of the p53R2 dimer with the R1 dimer because the heptapeptide needed for the interaction is missing33,34 (Figure 3E). Because the mutant protein causes a dominant trait, it is likely to compete with the wild-type p53R2 in binding the heterotetramer, leading to a gain-of-function (competitive binding of mutant p53R2 altering the function of the heterotetramer) or a dominant-negative effect (competitive binding inactivating the heterotetramer) on RNR function. Both models fit with the fact that the dysfunction is fairly subtle and that the outcome is adult-onset and muscle-restricted mtDNA deletions instead of the fatal multisystem mtDNA depletion that is caused by recessive RRM2B mutations.
Alterations in nucleotide pools can cause increased mutagenesis.35,36 We also determined the mtDNA point mutation load in the skeletal muscle of an adPEO patient (family 1, IV:6) as described37,38 but did not identify increased numbers of mtDNA point mutations (Figure 2D). The muscle mtDNA of this patient (age 36 years) had accumulated 0.96 point mutations per 10 kb in the control region of mtDNA and 0.34 mutations per 10 kb in the MT-CYB gene region. Approximately 30,000 nucleotides per mtDNA region were sequenced. Previous studies have shown that this mutation load is in the same range as that of the skeletal muscle of healthy control individuals (∼0.2–2 mutations per 10 kb in the control region and ∼0.2–1 mutations per 10 kb in the MT-CYB gene region).38 The R327X variant of p53R2 thus does not cause excessive accumulation of mtDNA point mutations but instead results in multiple mtDNA deletions. Our patients had no history of tumors, suggesting that RNR mutations are not involved in nuclear mutagenesis and induction of malignancies. Multiple mtDNA deletions in adPEO that are caused by defects in ANT1 are also thought to result from unbalanced dNTP pools. Furthermore, multiple deletions are also found in patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), which is caused by homozygous mutations in the TYMP gene encoding cytoplasmic thymidine phosphorylase (TP) that result in an excess of thymidine and deoxyuridine.39 Our results show that dominant defects in proteins involved in dNTP pool regulation are candidates for adult-onset mitochondrial myopathies.
In conclusion, we have identified a nonsense mutation in RRM2B as a cause of autosomal-dominant progressive external ophthalmoplegia. Our study shows that a defect in p53R2 can induce a mild muscle disease of adult onset through disturbance of mitochondrial homeostasis but that this defect does not appear to be oncogenic. Our finding extends the list of genes that can cause both dominant PEO and recessive MDS when mutated and suggests that other MDS genes should be considered for mutation analysis in patients with multiple mtDNA deletions. The mechanism of mtDNA deletion formation that is caused by a dominant-negative or gain-of-function alteration of truncated p53R2 remains to be determined.
Acknowledgments
The authors would like to thank families 1 and 2 for participation in the study; the Finnish Genome Center for performing the SNP chip protocol; and K. Ayyad, N. Romaine, and A. Harju for laboratory assistance. V. Bianchi and P. Reichard are acknowledged for valuable comments. This study was supported by the Academy of Finland (H.T. and A.S.), the Sigrid Juselius Foundation (A.S.), University of Helsinki (A.S.), and a VA Merit Review grant (R.G.H.).
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Zeviani M., Servidei S., Gellera C., Bertini E., DiMauro S., DiDonato S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature. 1989;339:309–311. doi: 10.1038/339309a0. [DOI] [PubMed] [Google Scholar]
- 2.Luoma P., Melberg A., Rinne J.O., Kaukonen J.A., Nupponen N.N., Chalmers R.M., Oldfors A., Rautakorpi I., Peltonen L., Majamaa K. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: Clinical and molecular genetic study. Lancet. 2004;364:875–882. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- 3.Melberg A., Lundberg P.O., Henriksson K.G., Olsson Y., Stalberg E. Muscle-nerve involvement in autosomal dominant progressive external ophthalmoplegia with hypogonadism. Muscle Nerve. 1996;19:751–757. doi: 10.1002/(SICI)1097-4598(199606)19:6<751::AID-MUS10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 4.Servidei S., Zeviani M., Manfredi G., Ricci E., Silvestri G., Bertini E., Gellera C., Di Mauro S., Di Donato S., Tonali P. Dominantly inherited mitochondrial myopathy with multiple deletions of mitochondrial DNA: Clinical, morphologic, and biochemical studies. Neurology. 1991;41:1053–1059. doi: 10.1212/wnl.41.7.1053. [DOI] [PubMed] [Google Scholar]
- 5.Suomalainen A., Majander A., Haltia M., Somer H., Lonnqvist J., Savontaus M.L., Peltonen L. Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J. Clin. Invest. 1992;90:61–66. doi: 10.1172/JCI115856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suomalainen A., Majander A., Wallin M., Setala K., Kontula K., Leinonen H., Salmi T., Paetau A., Haltia M., Valanne L. Autosomal dominant progressive external ophthalmoplegia with multiple deletions of mtDNA: Clinical, biochemical, and molecular genetic features of the 10q-linked disease. Neurology. 1997;48:1244–1253. doi: 10.1212/wnl.48.5.1244. [DOI] [PubMed] [Google Scholar]
- 7.Kaukonen J., Juselius J.K., Tiranti V., Kyttala A., Zeviani M., Comi G.P., Keranen S., Peltonen L., Suomalainen A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 8.Longley M.J., Clark S., Yu Wai Man C., Hudson G., Durham S.E., Taylor R.W., Nightingale S., Turnbull D.M., Copeland W.C., Chinnery P.F. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am. J. Hum. Genet. 2006;78:1026–1034. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spelbrink J.N., Li F.Y., Tiranti V., Nikali K., Yuan Q.P., Tariq M., Wanrooij S., Garrido N., Comi G., Morandi L. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 10.Van Goethem G., Dermaut B., Lofgren A., Martin J.J., Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- 11.Hudson G., Amati-Bonneau P., Blakely E.L., Stewart J.D., He L., Schaefer A.M., Griffiths P.G., Ahlqvist K., Suomalainen A., Reynier P. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: A novel disorder of mtDNA maintenance. Brain. 2008;131:329–337. doi: 10.1093/brain/awm272. [DOI] [PubMed] [Google Scholar]
- 12.Korhonen J.A., Pham X.H., Pellegrini M., Falkenberg M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004;23:2423–2429. doi: 10.1038/sj.emboj.7600257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goffart S., Cooper H.M., Tyynismaa H., Wanrooij S., Suomalainen A., Spelbrink J.N. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum. Mol. Genet. 2009;18:328–340. doi: 10.1093/hmg/ddn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hakonen A.H., Isohanni P., Paetau A., Herva R., Suomalainen A., Lonnqvist T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain. 2007;130:3032–3040. doi: 10.1093/brain/awm242. [DOI] [PubMed] [Google Scholar]
- 15.Palmieri L., Alberio S., Pisano I., Lodi T., Meznaric-Petrusa M., Zidar J., Santoro A., Scarcia P., Fontanesi F., Lamantea E. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum. Mol. Genet. 2005;14:3079–3088. doi: 10.1093/hmg/ddi341. [DOI] [PubMed] [Google Scholar]
- 16.Sarzi E., Goffart S., Serre V., Chretien D., Slama A., Munnich A., Spelbrink J.N., Rotig A. Twinkle helicase (PEO1) gene mutation causes mitochondrial DNA depletion. Ann. Neurol. 2007;62:579–587. doi: 10.1002/ana.21207. [DOI] [PubMed] [Google Scholar]
- 17.Van Goethem G., Luoma P., Rantamaki M., Al Memar A., Kaakkola S., Hackman P., Krahe R., Lofgren A., Martin J.J., De Jonghe P. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology. 2004;63:1251–1257. doi: 10.1212/01.wnl.0000140494.58732.83. [DOI] [PubMed] [Google Scholar]
- 18.Luoma P.T., Luo N., Loscher W.N., Farr C.L., Horvath R., Wanschitz J., Kiechl S., Kaguni L.S., Suomalainen A. Functional defects due to spacer-region mutations of human mitochondrial DNA polymerase in a family with an ataxia-myopathy syndrome. Hum. Mol. Genet. 2005;14:1907–1920. doi: 10.1093/hmg/ddi196. [DOI] [PubMed] [Google Scholar]
- 19.Bourdon A., Minai L., Serre V., Jais J.P., Sarzi E., Aubert S., Chretien D., de Lonlay P., Paquis-Flucklinger V., Arakawa H. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 20.Nordlund P., Reichard P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 21.Guittet O., Hakansson P., Voevodskaya N., Fridd S., Graslund A., Arakawa H., Nakamura Y., Thelander L. Mammalian p53R2 protein forms an active ribonucleotide reductase in vitro with the R1 protein, which is expressed both in resting cells in response to DNA damage and in proliferating cells. J. Biol. Chem. 2001;276:40647–40651. doi: 10.1074/jbc.M106088200. [DOI] [PubMed] [Google Scholar]
- 22.Hakansson P., Hofer A., Thelander L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J. Biol. Chem. 2006;281:7834–7841. doi: 10.1074/jbc.M512894200. [DOI] [PubMed] [Google Scholar]
- 23.Pontarin G., Ferraro P., Hakansson P., Thelander L., Reichard P., Bianchi V. p53R2-dependent ribonucleotide reduction provides deoxyribonucleotides in quiescent human fibroblasts in the absence of induced DNA damage. J. Biol. Chem. 2007;282:16820–16828. doi: 10.1074/jbc.M701310200. [DOI] [PubMed] [Google Scholar]
- 24.Pontarin G., Fijolek A., Pizzo P., Ferraro P., Rampazzo C., Pozzan T., Thelander L., Reichard P.A., Bianchi V. Ribonucleotide reduction is a cytosolic process in mammalian cells independently of DNA damage. Proc. Natl. Acad. Sci. USA. 2008;105:17801–17806. doi: 10.1073/pnas.0808198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakano K., Balint E., Ashcroft M., Vousden K.H. A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene. 2000;19:4283–4289. doi: 10.1038/sj.onc.1203774. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka H., Arakawa H., Yamaguchi T., Shiraishi K., Fukuda S., Matsui K., Takei Y., Nakamura Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 27.Byun D.S., Chae K.S., Ryu B.K., Lee M.G., Chi S.G. Expression and mutation analyses of P53R2, a newly identified p53 target for DNA repair in human gastric carcinoma. Int. J. Cancer. 2002;98:718–723. doi: 10.1002/ijc.10253. [DOI] [PubMed] [Google Scholar]
- 28.Deng Z.L., Xie D.W., Bostick R.M., Miao X.J., Gong Y.L., Zhang J.H., Wargovich M.J. Novel genetic variations of the p53R2 gene in patients with colorectal adenoma and controls. World J. Gastroenterol. 2005;11:5169–5173. doi: 10.3748/wjg.v11.i33.5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smeds J., Berggren P., Ma X., Xu Z., Hemminki K., Kumar R. Genetic status of cell cycle regulators in squamous cell carcinoma of the oesophagus: The CDKN2A (p16(INK4a) and p14(ARF)) and p53 genes are major targets for inactivation. Carcinogenesis. 2002;23:645–655. doi: 10.1093/carcin/23.4.645. [DOI] [PubMed] [Google Scholar]
- 30.Chang L., Zhou B., Hu S., Guo R., Liu X., Jones S.N., Yen Y. ATM-mediated serine 72 phosphorylation stabilizes ribonucleotide reductase small subunit p53R2 protein against MDM2 to DNA damage. Proc. Natl. Acad. Sci. USA. 2008;105:18519–18524. doi: 10.1073/pnas.0803313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kimura T., Takeda S., Sagiya Y., Gotoh M., Nakamura Y., Arakawa H. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat. Genet. 2003;34:440–445. doi: 10.1038/ng1212. [DOI] [PubMed] [Google Scholar]
- 32.Bornstein B., Area E., Flanigan K.M., Ganesh J., Jayakar P., Swoboda K.J., Coku J., Naini A., Shanske S., Tanji K. Mitochondrial DNA depletion syndrome due to mutations in the RRM2B gene. Neuromuscul. Disord. 2008;18:453–459. doi: 10.1016/j.nmd.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fisher A., Yang F.D., Rubin H., Cooperman B.S. R2 C-terminal peptide inhibition of mammalian and yeast ribonucleotide reductase. J. Med. Chem. 1993;36:3859–3862. doi: 10.1021/jm00076a015. [DOI] [PubMed] [Google Scholar]
- 34.Shao J., Zhou B., Zhu L., Qiu W., Yuan Y.C., Xi B., Yen Y. In vitro characterization of enzymatic properties and inhibition of the p53R2 subunit of human ribonucleotide reductase. Cancer Res. 2004;64:1–6. doi: 10.1158/0008-5472.can-03-3048. [DOI] [PubMed] [Google Scholar]
- 35.Kunz B.A., Kohalmi S.E., Kunkel T.A., Mathews C.K., McIntosh E.M., Reidy J.A. Deoxyribonucleoside triphosphate levels: A critical factor in the maintenance of genetic stability. Mutat. Res. 1994;318:1–64. doi: 10.1016/0165-1110(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 36.Mathews C.K. DNA precursor metabolism and genomic stability. FASEB J. 2006;20:1300–1314. doi: 10.1096/fj.06-5730rev. [DOI] [PubMed] [Google Scholar]
- 37.Hakonen A.H., Goffart S., Marjavaara S., Paetau A., Cooper H., Mattila K., Lampinen M., Sajantila A., Lonnqvist T., Spelbrink J.N. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Hum. Mol. Genet. 2008;17:3822–3835. doi: 10.1093/hmg/ddn280. [DOI] [PubMed] [Google Scholar]
- 38.Wanrooij S., Luoma P., van Goethem G., van Broeckhoven C., Suomalainen A., Spelbrink J.N. Twinkle and POLG defects enhance age-dependent accumulation of mutations in the control region of mtDNA. Nucleic Acids Res. 2004;32:3053–3064. doi: 10.1093/nar/gkh634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishino I., Spinazzola A., Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.