

Abstract
Based on histological and immunohistochemical examination of various organs of patients with autoimmune pancreatitis (AIP), a novel clinicopathological entity of IgG4-related sclerosing disease has been proposed. This is a systemic disease that is characterized by extensive IgG4-positive plasma cells and T-lymphocyte infiltration of various organs. Clinical manifestations are apparent in the pancreas, bile duct, gallbladder, salivary gland, retroperitoneum, kidney, lung, and prostate, in which tissue fibrosis with obliterative phlebitis is pathologically induced. AIP is not simply pancreatitis but, in fact, is a pancreatic disease indicative of IgG4-related sclerosing diseases. This disease includes AIP, sclerosing cholangitis, cholecystitis, sialadenitis, retroperitoneal fibrosis, tubulointerstitial nephritis, interstitial pneumonia, prostatitis, inflammatory pseudotumor and lymphadenopathy, all IgG4-related. Most IgG4-related sclerosing diseases have been found to be associated with AIP, but also those without pancreatic involvement have been reported. In some cases, only one or two organs are clinically involved, while in others, three or four organs are affected. The disease occurs predominantly in older men and responds well to steroid therapy. Serum IgG4 levels and immunostaining with anti-IgG4 antibody are useful in making the diagnosis. Since malignant tumors are frequently suspected on initial presentation, IgG4-related sclerosing disease should be considered in the differential diagnosis to avoid unnecessary surgery.
Keywords: Autoimmune pancreatitis, IgG4, IgG4-related sclerosing disease, Retroperitoneal fibrosis, Sclerosing cholangitis
INTRODUCTION
Since Yoshida et al[1] proposed the concept of autoimmune pancreatitis (AIP) in 1995, many cases have been reported in Western countries, as well as in Japan, and AIP has become a distinct entity recognized worldwide. Although the precise pathogenesis or pathophysiology of AIP remains unclear, many clinical, radiological, serological and histopathological characteristics are obvious. In patients with AIP, serum IgG4 levels are frequently and significantly elevated, and various extrapancreatic lesions are present[2]. Based on histological and immunohistochemical examination of various organs of AIP patients, we have found dense infiltration of IgG4-positive plasma cells and CD4- or CD8-positive T lymphocytes, as well as fibrosis in the peripancreatic retroperitoneal tissue, bile duct wall, gallbladder wall, periportal area of the liver, salivary glands, and the pancreas. Furthermore, all of the extrapancreatic lesions associated with AIP, such as sclerosing cholangitis, sclerosing sialadenitis, and retroperitoneal fibrosis, show infiltration of abundant IgG4-positive plasma cells[2–5]. Both the pancreatic and extrapancreatic lesions of AIP respond well to steroid therapy[6–8].
Therefore, we proposed the existence of a novel clinicopathological entity, IgG4-related sclerosing disease, and suggested that AIP is a pancreatic lesion of this systemic disease. Many recent reports of multiorgan, inflammatory, mass-forming lesions with increased numbers of IgG4-positive plasma cells affirm that AIP may have a systemic component[2,3,8]. On 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) performed in AIP patients, abnormal FDG uptake has been observed in various extrapancreatic lesions[9]. Furthermore, many IgG4-related sclerosing diseases of organs other than the pancreas have been recently reported. Although the nomenclature differs, IgG4-related sclerosing disease has been noted in hepatology, cholangiology, rheumatology, urology, nephrology, respirology, endocrinology, pathology, and radiology, as well as pancreatology. Based on our experience with 50 AIP patients, this review focuses on the clinical, laboratory, imaging, and histopathological features of IgG4-related sclerosing disease, including AIP.
IgG4-RELATED SCLEROSING DISEASE
IgG4-related sclerosing disease is a systemic disease characterized by extensive IgG4-positive plasma cells and T-lymphocyte infiltration of various organs. Clinical manifestations are apparent in the pancreas, bile duct, gallbladder, salivary gland, retroperitoneum, kidney, lung and prostate, in which tissue fibrosis with obliterative phlebitis is pathologically induced (Table 1). AIP is not simply pancreatitis, but it is a pancreatic disease that is indicative of IgG4-related sclerosing disease. Most IgG4-related sclerosing diseases have been found to be associated with AIP, but IgG4-related sclerosing diseases without pancreatic involvement have been reported. Some inflammatory pseudotumors may be involved in this disease. In some cases, only one or two organs are clinically involved, while in others, three or four organs are affected (Figure 1). The disease occurs predominantly in older men, is frequently associated with lymphadenopathy, and responds well to steroid therapy. Serum IgG4 levels and immunostaining with anti-IgG4 antibody are useful in making the diagnosis. The precise pathogenesis and pathophysiology of IgG4-related sclerosing disease remain unclear. Since malignant tumors are frequently suspected on initial presentation, IgG4-related sclerosing disease should be considered in the differential diagnosis to avoid unnecessary surgery[2,3,8].
Table 1.
Clinicopathological findings of IgG4-related sclerosing disease
| Clinicopathological findings | |
| Systemic disease characterized histopathologically by extensive IgG4-positive plasma cell infiltration of various organs together with T lymphocytes | |
| Major clinical manifestations are apparent in the organs in which tissues fibrosis with obstructive phlebitis is pathologically induced | |
| Pancreas | Autoimmune pancreatitis |
| Bile duct | IgG4-related sclerosing cholangitis |
| Gallbladder | IgG4-related sclerosing cholangitis |
| Salivary gland | IgG4-related sclerosing cholangitis |
| Retroperitoneum | IgG4-related retroperitoneal fibrosis |
| Kidney | IgG4-related tubulointerstitial nephritis |
| Lung | IgG4-related interstitial pneumonia |
| Prostate | IgG4-related prostatitis |
| Some inflammatory pseudotumors (liver, lung and hypophysis) may be involved in this disease | |
| Occasional association with lymphadenopathy | |
| Elderly male preponderance | |
| Frequent elevation of serum IgG4 levels | |
| Favorite response to steroid therapy | |
| Differentiation from malignant tumor is important | |
| Precise pathogenesis and pathophysiology remain unclear | |
Figure 1.

Schematic illustration showing the relationship between IgG4-related sclerosing disease, AIP, IgG4-related sclerosing cholangitis, IgG4-related sclerosing sialadenitis, IgG4-related retroperitoneal fibrosis, and IgG4-related pseudotumors.
Multifocal fibrosclerosis is an uncommon fibroproliferative systemic disorder with multiple manifestations, including sclerosing cholangitis, salivary gland fibrosis, retroperitoneal fibrosis, Riedel’s thyroiditis, and fibrotic orbital pseudotumor[10]. As the histopathological findings of these disorders are similar, fibrotic changes with lymphoplasmacytic infiltration and occasional phlebitis, it is suggested that they are all interrelated and probably different manifestations of a common disorder of fibroblastic proliferation. Several cases of pancreatic pseudotumor or chronic pancreatitis associated with multifocal fibrosclerosis have been reported. The histopathology of the extrapancreatic lesions associated with AIP strongly suggests that multifocal fibrosclerosis is an IgG4-related sclerosing disease[5].
AIP
Concept and clinical features
AIP is a unique form of pancreatitis in which auto-immune mechanisms are suspected to be involved in the pathogenesis. The histopathological findings of AIP include marked lymphoplasmacytic infiltration with fibrosis in the pancreas, known as lymphoplasmacytic sclerosing pancreatitis. AIP occurs more commonly in older men. In our series, the mean age of the patients was 66.5 years (range, 25-83 years), and the male-to-female ratio was 4:1. The major clinical symptom is obstructive jaundice, due to associated sclerosing cholangitis (74% in our series). Failure of pancreatic exocrine or endocrine function is frequently seen. Up to 50% of AIP patients present with glucose intolerance. Diabetes mellitus and AIP are simultaneously diagnosed in many cases, but some cases show exacerbation of pre-existing diabetes mellitus with the onset of AIP[11].
Recently, AIP with neutrophilic infiltration of the pancreatic duct epithelium has been reported by American[12] and European[13] pathologists. Their patients showed different clinicopathological features from AIP to those defined in Japan, as follows: no predilection for older men, a frequent association with inflammatory bowel disease, and a weaker association with other sclerosing diseases. In Japan, young AIP patients are more likely to have abdominal pain and serum amylase elevation than middle-aged or elderly patients[14]. Further, international surveys of a larger series of clinically relevant AIP subtypes are needed.
Pathogenesis
Levels of serum IgG4 are frequently elevated, and they are particularly high in AIP. Dense infiltration of IgG4-positive plasma cells is seen in various organs of AIP patients. These findings suggest that IgG4 plays a major role in the pathogenesis of AIP, although the trigger for the IgG4 elevation or its pathogenetic role in AIP has not been clearly established.
Although the actual effector cells of AIP have not been clearly delineated, there are an increased numbers of activated CD4- and CD8-positive T cells bearing HLA-DR among the peripheral blood lymphocytes and in the pancreas of AIP patients. In 2001, Okazaki et al[15] reported that the number of CD4+ T cells producing interferon-γ in peripheral blood and its secreted level were significantly higher in AIP patients than in controls, whereas the number of interleukin (IL)-4-producing CD4+ cells was not increased in AIP patients. They concluded that AIP may be mediated by a Th1-predominant immune reaction, similar to that in Sjogren’s syndrome or primary sclerosing cholangitis (PSC).
There are many findings that support the involvement of immunological mechanisms in AIP, but target antigens for AIP have not been detected. Given the preponderance of the disease amongst elderly males and the marked, dramatic response to oral steroid therapy, the pathogenesis of AIP may not involve an autoimmune mechanism, but other mechanisms, such as an allergic reaction. Zen et al[16] have reported that the expression of Th2 cytokines (IL-4, IL-5, and IL-13) and regulatory cytokines (IL-10 and transforming growth factor-β) was up-regulated in the affected tissues of patients with IgG4-related sclerosing pancreatitis and cholangitis. They have suggested that the predominant Th2 and regulatory immune reactions in this disease might reflect an allergic mechanism in its pathogenesis.
Diagnosis
It is of utmost importance that AIP be differentiated from pancreatic cancer, as some patients with AIP in whom pancreatic cancer is suspected undergo unnecessary laparotomy or pancreatic resection. Since there is currently no diagnostic serological marker for AIP, diagnosis should be on the basis of the presence of a combination of abnormalities unique to AIP. In 2002, the Japan Pancreas Society established the Diagnostic Criteria for AIP[17,18], which were revised in 2006[19]. In 2006, two new sets of diagnostic criteria for AIP were proposed, one in Korea and one in the USA.
Japanese criteria are based on the minimum consensus features of AIP, to minimize the risk of misdiagnosing pancreatic cancer. Revised criteria consist of three items: (1) radiological imaging showing diffuse or segmental narrowing of the main pancreatic duct with irregular walls and diffuse or localized enlargement of the pancreas; (2) laboratory data demonstrating abnormally elevated levels of serum gammaglobulin, IgG, or IgG4, or the presence of autoantibodies; and (3) histological examination of the pancreas showing lymphoplasmacytic infiltration and fibrosis. The diagnosis of AIP is made when either all three criteria are present, or criterion 1 together with either 2 or 3 is present. In addition to the Japanese criteria, the Korean criteria include the patient’s response to steroid therapy and the presence of extrapancreatic lesions.
Radiologically, pancreatic enlargement is usually hypoechoic, sometimes with scattered hyperechoic spots on ultrasonography (Figure 2A). On dynamic computed tomography (CT), there is delayed enhancement of the enlarged pancreatic parenchyma. Typical AIP cases show diffuse enlargement of the pancreas, the so-called sausage-like appearance. Since inflammatory and fibrous changes involve the peripancreatic adipose tissue, a capsule-like rim surrounding the pancreas, which appears as a low density on CT, is detected in some cases. Pancreatic calcification or a pseudocyst is rarely seen. Cases of focal enlargement of the pancreas are sometimes difficult to differentiate from pancreatic cancer. Endoscopic retrograde cholangiopancreatography discloses an irregular, narrow (< 3 mm in diameter) main pancreatic duct. In patients with segmental narrowing, absence of upstream dilatation of the main pancreatic duct is characteristic[20].
Figure 2.

AIP. (A) Diffuse hypoechoic enlargement of the pancreas on ultrasonography; (B) Dense infiltration of IgG4-positive plasma cells in the pancreas.
In our series of AIP patients, hypergammaglobulinemia (> 2.0 g/dL) and elevated serum IgG levels (> 1800 mg/dL) were detected in 34% and 56%, respectively, while autoantibodies, including antinuclear antibody and rheumatoid factor, were present in 44% and 16%.
Serum IgG4 levels are significantly and specifically high (> 135 mg/dL) in AIP patients. The sensitivity of elevated serum IgG4 levels is reported to be 67%-95%[21,22]. Although the measurement of serum IgG4 levels is useful for differentiating between AIP and pancreatic cancer, it should be noted that some patients with pancreatic cancer also have elevated serum IgG4 levels. Ghazale et al[23] have reported that serum IgG4 levels were elevated above 140 mg/dL in 13/135 (10%) patients with pancreatic cancer.
Histologically, dense lymphoplasmacytic infiltration with fibrosis is detected in the pancreas of AIP patients. Immunohistochemically, infiltrating inflammatory cells in the pancreas consist of CD4- or CD8-positive T lymphocytes and IgG4-positive plasma cells (Figure 2B). Lymphoid follicles are occasionally formed. The pancreatic duct is narrowed by periductal fibrosis and lymphoplasmacytic infiltration. Another characteristic histological finding is obliterative phlebitis that involves minor and major veins, including the portal vein. Such an inflammatory process extensively and markedly involves the contiguous soft tissue and peripancreatic retroperitoneal tissues.
Treatment and prognosis
The dramatic response to corticosteroids is well-known in AIP, but a standard steroid therapy regimen has not been established. Before steroid therapy is started, endoscopic or percutaneous transhepatic biliary drainage must be carried out in cases with obstructive jaundice, and glucose levels must be controlled in cases with diabetes mellitus. Oral prednisolone is usually started at 30-40 mg/d, and then it is tapered by 5 mg every 1-2 wk. Serological and imaging tests are followed periodically after commencement of steroid therapy. Usually, pancreatic size is normalized within a few weeks, and biliary drainage becomes unnecessary after 1-2 mo. Patients in whom complete radiological improvement is documented can stop their medication. To prevent relapse without complete discontinuation of steroids, continued maintenance therapy with prednisolone 5 mg/d is sometimes required. In half of steroid-treated patients, impaired exocrine or endocrine function improves. Some AIP patients relapse during maintenance therapy or after steroid medication is stopped; they should be retreated with high-dose steroid therapy. The indications for steroid therapy in AIP include obstructive jaundice due to stenosis of the bile duct, or the presence of other associated systemic diseases, such as retroperitoneal fibrosis[24–26].
The long-term prognosis of AIP is not well known. Recurrent attacks of AIP resulting in pancreatic stone formation have been reported[27,28].
IgG4-RELATED SCLEROSING CHOLANGITIS
Sclerosing cholangitis is a heterogeneous disease that may be associated with choledocholithiasis, biliary tumor, or infection. Sclerosing cholangitis of unknown origin is called PSC. PSC is progressive despite conservative therapy, and it involves the intra- and extrahepatic bile ducts, and results in liver cirrhosis. The effect of steroid therapy is questionable, and liver transplantation currently provides the greatest hope for a possible cure. PSC occurs from 30 to 40 years of age and is frequently associated with inflammatory bowel disease[29,30]. Pancreatography is not abnormal in most PSC cases[31].
IgG4-related sclerosing cholangitis is included within the sclerosing cholangitis group. This form is frequently associated with AIP and occasionally with other diseases such as sclerosing sialadenitis, all of which fall within the category of IgG4-related sclerosing disease. However, IgG4-related sclerosing cholangitis is not associated with inflammatory bowel disease. In many AIP cases, stenosis is located in the lower part of the common bile duct, but thickening of the common bile duct wall is sometimes detected even in the segment in which abnormalities are not clearly observed upon cholangiography. When stenosis is found in the intrahepatic or the hilar hepatic bile duct, the cholangiographic appearance is very similar to that of PSC (Figure 3A)[31,32]. Many cases of IgG4-related sclerosing cholangitis have been described with isolated biliary tract involvement in the absence of pancreatic disease[33,34]. Elevation of serum IgG4 is frequently observed in patients with IgG4-related sclerosing cholangitis, and it responds dramatically to steroid therapy, unlike PSC. Clinically, patients with IgG4-related sclerosing cholangitis are older at diagnosis than those with PSC. Patients with IgG4-related sclerosing cholangitis present more abruptly with obstructive jaundice, whereas obstructive jaundice is rarely present in PSC patients. The histological appearance is similar to that in the pancreas of AIP patients: transmural fibrosis, dense lymphoplasmacytic infiltration of the bile duct wall, along with lymphoplasmacytic infiltration and fibrosis in the periportal area of the liver, and obliterative phlebitis. Despite the dense periluminal inflammation, the biliary epithelium is usually intact, in contrast to PSC, in which mucosal erosion is often present. Furthermore, unlike in PSC, the inflammatory process is often more dense at the periphery of the duct. Neutrophils, commonly seen in PSC, are not a feature of this disease. Immunohistochemically, infiltration of abundant IgG4-positive plasma cells is detected in the bile duct wall (Figure 3B), in contrast to PSC. Given the age at onset, associated diseases, pancreatographic findings, response to steroid therapy, prognosis, and IgG4-related serological and immunohistochemical data, it can be said that IgG4-related sclerosing cholangitis is a different disease, and distinct from PSC[35,36].
Figure 3.
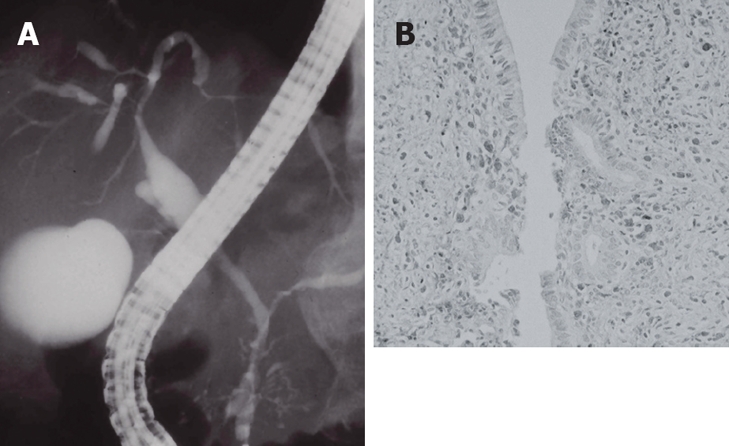
IgG4-related sclerosing cholangitis. (A) Stenosis of the intrahepatic bile duct, similar to that in PSC; (B) Dense infiltration of IgG4-positive plasma cells in the bile duct wall.
IgG4-RELATED SCLEROSING SIALADENITIS
Sclerosing sialadenitis has been referred to as Kuttner’s tumor, due to its presentation as a firm swelling of the salivary gland that is difficult to differentiate from a neoplasm[37]. Mikulicz’s disease is a unique condition that refers to bilateral, painless and symmetrical swelling of the lacrimal, parotid and submandibular glands[38]. Although Mikulicz’s disease has been considered a subtype of Sjogren’s syndrome, there are several differences between the two diseases. Patients with Mikulicz’s disease lack anti-SS-A and anti-SS-B antibodies, but frequently have elevated serum IgG4 levels. Infiltration of many IgG4-positive plasma cells into the lacrimal and salivary glands has been detected in Mikulicz’s disease.
Swelling of the salivary glands was present in 24% of our AIP patients (Figure 4A), and it was associated with cervical or mediastinal lymphadenopathy. Swelling of the salivary glands and the lymph nodes improves after steroid therapy. In the salivary glands of these patients, dense infiltration of IgG4-positive plasma cells and fibrosis were detected (Figure 4B). Kitagawa et al[39] have reported that dense infiltration of IgG4-positive plasma cells was detected in the salivary glands of 12 patients with sclerosing sialadenitis (Kuttner’s tumor). Five of these patients had associated sclerosing lesions in extrasalivary glandular tissue, such as in AIP, while the remaining seven patients had only salivary gland involvement. Thus, many cases of sclerosing sialadenitis, including Kuttner’s tumor and Mikulicz’s disease, may be salivary gland lesions of IgG4-related systemic disease[40,41].
Figure 4.
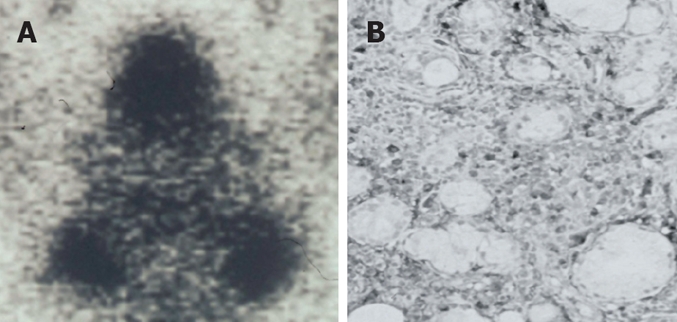
IgG4-related sclerosing sialadenitis. (A) Bilateral swelling of submandibular glands (Gallium scintigraphy); (B) Dense infiltration of IgG4-positive plasma cells in the salivary gland.
IgG4-RELATED RETROPERITONEAL FIBROSIS
Retroperitoneal fibrosis generally presents in a non-specific manner with malaise, fatigue, fever and weight loss. The pathognomonic feature of retroperitoneal fibrosis is a thick retroperitoneal fibrotic mass that covers the abdominal aorta and compresses the ureters[42]. The process of fibrosis can result in obstruction of the ureters and renal failure, or signs and symptoms may be related to the encasement or entrapment of other structures by the inflammatory mass, such as hydronephrosis. Retroperitoneal fibrosis has many causes, although in about 70% of cases, the cause is unknown[43].
Retroperitoneal fibrosis was present simultaneously or metachronously in 8% of our AIP patients. Dense infiltration of IgG4-positive plasma cells and obliterative phlebitis were found in the pancreas and the retroperitoneal fibrous mass. Both the retroperitoneal fibrosis and AIP resolved after steroid therapy[44]. Neild et al[43] have reported the histological findings of 12 patients with idiopathic retroperitoneal fibrosis, which showed, to varying degrees, fibrosis and intense inflammatory cell infiltration with T lymphocytes and IgG4-positive plasma cells, as well as venulitis and obliterative arteritis. Biopsy of the mass after steroid therapy revealed decreased infiltration of IgG4-positive plasma cells. Some cases of retroperitoneal fibrosis are the retroperitoneal lesions of IgG4-related systemic disease. Recently, a 52-year-old man with retroperitoneal and mediastinal fibrosis without AIP was reported to have elevated serum IgG4 levels[45].
IgG4-RELATED TUBULOINTERSTITIAL NEPHRITIS
Tubulointerstitial nephritis may be related to various causative factors, including infection, and allergic, toxic and autoimmune conditions. Tubulointerstitial nephritis is sometimes associated with AIP, but there have been a few cases of tubulointerstitial nephritis showing high serum IgG4 levels without notable pancreatic lesions[46–50]. Some AIP cases show several nodular lesions in the kidney that mimic metastatic tumors[46,47]. Takahashi et al[48] have reported that, in 40 AIP patients, 14 (35%) had renal involvement (12 with parenchymal involvement and five with extraparenchymal involvement). The renal lesions regressed after steroid therapy, but they progressed without steroid therapy. Immunohistochemically, dense infiltration of IgG4-positive plasma cells was detected in the renal interstitium. Furthermore, in some AIP cases, membranous nephropathy that showed IgG4-positive deposits in the glomeruli or tubular basement membrane have been reported[49]. These IgG4-positive lesions decreased with improvement of renal function after steroid therapy.
IgG4-RELATED INTERSTITIAL PNEUMONIA
Interstitial pneumonia, with X-ray findings such as an interstitial pattern, ground-glass appearance, and honeycombing, is sometimes associated with AIP[51–53]. On high-resolution CT of the lung, dense alveolar consolidation and air bronchograms in bilateral perihilar regions have been reported[51]. Some cases have respiratory failure, and steroid therapy is effective in improving respiratory function and radiological findings. In biopsy specimens, dense infiltration of IgG4-positive plasma cells is detected in the thickened alveolar septum[51–53]. Hirano et al[53] have reported that 4/30 AIP patients had pulmonary involvement during follow-up.
IgG4-RELATED SCLEROSING CHOLECYSTITIS
In our series, thickening of the gallbladder was detected on US and/or CT in 32% of AIP patients. Dense infiltration of IgG4-positive plasma cells and lymphocytes, as well as transmural fibrosis, was detected in the gallbladder wall of 6/8 patients examined[54].
IgG4-RELATED PROSTATITIS
IgG4-related prostatitis has recently been reported in patients with and without AIP. Patients show lower urinary tract symptoms, and prostate enlargement is evident on digital rectal examination. Significant FDG uptake by the prostate has been demonstrated. The symptoms and radiological findings improve after steroid therapy. Histologically, the prostate shows dense infiltration of IgG4-positive plasma cells and lymphocytes, obliterative phlebitis, and gland atrophy with dense fibrosis[55–57].
IgG4-RELATED INFLAMMATORY PSEUDOTUMOR OF THE LIVER, LUNG AND HYPOPHYSIS
Inflammatory pseudotumors occur in almost all major organs. An inflammatory pseudotumor is characterized histologically as an irregular proliferation of myofibroblasts intermixed with an infiltrate of inflammatory cells, mainly lymphocytes and plasma cells. One inflammatory pseudotumor is categorized as a plasma cell-rich type, plasma cell granuloma[58]. Although its pathogenesis is not known, a close relationship between plasma cell granuloma and IgG4-positive plasma cells has recently become obvious[5].
Some IgG4-related inflammatory pseudotumors of the liver[34,59,60] and lung[61], which are characterized by dense infiltration of IgG4-positive plasma cells and lymphocytes intermixed with fibrosis and obliterative phlebitis, have been recently reported in patients with or without AIP. Four cases of hypophysitis in association with AIP have been reported; they all presented with hypopituitarism and swelling of the pituitary lesion[62–65]. In one case[62], abundant infiltration of IgG4-positive plasma cell was demonstrated in the pituitary tumor. Steroid therapy is effective for these IgG4-related inflammatory pseudotumors.
IgG4-RELATED LYMPHADENOPATHY
In a study using gallium-67 scintigraphy, pulmonary hilar gallium-67 uptake was found in 41/51 (80.4%) patients with AIP[66]. In our series, abdominal lymphadenopathy of up to 2 cm in diameter was observed in 5/8 patients at laparotomy, and cervical or mediastinal lymphadenopathy of up to 1.5 cm in diameter was observed on CT in 33% of our AIP patients. In all these cases, the lymphadenopathy disappeared after steroid therapy. Dense infiltration of IgG4-positive plasma cells was detected in all abdominal and cervical lymph nodes. When bilateral hilar lymphadenopathy is marked, sarcoidosis is suspected clinically[4].
Other reported lesions associated with AIP are immune thrombocytopenic purpura[67,68], autoimmune sensorineural hearing loss[68], hypothyroidism[69], anosmia[70], and loss of taste[70].
CONCLUSION
IgG4-related sclerosing disease is a new clinicopathological systemic entity. It is characterized by extensive IgG4-positive plasma cells and T-lymphocyte infiltration of various organs, and major clinical manifestations are apparent in the organs, in which tissues fibrosis with obliterative phlebitis is pathologically induced. As steroid therapy is effective, accurate diagnosis is necessary.
Peer reviewers: Parimal Chowdhury, Professor, Department of Physiology and Biophysics, College of Medicine University of Arkansas for Medical Sciences, 4301 W Markham Street Little Rock, Arkansas 72205, United States; Karel van Erpecum, Department of Gastroenterology and Hepatology, University Hospital Utrecht, PO Box 855003508 GA, Utrecht, Netherlands
S- Editor Li DL L- Editor Kerr C E- Editor Ma WH
References
- 1.Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40:1561–1568. doi: 10.1007/BF02285209. [DOI] [PubMed] [Google Scholar]
- 2.Kamisawa T, Okamoto A. Autoimmune pancreatitis: proposal of IgG4-related sclerosing disease. J Gastroenterol. 2006;41:613–625. doi: 10.1007/s00535-006-1862-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, Okamoto A, Egawa N, Nakajima H. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38:982–984. doi: 10.1007/s00535-003-1175-y. [DOI] [PubMed] [Google Scholar]
- 4.Kamisawa T, Egawa N, Nakajima H. Autoimmune pancreatitis is a systemic autoimmune disease. Am J Gastroenterol. 2003;98:2811–2812. doi: 10.1111/j.1572-0241.2003.08758.x. [DOI] [PubMed] [Google Scholar]
- 5.Kamisawa T, Funata N, Hayashi Y, Tsuruta K, Okamoto A, Amemiya K, Egawa N, Nakajima H. Close relationship between autoimmune pancreatitis and multifocal fibrosclerosis. Gut. 2003;52:683–687. doi: 10.1136/gut.52.5.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A, Hayashi Y, Funata N. Gastrointestinal findings in patients with autoimmune pancreatitis. Endoscopy. 2005;37:1127–1130. doi: 10.1055/s-2005-870369. [DOI] [PubMed] [Google Scholar]
- 7.Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Extrapancreatic lesions in autoimmune pancreatitis. J Clin Gastroenterol. 2005;39:904–907. doi: 10.1097/01.mcg.0000180629.77066.6c. [DOI] [PubMed] [Google Scholar]
- 8.Kamisawa T, Nakajima H, Egawa N, Funata N, Tsuruta K, Okamoto A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology. 2006;6:132–137. doi: 10.1159/000090033. [DOI] [PubMed] [Google Scholar]
- 9.Nakajo M, Jinnouchi S, Fukukura Y, Tanabe H, Tateno R, Nakajo M. The efficacy of whole-body FDG-PET or PET/CT for autoimmune pancreatitis and associated extrapancreatic autoimmune lesions. Eur J Nucl Med Mol Imaging. 2007;34:2088–2095. doi: 10.1007/s00259-007-0562-7. [DOI] [PubMed] [Google Scholar]
- 10.Comings DE, Skubi KB, Van Eyes J, Motulsky AG. Familial multifocal fibrosclerosis. Findings suggesting that retroperitoneal fibrosis, mediastinal fibrosis, sclerosing cholangitis, Riedel's thyroiditis, and pseudotumor of the orbit may be different manifestations of a single disease. Ann Intern Med. 1967;66:884–892. doi: 10.7326/0003-4819-66-5-884. [DOI] [PubMed] [Google Scholar]
- 11.Kamisawa T, Egawa N, Inokuma S, Tsuruta K, Okamoto A, Kamata N, Nakamura T, Matsukawa M. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas. 2003;27:235–238. doi: 10.1097/00006676-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol. 2003;27:1119–1127. doi: 10.1097/00000478-200308000-00009. [DOI] [PubMed] [Google Scholar]
- 13.Zamboni G, Luttges J, Capelli P, Frulloni L, Cavallini G, Pederzoli P, Leins A, Longnecker D, Kloppel G. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch. 2004;445:552–563. doi: 10.1007/s00428-004-1140-z. [DOI] [PubMed] [Google Scholar]
- 14.Kamisawa T, Wakabayashi T, Sawabu N. Autoimmune pancreatitis in young patients. J Clin Gastroenterol. 2006;40:847–850. doi: 10.1097/01.mcg.0000225604.10611.61. [DOI] [PubMed] [Google Scholar]
- 15.Okazaki K, Uchida K, Ohana M, Nakase H, Uose S, Inai M, Matsushima Y, Katamura K, Ohmori K, Chiba T. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology. 2000;118:573–581. doi: 10.1016/s0016-5085(00)70264-2. [DOI] [PubMed] [Google Scholar]
- 16.Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, Nakanuma Y. Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology. 2007;45:1538–1546. doi: 10.1002/hep.21697. [DOI] [PubMed] [Google Scholar]
- 17.Members of the Criteria Committee for Autoimmune Pancreatitis of the Japan Pancreas Society. Diagnostic criteria for autoimmune pancreatitis by the Japan Pancreas Society (in Japanese) Suizou (J Jpn Pan Soc) 2002;17:585–587. [Google Scholar]
- 18.Pearson RK, Longnecker DS, Chari ST, Smyrk TC, Okazaki K, Frulloni L, Cavallini G. Controversies in clinical pancreatology: autoimmune pancreatitis: does it exist? Pancreas. 2003;27:1–13. doi: 10.1097/00006676-200307000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Okazaki K, Kawa S, Kamisawa T, Naruse S, Tanaka S, Nishimori I, Ohara H, Ito T, Kiriyama S, Inui K, et al. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol. 2006;41:626–631. doi: 10.1007/s00535-006-1868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A, Kamata N. Clinical difficulties in the differentiation of autoimmune pancreatitis and pancreatic carcinoma. Am J Gastroenterol. 2003;98:2694–2699. doi: 10.1111/j.1572-0241.2003.08775.x. [DOI] [PubMed] [Google Scholar]
- 21.Kamisawa T, Okamoto A, Funata N. Clinicopathological features of autoimmune pancreatitis in relation to elevation of serum IgG4. Pancreas. 2005;31:28–31. doi: 10.1097/01.mpa.0000167000.11889.3a. [DOI] [PubMed] [Google Scholar]
- 22.Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 23.Ghazale A, Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Clain JE, Pearson RK, Pelaez-Luna M, Petersen BT, et al. Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol. 2007;102:1646–1653. doi: 10.1111/j.1572-0241.2007.01264.x. [DOI] [PubMed] [Google Scholar]
- 24.Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Morphological changes after steroid therapy in autoimmune pancreatitis. Scand J Gastroenterol. 2004;39:1154–1158. doi: 10.1080/00365520410008033. [DOI] [PubMed] [Google Scholar]
- 25.Kamisawa T, Yoshiike M, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Treating patients with autoimmune pancreatitis: results from a long-term follow-up study. Pancreatology. 2005;5:234–238; discussion 238-240. doi: 10.1159/000085277. [DOI] [PubMed] [Google Scholar]
- 26.Kamisawa T, Okamoto A, Wakabayashi T, Watanabe H, Sawabu N. Appropriate steroid therapy for autoimmune pancreatitis based on long-term outcome. Scand J Gastroenterol. 2008;43:609–613. doi: 10.1080/00365520701731263. [DOI] [PubMed] [Google Scholar]
- 27.Takayama M, Hamano H, Ochi Y, Saegusa H, Komatsu K, Muraki T, Arakura N, Imai Y, Hasebe O, Kawa S. Recurrent attacks of autoimmune pancreatitis result in pancreatic stone formation. Am J Gastroenterol. 2004;99:932–937. doi: 10.1111/j.1572-0241.2004.04162.x. [DOI] [PubMed] [Google Scholar]
- 28.Kamisawa T, Okamoto A. Prognosis of autoimmune pancreatitis. J Gastroenterol. 2007;42 Suppl 18:59–62. doi: 10.1007/s00535-007-2052-x. [DOI] [PubMed] [Google Scholar]
- 29.LaRusso NF, Wiesner RH, Ludwig J, MacCarty RL. Current concepts. Primary sclerosing cholangitis. N Engl J Med. 1984;310:899–903. doi: 10.1056/NEJM198404053101407. [DOI] [PubMed] [Google Scholar]
- 30.Wiesner RH, Grambsch PM, Dickson ER, Ludwig J, MacCarty RL, Hunter EB, Fleming TR, Fisher LD, Beaver SJ, LaRusso NF. Primary sclerosing cholangitis: natural history, prognostic factors and survival analysis. Hepatology. 1989;10:430–436. doi: 10.1002/hep.1840100406. [DOI] [PubMed] [Google Scholar]
- 31.Nakazawa T, Ohara H, Sano H, Ando T, Aoki S, Kobayashi S, Okamoto T, Nomura T, Joh T, Itoh M. Clinical differences between primary sclerosing cholangitis and sclerosing cholangitis with autoimmune pancreatitis. Pancreas. 2005;30:20–25. [PubMed] [Google Scholar]
- 32.Kamisawa T, Egawa N, Tsuruta K, Okamoto A, Funata N. Primary sclerosing cholangitis may be overestimated in Japan. J Gastroenterol. 2005;40:318–319. doi: 10.1007/s00535-004-1543-2. [DOI] [PubMed] [Google Scholar]
- 33.Hamano H, Kawa S, Uehara T, Ochi Y, Takayama M, Komatsu K, Muraki T, Umino J, Kiyosawa K, Miyagawa S. Immunoglobulin G4-related lymphoplasmacytic sclerosing cholangitis that mimics infiltrating hilar cholangiocarcinoma: part of a spectrum of autoimmune pancreatitis? Gastrointest Endosc. 2005;62:152–157. doi: 10.1016/s0016-5107(05)00561-4. [DOI] [PubMed] [Google Scholar]
- 34.Zen Y, Harada K, Sasaki M, Sato Y, Tsuneyama K, Haratake J, Kurumaya H, Katayanagi K, Masuda S, Niwa H, et al. IgG4-related sclerosing cholangitis with and without hepatic inflammatory pseudotumor, and sclerosing pancreatitis-associated sclerosing cholangitis: do they belong to a spectrum of sclerosing pancreatitis? Am J Surg Pathol. 2004;28:1193–1203. doi: 10.1097/01.pas.0000136449.37936.6c. [DOI] [PubMed] [Google Scholar]
- 35.Nakanuma Y, Zen Y. Pathology and immunopathology of immunoglobulin G4-related sclerosing cholangitis: The latest addition to the sclerosing cholangitis family. Hepatol Res. 2007;37 Suppl 3:S478–S486. doi: 10.1111/j.1872-034X.2007.00243.x. [DOI] [PubMed] [Google Scholar]
- 36.Ghazale A, Chari ST, Zhang L, Smyrk TC, Takahashi N, Levy MJ, Topazian MD, Clain JE, Pearson RK, Petersen BT, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–715. doi: 10.1053/j.gastro.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 37.Kuttner H. Ueber entzundliche tumoren der submaxillar speicheldruse. Bruns Beitr Klin Chir. 1886;8:815–828. [Google Scholar]
- 38.Schaffer AJ, Jacobsen AW. Mikulicz’s syndrome: a report of ten cases. Am J Dis Child. 1927;34:327–346. [Google Scholar]
- 39.Kitagawa S, Zen Y, Harada K, Sasaki M, Sato Y, Minato H, Watanabe K, Kurumaya H, Katayanagi K, Masuda S, et al. Abundant IgG4-positive plasma cell infiltration characterizes chronic sclerosing sialadenitis (Kuttner's tumor) Am J Surg Pathol. 2005;29:783–791. doi: 10.1097/01.pas.0000164031.59940.fc. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto M, Harada S, Ohara M, Suzuki C, Naishiro Y, Yamamoto H, Takahashi H, Imai K. Clinical and pathological differences between Mikulicz's disease and Sjogren's syndrome. Rheumatology (Oxford) 2005;44:227–234. doi: 10.1093/rheumatology/keh447. [DOI] [PubMed] [Google Scholar]
- 41.Kamisawa T, Nakajima H, Hishima T. Close correlation between chronic sclerosing sialadenitis and immunoglobulin G4. Intern Med J. 2006;36:527–529. doi: 10.1111/j.1445-5994.2006.01119.x. [DOI] [PubMed] [Google Scholar]
- 42.Ormond JK. Bilateral ureteral obstruction due to envelo-pment and compression by an inflammatory retroperitoneal process. J Urol. 1948;59:950–954. doi: 10.1016/S0022-5347(17)69482-5. [DOI] [PubMed] [Google Scholar]
- 43.Neild GH, Rodriguez-Justo M, Wall C, Connolly JO. Hyper-IgG4 disease: report and characterisation of a new disease. BMC Med. 2006;4:23. doi: 10.1186/1741-7015-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamisawa T, Chen PY, Tu Y, Nakajima H, Egawa N. Autoimmune pancreatitis metachronously associated with retroperitoneal fibrosis with IgG4-positive plasma cell infiltration. World J Gastroenterol. 2006;12:2955–2957. doi: 10.3748/wjg.v12.i18.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zen Y, Sawazaki A, Miyayama S, Notsumata K, Tanaka N, Nakanuma Y. A case of retroperitoneal and mediastinal fibrosis exhibiting elevated levels of IgG4 in the absence of sclerosing pancreatitis (autoimmune pancreatitis) Hum Pathol. 2006;37:239–243. doi: 10.1016/j.humpath.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 46.Rudmik L, Trpkov K, Nash C, Kinnear S, Falck V, Dushinski J, Dixon E. Autoimmune pancreatitis associated with renal lesions mimicking metastatic tumours. CMAJ. 2006;175:367–369. doi: 10.1503/cmaj.051668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murashima M, Tomaszewski J, Glickman JD. Chronic tubulointerstitial nephritis presenting as multiple renal nodules and pancreatic insufficiency. Am J Kidney Dis. 2007;49:e7–e10. doi: 10.1053/j.ajkd.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi N, Kawashima A, Fletcher JG, Chari ST. Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings. Radiology. 2007;242:791–801. doi: 10.1148/radiol.2423060003. [DOI] [PubMed] [Google Scholar]
- 49.Watson SJ, Jenkins DA, Bellamy CO. Nephropathy in IgG4-related systemic disease. Am J Surg Pathol. 2006;30:1472–1477. doi: 10.1097/01.pas.0000213308.43929.97. [DOI] [PubMed] [Google Scholar]
- 50.Saeki T, Nishi S, Ito T, Yamazaki H, Miyamura S, Emura I, Imai N, Ueno M, Saito A, Gejyo F. Renal lesions in IgG4-related systemic disease. Intern Med. 2007;46:1365–1371. doi: 10.2169/internalmedicine.46.0183. [DOI] [PubMed] [Google Scholar]
- 51.Kobayashi H, Shimokawaji T, Kanoh S, Motoyoshi K, Aida S. IgG4-positive pulmonary disease. J Thorac Imaging. 2007;22:360–362. doi: 10.1097/RTI.0b013e31813fab9f. [DOI] [PubMed] [Google Scholar]
- 52.Takato H, Yasui M, Ichikawa Y, Fujimura M, Nakao S, Zen Y, Minato H. Nonspecific interstitial pneumonia with abundant IgG4-positive cells infiltration, which was thought as pulmonary involvement of IgG4-related autoimmune disease. Intern Med. 2008;47:291–294. doi: 10.2169/internalmedicine.47.0411. [DOI] [PubMed] [Google Scholar]
- 53.Hirano K, Kawabe T, Komatsu Y, Matsubara S, Togawa O, Arizumi T, Yamamoto N, Nakai Y, Sasahira N, Tsujino T, et al. High-rate pulmonary involvement in autoimmune pancreatitis. Intern Med J. 2006;36:58–61. doi: 10.1111/j.1445-5994.2006.01009.x. [DOI] [PubMed] [Google Scholar]
- 54.Kamisawa T, Tu Y, Nakajima H, Egawa N, Tsuruta K, Okamoto A, Horiguchi S. Sclerosing cholecystitis associated with autoimmune pancreatitis. World J Gastroenterol. 2006;12:3736–3739. doi: 10.3748/wjg.v12.i23.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoshimura Y, Takeda S, Ieki Y, Takazakura E, Koizumi H, Takagawa K. IgG4-associated prostatitis complicating autoimmune pancreatitis. Intern Med. 2006;45:897–901. doi: 10.2169/internalmedicine.45.1752. [DOI] [PubMed] [Google Scholar]
- 56.Nishimori I, Kohsaki T, Onishi S, Shuin T, Kohsaki S, Ogawa Y, Matsumoto M, Hiroi M, Hamano H, Kawa S. IgG4-related autoimmune prostatitis: two cases with or without autoimmune pancreatitis. Intern Med. 2007;46:1983–1989. doi: 10.2169/internalmedicine.46.0452. [DOI] [PubMed] [Google Scholar]
- 57.Uehara T, Hamano H, Kawakami M, Koyama M, Kawa S, Sano K, Honda T, Oki K, Ota H. Autoimmune pancreatitis-associated prostatitis: distinct clinicopathological entity. Pathol Int. 2008;58:118–125. doi: 10.1111/j.1440-1827.2007.02199.x. [DOI] [PubMed] [Google Scholar]
- 58.Anthony PP. Inflammatory pseudotumour (plasma cell granuloma) of lung, liver and other organs. Histopathology. 1993;23:501–503. doi: 10.1111/j.1365-2559.1993.tb00508.x. [DOI] [PubMed] [Google Scholar]
- 59.Kanno A, Satoh K, Kimura K, Masamune A, Asakura T, Unno M, Matsuno S, Moriya T, Shimosegawa T. Autoimmune pancreatitis with hepatic inflammatory pseudotumor. Pancreas. 2005;31:420–423. doi: 10.1097/01.mpa.0000179732.46210.da. [DOI] [PubMed] [Google Scholar]
- 60.Uchida K, Satoi S, Miyoshi H, Hachimine D, Ikeura T, Shimatani M, Matsushita M, Takaoka M, Takai S, Ashida K, et al. Inflammatory pseudotumors of the pancreas and liver with infiltration of IgG4-positive plasma cells. Intern Med. 2007;46:1409–1412. doi: 10.2169/internalmedicine.46.6430. [DOI] [PubMed] [Google Scholar]
- 61.Zen Y, Kitagawa S, Minato H, Kurumaya H, Katayanagi K, Masuda S, Niwa H, Fujimura M, Nakanuma Y. IgG4-positive plasma cells in inflammatory pseudotumor (plasma cell granuloma) of the lung. Hum Pathol. 2005;36:710–717. doi: 10.1016/j.humpath.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 62.Wong S, Lam WY, Wong WK, Lee KC. Hypophysitis presented as inflammatory pseudotumor in immunoglobulin G4-related systemic disease. Hum Pathol. 2007;38:1720–1723. doi: 10.1016/j.humpath.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 63.van der Vliet HJ, Perenboom RM. Multiple pseudotumors in IgG4-associated multifocal systemic fibrosis. Ann Intern Med. 2004;141:896–897. doi: 10.7326/0003-4819-141-11-200412070-00033. [DOI] [PubMed] [Google Scholar]
- 64.Tanabe T, Tsushima K, Yasuo M, Urushihata K, Hanaoka M, Koizumi T, Fujimoto K, Kubo K, Uehara T, Shigematsu S, et al. IgG4-associated multifocal systemic fibrosis complicating sclerosing sialadenitis, hypophysitis, and retroperitoneal fibrosis, but lacking pancreatic involvement. Intern Med. 2006;45:1243–1247. doi: 10.2169/internalmedicine.45.1759. [DOI] [PubMed] [Google Scholar]
- 65.Yamamoto M, Takahashi H, Ohara M, Suzuki C, Naishiro Y, Yamamoto H, Shinomura Y, Imai K. A case of Mikulicz's disease (IgG4-related plasmacytic disease) complicated by autoimmune hypophysitis. Scand J Rheumatol. 2006;35:410–411. doi: 10.1080/03009740600758110. [DOI] [PubMed] [Google Scholar]
- 66.Saegusa H, Momose M, Kawa S, Hamano H, Ochi Y, Takayama M, Kiyosawa K, Kadoya M. Hilar and pancreatic gallium-67 accumulation is characteristic feature of autoimmune pancreatitis. Pancreas. 2003;27:20–25. doi: 10.1097/00006676-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 67.Nakamura A, Funatomi H, Katagiri A, Katayose K, Kitamura K, Seki T, Yamamura F, Aoyagi Y, Nishida H, Mitamura K. A case of autoimmune pancreatitis complicated with immune thrombocytopenia during maintenance therapy with prednisolone. Dig Dis Sci. 2003;48:1968–1971. doi: 10.1023/a:1026170304531. [DOI] [PubMed] [Google Scholar]
- 68.Ohara H, Nakazawa T, Sano H, Ando T, Okamoto T, Takada H, Hayashi K, Kitajima Y, Nakao H, Joh T. Systemic extrapancreatic lesions associated with autoimmune pancreatitis. Pancreas. 2005;31:232–237. doi: 10.1097/01.mpa.0000175178.85786.1d. [DOI] [PubMed] [Google Scholar]
- 69.Komatsu K, Hamano H, Ochi Y, Takayama M, Muraki T, Yoshizawa K, Sakurai A, Ota M, Kawa S. High prevalence of hypothyroidism in patients with autoimmune pancreatitis. Dig Dis Sci. 2005;50:1052–1057. doi: 10.1007/s10620-005-2703-9. [DOI] [PubMed] [Google Scholar]
- 70.Dooreck BS, Katz P, Barkin JS. Autoimmune pancreatitis in the spectrum of autoimmune exocrinopathy associated with sialoadenitis and anosmia. Pancreas. 2004;28:105–107. doi: 10.1097/00006676-200401000-00018. [DOI] [PubMed] [Google Scholar]