

Abstract
AIM: To investigate the effect of lithium on proliferation of esophageal cancer (EC) cells and its preliminary mechanisms.
METHODS: Eca-109 cells were treated with lithium chloride, a highly selective inhibitor of glycogen synthase kinase 3β (GSK-3β), at different concen-trations (2-30 mmol/L) and time points (0, 2, 4, 6 and 24 h). Cell proliferative ability was evaluated by 3-(4,5-dimethylthiazole-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay, and cell cycle distribution was examined by flow cytometry. Expressions of p-GSK-3β, β-catenin, cyclin B1, cdc2 and cyclin D1 protein were detected by Western blotting, and the subcellular localization of β-catenin was determined by immunofluorescence. The mRNA level of cyclin B1 was detected by reverse transcription polymerase chain reaction (RT-PCR).
RESULTS: Lithium could inhibit the proliferation of Eca-109 cells. Lithium at a concentration of 20 mmol/L lithium for 24 h produced obvious changes in the distribution of cell cycle, and increased the number of cells in G2/M phase (P < 0.05 vs control group). Western blotting showed that lithium inhibited GSK-3β by Ser-9 phosphorylation and stabilized free β-catenin in the cytoplasm. Immunofluorescence further confirmed that free β-catenin actively translocated to the nucleus. Moreover, lithium slightly elevated cyclin D1 protein expression, whereas lowered the cyclin B1 expression after 24 h lithium exposure and no obvious change was observed for cdc2 protein.
CONCLUSION: Lithium can inhibit the proliferation of human esophageal cancer cell line Eca-109 by inducing a G2/M cell cycle arrest, which is mainly mediated through the inhibition of lithium-sensitive molecule, GSK-3β, and reduction of cyclin B1 expression.
Keywords: Lithium, Esophageal cancer, Cell cycle, Glycogen synthase kinase 3β
INTRODUCTION
Esophageal cancer (EC) is prevalent in some regions of the world, and occurs at a very high frequency in certain parts of China and the mortality rate ranked the fourth among cancer-related death[1,2]. However, the molecular basis of esophageal carcinogenesis remains poorly understood.
Glycogen synthase kinase-3β (GSK-3β) is a serine/threonine kinase that controls cell survival and cell fate through its involvement in multiple signaling pathways[3]. Recent studies in colorectal cancer, pancreatic cancer, hepatocellular carcinoma and ovarian cancer[4–7] demonstrate that GSK-3β is involved in the process of tumorigenesis. Inhibition of the expression and activity of GSK-3β attenuates cell proliferation and causes apoptosis in colorectal, pancreatic and ovarian cancer cells[4,5,7].
Lithium has been shown to be a specific and noncompetitive inhibitor of GSK-3β activity in vitro[8,9] and in vivo[10]. It has been proved that lithium could promote[11–14] or inhibit[7,15–17] cell cycle transition and proliferation of some primary cultures or cell lines by inhibiting GSK-3β, depending on the cell type. However, whether lithium influences the growth and proliferation of EC cells remains unknown to date.
In this report, we chose Eca-109 cells as a model, and observed the potential role of lithium in cell cycle progression and growth of EC cells and investigated its preliminary mechanisms.
MATERIALS AND METHODS
Cell culture and treatment
Human esophageal squamous cell carcinoma cell line Eca-109 was obtained from Institute of Biochemistry and Cell Biology, Shanghai, China and maintained in RPMI 1640 medium (Gibco Biocult, Paisley, UK) supplemented with 10% calf bovine serum (Sijiqing Biotechnology, China), 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in a water-saturated atmosphere of 5% CO2 in air. For G0/G1 synchronization, when Eca-109 cells grew to 70% confluence, the routine medium was removed and replaced by free-serum medium for 24 h. Then these cells were cultured in the free-serum medium supplemented with 2-30 mmol/L lithium (Alexis, USA) for indicated times.
3-(4,5-dimethylthiazole-2-yl) 2,5-diphenyl-tetrazolium bromide (MTT) assay
The IC50 of lithium on Eca-109 cells was measured by MTT assay, which was conducted as described before[18], and was calculated by Logit method. Briefly, one thousand Eca-109 cells (5 × 103/mL) were seeded in 96-well plates and cultured for 12 h. When they were adhesive, these Eca-109 cells were exposed to a range of concentrations of lithium from 2 to 30 mmol/L for 72 h, respectively. The Eca-109 cells treated with routine medium served as negative control. All exposures were performed in six wells. At the end of exposure, 20 μL of MTT (Sigma, USA) stock solution (5 mg/mL) was added to 200 μL of medium in each well and plates were incubated for 4 h at 37°C, and subsequently 150 μL of dimethyl sulfoxide (DMSO) was added to each well. The plates were incubated about 10 min at room temperature and read with enzyme-linked immunosorbent assay (490 nm) to determine absorbance values (A). The rate of inhibition was calculated by the following equation:
Rate of growth inhibition (%) = (1 - Atreated/Acontrol) × 100%.
Flow cytometry
Before the analysis was conducted by FCM, Eca-109 cells were exposed to lithium at a concentration of 20 mmol/L for 0, 2, 4, 6 and 24 h, respectively. According to the routine method, 1.0 × 106 Eca-109 cells from the control and treated groups were harvested by trypsinization and centrifugation, washed twice with ice-cold PBS and resuspended in PBS containing 10 mg/L propidium iodide (Sigma, USA) and 100 mg/L RNase A (Huamei Biotechnology, China) and then incubated at 25°C in the dark for at least 30 min. The percentage of cell population in each phase of the cell cycle was measured using FACStar and the results were analyzed with the software CELLQUEST. All measurements were carried out with the same instrument under the same experimental conditions.
Reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from specimens using Trizol reagent (Invitrogene, USA) and treated with DNase I (Tiangen Biotechnology, China). cDNA was synthesized from 2 μg of total RNA according to the manufacturer’s instruction (Fermentas, USA), and negative control reactions were run without reverse transcriptase. An equal volume of product was subjected to PCR. The levels of gene transcripts were quantified as the ratio of the intensity of the target gene to the intensity of β-actin. The PCR primers used in this study were as follows: forward primer 5’-CCATTATTGATCGGTTCATGCAGA-3’ and reverse primer 5’-CTAGTGCAGAATTCAGCTGTGGTA-3’ for cyclin B1 (585 bp)[19]. β-actin was amplified as internal control using the following primers: forward primer 5’-AGTTGCGTTACACCCTTTCTTG-3’ and reverse primer 5’-TCACCTTCACCGTTCCAGTTT-3’, with a 150 bp fragment product. The amplification conditions were 30 cycles of 94°C for 50 s, 55°C for 40 s, 72°C for 40 s for cyclin D1, and 30 cycles of 94°C for 50 s, 59°C for 40 s, 72°C for 40 s for cyclin B1 and β-actin. PCR products were run on 1.6% agarose gel and results were analyzed using Image Tool 3.0.
Extraction of nuclear protein
Cells were lysed in 400 μL of ice-cold buffer A [10 mmol/L HEPES (pH 7.9), 10 mmol/L KCl, 0.1 mmol/L ethylene diamine tetraacetic acid (EDTA), 0.1 mmol/L EGTA, 1 mmol/L DTT, 0.5 mmol/L PMSF] by gentle pipetting. The cells were allowed to swell on ice for 15 min, then 40 μL of a 10% solution of Nonidet P-40 was added and the tube was vigorously vortexed for 10 s. The homogenate was centrifuged for 30 s in a microcentrifuge. The supernatant was transferred and the nuclear pellet was lysed with 50 μL of buffer B [20 mmol/L HEPES (pH 7.9), 0.42 mol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L DTT, 1 mmol/L PMSF] and the tube was vigorously rocked at 4°C for 15 min on a shaking platform. The nuclear extract was centrifuged for 5 min in a microcentrifuge at 4°C. The supernatant containing nuclear protein was stored at -70°C.
Western blotting
Cells were washed twice with ice-cold PBS, collected by adding 0.25% trypsin and lysed in buffer [50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L ethylene diamine tetraacetic acid (EDTA), 0.25% sodium deoxycholate, 1% TritonX-100, 0.1% sodium dodecyl sulfate (SDS), 1 mmol/L NaF, 1 mmol/L Na3VO4], protease inhibitors (10 mg/L aprotinin and 1 mmol/L phenylmethylsulfonyl fluoride) were added to obtain whole cell protein. Equal amounts of cell protein, quantified by BCA protein assay kit (Pierce Biotechnology, Rockford, IL), were subjected to 10% SDS-polyacrylamide gel electrophoresis, and then transferred to polyvinylidene difluoride membrane. The membranes were blocked with 5% non-fat milk in TBST [50 mmol/L Tris-HCl (pH 7.6), 150 mmol/L NaCl, 0.1% Tween 20] for 2 h at room temperature, and subsequently incubated with primary antibody (anti-p-GSK3β, 1:400, anti-cyclin D1, 1:200, anti-cyclin B1, 1:400, anti-cdc2, 1:500, β-actin, 1:2000, were all purchased from Sant Cruz Biotechnology, USA, and anti-β-catenin, 1:400, was purchased from Chemicion Biotech, USA) in blocking buffer at 4°C overnight. Following a wash with TBST, the membranes were incubated with horseradish peroxidase conjugated rabbit anti-mouse secondary antibody (1:1000, Dako, Denmark) for 2 h at room temperature. The membranes were washed with TBST and protein bands were visualized by enhanced chemiluminescence according to the manufacturer’s instructions (KPL, USA). The β-actin bands were taken as loading control. The protein quantity was analyzed by UTHSCSA Image Tool 3.0. The target protein expression was evaluated by the relative intensity ratio of target protein/loading control.
Immunofluorescence
Cells were plated on the coverslips which had been put into the six-well plates in advance. After being treated with 20 mmol/L lithium for the indicated time as described above, cells were washed with ice-cold phosphate-buffered saline (PBS) prior to fixing with 4% paraformaldehyde for 15 min at room temperature. Next, cells were treated with 1 g/L TritonX-100 for 30 min and subsequently incubated with a blocking solution (10% normal goat serum in PBS) for 20 min, anti-β-catenin monoclonal antibody in PBS (1: 100, Chemicion, USA) overnight at 4°C and followed by fluorescein isothiocyanate-labeled goat anti-mouse IgG (Dako Biotechnology, Denmark) for 1 h at room temperature. Finally, the cells were visualized and photographed with an Olympus fluorescence microscope.
Statistical analysis
The data were obtained from at least three repeated experiments and expressed as mean ± SD. In order to compare the data between the treated groups and the control group, statistical significance was analyzed through analysis of variance (ANOVA) and values of P < 0.05 were considered significant.
RESULTS
Lithium inhibits proliferation of Eca-109 cells
The mean IC50 of lithium was 21.7 ± 0.5 mmol/L. The effect of inhibition of lithium on Eca-109 cells could be observed at a concentration of 2 mmol/L. From 2 to 30 mmol/L, the inhibitory effect was enhanced along with the increased concentration of lithium. The exhibition of dose-response relationship could be observed (Figure 1).
Figure 1.
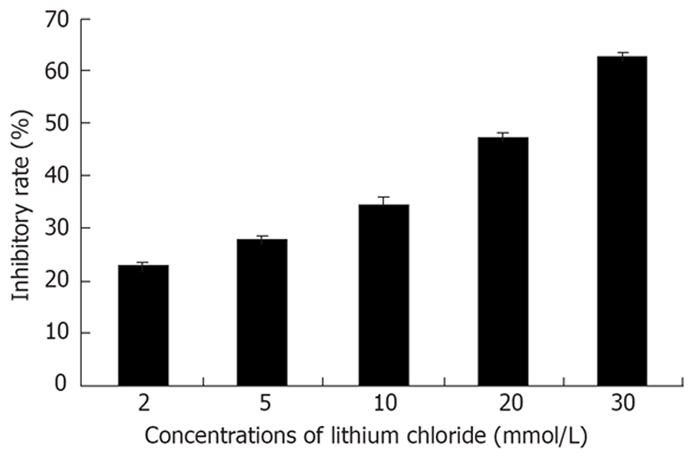
Lithium could impose inhibition on Eca-109 cells in day 3. With the increased concentration of lithium, the inhibitory effect was enhanced. Each bar came from six wells of 96-well plate and represented mean ± SD.
The growth curves of Eca-109 cells within 5 d after treatment with lithium were also plotted according to the data obtained from the MTT assay (Figure 2). The Eca-109 cells treated with different concentrations of lithium showed much slower growth than the untreated cells. The results indicated that the proliferation of Eca-109 cells in vitro was inhibited compared to that of the untreated cells.
Figure 2.
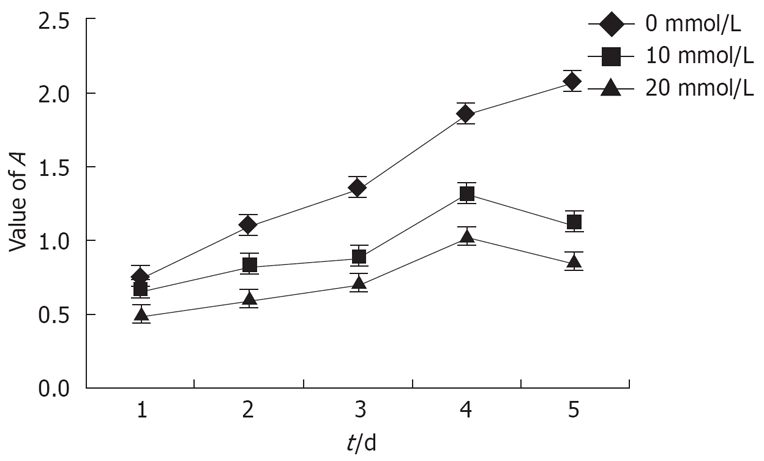
Growth curves of Eca-109 cells plotted by MTT assay. Each point was the mean ± SD from six independent experiments.
Lithium induces a G2/M cell cycle arrest in Eca-109 cells
To determine whether the lithium-induced inhibition of proliferation of Eca-109 cells was due to altered cell cycle regulation, Eca-109 cells were treated with lithium for various times, and cell cycle profiles were monitored by flow cytometric analysis of DNA content (Figure 3 and Table 1). The percentage of cells in G0/G1 phase decreased with the length of treatment from 59.6% at 0 h to 22.1% at 24 h, and the percentage of cells in S phase increased accordingly from 28.5% at 0 h to 42.5% at 6 h. At 24 h, while the cells were entering from S phase to G2/M phase, the percentage of cells in S phase decreased from 42.5% at 6 h to 31.5% at 24 h, but the percentage of cells in G2/M phase was increased markedly. The distribution in the phases of cell cycle indicated that lithium could promote Eca-109 cells entering into S phase and then G2/M, in which the population was increased by approximate 4 folds compared with that of the untreated group at 24 h. These results suggested that the growth-inhibitory effect of lithium on Eca-109 cells might be partly due to its ability to induce G2/M cell cycle arrest.
Figure 3.

Lithium inhibited the proli-feration of Eca-109 cells by an arrest in G2/M phase. Eca-109 cells were treated with lithium (20 mmol/L) for 0 h (A) and 24 h (B). Eca-109 cells were harvested and cell cycle profiles were obtained by staining with propidium iodide (PI).
Table 1.
Lithium affects cell cycle progression in Eca-109 cells
| Groups |
Distribution in cell cycle (%)1 |
||
| G0/G1 | S | G2/M | |
| 0 h | 59.6 | 28.5 | 11.9 |
| 2 h | 53.6 | 33.6 | 12.8 |
| 4 h | 47.4a | 39.4a | 13.2 |
| 6 h | 43.1a | 42.5a | 14.5 |
| 24 h | 22.1a | 31.5 | 46.4a |
Lithium reduced the G0/G1 phase population sharply and increased the population in G2/M phase accordingly.
P < 0.05 vs the control group.
Lithium induces a G2/M arrest by decreased cyclin B1 expression
Having established that lithium induces a G2/M cell cycle arrest, we attempted to characterize, at the molecular level, the mechanisms by which this effect is achieved. Since the cyclin B1/cdc2 complex is a master intracellular regulator entry into mitosis[20], we therefore investigated the effects of lithium in Eca-109 cells on key regulators of the G2 to M phase transition, cdc2 and cyclin B1 using Western blotting. Treatment for 24 h with lithium reduced the protein expression of cyclin B1 (Figure 4A). Moreover, down-regulation of cyclin B1 expression at 24 h was also confirmed at the mRNA levels by RT-PCR (Figure 4B). Similar to the untreated cells, the total levels of cdc2 protein remained stable upon lithium treatment (Figure 4C). These findings suggested that the lithium-induced G2/M arrest was probably due to decreased expression of cyclin B1, and had nothing to do with the expression of cdc2 protein.
Figure 4.

Effects of lithium on cell cycle regulatory molecules. A: Western blotting analysis for cyclin B1 protein levels after treatment with 20 mmol/L lithium; B: RT-PCR analysis for quantification of the relative mRNA abundance of cyclin B1 after treatment with 20 mmol/L lithium; C: Western blotting analysis for cdc2 protein levels after treatment with 20 mmol/L lithium. bP < 0.01 vs the control group.
Lithium induces β-catenin stabilization via inhibition of GSK-3β
Inhibition of GSK-3β (GSK-3β Ser-9 phosphorylation) was assessed by Western blotting using a phospho-Ser-9-specific antibody. As shown in Figure 5A, 20 mmol/L lithium increased the phosphorylated inactive form of GSK-3β in Eca-109 cells. Moreover, at a range of 2-30 mmol/L, this effect of lithium was dose-dependent (data not shown). β-catenin, a signaling molecule in the Wnt/β-catenin signaling pathway, is a target of GSK-3β. Inactivation of GSK-3β by Wnt signaling or by lithium leads to stabilization and nuclear translocation of β-catenin[21]. Here, we investigated the expression of β-catenin by Western blotting of total and nuclear extracts from Eca-109 cells treated with lithium. Results indicated that the intracellular total protein concentrations of β-catenin were increased following stimulation with 20 mmol/L lithium (Figure 5A). An increase of β-catenin nuclear pool was also observed in Eca-109 cells, exhibiting the similar trend (Figure 5B). In contrast to the untreated cells, in which β-catenin was primarily located in the cytoplasm, β-catenin was predominantly located in the nuclear of the lithium-treated Eca-109 cells (Figure 5C). These results showed that lithium could induce β-catenin nuclear translocation by inhibition of GSK-3β activity.
Figure 5.
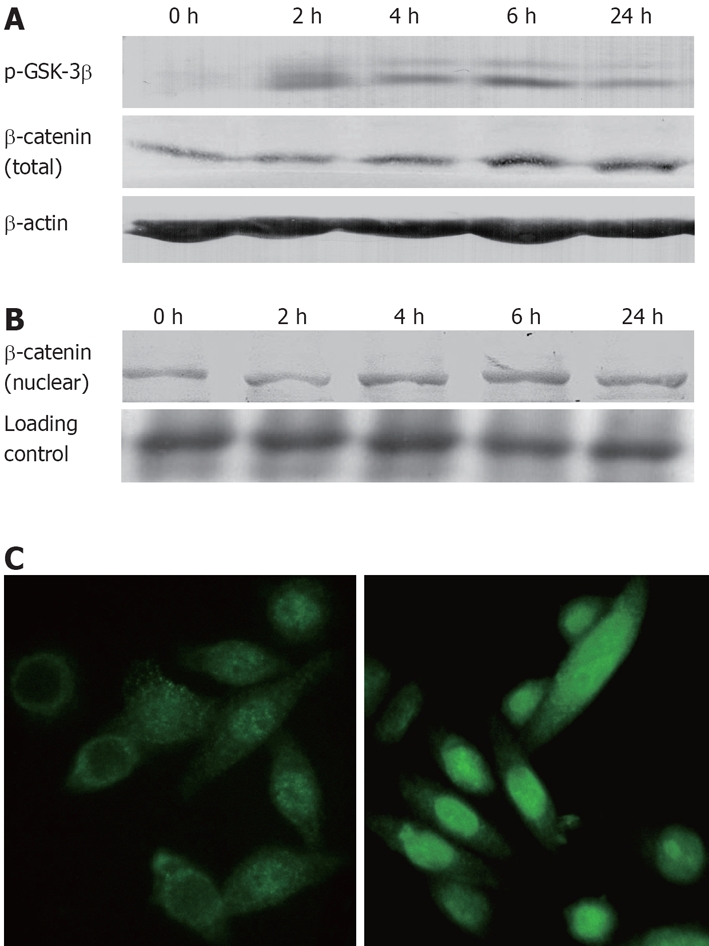
Lithium induced β-catenin stabilization via inhibition of GSK-3β in vitro. A: Eca-109 cells were incubated with 20 mmol/L lithium for 0-24 h. Expressions of p-GSK-3β and total β-catenin protein were analyzed by Western blotting. The untreated Eca-109 cells (0 h) served as negative control; B: Western blotting analysis for β-catenin protein level of nuclear lysates; C: Immunofluorescence demonstrated a predominant cytoplasm localization of β-catenin in the untreated cells (left) whereas after incubation with lithium (20 mmol/L) for 6 h, β-catenin was predominantly located in the nucleus of Eca-109 cells (right).
Lithium up-regulates intranuclear cyclin D1 levels
GSK-3β has been shown to regulate cyclin D1 proteolysis by directly phosphorylating cyclin D1 at Thr-286[22,23]. We hypothesized that inhibition of GSK-3β by lithium would be associated with an increase in cyclin D1 protein. As shown in Figure 6, lithium treatment induced a slight increase in the cyclin D1 protein levels by 1.3-fold at 4 h and 1.8-fold at 6 h of lithium treatment as compared with control cells, and the cyclin D1 protein levels diminished to the normal level at 24 h, which may be attributed to the decrease of cells in G0/G1 phase.
Figure 6.

Lithium induced cyclin D1 stabilization via inhibition of GSK-3β in vitro. Eca-109 cells were incubated with 20 mmol/L lithium for 0-24 h. Expression of cyclin D1 was analyzed by Western blotting. The untreated Eca-109 cells (0 h) served as negative control.
DISCUSSION
In this report, we treated EC cell line Eca-109 cells with different concentrations of lithium and in different time points, and observed the effect of lithium on EC cells. Results indicated that lithium inhibited the proliferation of Eca-109 cells (Figures 1 and 2). We performed flow cytometry to assess cell cycle progression of Eca-109 cells treated with lithium. Analysis of cell cycle distribution showing an increase of cells in G2/M phase and a decrease in G0/G1 phase indicated that lithium could lead to a G2/M cell cycle arrest (Figure 3 and Table 1), which is the common characteristic of cell cycle arrest induced by lithium. However, different cell types have their own doses of sensitivity[15,17]. In the report of Mao et al[17], 5 mmol/L lithium was enough to induce a G2/M phase arrest of bovine aortic endothelial cells. In our study, the mean IC50 of lithium on Eca-109 cells growth was 21.7 ± 0.5 mmol/L by MTT assay (Figure 1). Therefore, 20 mmol/L lithium was used to treat EC cells for indicated times for correlation detections.
Inhibition of cell proliferation can occur through activation of several possible checkpoints during cell cycle progression. Lithium is a highly selective inhibitor of GSK-3β, a multifunctional serine/threonine kinase which has a variety of putative substrates including cyclin D1, p21Waf1/Cip1, and transcription factors like c-myc, c-jun and β-catenin[24], which are implicated in the regulation of cell proliferation. It is well known that intracellular β-catenin levels are regulated through GSK-3β mediated phosphorylations on its serine and threonine residues (at Ser-33, Ser-37 and Thr-41)[25]. Inhibition of GSK-3β activity by lithium can result in β-catenin stabilization and its accumulation in the nucleus[26]. Our Immunoblot results showed that in Eca-109 cells lithium inhibited GSK-3β by Ser-9 phosphorylation and stabilized free β-catenin in the cytoplasm (Figure 5A). An increase of β-catenin nuclear pool was also observed (Figure 5B). Immunofluorescence studies further confirmed that free β-catenin translocated to the nucleus where β-catenin was transcriptionally active (Figure 5C). Although inhibition of cell proliferation by β-catenin signaling has not been described to date, β-catenin may be the signal for cell cycle arrest. Orford et al[27] showed that the nuclear localization of β-catenin was cell cycle regulated in the epithelial Madin-Darby canine kidney cells with a peak during the S phase. Interestingly, with the percentage of cells in S phase increased, β-catenin expression increased accordingly in our experiments (Table 1 and Figure 5). Damalas et al[28] reported that the accumulation of p53 in mouse fibroblasts NIH3T3 overexpressed a stable form of β-catenin (S37A). Stabilization of p21Waf1/Cip1 and induction of G2/M cell cycle arrest by lithium was demonstrated in bovine aortic endothelial cells, where lithium up-regulated p21Waf1/Cip1 protein level through activation and stabilization of p53[17], an effect of lithium possibly associated with GSK3β, as p53 was recently shown to be a substrate of this kinase[29]. Furthermore, lithium was shown to stabilize p21Waf1/Cip1 by inhibiting GSK-3β activity, and induce G2/M cell cycle arrest in human umbilical vein endothelial cells[30]. It is thus possible that a sustained retention of β-catenin in the nucleus during G2 or G2/M transition can be a signal for p53 induction and cell cycle arrest.
The cyclin B1/cdc2 complex is a master intracellular regulator entry into mitosis[20], G2/M cell cycle arrest may be associated with decreased expression or activity of this complex. Recently, Smits et al[15] observed a G2/M cell cycle arrest in various transformed (P19 embryonal carcinoma, U2OS osteosarcoma, and SK-N-MC neuroepithelioma) or immortalized (NIH3T3) cell lines after lithium treatment. They found that the activity of the cyclin B1/cdc2 complex was impaired after lithium treatment due to sustained phosphorylation of cdc2 on tyrosine residue 15, and the reduction in cyclin B1/cdc2 kinase activity was not caused by the reduction of cyclin B1 expression, since cyclin B1 protein levels were not influenced by lithium treatment. We detected the expression of cyclin B1 and cdc2 protein, and found that lithium lowered protein levels of cyclin B1 (Figure 3A), which is also a signal for cell cycle arrest in G2/M. The mRNA levels of cyclin B1 exhibited the similar results (Figure 3B). The above results suggested that lithium could influence the expression of cyclin B1 at the levels of both transcription and translation in Eca-109 cells. Chen et al[31] observed that lithium treatment caused a cell cycle arrest at G2/M phase in pig airway epithelial cells, thus inhibiting proliferation of the cells. However, the increased expression of cyclin B1 was observed, which may be attributed to the accumulation of cells in G2 phase. Our results indicated that G2/M cell cycle arrest was partly due to the reduction of cyclin B1 expression. However, further research is needed to clarify the mechanisms of cyclin B1 expression induced by lithium. Moreover, lithium treatment produced no obvious influences on cdc2 protein (Figure 3C). As we did not investigate the activity of the cyclin B1/cdc2 kinase, we could not know the change of cyclin B1/cdc2 kinase activity.
Previously, it was reported that phosphorylation of cyclin D1 at Thr-286 by GSK-3β regulates positively proteasomal degradation of cyclin D1[22]. Therefore, inactivation of GSK-3β was expected to lead to an increase of cyclin D1 protein. Our results agreed well with this rationale. Lithium treatment resulted in the inactivation of GSK-3β and induced a slight increase of cyclin D1 protein at 4 and 6 h (Figure 6). These results indicated that cyclin D1 might be involved in the transition of cell cycle from G1 to S phase in Eca-109 cells induced by lithium. Chen et al[31] observed that lithium treatment increased cyclin D1 expression and induced a G2/M cell cycle arrest in pig airway epithelial cells, which was consistent with our results. However, they failed to find evidence of G1/S transition. Mao et al[17] also observed a slight increase of cyclin D1 and G2/M cell cycle arrest in lithium-treated bovine aortic endothelial cells. On the contrary, a recent investigation on NIH3T3 cells indicated that the change of GSK-3β activity by lithium had no effect on cyclin D1 expression[32]. The causes of the discrepancy from these data might lie in the different experimental methods and different cell lines as well.
In conclusion, our present study demonstrated, for the first time, that lithium can arrest the growth of EC cells and induce a G2/M cell cycle arrest, which is mainly mediated through the inhibition of lithium-sensitive molecule, GSK-3β, and reduction of cyclin B1 expression. However, further studies are needed to determine the precise mechanisms that contribute to the regulation.
COMMENTS
Background
Esophageal squamous cell carcinoma (ESCC) is one of the leading causes of cancer-related death in certain parts of China, and the molecular basis of esophageal carcinogenesis remains poorly understood. It is necessary to develop effective new strategies for the treatment of esophageal cancer (EC).
Research frontiers
Recent researches have proved that lithium could promote or inhibit cell cycle transition and proliferation of certain primary cultures or cell lines by inhibiting GSK-3β, depending on the cell type. However, whether lithium influences the growth and proliferation of EC cells remains unknown to date.
Innovations and breakthroughs
This study demonstrated for the first time that lithium can inhibit the proliferation of esophageal squamous cell carcinoma cells (Eca-109), which is mainly mediated by the inhibition of GSK-3β and reduction of cyclin B1 expression.
Applications
This study indicates the possibility for the treatment of the esophageal squamous cell carcinoma with lithium cloride.
Terminology
Lithium is an inhibitor of GSK-3β activity. Lithium has been reported to reduce GSK-3β activity in two ways, both directly and by increasing the inhibitory phosphorylation of GSK-3β. These dual effects can act in concert to magnify the influence of lithium on crucial GSK-3β-regulated functions (gene expression, cell structure and survival).
Peer review
The purpose of this study is reasonable and results are clearly demonstrated, however, further studies are needed to determine the precise mechanisms that contribute to the regulation.
Supported by The Innovation Project of Central South University, No. 2340-76208
Peer reviewer: Satoshi Osawa, MD, First Department of Medicine, Hamamatsu University School of Medicine, 1-20-1 Handayama, Hamamatsu 431-3192, Japan
S- Editor Li DL L- Editor Ma JY E- Editor Yin DH
References
- 1.Parkin DM, Pisani P, Ferlay J. Global cancer statistics. CA Cancer J Clin. 1999;49:33–64, 1. doi: 10.3322/canjclin.49.1.33. [DOI] [PubMed] [Google Scholar]
- 2.Zou XN, Taylor PR, Mark SD, Chao A, Wang W, Dawsey SM, Wu YP, Qiao YL, Zheng SF. Seasonal variation of food consumption and selected nutrient intake in Linxian, a high risk area for esophageal cancer in China. Int J Vitam Nutr Res. 2002;72:375–382. doi: 10.1024/0300-9831.72.6.375. [DOI] [PubMed] [Google Scholar]
- 3.Woodgett JR. Regulation and functions of the glycogen synthase kinase-3 subfamily. Semin Cancer Biol. 1994;5:269–275. [PubMed] [Google Scholar]
- 4.Shakoori A, Ougolkov A, Yu ZW, Zhang B, Modarressi MH, Billadeau DD, Mai M, Takahashi Y, Minamoto T. Deregulated GSK3beta activity in colorectal cancer: its association with tumor cell survival and proliferation. Biochem Biophys Res Commun. 2005;334:1365–1373. doi: 10.1016/j.bbrc.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 5.Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005;65:2076–2081. doi: 10.1158/0008-5472.CAN-04-3642. [DOI] [PubMed] [Google Scholar]
- 6.Beurel E, Kornprobst M, Blivet-Van Eggelpoel MJ, Cadoret A, Capeau J, Desbois-Mouthon C. GSK-3beta reactivation with LY294002 sensitizes hepatoma cells to chemotherapy-induced apoptosis. Int J Oncol. 2005;27:215–222. [PubMed] [Google Scholar]
- 7.Cao Q, Lu X, Feng YJ. Glycogen synthase kinase-3beta positively regulates the proliferation of human ovarian cancer cells. Cell Res. 2006;16:671–677. doi: 10.1038/sj.cr.7310078. [DOI] [PubMed] [Google Scholar]
- 8.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lucas FR, Goold RG, Gordon-Weeks PR, Salinas PC. Inhibition of GSK-3beta leading to the loss of phosphorylated MAP-1B is an early event in axonal remodelling induced by WNT-7a or lithium. J Cell Sci. 1998;111(Pt 10):1351–1361. doi: 10.1242/jcs.111.10.1351. [DOI] [PubMed] [Google Scholar]
- 10.Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- 11.Ohteki T, Parsons M, Zakarian A, Jones RG, Nguyen LT, Woodgett JR, Ohashi PS. Negative regulation of T cell proliferation and interleukin 2 production by the serine threonine kinase GSK-3. J Exp Med. 2000;192:99–104. doi: 10.1084/jem.192.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao AS, Kremenevskaja N, Resch J, Brabant G. Lithium stimulates proliferation in cultured thyrocytes by activating Wnt/beta-catenin signalling. Eur J Endocrinol. 2005;153:929–938. doi: 10.1530/eje.1.02038. [DOI] [PubMed] [Google Scholar]
- 13.Smith E, Coetzee GA, Frenkel B. Glucocorticoids inhibit cell cycle progression in differentiating osteoblasts via glycogen synthase kinase-3beta. J Biol Chem. 2002;277:18191–18197. doi: 10.1074/jbc.M109708200. [DOI] [PubMed] [Google Scholar]
- 14.Hamelers IH, van Schaik RF, Sipkema J, Sussenbach JS, Steenbergh PH. Insulin-like growth factor I triggers nuclear accumulation of cyclin D1 in MCF-7S breast cancer cells. J Biol Chem. 2002;277:47645–47652. doi: 10.1074/jbc.M208727200. [DOI] [PubMed] [Google Scholar]
- 15.Smits VA, Essers MA, Loomans DS, Klompmaker R, Rijksen G, Medema RH. Inhibition of cell proliferation by lithium is associated with interference in cdc2 activation. FEBS Lett. 1999;457:23–27. doi: 10.1016/s0014-5793(99)01002-9. [DOI] [PubMed] [Google Scholar]
- 16.Madiehe AM, Mampuru LJ, Tyobeka EM. Induction of apoptosis in HL-60 cells by lithium. Biochem Biophys Res Commun. 1995;209:768–774. doi: 10.1006/bbrc.1995.1565. [DOI] [PubMed] [Google Scholar]
- 17.Mao CD, Hoang P, DiCorleto PE. Lithium inhibits cell cycle progression and induces stabilization of p53 in bovine aortic endothelial cells. J Biol Chem. 2001;276:26180–26188. doi: 10.1074/jbc.M101188200. [DOI] [PubMed] [Google Scholar]
- 18.Hu ZL, Wen JF, Xiao DS, Zhen H, Fu CY. Effects of transforming growth interacting factor on biological behaviors of gastric carcinoma cells. World J Gastroenterol. 2005;11:84–88. doi: 10.3748/wjg.v11.i1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bürger C, Wick M, Müller R. Lineage-specific regulation of cell cycle gene expression in differentiating myeloid cells. J Cell Sci. 1994;107(Pt 7):2047–2054. doi: 10.1242/jcs.107.7.2047. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Mizu H, Yamauchi H. Molecular response to phototoxic stress of UVB-irradiated ketoprofen through arresting cell cycle in G2/M phase and inducing apoptosis. Biochem Biophys Res Commun. 2007;364:650–655. doi: 10.1016/j.bbrc.2007.10.046. [DOI] [PubMed] [Google Scholar]
- 21.Barker N, Clevers H. Catenins, Wnt signaling and cancer. Bioessays. 2000;22:961–965. doi: 10.1002/1521-1878(200011)22:11<961::AID-BIES1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 22.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- 24.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hatsell S, Rowlands T, Hiremath M, Cowin P. Beta-catenin and Tcfs in mammary development and cancer. J Mammary Gland Biol Neoplasia. 2003;8:145–158. doi: 10.1023/a:1025944723047. [DOI] [PubMed] [Google Scholar]
- 27.Orford K, Orford CC, Byers SW. Exogenous expression of beta-catenin regulates contact inhibition, anchorage-independent growth, anoikis, and radiation-induced cell cycle arrest. J Cell Biol. 1999;146:855–868. doi: 10.1083/jcb.146.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damalas A, Ben-Ze'ev A, Simcha I, Shtutman M, Leal JF, Zhurinsky J, Geiger B, Oren M. Excess beta-catenin promotes accumulation of transcriptionally active p53. EMBO J. 1999;18:3054–3063. doi: 10.1093/emboj/18.11.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qu L, Huang S, Baltzis D, Rivas-Estilla AM, Pluquet O, Hatzoglou M, Koumenis C, Taya Y, Yoshimura A, Koromilas AE. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 2004;18:261–277. doi: 10.1101/gad.1165804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rössig L, Badorff C, Holzmann Y, Zeiher AM, Dimmeler S. Glycogen synthase kinase-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. J Biol Chem. 2002;277:9684–9689. doi: 10.1074/jbc.M106157200. [DOI] [PubMed] [Google Scholar]
- 31.Chen W, Wu R, Wang X, Li Y, Hao T. Effect of lithium on cell cycle progression of pig airway epithelial cells. J Huazhong Univ Sci Technolog Med Sci. 2004;24:318–321. doi: 10.1007/BF02861857. [DOI] [PubMed] [Google Scholar]
- 32.Yang K, Guo Y, Stacey WC, Harwalkar J, Fretthold J, Hitomi M, Stacey DW. Glycogen synthase kinase 3 has a limited role in cell cycle regulation of cyclin D1 levels. BMC Cell Biol. 2006;7:33. doi: 10.1186/1471-2121-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]